
Association of APOE ɛ4 and Plasma p-tau181 with Preclinical Alzheimer’s Disease and Longitudinal Change in Hippocampus Function
Abstract
Background:
The Apolipoprotein E (APOE) ɛ4 allele has been linked to increased tau phosphorylation and tangle formation. APOE ɛ4 carriers with elevated tau might be at the higher risk for Alzheimer’s disease (AD) progression. Previous studies showed that tau pathology begins early in areas of the medial temporal lobe. Similarly, APOE ɛ4 carriers showed altered hippocampal functional integrity. However, it remains unknown whether the influence of elevated tau accumulation on hippocampal functional changes would be more pronounced for APOE ɛ4 carriers.
Objective:
We related ɛ4 carriage to levels of plasma phosphorylated tau (p-tau181) up to 15 years prior to AD onset. Furthermore, elevated p-tau181 was explored in relation to longitudinal changes in hippocampal function and connectivity.
Methods:
Plasma p-tau181 was analyzed in 142 clinically defined AD cases and 126 matched controls. The longitudinal analysis involved 87 non-demented individuals (from population-based study) with two waves of plasma samples and three waves of functional magnetic resonance imaging during rest and memory encoding.
Results:
Increased p-tau181 was observed for both ɛ4 carriers and non-carriers close to AD onset, but exclusively for ɛ4 carriers in the early preclinical groups (7- and 13-years pre-AD). In ɛ4 carriers, longitudinal p-tau181 increase was paralleled by elevated local hippocampal connectivity at rest and subsequent reduction of hippocampus encoding-related activity.
Conclusion:
Our findings support an association of APOE ɛ4 and p-tau181 with preclinical AD and hippocampus functioning.
INTRODUCTION
The ɛ4 allele of the apolipoprotein E (APOE) gene is the major genetic risk factor for late-onset Alzheimer’s disease (AD) [1]. A likely mechanism by which the APOE ɛ4 allele increases risk for AD is through influencing amyloid-β (Aβ) deposition [2]. Independently of Aβ, the elevated risk of developing AD that is conferred by the APOE ɛ4 genotype may also involve mechanisms associated with tau aggregation [3, 4]. Importantly, APOE ɛ4 is a risk factor, so not all ɛ4 carriers will present with AD pathology [5], and conversely 30–40% of AD cases do not possess the ɛ4 allele. Thus, in the preclinical phase, APOE ɛ4 carriers with elevated Aβ and tau might be at the highest risk for AD progression. Although elevated Aβ and tau levels in the clinical phases are well documented relative to controls, there is less consensus about what constitutes elevated Aβ and tau levels in the preclinical phase. Longitudinal studies of population-based non-clinical samples can provide a unique opportunity to explore accumulation of Aβ and tau over several years. Here we present results from the Betula longitudinal study on aging, memory, and dementia, comprising 4,425 individuals randomly sampled from the population registry and examined every five years for up to 30 years [6]. All enrolled participants were judged free of AD when included into the study. Blood was sampled at each test wave, and plasma-based measures of phosphorylated tau (p-tau181) were quantified [7]. In a case-control setting, we established p-tau181 cut-offs indicative of AD pathology in clinical as well as in early and late preclinical phases. We predicted group differences in p-tau level during both clinical and pre-clinical phases, such that clinically defined AD cases would exhibit elevated p-tau compared to the control group in clinical phase as well as years before clinical onset. Moreover, elevated p-tau was expected to be more pronounced among APOE ɛ4 carriers (c.f. [8]).
A second aim of the study was to relate longitudinal trajectories of p-tau181 levels to hippocampus functioning in APOE ɛ4 carriers and non-carriers. Both postmortem and in vivo studies using positron emission tomography (PET) suggest that tau pathology begins early in areas of the medial temporal lobes [9, 10], indicating that tau could impact hippocampal functional integrity. Indeed, studies have linked higher tau accumulation to increased hippocampal connectivity at rest [11] and hyperactivity during memory encoding [12]. Moreover, previous genetic studies showed that APOE ɛ4 carriers exhibit elevated connectivity and task activation compared with non-carriers [13, 14]. Based on these and related findings, we predicted that the effect of longitudinal tau181 accumulation on hippocampal function would be more pronounced for ɛ4 carriers than for non-carriers.
METHODS
Betula study design
Data were obtained from the Swedish longitudinal Betula study [6] (http://www.umu.se/en/betula).
