
NLRP3 Inflammasome: A Starring Role in Amyloid-β- and Tau-Driven Pathological Events in Alzheimer’s Disease
Abstract
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease commonly diagnosed among the elderly population. AD is characterized by the loss of synaptic connections, neuronal death, and progressive cognitive impairment, attributed to the extracellular accumulation of senile plaques, composed by insoluble aggregates of amyloid-β (Aβ) peptides, and to the intraneuronal formation of neurofibrillary tangles shaped by hyperphosphorylated filaments of the microtubule-associated protein tau. However, evidence showed that chronic inflammatory responses, with long-lasting exacerbated release of proinflammatory cytokines by reactive glial cells, contribute to the pathophysiology of the disease. NLRP3 inflammasome (NLRP3), a cytosolic multiprotein complex sensor of a wide range of stimuli, was implicated in multiple neurological diseases, including AD. Herein, we review the most recent findings regarding the involvement of NLRP3 in the pathogenesis of AD. We address the mechanisms of NLRP3 priming and activation in glial cells by Aβ species and the potential role of neurofibrillary tangles and extracellular vesicles in disease progression. Neuronal death by NLRP3-mediated pyroptosis, driven by the interneuronal tau propagation, is also discussed. We present considerable evidence to claim that NLRP3 inhibition, is undoubtfully a potential therapeutic strategy for AD.
ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease among the elderly population without approved therapies to halt or reverse its progression [1]. AD is associated with a specific pattern of pathological changes in the brain that result in neurodegeneration, loss of synaptic connections, and progressive memory deficits and cognitive impairment [2].
AD can be divided into different types, based on the age of onset and genetic predisposition, which are comparable in most clinical aspects [2]. Sporadic or late onset AD accounts for over 90%of AD cases and begins after the age of 65 years. Familial AD, a small subset of AD cases (<10%), is caused by genetic mutations and has an early-onset (fourth to sixth decade of life) [1].
PROTEIN AGGREGATES IN AD
One of the pathological hallmarks of AD is the extracellular accumulation of senile plaques in the brain. Senile plaques are composed of insoluble aggregates of amyloid-β (Aβ) peptides, firstly described by Alois Alzheimer in 1907 as a “(. . .) deposition of a pathological metabolic substance in the neuron” [3]. Alzheimer also described “(. . .) the nucleus and the cell itself disintegrate and only a tangle of fibrils indicates the place where a neuron was previously located.” [3], referring to what is now recognized as the intracellular formation of neurofibrillary tangles (NFTs), consisting of hyperphosphorylated twisted filaments of the microtubule-associated protein tau [4]. It was also mentioned that “Many neurons, especially the ones in the upper layer, have completely disappeared.” [3], which corroborates the observed progressive loss of synapses and neuronal death [2], leading to an atrophy of the brain confirmed in postmortem samples [3]. In AD, this phenomenon starts to affect tissue from the frontal and temporal lobes progressing to other areas of the neocortex [2].
Accumulation of abnormally folded proteins is a key histopathological feature of many neurodegenerative diseases. In Huntington’s disease, the polyglutamine protein huntingtin occurs in intranuclear inclusions; in prion disease, prion particles bind to the membrane-bound protein and in Parkinson’s disease (PD), aggregates of α-synuclein comprise the Lewy bodies present in the cytoplasm of neurons. In this sense, AD is rather unusual, since both intracellular and extracellular aggregates are observed—intraneuronal tangles of hyperphosphorylated tau and extracellular amyloid plaques consisting of aggregated Aβ peptides—which can potentially result in neuronal dysfunction and death. Indeed, the symptomatic phases of the disease have the duration of 8–10 years and are preceded by prodromal and asymptomatic stages that can last for two decades, where the formation of Aβ plaques and NFTs starts [2].
Formation and toxicity of Aβ species
AD pathology is dominantly explained by the “amyloid cascade hypothesis”, which suggests Aβ as the initiator of all subsequent events that drive the progressive neuronal damage and cognitive dysfunction characteristic of the disease [5].
Aβ is a peptide produced in all neurons throughout life [2]. At physiological levels, Aβ is a normal, soluble product of neuronal metabolism, present in the brain of healthy people where it has important physiological functions, such as regulator of synaptic plasticity and enhancer of learning and memory, as well as antioxidant and metal chelator [6, 7]. Evidence indicates that synaptic activity directly evokes the release of Aβ at the synapse [6].
Aβ is produced through sequential proteolytic cleavage of amyloid-β protein precursor (AβPP), a type-1 transmembrane glycoprotein, by two enzymatic complexes: the β- and γ-secretases [7]. The function of AβPP remains unknown, although involvement in synaptic transmission and plasticity was hypothesized [8]. Recently, it was shown that full length AβPP has cell adhesion properties, mediating the stability of synaptic structures and maintaining adequate spine numbers [9]. Most of intracellular Aβ is distributed in the neuronal cytosol, colocalizing with different organelles dependent on where AβPP, β- and γ-secretases occur. It has been reported to be produced in the secretory pathway related organelles (endoplasmic reticulum, medial Golgi and trans-Golgi network), and to be associated with mitochondria [10].
AβPP can be processed through two enzymatic pathways: the amyloidogenic pathway and the non-amyloidogenic one. In the amyloidogenic pathway, the process of Aβ biogenesis, AβPP is first cleaved by the β-secretase, generating a soluble fragment from the N-terminal domain of AβPP and the β-cleaved C-terminal fragment, which is then processed by γ-secretase complex releasing the AβPP intracellular domain and Aβ [7]. Aβ is a heterogeneous mixture of peptides, composed of 37–43 amino acids, with different solubility, stability, biological and toxic properties [11]. The major isoforms of Aβ are peptides with 40 amino acids in length (Aβ40), and 42 amino acid species (Aβ42) [6]. Aβ40 is continuously and abundantly produced in both healthy and AD-affected brain tissue, whereas other Aβ peptides are continuously produced at lower levels [11]. In physiological conditions, more than 90%of Aβ is in the form of Aβ40, while less than 5%is generated as Aβ42 [10]. The latter shows a higher propensity to aggregate into different states ranging from dimers to soluble oligomers to insoluble aggregates of fibrils that develop into plaques [6, 10]. However, one must not forget that aggregation and toxic properties were mainly studied with solutions containing a single type of Aβ, and these properties are strongly influenced by the presence of other Aβ species [11]. Indeed, polymerization of Aβ is complex and occurs via a process called “nucleated conformational conversion”, which involves several metastable intermediates. So, not only quantitative increases of individual Aβ peptides, but also qualitative changes in the spectrum of Aβ peptides, can determine the biophysical and pathological properties of Aβ [11].
In AD patients, Aβ42 is present at a higher percentage and, in the classic view, its aggregation is considered the primary cause for the toxic Aβ effects, such as synaptic dysfunction, excitotoxicity, mitochondrial dysfunction, oxidative stress, and alteration of cell signaling pathways [7]. The most toxic forms are the high β-sheet structures, such as oligomers and fibrils [12, 13]. Thus, Aβ accumulation in the brain and its conformational changes are considered to play a pivotal role in the pathogenesis of AD [14]. At the diagnostic level, Aβ42/Aβ40 and Aβ42/Aβ38 ratios in the cerebrospinal fluid (CSF) are considered significantly better biomarkers for AD than Aβ42 alone [15]. Indeed, decreased Aβ42/Aβ40 and Aβ42/Aβ38 ratios in CSF reflect AD pathology better, whereas decline in Aβ42 alone is also associated with non-AD dementias [15].
The non-amyloidogenic pathway is the innate mechanism which is thought to antagonize Aβ generation, as AβPP is firstly proteolyzed by α-secretase within Aβ domain, generating big soluble AβPP fragments, further cleaved by γ-secretase, and producing non-toxic products [10].
It is well accepted that limiting the concentration of Aβ in the brain tissue, either by reducing Aβ production or enhancing Aβ clearance, is a putative way to inhibit the cascade of Aβ-induced pathological events [10]. Given the crucial role of β- and γ-secretases in Aβ production, manipulation of these secretases disturb the generation of Aβ [10]. Proteolytic degradation of Aβ into shorter soluble fragments, devoid of toxic effect, prevents Aβ aggregation or deposition into plaques. Lysosome-mediated endocytosis of Aβ also contributes to the clearance of toxic Aβ peptide and Aβ deposits [10].
In sporadic AD, the balance between the amyloidogenic and the non-amyloidogenic pathways becomes altered. There is an excessive production of Aβ, via the amyloidogenic pathway, and a lower degradation due to a decreased activity of the degrading enzymes, such as neprilysin, insulin-degrading enzyme, and angiotensin converting enzyme I, thus causing impaired clearance mechanisms of Aβ from the brain [16]. The familial AD form is caused by autosomal dominant mutations in genes related to AβPP processing, leading to Aβ overproduction [2].
Tau aggregation in NFTs
Human tau protein can display six isoforms, of 37–46 kDa, which result from alternative splicing of a 6 Kb mRNA transcript originated from the transcription of the Microtubule Associated Protein Tau gene, located on chromosome 17 [17]. Regardless of the isoform, this protein can be divided in four main domains: a N-terminal acidic projection domain; a proline-rich domain; a microtubule binding domain and, finally, a C-terminal tail [18].
Tau is a soluble protein acting as a microtubule stabilizer in neuronal cells [19]. It participates in the stabilization of microtubules, by binding to their surface, and fosters microtubules’ self-assembly [18]. Although this is the most prominent function of tau in healthy brain cells, it has also been reported that tau interacts with constituents of the neural plasma membrane, regulates signal transduction, and can interact with DNA and RNA [20]. The hydrophilic quality renders tau to be a “natively unfolded” protein with high mobility and flexibility. However, it displays intradomain interactions forming a “paperclip” like shape where the N-terminal, the microtubule binding domain and the C-terminal are brought close to each other [21].
Tau can occur in several forms, such as monomers (highly soluble proteins of 55–74 kDa), dimers (composed of two tau monomers in anti-parallel orientation linked by disulfide bonds), small soluble oligomers, granular tau oligomers (composed of an average of 40 densely packed tau monomers) and filamentous forms (such as the paired helical filaments and the straight filaments) [22]. Under pathological conditions tau becomes hyperphosphorylated and eventually forms intracellular aggregates. Tau aggregation is a hallmark of AD and other tauopathies, such as some forms of frontotemporal dementia and Pick’s disease, among others [22].
Tau function is regulated by a broad range of posttranslational modifications, including phosphorylation, acetylation, glycation, isomerization, nitration, SUMOylation, and ubiquitination, and changes in these modifications can affect tau functions and lead to pathological events [20].
Tau phosphorylation is highly increased in AD and has gain a particular interest for disease pathology. The effect of increased phosphorylation varies depending on the site of phosphorylation, but overall, it reduces tau’s affinity for microtubules leading to cytoskeleton destabilization [20]. Moreover, the tau-tau interaction site is coincident with the tau-microtubule interaction site, so when tau is detached from microtubules it tends to aggregate [18]. Therefore, tau is shielded from aggregation when bound to microtubules, but loses this protection when it is detached from them due to phosphorylation [18]. Self-aggregation of tau and further recruitment of more tau monomers and dimers creates a nucleation center that harbors the process of oligomerization [23]. Tau oligomers then acquire a β-sheet structure, with subsequent aggregation into fibrils, culminating in the formation of NFTs [19, 20]. As observed for Aβ, intermediate species generated during the formation of tangles, are the most toxic ones affecting neuronal and synaptic function [20]. Hence, aberrant hyperphosphorylation of tau leads to the loss of tau’s biological activity, its dissociation from microtubules, and promotion of tau aggregation [20].
Tau is normally phosphorylated by regulation of both kinases and phosphatases [20]. Important tau kinases comprise the glycogen-synthase kinase-3β, cyclin-dependent protein kinase 5, cAMP-dependent protein kinase and stress-activated protein kinases, while protein phosphatase 2 has been highlighted as the most important tau phosphatase [24]. Tau kinase 5 was found to be upregulated in AD brain, whereas protein phosphatase 2 was downregulated. Moreover, decreased addition of a single N-acetyl-glucosamine residue to specific serine/threonine residues, a protein posttranslational modification known as O-GlcNAcylation, is observed in AD brains, and is correlated with tau hyperphosphorylation. The decreased O-GlcNAcylation may be caused by the compromised glucose metabolism observed in the AD brain [24]. Tau can also undergo proteolytic cleavage by caspases, generating products that not only are toxic for the cell, but also disturb the paperclip-like conformation of tau, triggering its aggregation [18].
In light of the above, it is crucial to understand how toxic tau species mediate dysfunction and degeneration in AD and other tauopathies [22].
NEUROINFLAMMATION
In many acute and chronic diseases of the CNS, the neural-centric vision fails to elucidate the mechanisms underlying disease onset and progression. Non-neuronal cells, such as astrocytes and microglia, which monitor brain parenchyma and are in constant communication with neurons, actively contribute to maintain brain homeostasis.
Definition
Neuroinflammation is defined as the reactive response of the CNS against elements that interfere with its homeostasis and this response is involved in all neurological diseases, including developmental, traumatic, ischemic, metabolic, infectious, toxic, neoplastic, and neurodegenerative diseases. Emerging evidence suggests that neuroinflammation is not only a consequence of neurodegenerative diseases, but also a risk factor for many of them [25, 26]. Thus, understanding and controlling neuroinflammation might be key for the prevention or delay of late onset CNS diseases.
Neuroinflammation is mediated by the production of cytokines, chemokines, reactive oxygen species (ROS), and secondary messengers. These mediators are mostly produced by activated microglia and astrocytes, the resident CNS immune cells, and by peripherally derived immune cells, in the case of increased blood-brain barrier (BBB) permeability. However, although microglia and astrocytes have a crucial role as effectors of neuroinflammation, other cells, including neurons, express receptors for cytokines and other inflammatory mediators and can therefore participate in the coordinated inflammatory response within the CNS [25, 26].
Neuroinflammation is often a term with negative connotation. However, the degree of neuroinflammation depends on the context, duration, and course of the primary stimulus or insult [27]. There is evidence of active microglia and production of cytokines in early brain development, providing support, and ensuring synaptic pruning within the CNS. Furthermore, neuroinflammation is a protective response essential for neural tissue repair after injury, infection or trauma [28]. It helps to eliminate the cause of injury, and can fix and prevent further tissue damage [29, 30]. Such degree of neuroinflammation influences cellular biochemistry, physiology, and development, and is beneficial [27]. On the other hand, chronic neuroinflammation, characterized by excessive and sustained neuroinflammatory events, is detrimental, perpetuates tissue injury and neural dysfunction and, ultimately, impairs neuronal regeneration [31–34].
Glial cells: key players in neuroinflammation
Although there is irrefutable evidence for a neurotoxic role of Aβ in AD, neuroinflammatory cascades and long-lasting activation of microglia and astrocytes promote alterations in the crosstalk between glial cells and neurons which, together with the sustained exposure of neurons to pro-inflammatory mediators, can cause neuronal dysfunction and contribute to cell death. Indeed, glia-based neuroinflammatory events driven by Aβ are nowadays considered a major contributing factor for the pathogenesis of AD [35–37]. Furthermore, activation of the NLRP3 inflammasome (NLRP3), a cytosolic multiprotein complex mediator of inflammation, is believed to have a leading role in the process [38–42].
In physiological conditions, the Aβ peptide is produced continuously, undergoes degradation and is transferred to the CSF and local blood vessels [13]. However, in pathological conditions there is an abnormal accumulation and consequent aggregation of Aβ in the brain, which leads to the formation of aggregates capable of triggering a chronic activation of the CNS immune system [13, 43].
Microglia
Microglia are the first line of defense of the brain’s immune system [44], and due to this immunological function, they are considered the macrophage-like cells of the brain [45]. These cells originate from primitive macrophages that exist in the yolk sac and colonize the CNS very early in development [46]. In the normal physiological state, microglia surveil the diverse brain regions being involved in multiple developmental events, such as the establishment of neuronal circuits, synaptic pruning and remodeling, keeping the homeostasis and assuring the proper development and neuroprotection of the CNS [47]. As an immune cell, microglia act as sentinels, detecting the first signs of invasion of pathogens or tissue damage.
When microglia survey the microenvironment—resting state—they have a ramified appearance with multiple branches and rapidly extend and retract their processes [48]. In the case of injury or infection, microglia become activated, adopt different morphologies and produce inflammatory mediators [49]. Based on gene expression profiles, activated microglia can be divided into several different populations, with the M1 and M2 subtypes representing the extremes of the spectrum. Activated microglia is essential for the resolution of CNS damage, but under a long-lasting over-activation microglia excessively produces cytotoxic factors that perpetuate neuroinflammation [50]. Besides that, reactive microglia stop the surveillance of synapses leading to a disruption of neuronal circuits [50].
Astrocytes
Astrocytes derive from the neuroectoderm lineage and are the most abundant cells in the CNS parenchyma playing an essential role in the tripartite synapse [51]. In addition to participating in the fine tuning of synaptic transmission, structural support, glutamate uptake, metabolism of neurotransmitters, regulation of extracellular pH and K+ levels, synaptic pruning, and clearance of debris and dead cells [52], astrocytes are also of major importance for the maintenance of the BBB [53–55].
In the 21st century, astrocytes are increasingly viewed as having a pivotal role in neuroinflammation and as critical contributors to CNS pathologies [56]. Indeed, astrocytes also become reactive in response to noxious stimuli and, together with microglia, are important players in neuroinflammatory processes influencing aspects of inflammation and immune reactivity highly relevant to neurological diseases [57].
Reactive astrogliosis is a graded and non-homogeneous response that fluctuates according to the type, severity, time, and duration of the insult [54, 56, 58, 59]. It is defined as the process whereby astrocytes undergo changes in transcriptional regulation, as well as biochemical, morphological, metabolic, and physiological remodeling, leading to gain of new function(s) or loss/upregulation of homeostatic ones [56].
Reactive astrocytes undergo this remodeling in response to diverse pathologies and have the capacity to adopt distinct state(s), characterized by a specific molecular profile, specific functions, and distinct impact on diseases [56]. The spectrum of reactive astrocytes was recently considered to be between two extreme states: the neurotoxic A1, induced by neuroinflammatory insults, and the neuroprotective A2, associated with ischemia. However, these two distinct reactive states most likely co-exist, constituting a part of a heterogeneous population that contain both [56, 60, 61]. Thus, reactive astrogliosis can have a pro-inflammatory or an anti-inflammatory outcome, depending on the signaling pathways that prevail [40, 56]. Reactive astrogliosis and scar formation are essential for confining CNS inflammation to the lesion site [58]. However, under pathological conditions, reactive astrocytes are known to aggravate and perpetuate inflammatory responses, producing an exacerbated amount of pro-inflammatory mediators [40]. Furthermore, it was shown that microglia can induce the neurotoxic phenotype in astrocytes through the secretion of three cytokines (Interleukin (IL)-1α, Tumor Necrosis Factor (TNF)-α, and Complement component 1q) [61], which prompts the hypothesis that astrocytes are indeed crucial in neuroinflammatory processes. However, whether astrocytes can directly respond to an insult, or if they always require a priming by microglia is still debated.
Inflammasomes
The innate immune system comprises pattern recognition receptors (PRRs) that trigger signaling cascades and promote gene transcription through: 1) Nuclear factor kappa B (NF-κB) and activator protein 1, driving proinflammatory cytokine/chemokine production, and 2) members of the interferon regulatory factor, which mediate type I interferon-dependent antiviral responses [62, 63].
PRRs can be divided into two major classes based on their subcellular localization. Toll-like receptors (TLRs) and C-type lectin receptors are transmembrane proteins found in the plasma membrane and endosomes, where they can survey pathogen-associated molecular patterns (PAMPs) and damaged-associated molecular patterns (DAMPs) in the extracellular milieu [62]. A second class of PRRs resides in the intracellular compartment and includes the nucleic-acid sensing PRRs, such as the RNA-sensing Retinoic acid-inducible gene I-like receptors, and the DNA-sensing DNA receptor Absent in Melanoma 2-like receptors [62]. Another set of intracellular PRRs, distinct from those mentioned above, are the nucleotide-binding domain and leucine-rich repeat-containing (NLR) receptors, that recognize a wide range of PAMPs and DAMPs [62].
A subset of PRRs assemble into caspase-1-activating platforms called “inflammasomes”. These cytosolic multiprotein complexes were first described by Martinon and co-workers [64] and generally have three main components: the cytosolic PRR (the sensor domain), the adaptor domain and the cysteine protease pro-caspase-1 as the effector domain. Through canonical activation inflammasomes convert pro-caspase-1 into the catalytically active enzyme, and mediate immune responses through caspase-1, upstream the production of the pro-inflammatory cytokines IL-1β and IL-18 [62, 65, 66]. They can be activated by a wide range of exogenous stimuli, such as lipopolysaccharides (LPS) and viral RNA [67], and also by endogenous signals, such as ROS [65], urate crystals [68], cholesterol crystals [69], and aggregated forms of Aβ peptide [70, 71] and α-synuclein [72].
The NLR family of inflammasomes has been highly explored. It is characterized by the presence of a central nucleotide-binding and oligomerization domain, which is commonly flanked by a C-terminal leucine-rich repeats and a N-terminal caspase recruitment (CARD) or pyrin (PYD) domains [62].
NLRP3 inflammasome
The NLRP3 (nucleotide-binding oligomerization domain-, leucine-rich repeat-, and pyrin domain-containing 3) inflammasome is the most studied inflammasome in the CNS. It belongs to the NLR family, the sensor protein is the NLRP3 domain, and the adaptor protein is the apoptosis-associated speck-like protein (ASC) containing a CARD [40]. The NLRP3 domain itself is composed of three distinct subdomains: 1) a C-terminal leucine-rich repeat domain that mainly recognizes the activating signal and mediates the resulting interactions [73, 74]; 2) a central nucleotide-binding and oligomerization domain that has ATPase activity being necessary for inflammasome assembly [75]; and 3) a N-terminal PYD that enables homotypic interactions between NLRP3 and the adaptor ASC [76]. In turn, ASC is composed of two domains: a N-terminal PYD that binds to the N-terminal PYD of NLRP3 through a homotypic interaction resulting in ASC dimer assembly [77], and a C-terminal CARD that interacts with the CARD domain of the pro-caspase-1 [78].
Activation and consequent assembly of NLRP3 leads to proximity-induced autocatalysis of pro-caspase-1 producing mature caspase-1 which, in turn, cleaves the inactive pro-inflammatory cytokines—pro-IL-1β and pro-IL-18—in their secreted forms, IL-1β and IL-18, respectively [62, 65]. Caspase-1 also cleaves another substrate, Gasdermin D (GSDMD), generating a pore-forming fragment that translocates to the plasma membrane leading to a lytic form of cell death called pyroptosis (further detailed in a forthcoming section) [79].
The basal levels of NLRP3 domain in resting cells are considered not sufficient to activate the inflammasome, and it is nowadays accepted that the canonical NLRP3 activation requires two-signals: 1) a priming signal, which is provided by a NF-κB-activating stimuli to transcriptionally enhance the expression of NLRP3 domain and pro-IL-1β; and 2) an activating signal that promotes the assembly of the complex [62, 65, 67, 80].
The priming step enhances the levels of NLRP3 and pro-IL-1β, but not of ASC, pro-caspase-1 and pro-IL-18 [81], and besides promoting this de novo protein synthesis, controls NLRP3 domain post-translational modifications [82]. The NLRP3 domain is, normally, in an ubiquitinated state that prevents its oligomerization, and the priming event stimulates its deubiquitination allowing for NLRP3 to oligomerize [83]. Thus, there are two types of priming: the transcriptional priming and the post-transcriptional priming, also denominated the non-transcriptional priming [83]. Priming signals can be provided by TLRs’ ligands, as LPS, or endogenous molecules as TNF-α [84].
The second step is characterized by the assembly (oligomerization) and activation of the inflammasome, resulting in a ring-like multiprotein complex. A hallmark of inflammasome activation is the ASC speck, a micrometer-sized structure formed by filamentous clusters of the adaptor protein ASC [85]. ASC specks can be released and taken up by neighboring cells promoting ASC assembly in the recipient cells, and consequently providing another form of inflammasome amplification [86]. The numerous NLRP3-activating stimuli are highly different at the chemical and structural level, making the possibility of a direct interaction with each activator unlikely [67]. It was therefore assumed that NLRP3 either senses a common secondary signal downstream of these stimuli or responds to cellular stress associated with physiological damage [86]. ROS production, potassium (K+) efflux, calcium (Ca2 +) signaling disturbance, and lysosomal rupture were proposed as upstream signals required for NLRP3 activation [65, 67, 86]. Recently, ceramide was also found to trigger NLRP3 activation in microglia [87].