Participants
The participants are from two longitudinal samples [6, 15]; sample S1 (n = 1000) included at wave W1 (1988–1990), and sample S3 (n = 963) included at W2 (1993–1995). The route from the parent cohort to the two present study samples, are illustrated in Fig. 1a and 1b.
Fig. 1
a) Flowchart of cross-sectional AD-case –control study with Wave 3 (W3) as study baseline, and (b) longitudinal imaging study with Wave 5 as study baseline. –/–, not included in the present study; W, wave; S, sample.
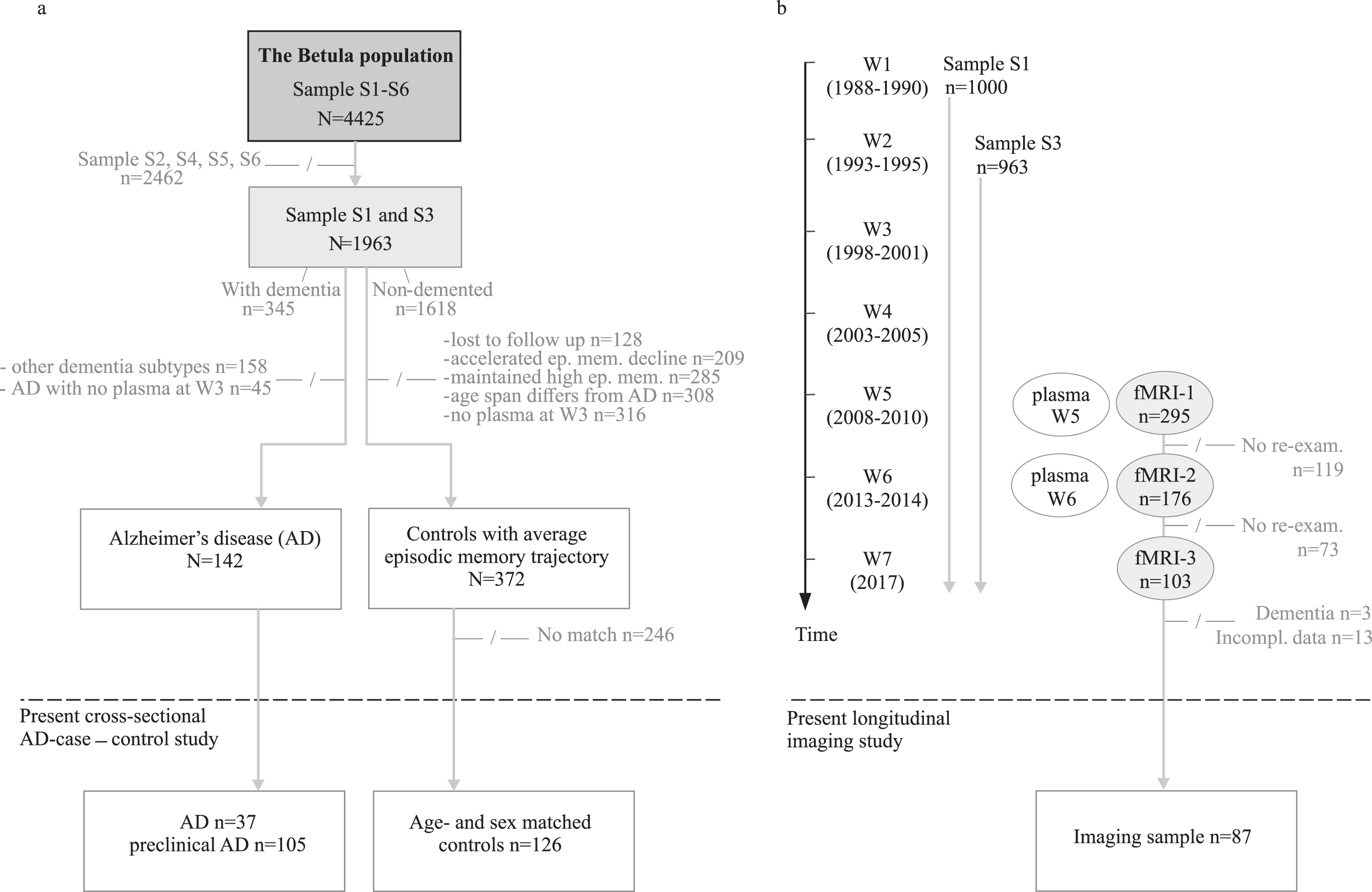
The cross-sectional AD case-control sample consists of n = 142 individuals with either manifest (n = 37) or preclinical AD (n = 105) and matched controls (n = 126). The clinical AD status is defined in relation to wave W3, the timepoint from which the plasma p-tau181 analyses are based. The AD cases had a clinical onset on average 2 years (range = 0–6 years) before W3, whereas preclinical AD subsequently developed AD on average 2 (range = 1–4 years), 7 (range = 5–9 years), and 13 (range = 10–15 years) years after W3. All cases with current or forthcoming AD with available W3 plasma defines the case population. The age- and sex-matched controls, selected from the same population and age cohorts as the cases, were classified as having an average episodic memory decline relative to their age [16], had sufficient W3 plasma, and remained non-demented throughout the studied period (1988–2017). For 16 cases, there were no eligible controls. Characteristics of the AD case and control sample are given in Table 1.
Table 1
Characteristics of the AD case-control sample at W3
| clinical AD | preclinical AD | controls | |||
| Sample char. | 13 (10–15 y) | 7 (5–9 y) | 2 (1–4 y) | ||
| N | 37 | 35 | 30 | 40 | 126 |
| Sex, % -female | 87% | 86% | 63% | 70% | 74% |
| Age, y | 83 [70–91] | 71 [60–81] | 72 [61–85] | 79 [65–91] | 76 [60–91] |
| Education, y | 7.8 [3–18] | 8.7 [4.5–17] | 7.7 [6–13] | 7.5 [5–17] | 7.7 [3–16] |
| APOE ɛ4 carrier | 43% | 54% | 57% | 58% | 17% |
| pτ181 (pg/ml) | 4.9 [0.9–33.1] | 3.0 [0.2–12.6] | 3.2 [1.1–29.4] | 4.9 [1.2–14.9>] | 2.9 [0.4–22.0] |
| EMC | 9 [4–25] | 34 [17–55] | 28 [12–44] | 17 [2–33] | 27 [7–50] |
| MMSE | 21 [9–27] | 28 [24–30] | 27 [23–30] | 26 [20–30] | 27 [14–30] |
For age and education, values are mean [range]. For pτ181 and memory scores, values are median [range]. EMC, Episodic Memory Composite score; a composite of five tasks (max score 76). Education data, EMC, and MMSE (Mini-Mental State Examination) were available for 86–97%. Of those with clinical AD, 51% contributed complete EMC data. Age, APOE ɛ4 carriership, and plasma pτ181 levels were available for all.
The longitudinal imaging sample comprises 98 non-demented study participants with imaging data obtained at W5 (2008–2010), W6 (2013–2014), and W7 (2017), with an average of 4 years between measurement points. Participants with complete p-tau181 data from waves 5–6 and fMRI observations at waves 5, 6, and 7 were included in the longitudinal analyses (n = 87). Seven of these also served as controls in the case-control study. The imaging study sample, in the larger context of the Betula longitudinal study design, is displayed in Fig. 1b and presented in full details elsewhere [6]. Sample characteristics are presented in Table 2.
Table 2
Characteristics of the longitudinal imaging sample at W5–W7
| Sample char. | high pτ181 ɛ4+ | high pτ181 ɛ4- | low pτ181 |
| N | 20 | 21 | 46 |
| Sex, % -female | 45% | 43% | 41% |
| Age W5, y | 65 [56–77] | 68 [56–81] | 64 [56–76] |
| Education, y | 14.6 [7–26] | 13.1 [6–19.5] | 13.6 [6–24.5] |
| EMC W5 | 43.5 [22–61] | 39 [32–56] | 42 [24–57] |
| EMC W6 | 39.5 [17–55] | 34 [25–52] | 43 [23–61] * |
| EM W7 | 12 [3–16] | 10 [2–17] | 10 [6–20] |
| MMSE W5 | 28 [25–30] | 28 [24–30] | 28 [24–30] |
| MMSE W6 | 28.5 [24–30] | 28 [25–30] | 28 [22–30] |
| MMSE W7 | 27 [24–30] | 27 [24–30] | 27 [21–30] |
| APOE ɛ4 carrier | 100% | 0% | 30% |
| pτ181 W5, (pg/ml) | 2.9 [1.4–6.0] | 3.08 [1.0–7.1] | 1.73 [0.5–4.3] |
| pτ181 W6, (pg/ml) | 5.5 [3.3–11.3] | 5.22 [3.7–9.4] | 2.19 [0.7–3.5] |
EM, summed score of the two EMC-free recall tasks that were included at W7 (max score 28); units and abbreviations for all other entries are the same as in Table 1. Age, Education data, APOE ɛ4 carriership, plasma pτ181 levels, EM, and MMSE were available for all. * One subject excluded from the EMC W6-calculation due to missing data.
The Betula study was approved by the Regional Ethical Review Board and carried out in accordance with the Declaration of Helsinki. Written consent for study participation was obtained from each participant.