Non-canonical NLRP3 activation involves the engagement of caspases-4/5 (in human) and caspase-11 (in murine) either directly by LPS or downstream of TLR4. In relation to caspase-1, these caspases display a lower affinity for pro-IL-1β and pro-IL-18, but a comparable affinity for GSDMD [63]. Initially caspases-4/5/11 activity promotes membrane pores without cytokine release. However, membrane pores induce ionic flux and subsequent NLRP3 assembly with caspase-1 activation. Thus, signaling by the non-canonical pathway indirectly leads to IL-1β and IL-18 secretion via caspase-1 [63, 88].
NLRP3 inflammasome regulated cytokines: IL-1β and IL-18
Cytokines are low molecular weight proteins or glycoproteins secreted by several cell types. As signaling molecules, cytokines provide communication between cells and play a crucial role in the modulation of the innate and adaptive immune response. Cytokines are classified into lymphokines (regulators of the immune response secreted by T cells), pro-inflammatory cytokines (which amplify and perpetuate the inflammatory process), anti-inflammatory cytokines (that negatively modulate the inflammatory process), growth factors (which promote cell survival) and chemokines (that coordinate cell migration) [89].
Of these, this review will focus on NLRP3 regulated cytokines IL-1β and IL-18, which have emerged as critical signaling pathways in the initiation and perpetuation of the inflammatory reactions in the CNS.
IL-1β and IL-18 bind to their respective receptors—IL-1R1 and IL-18R—on microglial cells, astrocytes, neurons, and endothelial cells, triggering NF-κB-dependent transcriptional events, which result in the generation of multiple inflammation-associated genes [90]. IL-1β is also associated with the integrity of the blood barriers modulating the infiltration of peripheral immune cells into the CNS and IL-18 regulates the function of interferon-γ in T cells and natural killer cells [90].
There is also evidence that increased IL-1β levels are implicated in the response to Aβ deposition [91] and high levels of IL-1β and IL-18 were found in the CSF, brain tissue and plasma of patients with CNS infections, brain injury and neurodegenerative diseases, such as AD [92]. These pro-inflammatory cytokines are also powerful regulators of synaptic function and have been shown to inhibit long-term potentiation, a form of neuronal plasticity believed to reproduce learning and memory [93].
It is worthwhile to emphasize that IL-1β and IL-18 maturation by caspase-1 is sufficient for slow, caspase-1/GSDMD-independent secretion of the active cytokines [94]. However, one cannot forget that caspase-1, besides processing the pro-IL-1β and pro-IL-18, also cleaves GSDMD generating membrane pores which boosts cytokine release, even in the absence of pyroptosis. Indeed, data suggest that cytokine secretion via GSDMD pores starts before cell rupture during pyroptosis [88]. When the membrane-repair mechanisms do not annihilate the GSDMD pore burden and the cell undergoes pyroptosis, the whole intracellular content is released to the surroundings, including IL-1β and IL-18.
In the non-canonical inflammasome activation, caspase-4/5/11 cleaves GSDMD and the occurrence of membrane pores causes ionic flux that triggers NLRP3 assembly and caspase-1 activation, leading to caspase-1–dependent IL-1β/IL-18 processing with enhanced secretion through GSDMD pores [63, 88].
The role of the blood-brain barrier
The brain is an immunoprivileged site, due to the existence of the BBB, a structure localized at the interface between the blood and the cerebral tissue, that regulates the exchange of substances and blood cells in and out of the brain [55, 95]. The BBB is mainly composed of endothelial cells, pericytes and astrocytes, that create the neurovascular unit with the adjacent neurons, protecting the brain and maintaining its homeostasis [96]. BBB disruption involves phenotypical changes in endothelial cells and astrocytes and allow for blood cells to enter the brain parenchyma and engage in a crosstalk with brain cells, contributing to the modulation of the immune responses in the nervous tissue [91]. IL-1β signaling plays a major role in the initiation and continuation of the inflammatory reactions in the CNS in response to various adverse stimuli. High levels of IL-1β, and other inflammatory mediators, are deleterious to BBB integrity, which prompts the infiltration of peripheral immune cells into the brain upon a chronic neuroinflammatory context. These cells can enter the brain through the defective BBB, proliferate at the site of inflammation and further enhance neuroinflammation, either directly or through glial and neuronal cells [34].
Moreover, the inflammasomes, and more specifically the NLRP3 inflammasome, is a crucial component of the peripheral immune response. It was already reported that Aβ oligomers can directly interact with the NLRP3 domain and ASC, present in the infiltrated macrophages and cause inflammasome activation, further contributing to the neuroinflammatory milieu [97].
NLRP3 INFLAMMASOME IN AD
The association between pro-inflammatory cytokines, NLRP3 and AD was mentioned in several reports. In 1995, high concentrations of IL-1β and IL-6 were observed in the CSF of patients with AD [98]. Years later, a study pointed to the induction of tau-hyperphosphorylation by high levels of IL-1β, aggravating AD pathogenesis [99]. In microglia cell cultures, NLRP3 was identified as a sensor for fibrillar Aβ in a process involving Aβ phagocytosis, lysosomal damage, and Cathepsin B release [70]. More recently, the in vivo relevance of the NLRP3 in AD was confirmed by Heneka and colleagues [38].
NLRP3 priming and activation by Aβ species
Several hypotheses on how Aβ species guides NLRP3 priming and activation in microglia are represented in Fig. 1.
Fig. 1
Schematic illustration for Aβ-mediated NLRP3 inflammasome priming and activation mechanisms described in microglia. Aβ species can work either as a priming stimulus (middle panel) or as an activating stimulus (left and right panels). As a priming signal, Aβ oligomers bind to the CD36 surface receptor, triggering the formation of a TLR4-TLR6 heterodimer. This heterodimer activates a cascade of signaling molecules resulting in the activation and nucleus translocation of the transcription factor NF-kB, that promotes the transcription of NLRP3 domain and of pro-IL-1β. Aβ plaques can act as an activating signal by two main mechanisms (right panel). Aβ is known to cause synaptic dysfunction and neuronal damage. Considering this, P2X7R is activated by ATP released from dying neurons and recruits the Pannexin-1 channel that allows the entrance of NLRP3 agonists to the cell. Also, ATP binding to the purinergic receptor induces K+ efflux and Ca2 + influx, known to promote NLRP3 activation. On the other hand, Aβ plaques can also be phagocytized and incorporated into lysosomes, boosting lysosomal destabilization and consequent content release. Cathepsin B, a lysosomal proteolytic enzyme, promotes the assembly of the inflammasome by a still unknown mechanism. Aβ oligomers are also able to activate NLRP3 through a mechanism that does not involve phagocytosis (left panel). Soluble oligomeric Aβ species can induce pore formation in the cell membrane and ROS production, which then promotes oxidation of K+ channels. These events might culminate in K+ efflux promoting the activation of the inflammasome. The assembly and activation of NLRP3, independently of the mechanism involved, ultimately results in the production and release of the inflammatory cytokines IL-1β and IL-18. Created with BioRender.com.
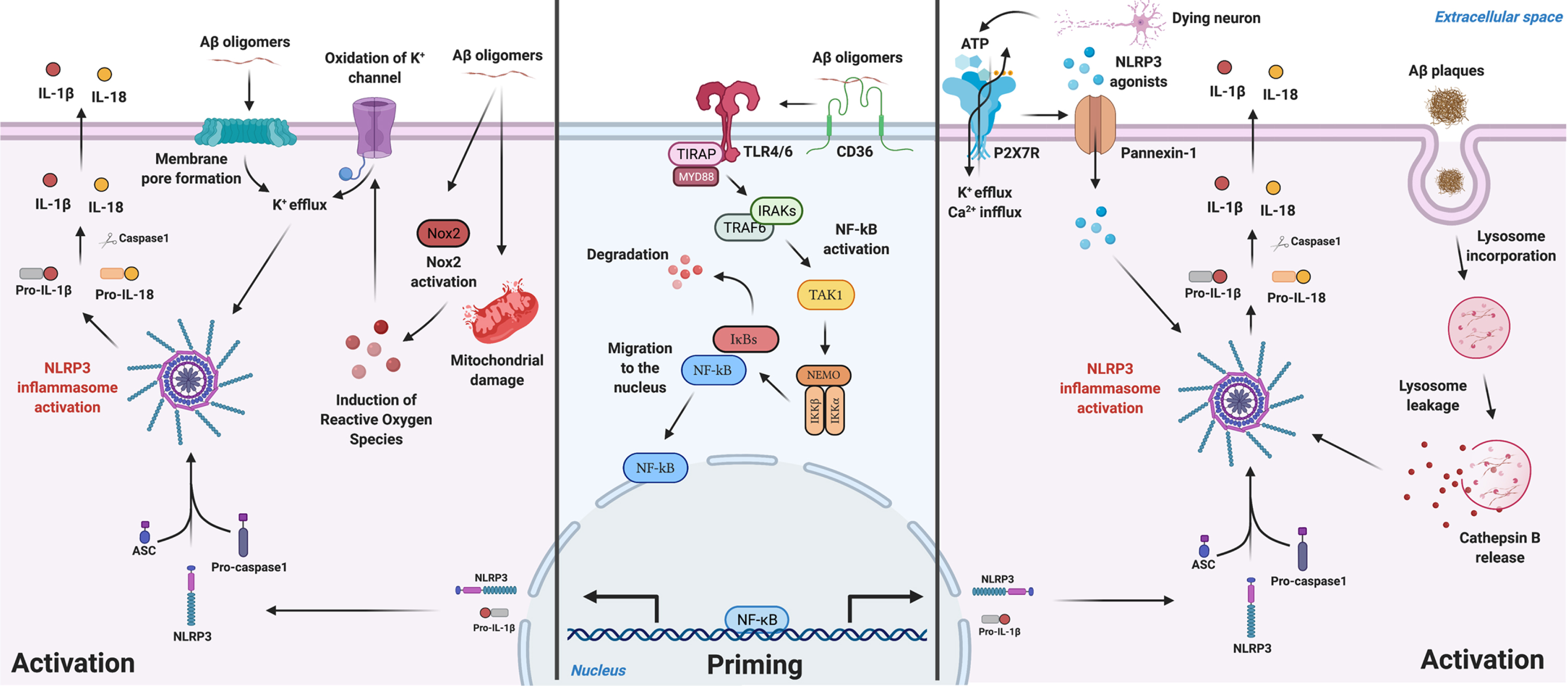
One of the hypotheses relates to its role as a priming stimulus to NLRP3 (middle panel in Fig. 1). This happens by the postulated connection Aβ-CD36 [100], promoting the formation of a TLR4-TLR6 heterodimer, forming then a receptor complex composed by the previous three: CD36/TLR4/TLR6. This leads to the translocation of NF-κB to the nucleus, promoting the transcription of the NLRP3 domain of the inflammasome and pro-IL-1β [101]. Then, a second signal is needed to promote the oligomerization of the NLRP3 domain and assembly with ASC which recruits caspase-1 through its CARD, leading to the assembly of the inflammasome [78].
Regarding the Aβ-mediated NLRP3 activation, one mechanism that has gained increasing interest, is the phagocytosis of the peptide (right panel in Fig. 1). The peptide is incorporated in lysosomes promoting its destabilization, activation and content release, namely Cathepsin B, a lysosomal proteolytic enzyme [70]. The latter promotes the assembly of NLRP3, by a still unknown mechanism, and consequently, maturation and release of IL-1β and IL-18 [70]. Heneka’s work [26] mentioned that NLRP3-ASC oligomerization pathway together with CD36/TLR4/TLR6 complex formation and lysosomal damage were necessary for aberrant host proteins to promote the assembly of NLRP3. Another possibility of Aβ-mediated activation of the NLRP3 complex involves the release of ATP by dying neurons which, in turn, activates the purinergic receptor P2X7 [102, 103], predominantly expressed by microglia and in lesser amounts by astrocytes [104], causing K+ efflux and inducing the recruitment of the membrane pore pannexin-1 [105]. This mechanism postulates that extracellular molecules/components can enter the cell through the hemichannel pannexin-1 causing homeostasis alterations in the cytosol and activating NLRP3 [105]. To note that the expression of P2X7 receptor is increased in human brains with AD [106].
It was also shown [107] that soluble oligomeric Aβ species were able to activate NLRP3 through a mechanism that does not involve phagocytosis of the peptide (left panel in Fig. 1). Soluble oligomeric Aβ species were described to induce pore formation in the cell membrane [108], and were shown to induce ROS production in microglia [109], which then promotes oxidation of K+ channels [110]. These events might culminate in K+ efflux promoting the activation of the inflammasome [107].
The sequence of events that culminate in the activation of the inflammasome are tightly regulated so that the inflammatory response proceeds its correct path. Some known regulators of the inflammasome are Double-stranded RNA-dependent protein kinase, Guanylate-Binding Protein 5 [111, 112], NLRP10 [113], and Nek7 [114]. The last one is the best characterized member of the Never in Mitosis Gene A (NIMA)-related kinases (NEK proteins) and the way Nek7 regulates NLRP3 has been well studied and characterized, being recognized that K+ efflux is involved by promoting conformational changes in the inflammasome allowing the connection of Nek7 to the catalytic domain of the leucine rich repeat domain, thus regulating the activation and oligomerization of NLRP3 [114].
NLRP10 is a negative regulator of NLRP3 inflammasome, once it interacts with ASC, decreasing its availability and preventing ASC oligomerization with the NLRP3 domain [113, 115]. The work of Murphy and colleagues [115] showed that in LPS-primed mixed glial cultures treated with aggregated Aβ42 and Aβ40, the NLRP10 inflammasome is degraded allowing ASC to assembly with the NLRP3 domain. This degradation was proposed to be via phagocytosis and consequent release of Cathepsins. In this manner, ASC is free to associate with the NLRP3 domain and form a functional inflammasome.
The role of microglia
Neuroinflammation in AD is primarily driven by the resident microglia population. Microglia express the NLRP3 components, namely NLRP3 domain, ASC and caspase-1 [116] and NLRP3 assembly, with the subsequent caspase-1 activation and IL-1β and IL-18 secretion, has been well explored in microglia. Several works point to a relevant role of these cells for AD progression through neuroinflammatory responses mediated by NLRP3 [38, 42, 70, 116, 117].
Under healthy conditions, microglia activation leads to engulfment and clearance of Aβ. However, prolonged exposure to Aβ species leads to a persistent priming and activation of NLRP3 in microglia [38] causing inflammasome components, such as the adaptor protein ASC, to be released to the extracellular space. There, Aβ clusters around exogenous ASC fibrils and forms ASC-Aβ composites which can boost NLRP3 activation in the surrounding microglia, reducing the capacity of microglia for Aβ clearance and leading to pyroptotic cell death. During pyroptosis, vast quantities of ASC are set free, starting the vicious cycle of ASC-Aβ composites all over [118, 119]. Upon Aβ accumulation, microglial cells become progressively impaired in Aβ clearance [120].
It is reported that microglia are recruited to plaques [121] and can phagocytose Aβ [117], starting an immunological response towards it [122]. When phagocytosed by microglial cells, Aβ induce lysosomal swelling and loss of lysosomal integrity, triggering the release of the lysosomal contents, as Cathepsin B, into the cytosol, that activates NLRP3 and initiates the IL-1β pathway in microglial cells. In fact, an increased expression of this cytokine was detected in microglia surrounding the Aβ plaques in patients with AD [117]. Released IL-1β is then capable of stimulating the production of other pro-inflammatory molecules [70], and the excessive production of these molecules may lead to neuronal damage, by activating harmful signaling cascades [123]. It was also reported that microglial cells are incapable of efficiently degrading fibrillar Aβ for a period of weeks after its internalization [124], which can be explained by the structural damage of Aβ-containing lysosomes mentioned above.
Heneka and co-workers [38] deeply assessed the involvement of NLRP3 in the Aβ-mediated inflammatory responses in vivo. These authors showed a significant increase in cleaved caspase-1 in the brain lysates from hippocampus and frontal cortex derived from AD patients. This increase, as well as an increase in IL-1β in the tissue, was replicated in the APP/PS1 mice model of AD, that carry mutations in AβPP and presenilin-1, together with the chronic deposition of Aβ, neuroinflammation, and cognitive impairment. Furthermore, NLRP3 or caspase-1 deficiency protected APP/PS1 mice from memory deficits, long-term potentiation suppression, and reduction in spine density in the pyramidal neurons, supporting a fundamental role of the NLRP3/caspase-1 axis in behavioral and cognitive dysfunction in AD. Additionally, APP/PS1 mice deficient for NLRP3 or caspase-1 revealed a considerable reduction in brain concentrations of aggregated forms of Aβ, further suggesting that NLRP3 acts through caspase-1 to exert the observed effects. It was also reported that NLRP3 activation negatively impacts the microglial Aβ clearance function in AD. Prolonged exposure to Aβ leads to persistent activation of microglial cells and it was observed that the M1 phenotype was reduced in NLRP3- or caspase-1- deficient APP/PS1 mice, while markers of M2-like microglia were increased. This led to the hypothesis that impairments in NLRP3/caspase-1 cascade promotes a switch of activated microglial cells toward an M2-like state, characterized by increased Aβ clearance and enhanced tissue remodeling.
The contribution of astrocytes
NF-kB pathway is associated with neuroinflammation, controlling cytokine production, and reactivity in glial cells, and it is strongly activated during neurodegeneration [125–127]. In animal models of AD, studies have demonstrated activation of this pathway in astrocytes, proving that these cells play a role in neuroinflammation in this context [122]. Furthermore, astrocytes express several proteases and receptors involved in the biogenesis of Aβ, which includes neprilysin, insulin-degrading enzyme and endothelin-converting enzyme 1/2, being involved in the degradation and clearance of the peptide [128, 129]. These cells are also the major producers, in the brain, of Apolipoprotein E, one of the major risk factors of late-onset AD [130]. Moreover, the neurotoxic phenotype of astrocytes is characterized by the upregulation of NF-kB and, consequently, of the complement component 3 (C3) [60, 131], that was shown to be significantly increased in the brain synapses and in the CSF of AD patients [132]. These cells secreted C3 in response to Aβ, which resulted in the shrinkage of some synapses and increased activity of others, process that was associated with NF-κB signaling [133]. Besides, C3 was shown to bind to C3a receptor in microglia, inhibiting microglia’s ability to phagocytose Aβ [134].
Recently, several astrocytic receptors and transporters were proposed to be involved in the pathogenesis of AD and its targeting was considered a viable approach for treating this pathology [135].
It is not well established the neuroinflammatory response of astrocytes towards Aβ [130], but evidence suggests an Aβ-induced pro-inflammatory profile in astrocytes [136]. It was reported that, in the presence of Aβ, astrocytes adopt a phagocytic phenotype mediated by CD36, CD47 and RAGE [137]. In fact, the RAGE/NF-kB pathway seems to be an important contributor to the inflammation observed in AD [130]. NF-kB, as mentioned earlier, is involved in NLRP3 priming and, in rats treated with Aβ1–42 oligomers, IL-1β was shown to be expressed in reactive astrocytes that surrounded the Aβ injection site [138]. Given the relevance of NLRP3 signaling in IL-1β maturation and release, it can be hypothesized that astrocytes’ contribution to the inflammatory milieu observed in AD is NLRP3-dependent. However, this is not entirely clear, with studies stating opposite results regarding the expression of NLRP3 components and its function in these cells.
Primary cultures of astrocytes primed with LPS and other classical NLRP3 stimuli did not result in significant expression of NLRP3 domain, ASC and IL-1β, pointing to a lack of a functional inflammasome in astrocytes [116]. The authors postulated that this could be due to a weak expression of NLRP3 domain and to the lack of ASC in astrocytes. This observation was also corroborated in neurosphere-derived astrocytes [139]. This study tested the potential of Aβ25–35 of inducing the expression of NLRP3 in astrocytes, obtaining similar results to the previous ones, and suggesting that astrocytes do not react to Aβ25–35, at least not in in vitro. Also, IL-1β mRNA was not translated into protein by human astrocytes [139]. These authors have excluded the possibility of a rapid protein degradation rate and postulated the occurrence of a highly unusual repression of the translational mechanism.
However, in the transgenic Tg2576 mice that develops Aβ plaques in the aged brain, a strong expression of IL-1β in the reactive astrocytes that surrounded the Aβ deposits was shown [140]. Corroborating this study, others [141] demonstrated that astrocytes express all NLRP3 components (NLRP3, ASC, and caspase-1) and can produce IL-1β in several CNS lesions, like active demyelinating lesions of multiple sclerosis, active necrotic lesions of neuromyelitis optica, and acute necrotic lesions of cerebral infarction. The expression of all NLRP3 components was also confirmed in cortical astrocytes in a mouse model of traumatic brain injury [142]. Recently, Johann and colleagues [143] examined human samples of sporadic amyotrophic lateral sclerosis and found that human astrocytes were the major cell type expressing NLRP3 and ASC domains, inferring that astrocytes were involved in motoneuron degeneration via NLRP3 activation.
Altogether, the studies suggest that, as happen in other CNS pathologies (multiple sclerosis, traumatic brain injury, amyotrophic lateral sclerosis), the expression and activation of NLRP3 in astrocytes and the subsequent release of IL-1β and IL-18, might play a role in the neuroinflammatory milieu observed in AD, aggravating, and perpetuating the damage made by the primary line of defense in the CNS: the microglia. This controversial NLRP3 activation in astrocytes certainly deserves to be elucidated in the context of AD, both in animal models and human samples of the disease.
CELL DEATH BY NLRP3 INFLAMMASOME-MEDIATED PYROPTOSIS
Many physiological processes require cell death, as happen during embryonic development, and this mechanism occurs efficiently under normal conditions [144]. However, cell death is also the final solution for a cell when numerous stresses are accumulated up to a level beyond its recovery capacity, a situation depicted in neurodegenerative diseases [145].
For a long time, cell death was discussed dichotomously as either necrosis or apoptosis, and these were believed to be the two major death pathways for neurons in brain pathologies [145]. However, the knowledge of cell death has changed a lot throughout decades, and several other types of cell death were described, and associated to neurodegenerative diseases [145, 146]. Nowadays, the best-studied forms of programmed cell death are apoptosis, necroptosis, and pyroptosis [144].
Necrosis is defined as a passive, accidental cell death arising from environmental perturbations with uncontrolled release of cellular contents and thus highly inflammatory, while apoptosis is an active, programmed process of autonomous cellular disassembling that does not elicit inflammation, thus immunologically silent [144, 147].
Necroptosis is an alternative mode of regulated cell death that mimics features of apoptosis and necrosis [148]. Although TNF-α has long been considered an inducer of apoptosis, necroptosis also occurs following the activation of the TNF receptor-1 by TNF-α and requires protein receptor-interacting protein kinase 3 (RIPK3) [144, 148]. Multiple studies have focused on TNF-α, RIPK3, and caspase-8 to understand the molecular mechanism of necroptosis and it was shown that inhibition of caspase-8 shifts apoptosis to necroptosis due to activation of RIPK3 and its ligand [148]. Once activated, necroptosis triggers a process of cellular auto destruction, resulting in passive release of cytokines and other DAMPs to the extracellular space leading to robust inflammation [144, 148].
Pyroptosis is the newest form of programmed cell death inherently associated with inflammation. It was long regarded as caspase-1-mediated cell death but was redefined as a GSDMD-dependent and programmed necrosis [149]. Recent observations confirmed that the pyroptosis executioner, GSDMD, is also a substrate of inflammatory caspases-4/5/11 [150]. Gasdermins, a protein family bearing membrane pore-forming activity, were associated with various genetic diseases, but their cellular function and mechanism of activation, except for GSDMD, are still under study [150].
GSDMD is constitutionally auto-inhibited by the binding of the C-terminal repressor domain (GSDMD-CT) to the N-terminal pore-forming domain (GSDMD-NT). Full-length GSDMD is inactive, but inflammatory caspases relieve this inhibition by catalyzing the proteolytic cleavage of the interdomain loop and promoting the release of the pyroptosis inducer GSDMD-NT [149, 150]. GSDMD-NT can translocate to the inner leaflet of the plasma membrane (or the bacterial plasma membrane), where it binds with high specificity to selected phosphoinositides (or cardiolipin) and oligomerizes, generating membrane pores. The membrane repair mechanisms, through the endosomal sorting complexes required for transport machinery, first attempts to contain the damage inflicted by GSDMD pores. However, if the membrane repair mechanisms are not able to overcome the GSDMD pore burden in the plasma membrane, the cell will die by pyroptosis [88, 144]. The outer membrane of endosomes and phagosomes contain the same phospholipids, suggesting that GSDMD-NT may also bind to these organelles. To note, that because of its lipid-binding preferences, GSDMD-NT kills from within the cell, and does not harm neighboring cells when it is released during pyroptosis [79, 150].