Diagnostic assessment
The diagnostic protocol was reported in full details elsewhere [6] and was based on repeated evaluations of multi-disciplinary clinical documentation, further supplemented by outcomes of the Betula study health- and memory assessments [6]. The diagnostic procedure allowed individual health trajectories, throughout the study period, to be followed at a symptom- and functional level and considered also other clinician’s assessments in the diagnostic decision. Participants receiving an AD diagnosis exhibited an insidious onset and progressive cognitive decline as well as other symptoms typically attributable to clinical AD. Individuals with cardiovascular insults together with neurological signs, a fluctuating symptomatology, and stepwise progression of cognitive deficiencies received a clinical diagnosis of vascular dementia. Individuals with complex diagnostic features or insufficient clinical information were classified as dementia not otherwise specified. Less common dementia disorders such as frontotemporal dementia, Parkinson dementia, Lewy body dementia, cortico-basal syndrome, and progressive supranuclear palsy were thoroughly clinically investigated and diagnosed within the healthcare system. Individuals with cognitive impairment close to death, which was accompanied by severe somatic conditions, were not considered as demented, nor were individuals with neurocognitive deficits of non-progressive nature after, e.g., trauma, tumor, subarachnoid hemorrhage. The disease onset was determined as the year at which the clinical symptoms with sufficient severity elicit interference with social functioning and instrumental activities of daily living, i.e., when the core criteria of dementia were met [17]. The diagnostic assessments were coordinated by co-author R.A. throughout the study period, applying DSM-IV classification core criteria for dementia. Additional inclusion and exclusion criteria were applied, which increased the sensitivity and specificity [18–20]. To improve the reliability, the diagnostic procedure was blindly carried out every 5th year without information on the previously determined diagnostic status at hand. Any disparity in diagnosis and/or onset age were re-examined and solved.
Plasma p-tau181
Plasma p-tau181 levels were measured with a fully validated in-house ultrasensitive Simoa immunoassay, previously described in detail [21].
Statistical analyses
A cut-off for elevated p-tau181 levels of > 6 pg/ml was determined from the distribution of values in the control group, which approximately corresponded to 1 SD (2.6 pg/ml) above the Mean (3.3 pg/ml). In a sensitivity analysis of the relation between p-tau and APOE we also considered the stricter threshold of > 10 pg/ml. Furthermore, we considered p-tau 181 as a continuous variable in a conditional logistic regression approach to predict AD.
Comparison of p-tau levels between cases and controls within the dementia groups were performed using paired one-sided t-tests as well as Wilcoxon signed-rank tests. The treatment of matched case-control data using parametric and nonparametric tests for matched samples is in accordance with previous recommendations [22]. Cases where no control was identified were discarded in the main analysis. In a further analysis we performed imputation using the control age group median as the control observation for cases without controls, and then proceeded as stated above. For investigating the association between elevated p-tau levels and APOE ɛ4 carrier status, the Chi-square test for cross-tables was used. If there were expected cell counts below five, we performed a control analysis using Monte-Carlo simulation on the cross-tables to validate the obtained Chi-square p-value (the control analyses confirmed the main findings). A signed paired Wilcoxon’s rank test between APOE groups for AD cases at different stages were performed.
Neuroimaging: Pre- and postprocessing
The acquisition of T1-weighted images, T2-weighted images, EPI-series, and experiment setup has been thoroughly described 23]. The fMRI-sequences included a 10-min blocked face–name recognition task with 6 encoding blocks, 6 retrieval blocks, and 8 blocks of an active perceptual baseline task [23, 24].
Within each encoding and retrieval block (six blocks each), four stimuli were presented for 4 s each, with a randomized interstimulus interval of 1.5, 2.5, 3, or 4.5 s. Participants were instructed before each block for 4 s. The face-name stimuli were presented during the encoding blocks, and the participants were instructed to memorize face-name associates. A button press, using the index finger, was expected to indicate that a face-name pair was seen. During the retrieval blocks, each face was presented along with three letters of which one corresponded to the first letter of the previously encoded name. Subjects were expected to indicate the letter corresponding to the name which was previously encoded with the face. The top, middle, and bottom letters corresponded to the index, the ring, and the middle fingers, respectively. Respond by guessing was expected if participants could not remember the association. During the active baseline condition, a cross-hair was shown and this cross-hair was converted to a circle every now and then. The subjects were expected to press a button as soon as a conversion took place. This condition was used to dissociate different aspects of episodic memory from sensory motor components. The fMRI-part also included a resting-state (RS) sequence whose processing was previously described [25–28]. In brief, the T1-images were segmented, and the grey matter, white matter, and cerebrospinal fluid-likelihood-maps were used to generate, first, a subject-specific template, and then a group-specific template in Dartel-space. The fMRI-data was movement corrected, normalized to MNI (Montreal Neurological Institute) space by flow field files from the Dartel-processing, and smoothed. An adjustment for B0-inhomogeneities was performed by a B0-template calculated from W7.
For task-fMRI, a high pass filter was applied with a time constant of 200 s. A test of encoding versus baseline at W5 had revealed two peaks in left [22 –8 –16] and right [–20 –10 –16] anterior hippocampus [23], and here we extracted encoding-baseline contrast (all trials) values within two 5 mm ROIs around the peaks. We tested for between-groups differences in hippocampus activity (collapsed across the left and right hemisphere regions-of-interest) within each timepoint. At the group level, a 2-way ANOVA with the three groups as one factor and the three time points as another factor was conducted, followed by two-sample post-hoc t-tests when significant interactions were observed. A Pearson correlation test was performed between differences in anterior hippocampus activation and memory performance across test waves. No significant difference in time interval between T5 and T6 was observed between APOE groups (p = 0.44).
For RS-fMRI, linear and quadratic effects of average cerebrospinal fluid and white matter-specific time courses, a global signal using matter-specific probability maps (ps > 0.5) and a 24-parameter model (six motion parameters, six temporal derivatives, and their squares was used) were regressed out and then a high pass filter with a time constant of 130 s were applied. For each participant and time point, average resting-state time series were extracted from 264 brain regions (3 mm radius spheres) based on a commonly used functional connectivity parcellation [29]. Here, we focused on 58 cortical ROIs that compromise the default mode network (DMN), known to include hippocampus as an important subsystem [25, 26]. Since this functional parcellation does not include hippocampus regions, we added four anterior and posterior hippocampus regions (3 mm radius spheres; XYZ = –20 –10 –16; 22 –8 –16; –24 –30 –4; 26 –30 –2; Supplementary Figure 1) which previously showed reliable activations during a memory task [23]. To ensure that the extracted time series were derived from grey matter regions, only grey matter voxels were considered by eroding each sphere by a high probability grey matter mask (ps < 0.001).
The extracted time series were correlated to create a 62×62 connectivity matrix (58 DMN + 4 hippocampus ROIs) for each participant and time point using Pearson’s correlations, transformed to z-values using Fisher’s r-to-z transformation. Then, the averaged correlation among all 4 ROIs were computed as a measure of local hippocampal connectivity. In a control analysis, given potential concerns about the validity of negative correlations [30], we set all negative correlations to zero and found that the longitudinal increase in hippocampal FC remained significant. To explore hippocampal-DMN connectivity, we computed the mean of two values: first, the average of the left hippocampus connectivity with the left DMN ROIs and second, the average of right hippocampal connectivity with right DMN ROIs.
Preprocessing and analyses were made with the SPM12 software (https://www.fil.ion.ucl.ac.uk/spm/) using an inhouse program for batching and visualization (DataZ).
Data availability
Providing data transfer is in agreement with European Union legislation on the General Data Protection Regulation, anonymized data will be shared upon request from qualified investigators for the sole purpose of replication.