Summarizing, pyroptosis is a novel form of regulated cell death recently implicated in the pathogenesis of multiple neurological diseases [151, 152]. It is characterized by the formation of GSDMD-mediated membrane pores, downstream of canonical and non-canonical inflammasome activation [152]. The pore formation in the plasma membrane of pyroptotic cells is dependent on GSDMD cleavage by inflammatory caspases, resulting in the permeability of plasma membrane, water influx, cell swelling and osmotic lysis, prompting the release of pro-inflammatory cytokines and cytosolic content to the extracellular space [153, 154]. As happens in apoptosis, cells undergoing pyroptosis depict chromatin condensation and DNA fragmentation [153]. On the other hand, apoptosis is initiated by perturbations of the microenvironment, does not comprise osmotic lysis, involves packaging of cellular contents in apoptotic bodies, and caspase-3 is the main downstream executioner caspase that cleaves several other pro-caspases (as caspase-2, -6, -8 and -10) into their active forms, creating an apoptosis-amplifying cascade [144, 147, 152]. Furthermore, necrosis, necroptosis and pyroptosis result in the release of alarmins and other proinflammatory signals into the cellular surroundings, while apoptosis is considered “silent” and does not elicit subsequent immune responses [144]. Although cell death pathways have long been considered to act in parallel with little or no overlap, it is now clear that apoptosis, necroptosis and pyroptosis are closely related and can regulate each other [144].
NLRP3-mediated pyroptosis, mainly associated to microglia, was found to be critical in the pathogenesis of AD [42]. However, the expression of the inflammasome domains has also been reported in other cell types [41], including astrocytes, neurons, oligodendrocytes, and endothelial cells, implying that these cells can also undergo a NLRP3-mediated cell death by pyroptosis. Indeed, NLRP3 activation within dopaminergic neurons was recently proposed to contribute to neurodegeneration in a context of PD [155].
The impact of neuronal death by pyroptosis
AD is characterized by a progressive neuronal dysfunction, which, regardless of the contributing mechanisms, leads to neuronal death [145]. Neuronal death contributes to the neuroinflammatory milieu through the release of elements (ATP, pro-inflammatory cytokines, and other cytosolic components) that can recruit a NLRP3-mediated inflammatory response in neighboring glial cells and boost cytokine release. However, the mechanism under which NLRP3 activation may occur within neurons, in a context of AD, and the link between inflammasome activation in neurons and neuronal death by pyroptosis was not explored.
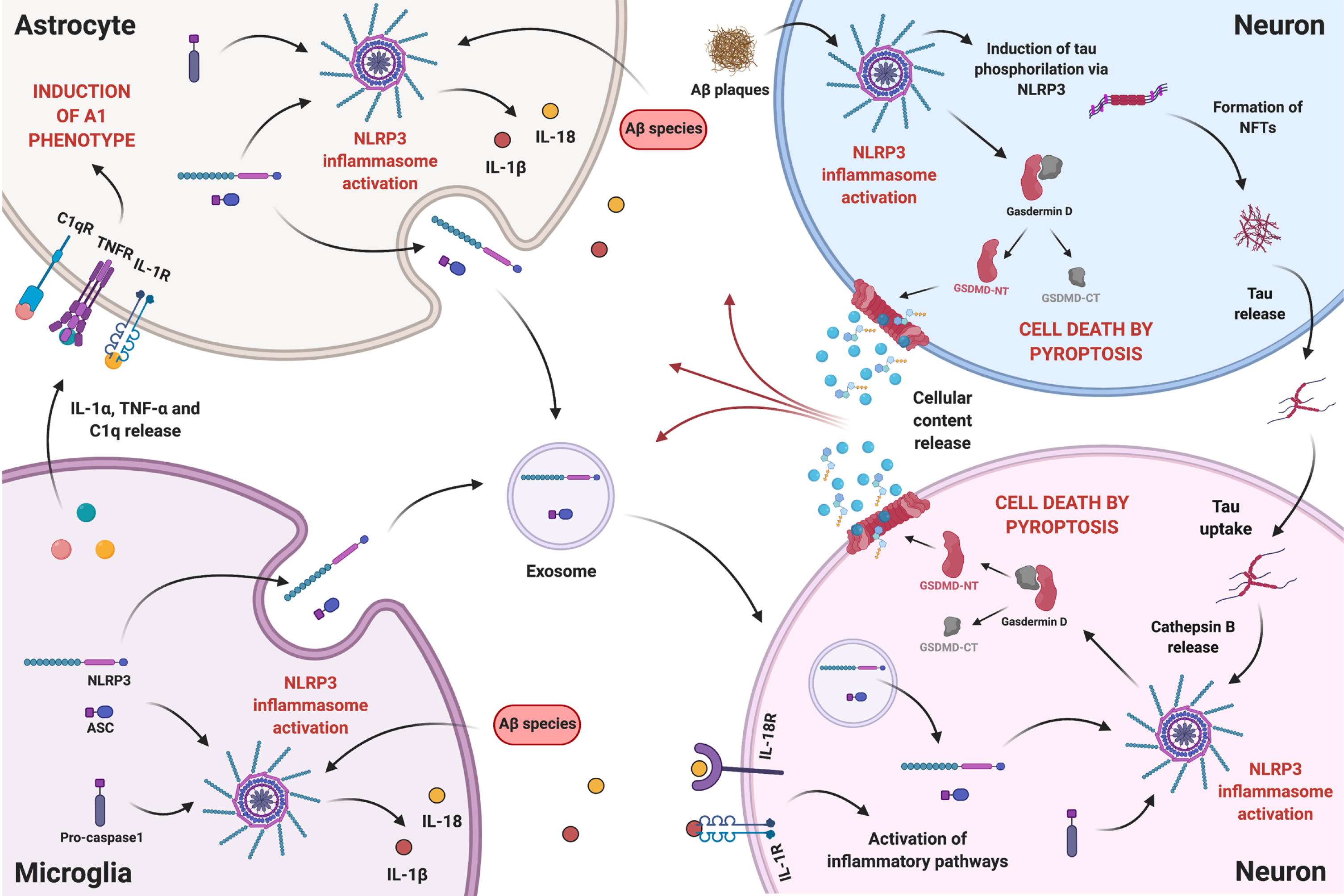
The next paragraphs argue that NLRP3-mediated pyroptosis in neurons may constitute a cell death mechanism that contributes to neurodegeneration in AD. The proposed intercellular communication among microglia, astrocytes, and neurons in the presence of Aβ species is illustrated in Fig. 2.
Fig. 2
The intercellular communication between neurons and glial cells in the presence of Aβ species is a complex and reciprocal process. Aβ-induced reactive microglia release IL-1α, TNF-α, and C1q, which can be received by resting astrocytes promoting the reactive A1 state. NLRP3 activation in reactive glial cells, boosts the release of pro-inflammatory cytokines IL-1β and IL-18, which can bind to their respective receptors in neurons activating inflammatory pathways. Reactive glial cells might also release NLRP3 components, namely NLRP3 domain and ASC, through exosomes that can be internalized by neurons. On the other hand, Aβ plaques can, in neighboring neurons, mediate the occurrence of pyroptosis and promote the development of NFTs, via NLRP3 activation. More importantly, tau can be released and taken up by other neurons, activating NLRP3 in the recipient cell, hypothetically through lysosomal destabilization and Cathepsin B release, and inducing pyroptosis. Therefore, proximity to Aβ plaques will not determine neuronal death by NLRP3-mediated pyroptosis, since, due to tau interneuronal propagation, pyroptosis can occur in neurons far from plaques. Ultimately, release of cellular content caused by pyroptosis will feed a self-propagating loop of neuroinflammatory mediators. Created with BioRender.com.
Tau–mediated neuronal pyroptosis
Extracellular accumulation of Aβ in senile plaques, together with the intraneuronal formation of NFTs, are considered hallmarks of AD, with plaque formation preceding NFTs. Although Aβ still is the major target for AD therapy, there is a growing focus on strategies aiming at other pathogenetic players in AD, as the tau protein [156].
Tau, the major component of NFTs, is a microtubule-associated protein mainly expressed in neurons. Under physiological conditions, tau is involved in polymerizing microtubules and maintain their stability. As aforementioned, tau function is regulated by a broad range of posttranslational modifications and changes in these modifications can affect tau functions and lead to pathological events [157].
A cross-sectional neuropathological study showed that the tau pathology of AD typically spreads from one area to another along a neural network [157]. Masters and co-workers [2] suggested that NFTs extended into the dendrites, where tau could be released and taken up by other neurons. The mechanism underlying the stereotypical progression of NFTs in AD has not been clarified, but recent studies indicate that tau spreads by interneuronal transfer of the pathological form of tau, named “tau propagation”. This process can be divided into three basic steps: 1) release of the intracellular pathological form of tau into the extracellular space by the donor cell, 2) uptake of tau by recipient cells, and 3) formation of new intracellular aggregates of tau in the recipient cells [157]. Passive leakage from degenerated cells and tau dissociation from tangles are likely to contribute to tau release into the extracellular space, although studies have suggested that active tau release could occur without neurodegeneration [157, 158]. Extracellular tau uptake can be mediated by endocytosis occurring on the cell surface, and studies have also suggested the involvement of exosomes in the mechanism of interneuronal tau transfer [157, 159, 160].
The association between Aβ and NLRP3 in microglia has been thoroughly explored over the years (this paper included) and is nowadays being extended to astrocytes. But the relationship between NLRP3 and tau was less examined. It was recently demonstrated that synthetic tau seeds, pre-aggregated tau fragments comprising the microtubule-binding domain of tau, induced IL-1β secretion in primed primary microglia, that was abolished by a specific NLRP3 inhibitor, indicating an inflammasome dependence [161]. These results were confirmed in TauP301S transgenic mice, fully proving the occurrence of tau-mediated NLRP3 activation. These authors further showed that tau seeds were incorporated by microglia and activated NLRP3 through lysosomal destabilization and Cathepsin B release. It was also described [2] that Aβ plaques induce tau aggregation in nearby neurons, mediating the development of NFTs by an unknown mechanism. Only recently, a study by Ising and co-workers [162] showed that NLRP3 acted as a link between Aβ plaques and NFTs formation. These authors demonstrated that the injection of Aβ-containing APP/PS1 brain homogenates induced tau hyperphosphorylation in Tau22 mice, but not in Tau22 mice deficient for NLRP3 or ASC, indicating that NLRP3 was an important mediator of Aβ-induced tau pathology. Altogether, this work placed Aβ upstream NFTs formation.
We herein propose that Aβ plaques mediate, in nearby neurons, the occurrence of pyroptosis and promote the development of NFTs, via NLRP3 activation (Fig. 2, top right blue neuron). On the other hand, tau species can be released and taken up by other neurons, activating NLRP3 in the recipient cell (Fig. 2, bottom right pink neuron), through lysosomal destabilization and Cathepsin B release, as described for tau seeds incorporated by microglia [161], and thus promote pyroptosis. Therefore, it is reasonable to anticipate that, through tau interneuronal propagation, NLRP3-mediated pyroptosis can contribute to neurodegeneration in neurons far from Aβ plaques.
The contribution of extracellular vesicles for neuronal pyroptosis
Extracellular vesicles (EVs) are lipid bound vesicles secreted by most cells into the extracellular space. The three main subtypes of EVs are microvesicles, exosomes, and apoptotic bodies, with a diameter size typically ranging from 0.1–1μm, 30–150 nm, and 50 nm–5μm, respectively [163]. EVs provide a new type of intercellular communication, which transfer a box of information from a cell (either proteins, lipids, or nucleic acids) to another [164]. Their production changes dynamically in number and content in response to specific environmental signals, and their content, or cargo, is released into the cytoplasm of receiving cells, where it mediates several functional effects [163, 164].
In the nervous system, EVs participate in neuron-glial crosstalk, a bidirectional communication important to preserve brain homeostasis and, when dysfunctional, involved in several CNS diseases. The impact of this type of intercellular communication and its relevance at the clinical level is being strongly recognized by the scientific community [165].