RESULTS
P-tau181 levels in clinical and preclinical AD in relation to APOE ɛ4
Figure 2a shows case-control comparisons of plasma p-tau181 levels as a function of clinical phase. Across phases, the cases had higher p-tau181 levels than controls, and the highest value was seen for the manifest AD-cases (ca two years post clinical AD onset). The case-control difference was significant in the clinical phase (p = 0.0033 for non-parametric and p = 0.0072 for parametric tests) and also in the late preclinical phase (ca 2 years pre-onset, p = 0.0041 for non-parametric and p = 0.033 for parametric tests). In the early preclinical phases (ca 13- and 7 years pre-onset, respectively), only a statistical trend at the group level was observed in the 7-years pre-onset group (p = 0.058, parametric, p = 0.097 non-parametric). The significant group difference at clinical and late clinical phases survived correction for multiple comparisons (0.05/4 = 0.0125). Further conditional logistic regression with p-tau181 as a continuous variable revealed that the odds ratio (OR) for AD associated with p-tau181 is 1.18 (if we transform p-tau181 to a z-score, the OR for AD = 1.88). Thus, one standard deviation increase in p-tau was associated with almost a doubling of the odds for AD.
Fig. 2
P-tau181 levels for cases and controls. a) Median p-tau181 levels as a function of time to/from AD onset. Number of subjects in each group is indicated. b) Median p-tau181 levels as a function of time to/from AD onset as a function of APOE status. Number of subjects in each group is indicated. c) Distribution of individual p-tau181 levels for AD cases and controls, where arrows indicate tentative cut-off for elevated values (see text). d) Percentage with p-tau181 levels exceeding the cut-off for controls and AD cases as a function of time to/from diagnosis. Error-bars are 25/75-percentile in (a) and (b). “*” denotes a group difference, p < 0.05.
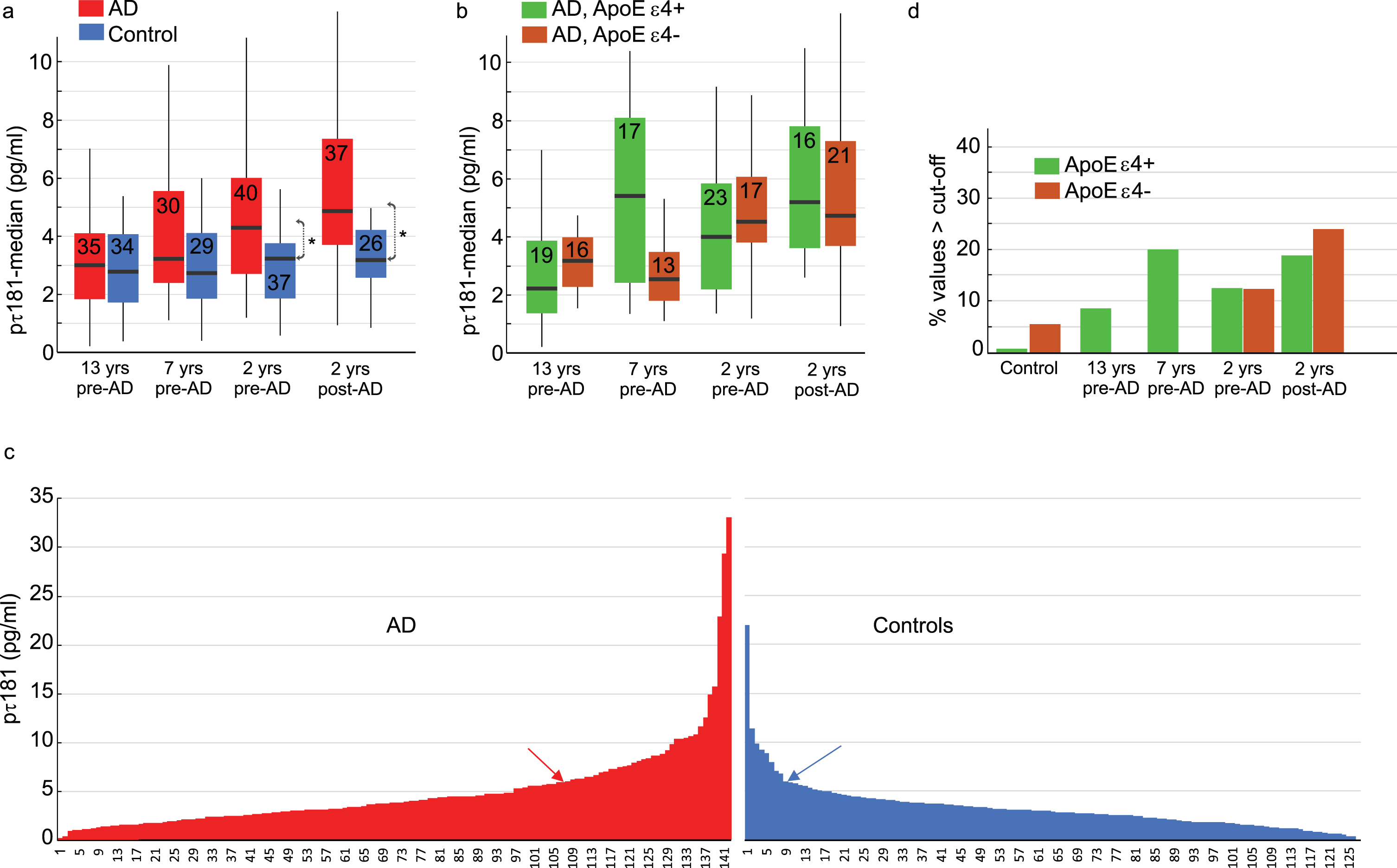
Next, we addressed the issue of elevated p-tau181 levels in relation to APOE. Of the 142 cases, 75 were ɛ4 carriers (53%), whereas only 21 of 126 controls were ɛ4 carriers (17%). Figure 2b plots ɛ4 status as a function of diagnostic stage for cases. There were no significant differences (ps > 0.15) between APOE ɛ4 carriers (ɛ4+) and non-carriers (ɛ4–) among the AD-cases at the different clinical stages, except for a trend (p = 0.072) at 7 years pre-AD towards elevated p-tau181 levels in ɛ4 carriers (Median = 5.41 pg/ml) compared to non-carriers (Median = 2.54 pg/ml).
Figure 2c shows the highly skewed distributions of p-tau181 levels in controls and cases. Of the controls, eight (6%) had a p-tau181 > 6 pg/ml (≅ M + 1 SD; blue arrow in Fig. 2c). Using this control-group derived cut-off for elevated p-tau181 levels, 34 AD cases (24%) had an elevated value (red arrow in Fig. 2c).
Figure 2d plots the p-tau181 elevated controls and AD cases as a function of clinical phase and APOE status. In the early preclinical phases (13 and 7 years), all cases with elevated values were APOE ɛ4 carriers, and there was a significant association between elevated p-tau181 and APOE genotype (χ2 = 8.42; p = 0.004). As expected, the percentages of individuals with elevated p-tau181 values were higher in closer proximity to AD onset, but at these stages there was no APOE effect (i.e., similar rates of ɛ4 carriers and non-carriers in the late preclinical phases; χ2 = 0.32; p = 0.57).