Growing evidence correlates inflammasome activity with enhanced secretion of exosomes and modulation of their protein cargo [166]. This inflammasome-mediated secretion of proteins represents a novel paradigm in inflammatory responses [166], and has recently received some attention.
In the CNS, microglia, astrocytes, oligodendrocytes, and neurons have been reported to secrete exosomes into the extracellular space, and it was suggested that exosomes can carry bioactive cytokines, such as IL-1β, as well as inflammasome components [167]. Indeed, it was shown that NLRP1 inflammasome domains were present in exosomes derived from CSF of spinal cord injury and traumatic brain-injured patients following trauma [168]. Also, Sarkar and co-workers [169] have shown that exposure to manganese, a transition metal linked to neurodegenerative disorders, such as AD and PD, activated NLRP3 in primed microglial cells in vitro and in vivo, with formation of ASC specks and enhanced exosomal release of ASC.
Given recent data [170] proving the occurrence of glia-to-neuron transfer of extracellular vesicles’ cargo, it is herein proposed that NLRP3 activation in glial cells by Aβ species can induce an exacerbated release of exosomes, carrying inflammasome domains—NLRP3 and ASC—, which can be transferred to neurons contributing to the levels of NLRP3 components in the receiving cell (Fig. 2). Presuming the tau interneuronal propagation hypothesis, this will strengthen the impact of tau-induced NLRP3 activation in neurons, as explained above.
NLRP3 INHIBITION AS A POTENTIAL THERAPEUTIC TARGET IN AD
NLRP3 is, by far, the most studied and the best characterized inflammasome. Aberrant activation of this complex and IL-1β/IL-18 secretion has been detected in various diseases [40], including inflammatory diseases, diabetes, atherosclerosis, and neurodegenerative diseases, evoking a substantial clinical interest in exploring the effectiveness of its inhibitors.
Studies have uncovered various inhibitors of the NLRP3 cascade, which were validated through in vitro studies and in vivo experiments in animal models of diseases. Pharmacological approaches to inhibit the NLRP3 inflammasome, either directly or indirectly, were exhaustedly reviewed by Zahid and coworkers [171], such as blocking the expression of NLRP3, ASC, caspase-1, and neutralizing the expression of inflammatory cytokines (JC124), inhibiting NLRP3 ATPase activity (CY-09, MNS and OLT1177), abolishing NLRP3-Nek7 interaction (Oridonin), disturbing ASC oligomerization (Glyburide and BHB), and inhibiting caspase-1 activation (VX-740 and VX-765), among others.
Nowadays, NLRP3 is considered a highly promising therapeutic target for many diseases and NLRP3 inhibitors have caught the attention of many pharmaceutical companies [172]. Academic chemists are still struggling with NLRP3 biology, as it is a multiprotein complex that requires priming and assembly before triggering downstream signaling, but even so a portfolio of NLRP3 inhibitors are already in preclinical and clinical trials [172].
The diarylsulfonylurea-containing small molecule, termed as MCC950, was found to block canonical and non-canonical NLRP3 activation with no effect on other inflammasomes [173]. Later, studies [174] indicated that MCC950 binds non-covalently to the Walker B motif within the NLRP3 domain, thereby blocking the ability of NLRP3 to hydrolyze ATP and inhibiting NLRP3 oligomerization and activation. Nowadays, MCC950 is considered a potent and selective NLRP3 inhibitor and chemists from many NLRP3-devoted companies used it as a starting point to design other molecules [172].
However, few studies have explored the effect of MCC950 within the context of AD. Dempsey and coworkers [175] showed that LPS-primed microglia were able to induce an increase in IL-1β release in the presence of ATP or Aβ, and that MCC950 attenuated the effect of both stimuli. Also, these authors demonstrated that inhibition of the inflammasome by MCC950 improved Aβ phagocytosis by cultured microglia. These results were corroborated in the APP/PS1 mouse model of AD, where Aβ-containing plaques were significantly reduced after MCC950 treatment, indicating an increased Aβ phagocytosis as happened in vitro. Overall, it was shown that MCC950 was able to reduce Aβ-guided pathologic events and enhance cognitive function. Also, chronic intracerebral administration of MCC950 in TauP301S transgenic mice was shown to inhibit exogenously seeded tau pathology [161].
Thus, MCC950 and MCC950-based compounds are a promising therapeutic approach in AD and should be thoroughly examined in various in vitro and in vivo models to fully understand their value in this disease.
SUMMARY
The so-called amyloid cascade hypothesis [176], which postulates that the accumulation and aggregation of Aβ, the main component of senile plaques, in the brain parenchyma is the initiator of the subsequent events that lead to AD, has dictated and guided much of the academic and pharmaceutical research over the past two decades. Fundamental knowledge on the role of Aβ in this disease arise from studies that focused on how Aβ forms senile plaques and how these abnormal structures induce neurodegeneration and neuronal loss [177, 178]. However, pharmaceutical drugs against Aβ failed in various phases of clinical trials, and the idea that tau, and not only Aβ, can be a relevant factor underlying the development and progression of AD, has emerged [156, 179].
Furthermore, neuroinflammation and inflammatory mediators are known to be involved in CNS diseases, and research on the interaction between inflammatory pathways, triggered in glial cells via NLRP3 activation, and disease progression, has recently boosted in AD.
Microglia is the “maestro” of the CNS immune response and was always at the core of Aβ-induced NLRP3 activation. Indeed, the mechanisms of NLRP3-mediated immune response triggered by Aβ species were deeply highlighted in microglia and only recently these studies were extended to astrocytes.
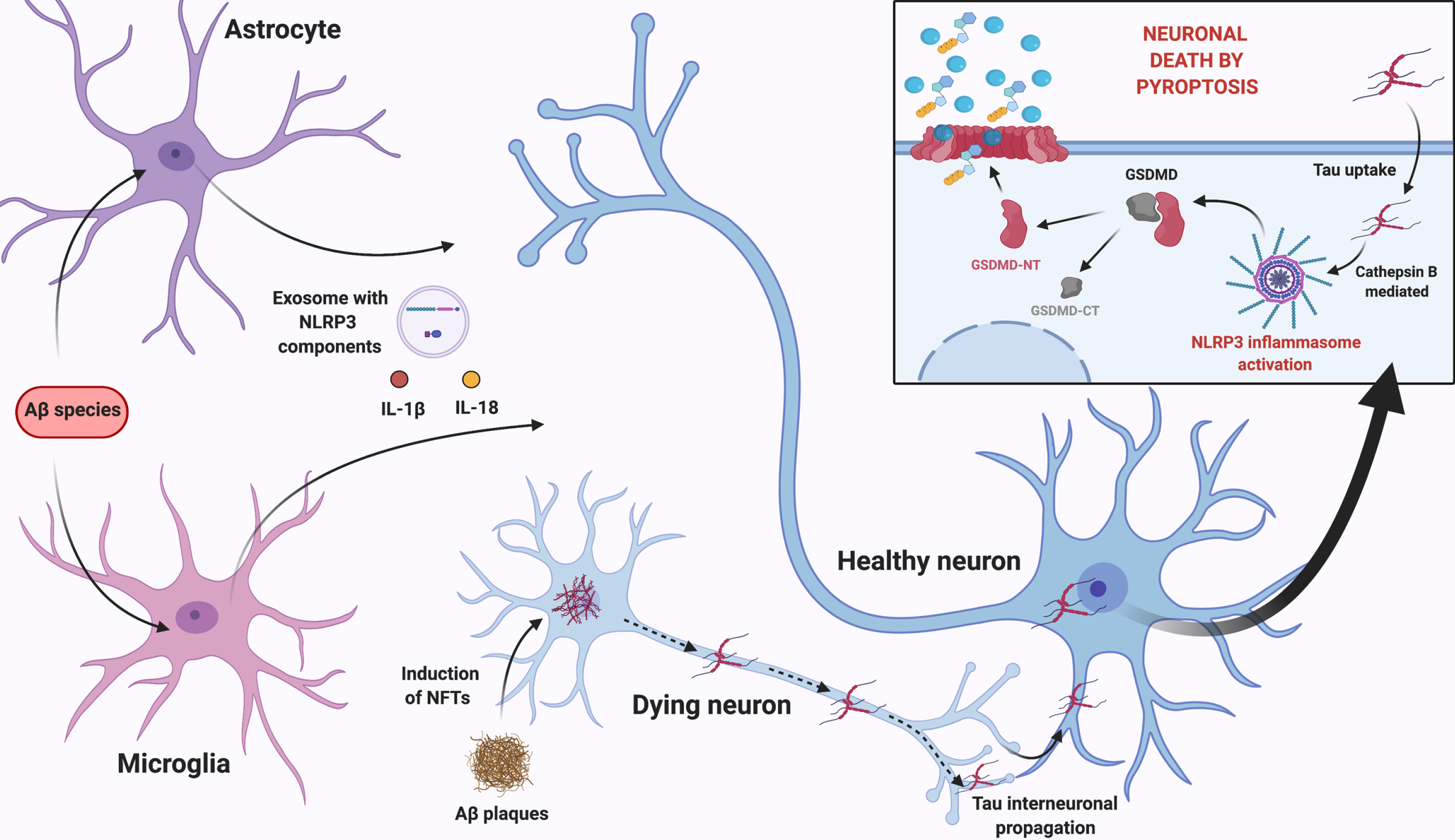
After reviewing the most recent data on this subject, we propose that the neuroinflammatory milieu arising from Aβ-induced NLRP3 activation in glial cells, prompts the release of pro-inflammatory cytokines and the exosomal release of NLRP3 domain and ASC. We suggest that these events can sum up to the neurotoxic effect of insoluble Aβ plaques that mediate the occurrence of pyroptosis and promote the development of NFTs, via NLRP3 activation. More importantly, considering the occurrence of tau interneuronal propagation, released tau can be taken up by neighboring neurons and activate NLRP3 in recipient cells causing neuronal death by pyroptosis. This multicellular feedback loop, summarized in Fig. 3, feeds on inflammasome activation in glial cells and neurons.
Fig. 3
Proposed multicellular feedback loop that feeds on NLRP3 inflammasome activation in glial cells and neurons. Aβ-induced NLRP3 activation in glial cells, prompts the release of pro-inflammatory cytokines, as well as exosomal release of NLRP3 domain and ASC. This neuroinflammatory milieu sums up to the neurotoxic effect of Aβ plaques in nearby neurons, which induce tau destabilization and NFTs formation, via NLRP3 activation. According to tau interneuronal propagation hypothesis, tau is then released and taken up by neighboring healthy neurons activating NLRP3 in these cells, most likely through lysosomal release of Cathepsin B, causing neuronal death by pyroptosis with cellular content release and perpetuating glial cells’ activation. Created with BioRender.com.
CONCLUSION
NLRP3 targeting was recently proposed as a therapeutic approach for AD [180]. Based on all the topics reviewed in this paper, we hereby claim that NLRP3 inhibition, due to its starring role in Aβ- and tau-driven pathological events, is undoubtfully a promising strategy for the development of therapies for this disease.
ABBREVIATIONS
Aβ, amyloid-β; AβPP, amyloid-β precursor protein; AD, Alzheimer’s disease; ASC, apoptosis-associated speck-like protein; ATP, adenosine triphosphate; BBB, blood brain barrier; CARD, caspase activating and recruitment domain; CNS, central nervous system; CSF, cerebrospinal fluid; C3, complement component 3; EVs, extracellular vesicles; GSDMD, gasdermin D; GSDMD-CT, C-terminal fragment of GSDMD; GSDMD-NT, N-terminal fragment of GSDMD; IL, Interleukin; LPS, lipopolysaccharides; NF-κB, Nuclear factor kappa B; NFTs, neurofibrillary tangles; NLRP3, NLRP3 inflammasome; PD, Parkinson’s disease; PRR, pattern recognition receptor; PYD, pyrin domain; RIPK3, receptor-interacting protein kinase 3; ROS, reactive oxygen species; TLR, Toll-Like Receptor; TNF, Tumour Necrosis Factor
ACKNOWLEDGMENTS
The work in the authors’ laboratory receives funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 952455, and Fundação para a Ciência e Tecnologia (FCT).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0268r1).