A similar analysis based on a stricter cut-off for defining elevated p-tau181 levels (> 10 pg/ml) yielded a similar outcome with regard to APOE. In the early preclinical AD phases (13 and 7 years), 4 individuals (6.2%) had values exceeding 10 pg/ml, and all were ɛ4 carriers. In the later preclinical phases (–2 years from diagnosis), 8 (10.5%) had values exceeding the stricter cut-off but only 2 were ɛ4 carriers. Only 2 controls (1.6%) had a value exceeding the stricter cut-off.
Hippocampus activation and connectivity in relation to p-tau181 and APOE ɛ4
In the imaging study, analyses were based on fMRI data from waves 5–7, and plasma samples from test waves 5 and 6 (Fig. 1b). The longitudinal tau-trajectories revealed that 46 individuals (53%) had low p-tau181 levels at both waves 5 and 6 (14 of these were ɛ4 carriers), whereas 41 (47%) had higher levels that increased from wave 5 to 6 (Fig. 3a). The latter 41 individuals could be subdivided into ɛ4 carriers (N = 20) and non-carriers (N = 21).
Fig. 3
Longitudinal brain-imaging study. a) Longitudinal change in p-tau181 levels in relation to APOE status. b) Longitudinal change in hippocampus activity and (c) connectivity as a function of p-tau181 levels (high/rising versus low) and APOE status (ɛ4 carrier, ɛ4+ versus non-carrier, ɛ4–). d) Change in memory performance between waves 6-7. Error-bars are standard error of mean in (b) and (c). Error-bars are standard deviation in (d). Error-base are 25/75-percentile in (a). “*” denotes a group difference with p < 0.05 in (a) and (b), and within group change with p < 0.05 in (c) and (d).
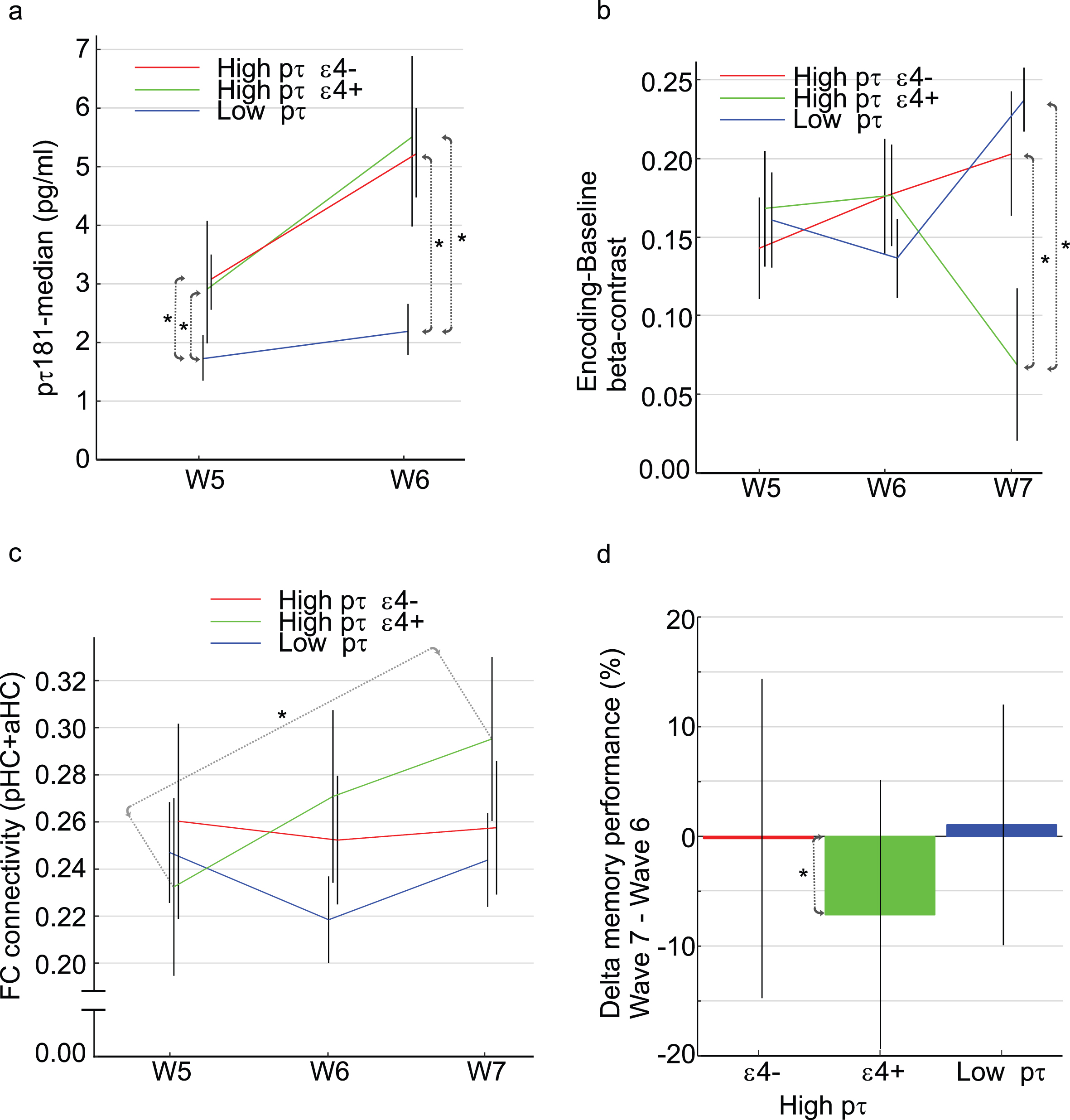
First, we examined whether brain activity in the anterior hippocampus during face-name encoding was related to tau accumulation across waves, and whether this relation differed for ɛ4 carriers and non-carriers. A repeated measure ANOVA revealed a significant group by time interaction [F(4,168) = 4.59 p = 0.0015]. Post-hoc t-tests revealed no group differences at waves 5 and 6 (Fig. 3b), but a marked reduction in bilateral hippocampus activity at wave 7 was evident in high p-tau ɛ4 carriers (green) relative to the low p-tau group (blue; p < 0.001) and also relative to the ɛ4 non-carriers high p-tau group (red; p = 0.018). In the low p-tau181 group, there was no difference in anterior hippocampus activity between ɛ4 carriers and non ɛ4 carriers at W7 [t(44) = 1.46, p = 0.15].
Next, in view of recent findings of elevated local hippocampal connectivity during resting state [11], we quantified bilateral hippocampal connectivity at each test wave for the three sub-groups. In the ɛ4 carriers with elevated p-tau levels, a gradual increase in hippocampus connectivity was observed across waves, and this increase was significant between waves 5 and 7 [Fig. 3c; t(18) = 2.20, p < 0.05]. No significant longitudinal change in hippocampal connectivity was seen in the other groups; thus, hippocampal hyperconnectivity was specifically seen for ɛ4 carriers with elevated p-tau181 levels. To explore whether elevated hippocampal connectivity was accompanied by decreased hippocampus-cortical connectivity, we examined the connectivity between the hippocampus and the DMN. We found a significant decline in hippocampus-DMN connectivity from wave 5 to wave 6 (t = 2.62, p = 0.0166) for ɛ4 carriers with elevated p-tau181 levels but not for the other two groups (ps > 0.05).
The change in face-name memory performance across waves were examined for the three groups. Only for ɛ4 carriers with elevated p-tau181 levels was a significant performance reduction from wave 6 to wave 7 observed [Fig. 3d; t(19) = 2.60, p = 0.018]. In further support of a relation between hippocampus activation and memory performance, across the sample, a significant change-change correlation was observed between the wave 6–7 differences in anterior hippocampus activation and memory performance [r(85) = 0.28, p = 0.0092].
DISCUSSION
P-tau as a preclinical AD-marker
Our population-based study approach, with dementia as an exclusion criterion at initial study enrolment, provided novel information by means of early collected (W3) plasma samples from age- and sex-matched controls who were longitudinally assessed for up to 20 years to confirm that they remained non-demented. Consistent with their non-AD status, the absolute majority of controls had low p-tau181 values. Preclinical AD as well as other pathologies or measurement error remain as possible sources of high values in a few controls, although re-assessment of those with the highest p-tau values revealed no clinical support for dementia progress during the follow-up period.
The control distribution offered a means to define a cut off for elevated p-tau181 level in preclinical and clinical AD cases. At the group level (Fig. 2a), significant case-control differences were seen close to disease onset (–2 years), and at the individual level, for both control-group derived cut-offs considered, the proportion of cases with elevated p-tau181 levels exceeded that for controls. Still, with a mean for diagnosed cases of 6.4 pg/ml and less than 50% of cases having values exceeding the lenient cut-off of 6 pg/ml, it should be noted that our clinically diagnosed cases had modest p-tau181 levels relative to previous studies of p-tau181 in blood across the AD spectrum [31]. For example, p-tau181 cut-offs of 14–18 pg/ml were determined in relation to defining Aβ status in the ADNI cohort [32], and p-tau181 values around 20–30 pg/ml were reported 8 years before postmortem [33]. While these differences in part might reflect different study populations, it should also be noted that the diagnostic assessment, implemented in the current study, was based on a clinical diagnostic approach [6] which together with the repeated follow-up assessments could have identified demented individuals at a quite early stage.
The present analyses converged to suggest that elevated plasma p-tau181 levels are indicative of forthcoming AD in preclinical APOE ɛ4 carriers. Specifically, while no significant effect of APOE was seen near AD onset, in the early preclinical phases all cases with elevated values were ɛ4 carriers. At the stage of clinically manifest AD, no influence of the genetic risk factor on tau burden is expected as all these individuals by definition have an active disease process [5]. In the preclinical phase, the APOE genotype could be regarded as a surrogate measure of time with Aβ pathology, with each ɛ4 allele being associated with ∼10 years earlier onset of Aβ deposition. By this view, compared with non-carriers, similarly aged APOE ɛ4 carriers with Aβ pathology would be further along in the AD process which could explain the positive association with p-tau181 levels. The observed association between APOE and p-tau in the preclinical phase could reflect an APOE influence on tau pathology, such that the deposition of neurofibrillary tangles begins earlier and/or is accumulating faster in ɛ4 carriers. In support of this interpretation, APOE ɛ4 has been linked to increased tau phosphorylation and tangle formation [34].
It should be noted that the lack of data on Aβ load prevented us from assessing potential contributions to the observed p-tau181 findings of amyloid pathology [35]. Abnormal p-tau secretion may occur concomitantly with, or in response to, brain Aβ pathology, including PHF-tau positive dystrophic neurites surrounding plaques, but recent studies suggest that APOE ɛ4 may have an Aβ-independent effect on tau-burden in the medial temporal cortex [36, 37]. Thus, speculatively, elevated p-tau181 levels close to disease onset in non-carriers would coincide with elevated Aβ load, whereas in the preclinical phase elevated tau in APOE ɛ4 carriers might precede overt amyloid pathology.
Hippocampus functioning in relation to p-tau and APOE
In the imaging study, the longitudinal design identified a sub-group of ɛ4 carriers with increasing p-tau181 levels across test waves. This sub-group could be compared with an APOE ɛ4 non-carrier group showing a similar longitudinal increase in p-tau levels, but also with a low p-tau181 group. Only for ɛ4 carriers was increasing p-tau181 related to subsequent reduction (between waves 6–7) in hippocampus encoding-related activity. This observation supports findings that presence of APOE ɛ4 and lower hippocampal fMRI activation is a predictor of future neurocognitive impairment [38] and extends previous reports by highlighting p-tau as a possible mechanism.
The finding that a longitudinal increase in p-tau levels translated into a reduced hippocampus activity during memory encoding is noteworthy, as several previous cross-sectional studies reported that APOE ɛ4 [13, 39, 40] and higher tau levels [11, 12, 41, 42] are related to higher hippocampal activity and connectivity. Here, too, we observed a longitudinal increase in resting-state hippocampus connectivity for ɛ4 carriers with high/rising p-tau levels. This constitutes novel, within-person evidence for the concept of hyperconnectivity. The trend for elevation in connectivity was seen already between waves 5–6, coinciding with tau elevation, whereas the reduction in task activity was seen at the final wave. This cascade of event is in line with previous observations that increased local hippocampal connectivity during rest translated into lower task-related hippocampus activation during memory encoding [25], and findings that tau deposition drives elevated local hippocampal connectivity and induces hippocampal disconnection from functional networks of relevance for episodic memory [11]. Strikingly, consistent with these findings, we observed a disconnection of the hippocampus from the DMN for ɛ4 carriers with high/rising p-tau levels with a significant connectivity decrease from wave 5 to 6 (i.e., the period during which elevated p-tau was observed). Thus, tau accumulation may induce increased local hippocampal synchrony, and eventually reduced ability to engage the hippocampus during active mnemonic processing. Although the longitudinal design is a strength of the current study, our small sample size and lack of p-tau at T7 limit the choice of analytical approach.
The apparent selectivity for ɛ4 carriers for rising p-tau181 levels could reflect an APOE effect on rate of tau accumulation, and it is also consistent with findings that APOE ɛ4 aggravates the negative influence of tau pathology on neuronal function [3]. Interaction of p-tau and APOE genotype on AD has been related to dysfunction of synaptic plasticity. Recent studies have shown that alterations in long-term potential plasticity are associated with cognitive decline [43] and elevated p-tau level [44]. Moreover, elevated p-tau was associated with impaired long-term potential plasticity and accelerated progress of disease in AD patients with ɛ4 carriage [8].
Conclusion
Our findings indicate an association of APOE ɛ4 and tau with preclinical AD and hippocampus functioning. In APOE ɛ4 carriers only, elevated plasma p-tau181 levels were indicative of forthcoming clinically-defined AD, and longitudinal p-tau181 increases were paralleled by increased hippocampus synchrony at rest and reduced hippocampus encoding-related activity. These findings support and extend prior observations of selective neurocognitive APOE effects in normal aging and age-related pathology [8, 45, 46].
ACKNOWLEDGMENTS
LN and the Betula study are supported by a Knut and Alice Wallenberg (KAW) Scholar grant. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), AD Strategic Fund and the Alzheimer’s Association (ADSF-21-831376-C, ADSF-21-831381-C and ADSF-21-831377-C), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. KB is supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), and European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236). MS is supported by the Knut and Alice Wallenberg Foundation (Wallenberg Centre for Molecular and Translational Medicine; KAW 2014.0363), the Swedish Research Council (#2017-02869) and the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-813971).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0673r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-210673.