REFERENCES
[1] | Bali J , Gheinani AH , Zurbriggen S , Rajendran L ((2012) ) Role of genes linked to sporadic Alzheimer’s disease risk in the production of β-amyloid peptides. Proc Natl Acad Sci U S A 109: , 15307–15311. |
[2] | Masters CL , Bateman R , Blennow K , Rowe CC , Sperling RA , Cummings JL ((2015) ) Alzheimer’s disease. Nat Rev Dis Prim 1: , 15056. |
[3] | Stelzmann RA , Norman Schnitzlein H , Reed Murtagh F ((1995) ) An English translation of Alzheimer’s 1907 paper, “über eine eigenartige erkankung der hirnrinde.”. Clin Anat 8: , 429–431. |
[4] | Querfurth HW , Laferla FM ((2010) ) Alzheimer’s disease. N Engl J Med 9: , 329–344. |
[5] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[6] | Parihar MS , Brewer GJ ((2010) ) Amyloid beta as a modulator of synaptic plasticity. J Alzheimers Dis 22: , 741–763. |
[7] | Carrillo-Mora P , Luna R , Colín-Barenque L ((2014) ) Amyloid beta: Multiple mechanisms of toxicity and only some protective effects? Oxid Med Cell Longev 2014: , 1–15. |
[8] | Korte M , Herrmann U , Zhang X , Draguhn A ((2012) ) The role of APP and APLP for synaptic transmission, plasticity, and network function: Lessons from genetic mouse models. Exp Brain Res 217: , 435–440. |
[9] | Ludewig S , Korte M ((2017) ) Novel insights into the physiological function of the APP (GENE) family and its proteolytic fragments in synaptic plasticity. Front Mol Neurosci 9: , 161. |
[10] | Sun X , Chen WD , Wang YD ((2015) ) β-amyloid: The key peptide in the pathogenesis of Alzheimer’s disease. Front Pharmacol 6: , 221. |
[11] | Benilova I , Karran E , De Strooper B ((2012) ) The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat Neurosci 15: , 349–357. |
[12] | Golde TE , Eckman CB , Younkin SG ((2000) ) Biochemical detection of Aβ isoforms: Implications for pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Biochim Biophys Acta 1502: , 172–187. |
[13] | Lučiūnaitė A , McManus RM , Jankunec M , Rácz I , Dansokho C , Dalgėdienė I , Schwartz S , Brosseron F , Heneka MT ((2020) ) Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J Neurochem 155: , 650–661. |
[14] | Holtzman DM , Bales KR , Paul SM , DeMattos RB ((2002) ) Aβ immunization and anti-Aβ antibodies: Potential therapies for the prevention and treatment of Alzheimer’s disease. Adv Drug Deliv Rev 38: , 1603–1613. |
[15] | Janelidze S , Zetterberg H , Mattsson N , Palmqvist S , Vanderstichele H , Lindberg O , van Westen D , Stomrud E , Minthon L , Blennow K , Hansson O ((2016) ) CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: Better diagnostic markers of Alzheimer disease. Ann Clin Transl Neurol 3: , 154–165. |
[16] | Folch J , Busquets O , Ettcheto M , Sánchez-López E , Castro-Torres RD , Verdaguer E , Garcia ML , Olloquequi J , Casadesús G , Beas-Zarate C , Pelegri C , Vilaplana J , Auladell C , Camins A ((2018) ) Memantine for the treatment of dementia: A review on its current and future applications. J Alzheimers Dis 62: , 1223–1240. |
[17] | Andreadis A ((2006) ) Misregulation of tau alternative splicing in neurodegeneration and dementia. Prog Mol Subcell Biol 44: , 89–107. |
[18] | Mandelkow EM , Mandelkow E ((2012) ) Biochemistry and cell biology of Tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2: , 1–25. |
[19] | Martinisi A , Flach M , Sprenger F , Frank S , Tolnay M , Winkler DT ((2021) ) Severe oligomeric tau toxicity can be reversed without long-term sequelae. Brain 144: , 963–974. |
[20] | Guo T , Noble W , Hanger DP ((2017) ) Roles of tau protein in health and disease. Acta Neuropathol 133: , 665–704. |
[21] | Jeganathan S , Von Bergen M , Brutlach H , Steinhoff HJ , Mandelkow E ((2006) ) Global hairpin folding of tau in solution. Biochemistry 45: , 2283–2293. |
[22] | Cowan CM , Mudher A ((2013) ) Are tau aggregates toxic or protective in tauopathies? Front Neurol 4: , 114. |
[23] | Barghorn S , Mandelkow E ((2002) ) Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry 41: , 14885–14896. |
[24] | Gong C-X , Iqbal K ((2008) ) Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr Med Chem 15: , 2321–2328. |
[25] | Lucas SM , Rothwell NJ , Gibson RM ((2006) ) The role of inflammation in CNS injury and disease. Br J Pharmacol 147: , 232–240. |
[26] | Heneka MT , Kummer MP , Latz E ((2014) ) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14: , 463–477. |
[27] | DiSabato D , Quan N , Godbout JP ((2016) ) Neuroinflammation: The devil is in the details. J Neurochem 139: , 136–153. |
[28] | Tahara K , Kim HD , Jin JJ , Maxwell JA , Li L , Fukuchi KI ((2006) ) Role of toll-like receptor signalling in Aβ uptake and clearance. Brain 129: , 3006–3019. |
[29] | Agostinho P , Cunha RA , Oliveira C ((2010) ) Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharmacutical Des 16: , 2766–2778. |
[30] | Ravizza T , Balosso S , Vezzani A ((2011) ) Inflammation and prevention of epileptogenesis. Neurosci Lett 497: , 223–230. |
[31] | Lazovic J , Basu A , Lin HW , Rothstein RP , Krady JK , Smith MB , Levison SW ((2005) ) Neuroinflammation and both cytotoxic and vasogenic edema are reduced in interleukin-1 type 1 receptor-deficient mice conferring neuroprotection. Stroke 36: , 2226–2231. |
[32] | Choi DY , Liu M , Hunter RL , Cass WA , Pandya JD , Sullivan PG , Shin EJ , Kim HC , Gash DM , Bing G ((2009) ) Striatal neuroinflammation promotes parkinsonism in rats. PLoS One 4: , e5482. |
[33] | Abo-ouf H , Hooper AWM , White EJ , Van Rensburg HJJ , Trigatti BL , Igdoura SA ((2013) ) Deletion of tumor necrosis factor-α ameliorates neurodegeneration in sandhoff disease mice. Hum Mol Genet 22: , 3960–3975. |
[34] | Kempuraj D , Thangavel R , Natteru PA , Selvakumar GP , Saeed D , Zahoor H , Zaheer S , Iyer SS , Zaheer A ((2016) ) Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine 1: , 1003. |
[35] | McGeer PL , Rogers J , McGeer EG ((2016) ) Inflammation, antiinflammatory agents, and Alzheimer’s disease: The last 22 years. J Alzheimers Dis 54: , 853–857. |
[36] | Zádori D , Veres G , Szalárdy L , Klivényi P , Vécsei L ((2018) ) Alzheimer’s disease: Recent concepts on the relation of mitochondrial disturbances, excitotoxicity, neuroinflammation, and kynurenines. J Alzheimers Dis 62: , 523–547. |
[37] | Yang qiao Q , Zhou wei J ((2018) ) Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 67: , 1017–1035. |
[38] | Heneka MT , Kummer MP , Stutz A , Delekate A , Saecker A , Griep A , Axt D , Remus A , Tzeng T , Gelpi E , Halle A , Korte M , Latz E , Golenbock D ((2013) ) NLRP3 is activated in AD and contributes to pathology in APP/PS1 mice. Nature 493: , 674–678. |
[39] | Olsen I , Singhrao SK ((2016) ) Inflammasome involvement in Alzheimer’s disease. J Alzheimers Dis 54: , 45–53. |
[40] | Song L , Pei L , Yao S , Wu Y , Shang Y ((2017) ) NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Aging Neurosci 11: , 63. |
[41] | Voet S , Srinivasan S , Lamkanfi M , Loo G ((2019) ) Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med 11: , 1–16. |
[42] | Hanslik KL , Ulland TK ((2020) ) The role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front Neurol 11: , e10248. |
[43] | Heneka MT , Golenbock DT , Latz E ((2015) ) Innate immunity in Alzheimer’s disease. Nat Immunol 16: , 229–236. |
[44] | Becher B , Prat A , Antel JP ((2000) ) Brain-immune connection: Immuno-regulatory properties of CNS-resident cells. Glia 29: , 293–304. |
[45] | Kierdorf K , Erny D , Goldmann T , Sander V , Schulz C , Perdiguero EG , Wieghofer P , Heinrich A , Riemke P , Hölscher C , Müller DN , Luckow B , Brocker T , Debowski K , Fritz G , Opdenakker G , Diefenbach A , Biber K , Heikenwalder M , Geissmann F , Rosenbauer F , Prinz M ((2013) ) Microglia emerge from erythromyeloid precursors via Pu.1-and Irf8-dependent pathways. Nat Neurosci 16: , 273–280. |
[46] | Ginhoux F , Greter M , Leboeuf M , Nandi S , See P , Mehler MF , Conway SJ , Ng LG , Stanley ER , Igor M , Merad M ((2010) ) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330: , 841–845. |
[47] | Paolicelli RC , Bolasco G , Pagani F , Maggi L , Scianni M , Panzanelli P , Giustetto M , Ferreira TA , Guiducci E , Dumas L , Ragozzino D , Gross CT ((2011) ) Synaptic pruning by microglia is necessary for normal brain development. Science 333: , 1456–1458. |
[48] | Rock RB , Gekker G , Hu S , Sheng WS , Cheeran M , Lokensgard JR , Peterson PK ((2004) ) Role of microglia in central nervous system infections. Clin Microbiol Rev 17: , 942–964. |
[49] | Helmut K , Hanisch UK , Noda M , Verkhratsky A ((2011) ) Physiology of microglia. Physiol Rev 91: , 461–553. |
[50] | Moss DW , Bates TE ((2001) ) Activation of murine microglial cell lines by lipopolysaccharide and interferon-γ causes NO-mediated decreases in mitochondrial and cellular function. Eur J Neurosci 13: , 529–538. |
[51] | Araque A , Parpura V , Sanzgiri RP , Haydon PG ((1999) ) Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci 22: , 208–215. |
[52] | Tasdemir-Yilmaz OE , Freeman MR ((2014) ) Astrocytes engage unique molecular programs to engulf pruned neuronal debris from distinct subsets of neurons. Genes Dev 28: , 20–33. |
[53] | Perea G , Navarrete M , Araque A ((2009) ) Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci 32: , 421–431. |
[54] | Allaman I , Bélanger M , Magistretti PJ ((2011) ) Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci 34: , 76–87. |
[55] | Sweeney MD , Zhao Z , Montagne A , Nelson AR , Zlokovic BV ((2019) ) Blood-brain barrier: From physiology to disease and back. Physiol Rev 99: , 21–78. |
[56] | Escartin C , Galea E , Lakatos A , O’Callaghan JP , Petzold GC , Serrano-Pozo A , Steinhäuser C , Volterra A , Carmignoto G , Agarwal A , Allen NJ , Araque A , Barbeito L , Barzilai A , Bergles DE , Bonvento G , Butt AM , Chen W-T , Cohen-Salmon M , Cunningham C , Deneen B , De Strooper B , Díaz-Castro B , Farina C , Freeman M , Gallo V , Goldman JE , Goldman SA , Götz M , Gutiérrez A , Haydon PG , Heiland DH , Hol EM , Holt MG , Iino M , Kastanenka K V , Kettenmann H , Khakh BS , Koizumi S , Lee CJ , Liddelow SA , MacVicar BA , Magistretti P , Messing A , Mishra A , Molofsky A V , Murai KK , Norris CM , Okada S , Oliet SHR , Oliveira JF , Panatier A , Parpura V , Pekna M , Pekny M , Pellerin L , Perea G , Pérez-Nievas BG , Pfrieger FW , Poskanzer KE , Quintana FJ , Ransohoff RM , Riquelme-Perez M , Robel S , Rose CR , Rothstein JD , Rouach N , Rowitch DH , Semyanov A , Sirko S , Sontheimer H , Swanson RA , Vitorica J , Wanner I-B , Wood LB , Wu J , Zheng B , Zimmer ER , Zorec R , Sofroniew M V , Verkhratsky A ((2021) ) Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 24: , 312–325. |
[57] | Dong Y , Benveniste EN ((2001) ) Immune function of astrocytes. Glia 36: , 180–190. |
[58] | Sofroniew M V. ((2009) ) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32: , 638–647. |
[59] | Sofroniew MV , Vinters HV ((2010) ) Astrocytes: Biology and pathology. Acta Neuropathol 119: , 7–35. |