REFERENCES
[1] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small GW , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[2] | Holtzman DM , Herz J , Bu G ((2012) ) Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006312. |
[3] | Shi Y , Yamada K , Liddelow SA , Smith ST , Zhao L , Luo W , Tsai RM , Spina S , Grinberg LT , Rojas JC , Gallardo G , Wang K , Roh J , Robinson G , Finn MB , Jiang H , Sullivan PM , Baufeld C , Wood MW , Sutphen C , McCue L , Xiong C , Del-Aguila JL , Morris JC , Cruchaga C , Alzheimer’s Disease Neuroimaging I, Fagan AM , Miller BL , Boxer AL , Seeley WW , Butovsky O , Barres BA , Paul SM , Holtzman DM ((2017) ) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549: , 523–527. |
[4] | Therriault J , Benedet AL , Pascoal TA , Mathotaarachchi S , Chamoun M , Savard M , Thomas E , Kang MS , Lussier F , Tissot C , Parsons M , Qureshi MNI , Vitali P , Massarweh G , Soucy JP , Rej S , Saha-Chaudhuri P , Gauthier S , Rosa-Neto P ((2020) ) Association of Apolipoprotein E epsilon4 with medial temporal tau independent of amyloid-beta. JAMA Neurol 77: , 470–479. |
[5] | Jack CR Jr. ((2020) ) Preclinical Alzheimer’s disease: A valid concept. Lancet Neurol 19: , 31. |
[6] | Nyberg L , Boraxbekk CJ , Sorman DE , Hansson P , Herlitz A , Kauppi K , Ljungberg JK , Lovheim H , Lundquist A , Adolfsson AN , Oudin A , Pudas S , Ronnlund M , Stiernstedt M , Sundstrom A , Adolfsson R ((2020) ) Biological and environmental predictors of heterogeneity in neurocognitive ageing: Evidence from Betula and other longitudinal studies. Ageing Res Rev 64: , 101184. |
[7] | Janelidze S , Mattsson N , Palmqvist S , Smith R , Beach TG , Serrano GE , Chai X , Proctor NK , Eichenlaub U , Zetterberg H , Blennow K , Reiman EM , Stomrud E , Dage JL , Hansson O ((2020) ) Plasma P-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med 26: , 379–386. |
[8] | Koch G , Di Lorenzo F , Loizzo S , Motta C , Travaglione S , Baiula M , Rimondini R , Ponzo V , Bonni S , Toniolo S , Sallustio F , Bozzali M , Caltagirone C , Campana G , Martorana A ((2017) ) CSF tau is associated with impaired cortical plasticity, cognitive decline and astrocyte survival only in APOE4-positive Alzheimer’s disease. Sci Rep 7: , 13728. |
[9] | Wu JW , Hussaini SA , Bastille IM , Rodriguez GA , Mrejeru A , Rilett K , Sanders DW , Cook C , Fu H , Boonen RA , Herman M , Nahmani E , Emrani S , Figueroa YH , Diamond MI , Clelland CL , Wray S , Duff KE ((2016) ) Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19: , 1085–1092. |
[10] | Gibbons GS , Lee VMY , Trojanowski JQ ((2019) ) Mechanisms of cell-to-cell transmission of pathological tau: A review. JAMA Neurol 76: , 101–108. |
[11] | Harrison TM , Maass A , Adams JN , Du R , Baker SL , Jagust WJ ((2019) ) Tau deposition is associated with functional isolation of the hippocampus in aging. Nat Commun 10: , 4900. |
[12] | Huijbers W , Schultz AP , Papp KV , LaPoint MR , Hanseeuw B , Chhatwal JP , Hedden T , Johnson KA , Sperling RA ((2019) ) Tau accumulation in clinically normal older adults is associated with hippocampal hyperactivity. J Neurosci 39: , 548–556. |
[13] | Filippini N , MacIntosh BJ , Hough MG , Goodwin GM , Frisoni GB , Smith SM , Matthews PM , Beckmann CF , Mackay CE ((2009) ) Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A 106: , 7209–7214. |
[14] | Westlye ET , Lundervold A , Rootwelt H , Lundervold AJ , Westlye LT ((2011) ) Increased hippocampal default mode synchronization during rest in middle-aged and elderly APOE epsilon4 carriers: Relationships with memory performance. J Neurosci 31: , 7775–7783. |
[15] | Nyberg L , Lundquist A , Nordin Adolfsson A , Andersson M , Zetterberg H , Blennow K , Adolfsson R ((2020) ) Elevated plasma neurofilament light in aging reflects brain white-matter alterations but does not predict cognitive decline or Alzheimer’s disease. Alzheimers Dement (Amst) 12: , e12050. |
[16] | Josefsson M , de Luna X , Pudas S , Nilsson LG , Nyberg L ((2012) ) Genetic and lifestyle predictors of 15-year longitudinal change in episodic memory. J Am Geriatr Soc 60: , 2308–2312. |
[17] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr. , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[18] | Roman GC , Tatemichi TK , Erkinjuntti T , Cummings JL , Masdeu JC , Garcia JH , Amaducci L , Orgogozo JM , Brun A , Hofman A , et al ((1993) ) Vascular dementia: Diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 43: , 250–260. |
[19] | Wiederkehr S , Simard M , Fortin C , van Reekum R ((2008) ) Comparability of the clinical diagnostic criteria for vascular dementia: A critical review. Part I. J Neuropsychiatry Clin Neurosci 20: , 150–161. |
[20] | Wiederkehr S , Simard M , Fortin C , van Reekum R ((2008) ) Validity of the clinical diagnostic criteria for vascular dementia: A critical review. Part II. J Neuropsychiatry Clin Neurosci 20: , 162–177. |
[21] | KarikariTK, PascoalTA, AshtonNJ, JanelidzeS, BenedetAL, RodriguezJL, ChamounM, SavardM, KangMS, TherriaultJ, SchöllM, MassarwehG, SoucyJ-P, HöglundK, BrinkmalmG, MattssonN, PalmqvistS, GauthierS, StomrudE, ZetterbergH, HanssonO, Rosa-NetoP, BlennowK ((2020) ) Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. The Lancet Neurology 19: , 422–433. |
[22] | Niven DJ , Berthiaume LR , Fick GH , Laupland KB ((2012) ) Matched case-control studies: A review of reported statistical methodology. Clin Epidemiol 4: , 99–110. |
[23] | NybergL, AnderssonM, LundquistA, SalamiA, WahlinA ((2019) ) Frontal contribution to hippocampal hyperactivity during memory encoding in aging. Front Mol Neurosci 12: , 229. |
[24] | Salami A , Eriksson J , Nyberg L ((2012) ) Opposing effects of aging on large-scale brain systems for memory encoding and cognitive control. J Neurosci 32: , 10749–10757. |
[25] | Salami A , Pudas S , Nyberg L ((2014) ) Elevated hippocampal resting-state connectivity underlies deficient neurocognitive function in aging. Proc Natl Acad Sci U S A 111: , 17654–17659. |
[26] | Salami A , Wåhlin A , Kaboodvand N , Lundquist A , Nyberg L ((2016) ) Longitudinal evidence for dissociation of anterior and posterior MTL resting-state connectivity in aging: Links to perfusion and memory. Cereb Cortex 26: , 3953–3963. |
[27] | Avelar-Pereira B , Backman L , Wahlin A , Nyberg L , Salami A ((2020) ) Increased functional homotopy of the prefrontal cortex is associated with corpus callosum degeneration and working memory decline. Neurobiol Aging 96: , 68–78. |
[28] | Pedersen R , Geerligs L , Andersson M , Gorbach T , Avelar-Pereira B , Wahlin A , Rieckmann A , Nyberg L , Salami A ((2021) ) When functional blurring becomes deleterious: Reduced system segregation is associated with less white matter integrity and cognitive decline in aging. Neuroimage 242: , 118449. |
[29] | Power JD , Cohen AL , Nelson SM , Wig GS , Barnes KA , Church JA , Vogel AC , Laumann TO , Miezin FM , Schlaggar BL , Petersen SE ((2011) ) Functional network organization of the human brain. Neuron 72: , 665–678. |
[30] | Murphy K , Birn RM , Handwerker DA , Jones TB , Bandettini PA ((2009) ) The impact of global signal regression on resting state correlations: Are anti-correlated networks introduced? Neuroimage 44: , 893–905. |
[31] | Moscoso A , Grothe MJ , Ashton NJ , Karikari TK , Rodriguez JL , Snellman A , Suarez-Calvet M , Zetterberg H , Blennow K , Scholl M , Alzheimer’s Disease Neuroimaging Initiative ((2021) ) Time course of phosphorylated-tau181 in blood across the Alzheimer’s disease spectrum. Brain 144: , 325–339. |
[32] | Karikari TK , Benedet AL , Ashton NJ , Lantero Rodriguez J , Snellman A , Suarez-Calvet M , Saha-Chaudhuri P , Lussier F , Kvartsberg H , Rial AM , Pascoal TA , Andreasson U , Scholl M , Weiner MW , Rosa-Neto P , Trojanowski JQ , Shaw LM , Blennow K , Zetterberg H , Alzheimer’s Disease Neuroimaging Initiative ((2021) ) Diagnostic performance and prediction of clinical progression of plasma phospho-tau181 in the Alzheimer’s Disease Neuroimaging Initiative. Mol Psychiatry 26: , 429–442. |
[33] | Lantero Rodriguez J , Karikari TK , Suarez-Calvet M , Troakes C , King A , Emersic A , Aarsland D , Hye A , Zetterberg H , Blennow K , Ashton NJ ((2020) ) Plasma p-tau181 accurately predicts Alzheimer’s disease pathology at least 8 years prior to post-mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol 140: , 267–278. |
[34] | Safieh M , Korczyn AD , Michaelson DM ((2019) ) ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med 17: , 64. |
[35] | Barthelemy NR , Bateman RJ , Hirtz C , Marin P , Becher F , Sato C , Gabelle A , Lehmann S ((2020) ) Cerebrospinal fluid phospho-tau T217 outperforms T181 as a biomarker for the differential diagnosis of Alzheimer’s disease and PET amyloid-positive patient identification. Alzheimers Res Ther 12: , 26. |
[36] | Sanchez JS , Becker JA , Jacobs HIL , Hanseeuw BJ , Jiang S , Schultz AP , Properzi MJ , Katz SR , Beiser A , Satizabal CL , O’Donnell A , DeCarli C , Killiany R , El Fakhri G , Normandin MD , Gomez-Isla T , Quiroz YT , Rentz DM , Sperling RA , Seshadri S , Augustinack J , Price JC , Johnson KA ((2021) ) The cortical origin and initial spread of medial temporal tauopathy in Alzheimer’s disease assessed with positron emission tomography. Sci Transl Med 13: , eabc0655. |
[37] | Weigand AJ , Thomas KR , Bangen KJ , Eglit GML , Delano-Wood L , Gilbert PE , Brickman AM , Bondi MW , Alzheimer’s Disease Neuroimaging Initiative ((2021) ) APOE interacts with tau PET to influence memory independently of amyloid PET in older adults without dementia. Alzheimers Dement 17: , 61–69. |
[38] | Woodard JL , Seidenberg M , Nielson KA , Smith JC , Antuono P , Durgerian S , Guidotti L , Zhang Q , Butts A , Hantke N , Lancaster M , Rao SM ((2010) ) Prediction of cognitive decline in healthy older adults using fMRI. J Alzheimers Dis 21: , 871–885. |
[39] | Bookheimer SY , Strojwas MH , Cohen MS , Saunders AM , Pericak-Vance MA , Mazziotta JC , Small GW ((2000) ) Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med 343: , 450–456. |
[40] | Persson J , Lind J , Larsson A , Ingvar M , Sleegers K , Van Broeckhoven C , Adolfsson R , Nilsson LG , Nyberg L ((2008) ) Altered deactivation in individuals with genetic risk for Alzheimer’s disease. Neuropsychologia 46: , 1679–1687. |
[41] | Marks SM , Lockhart SN , Baker SL , Jagust WJ ((2017) ) Tau and beta-amyloid are associated with medial temporal lobe structure, function, and memory encoding in normal aging. J Neurosci 37: , 3192–3201. |
[42] | Berron D , Cardenas-Blanco A , Bittner D , Metzger CD , Spottke A , Heneka MT , Fliessbach K , Schneider A , Teipel SJ , Wagner M , Speck O , Jessen F , Duzel E ((2019) ) Higher CSF tau levels are related to hippocampal hyperactivity and object mnemonic discrimination in older adults. J Neurosci 39: , 8788–8797. |
[43] | Di Lorenzo F , Ponzo V , Bonni S , Motta C , Negrao Serra PC , Bozzali M , Caltagirone C , Martorana A , Koch G ((2016) ) Long-term potentiation-like cortical plasticity is disrupted in Alzheimer’s disease patients independently from age of onset. Ann Neurol 80: , 202–210. |
[44] | Koch G , Di Lorenzo F , Del Olmo MF , Bonni S , Ponzo V , Caltagirone C , Bozzali M , Martorana A ((2016) ) Reversal of LTP-like cortical plasticity in Alzheimer’s disease patients with tau-related faster clinical progression. J Alzheimers Dis 50: , 605–616. |
[45] | GorbachT, PudasS, Bartres-FazD, BrandmaierAM, DuzelS, HensonRN, IdlandAV, LindenbergerU, Macia BrosD, MowinckelAM, Sole-PadullesC, SorensenO, WalhovdKB, WatneLO, WesterhausenR, FjellAM, NybergL ((2020) ) Longitudinal association between hippocampus atrophy and episodic-memory decline in non-demented APOE ɛ4 carriers. Alzheimers Dement 12: , e12110. |
[46] | MontagneA, NationDA, SagareAP, BarisanoG, SweeneyMD, ChakhoyanA, PachicanoM, JoeE, NelsonAR, D’OrazioLM, BuennagelDP, HarringtonMG, BenzingerTLS, FaganAM, RingmanJM, SchneiderLS, MorrisJC, ReimanEM, CaselliRJ, ChuiHC, TcwJ, ChenY, PaJ, ContiPS, LawM, TogaAW, ZlokovicBV ((2020) ) APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581: , 71–76. |