[60] | Liddelow SA , Barres BA ((2017) ) Reactive Astrocytes: Production, function, and therapeutic potential. Immunity 46: , 957–967. |
[61] | Liddelow SA , Guttenplan KA , Clarke LE , Bennett FC , Bohlen CJ , Schirmer L , Bennett ML , Münch AE , Chung WS , Peterson TC , Wilton DK , Frouin A , Napier BA , Panicker N , Kumar M , Buckwalter MS , Rowitch DH , Dawson VL , Dawson TM , Stevens B , Barres BA ((2017) ) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541: , 481–487. |
[62] | Schroder K , Tschopp J ((2010) ) The inflammasomes. Cell 140: , 821–832. |
[63] | Lamkanfi M , Dixit VM ((2014) ) Mechanisms and functions of inflammasomes. Cell 157: , 1013–1022. |
[64] | Martinon F , Burns K , Boveresses C , Epalinges C- ((2002) ) The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of pro IL-β . Mol Cell 10: , 417–426. |
[65] | Tschopp J , Schroder K ((2010) ) NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10: , 210–215. |
[66] | Rathinam VAK , Vanaja SK , Fitzgerald KA ((2012) ) Regulation of inflammasome signaling. Nat Immunol 13: , 333–342. |
[67] | He Y , Hara H , Núñez G ((2016) ) Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 41: , 1012–1021. |
[68] | Martinon F , Pétrilli V , Mayor A , Tardivel A , Tschopp J ((2006) ) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440: , 237–241. |
[69] | Duewell P , Kono H , Rayner KJ , Sirois CM , Bauernfeind FG , Abela GS , Franchi L , Nuñez G , Espevik T , Lien E , Fitzgerald KA , Rock KL , Vladimer G , Nunez G , Schnurr M , Moore KJ , Wright SD , Hornung V , Latz E ((2010) ) NLRP3 inflamasomes are required for atherogenesis and activated by cholesterol crystals that form early in disease. Nature 464: , 1357–1361. |
[70] | Halle A , Hornung V , Petzold GC , Stewart CR , Monks BG , Reinheckel T , Fitzgerald KA , Latz E , Moore KJ , Golenbock DT ((2008) ) The NALP3 inflammasome is involved in the innate immune response to amyloid-β . Nat Immunol 9: , 857–865. |
[71] | Martinon F , Mayor A , Tschopp J ((2009) ) The inflammasomes: Guardians of the body. Annu Rev Immunol 27: , 229–265. |
[72] | Wang X , Chi J , Huang D , Ding L , Zhao X , Jiang L , Yu Y , Gao F ((2020) ) α-synuclein promotes progression of Parkinson’s disease by upregulating autophagy signaling pathway to activate NLRP3 inflammasome. Exp Ther Med 19: , 931–938. |
[73] | Meng G , Grabiec A , Vallon M , Ebe B , Hampel S , Bessler W , Wagner H , Kirschning CJ ((2003) ) Cellular recognition of tri-/di-palmitoylated peptides is independent from a domain encompassing the N-terminal seven leucine-rich repeat (LRR)/LRR-like motifs of TLR2. J Biol Chem 278: , 39822–39829. |
[74] | Hoffman HM , Scott P , Mueller JL , Misaghi A , Stevens S , Yancopoulos GD , Murphy A , Valenzuela DM , Liu-Bryan R ((2010) ) Role of the leucine-rich repeat domain of cryopyrin/NALP3 in monosodium urate crystal-induced inflammation in mice. Arthritis Rheum 62: , 2170–2179. |
[75] | Duncan JA , Bergstralh DT , Wang Y , Willingham SB , Ye Z , Zimmermann AG , Ting JP ((2007) ) Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A 104: , 8041–8046. |
[76] | Liepinsh E , Barbals R , Dahl E , Sharipo A , Staub E , Otting G ((2003) ) The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J Mol Biol 332: , 1155–1163. |
[77] | Dowds TA , Masumoto J , Zhu L , Inohara N , Núñez G ((2004) ) Cryopyrin-induced interleukin 1β secretion in monocytic cells: Enhanced activity of disease-associated mutants and requirement for ASC. J Biol Chem 279: , 21924–21928. |
[78] | Srinivasula SM , Poyet JL , Razmara M , Datta P , Zhang Z , Alnemri ES ((2002) ) The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem 277: , 21119–21122. |
[79] | Liu X , Zhang Z , Ruan J , Pan Y , Magupalli VG , Wu H , Lieberman J ((2016) ) Inflammasome - activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: , 153–158. |
[80] | Sutterwala FS , Haasken S , Cassel SL ((2014) ) Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci 1319: , 82–95. |
[81] | Schroder K , Sagulenko V , Zamoshnikova A , Richards AA , Cridland JA , Irvine KM , Stacey KJ , Sweet MJ ((2012) ) Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 217: , 1325–1329. |
[82] | Yang J , Liu Z , Xiao TS ((2017) ) Post-translational regulation of inflammasomes. Cell Mol Immunol 14: , 65–79. |
[83] | Juliana C , Fernandes-Alnemri T , Kang S , Farias A , Qin F , Alnemri ES ((2012) ) Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287: , 36617–36622. |
[84] | Bauernfeind F , Horvath G , Stutz A , Alnemri ES , Speert D , Fernandes-alnemri T , Wu J , Brian G , Fitzgerald KA , Hornung V , Latz E ((2010) ) NF-kB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183: , 787–791. |
[85] | Stutz A , Horvath GL , Monks BG , Latz E ((2013) ) ASC speck formation as a readout for inflammasome activation. Methods Mol Biol 1040: , 91–101. |
[86] | Sharma D , Kanneganti TD ((2016) ) The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol 213: , 617–629. |
[87] | Scheiblich H , Schlütter A , Golenbock DT , Latz E , Martinez-Martinez P , Heneka MT ((2017) ) Activation of the NLRP3 inflammasome in microglia: The role of ceramide. J Neurochem 143: , 534–550. |
[88] | Chan AH , Schroder K ((2019) ) Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med 217: , e20190314. |
[89] | Ray A ((2016) ) Cytokines and their role in health and disease: A brief overview. MOJ Immunol 4: , 00121. |
[90] | Walsh JG , Muruve DA , Power C ((2014) ) Inflammasomes in the CNS. Nat Rev Neurol 15: , 84–97. |
[91] | Lucin KM , Wyss-Coray T ((2009) ) Immune activation in brain aging and neurodegeneration: Too much or too little? Neuron 64: , 110–122. |
[92] | Italiani P , Puxeddu I , Napoletano S , Scala E , Melillo D , Manocchio S , Angiolillo A , Migliorini P , Boraschi D , Vitale E , Di Costanzo A ((2018) ) Circulating levels of IL-1 family cytokines and receptors in Alzheimer’s disease: New markers of disease progression? J Neuroinflammation 15: , 342. |
[93] | Pickering M , O’Connor JJ ((2007) ) Pro-inflammatory cytokines and their effects in the dentate gyrus. Prog Brain Res 163: , 339–354. |
[94] | Monteleone M , Stanley AC , Chen KW , Brown DL , Bezbradica JS , von Pein JB , Holley CL , Boucher D , Shakespear MR , Kapetanovic R , Rolfes V , Sweet MJ , Stow JL , Schroder K ((2018) ) Interleukin-1β maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep 24: , 1425–1433. |
[95] | Pachter JS , De Vries HE , Fabry Z ((2003) ) The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol 62: , 593–604. |
[96] | Cabezas R , Ávila M , Gonzalez J , El-Bachá RS , Báez E , García-Segura LM , Coronel JCJ , Capani F , Cardona-Gomez GP , Barreto GE ((2014) ) Astrocytic modulation of blood brain barrier: Perspectives on Parkinson’s disease. Front Cell Neurosci 8: , 211. |
[97] | Hu MY , Lin YY , Zhang BJ , Lu DL , Lu ZQ , Cai W ((2019) ) Update of inflammasome activation in microglia/macrophage in aging and aging-related disease. CNS Neurosci Ther 25: , 1299–1307. |
[98] | Blum-Degen D , Muller T , Kuhn W , Gerlach M , Przuntek H , Riederer P ((1995) ) Interleukin-1β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett 202: , 17–20. |
[99] | Griffin WST , Liu L , Li Y , Mrak RE , Barger SW ((2006) ) Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation 3: , 5. |
[100] | Sheedy FJ , Grebe A , Rayner KJ , Kalantari P , Ramkhelawon B , Carpenter SB , Becker CE , Ediriweera HN , Mullick AE , Golenbock DT , Stuart LM , Latz E , Fitzgerald KA , Moore KJ ((2013) ) CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol 14: , 812–820. |
[101] | Stewart CR , Stuart LM , Wilkinson K , van Gils JM , Deng J , Halle A , Rayner KJ , Boyer L , Zhong R , Frazier WA , Lacy-Hulbert A , El Khoury J , Golenbock DT , Moore KJ. ((2010) ) CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 11: , 155–161. |
[102] | Mariathasan S , Weiss DS , Newton K , McBride J , O’Rourke K , Roose-Girma M , Lee WP , Weinrauch Y , Monack DM , Dixit VM ((2006) ) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: , 228–232. |
[103] | Kahlenberg JM , Dubyak GR , Michelle J , Mechanisms GRD ((2020) ) Mechanisms of caspase-1 activation by P2X 7 receptor-mediated+release. Am J Physiol Cell Physiol 44106: , 1100–1108. |
[104] | Yue N , Huang H , Zhu X , Han Q , Wang Y , Li B , Liu Q , Wu G , Zhang Y , Yu J ((2017) ) Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J Neuroinflammation 14: , 1–15. |
[105] | Kanneganti TD , Lamkanfi M , Kim YG , Chen G , Park JH , Franchi L , Vandenabeele P , Núñez G ((2007) ) Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of toll-like receptor signaling. Immunity 26: , 433–443. |
[106] | McLarnon JG , Ryu JK , Walker DG , Choi HB ((2006) ) Upregulated expression of purinergic P2X7 receptor in Alzheimer disease and amyloid-β peptide-treated microglia and in peptide-injected rat hippocampus. J Neuropathol Exp Neurol 65: , 1090–1097. |
[107] | Parajuli B , Sonobe Y , Horiuchi H , Takeuchi H , Mizuno T , Suzumura A ((2013) ) Oligomeric amyloid β induces IL-1β processing via production of ROS: Implication in Alzheimer’s disease. Cell Death Dis 4: , e975. |
[108] | Lashuel HA , Hartley D , Petre BM , Walz T , Lansbury PT ((2002) ) Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 418: , 291–292. |
[109] | Reddy PH , Beal MF ((2008) ) Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med 14: , 45–53. |
[110] | Sesti F , Liu S , Cai SQ ((2010) ) Oxidation of potassium channels by ROS: A general mechanism of aging and neurodegeneration? Trends Cell Biol 20: , 45–51. |
[111] | Lu B , Nakamura T , Inouye K , Li1 J , Tang Y , Lundbäck P , Valdes-Ferrer SI , Olofsson PS , Kalb T , Roth J , Zou Y , Erlandsson-Harris H , Yang H , Ting JP-Y , Wang H , Andersson U , Antoine DJ , Chavan SS , Hotamisligil GS , Tracey KJ ((2012) ) Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488: , 670–674. |
[112] | Shenoy AR , Wellington DA , Kumar P , Kassa H , Booth CJ , Cresswell P , MacMicking JD ((2012) ) GBP5 Promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336: , 481–485. |
[113] | Wang Y , Hasegawa M , Imamura R , Kinoshita T , Kondo C , Konaka K , Suda T ((2004) ) PYNOD, a novel Apaf-1/CED4-like protein is an inhibitor of ASC and caspase-1. Int Immunol 16: , 777–786. |
[114] | He Y , Zeng MY , Yang D , Motro B , Núñez G ((2016) ) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: , 354–357. |
[115] | Murphy N , Grehan B , Lynch MA ((2014) ) Glial uptake of amyloid beta induces NLRP3 inflammasome formation via cathepsin-dependent degradation of NLRP10. Neuromolecular Med 16: , 205–215. |
[116] | Gustin A , Kirchmeyer M , Koncina E , Felten P , Losciuto S , Heurtaux T , Tardivel A , Heuschling P , Dostert C ((2015) ) NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 10: , e0130624. |
[117] | Simard AR , Soulet D , Gowing G , Julien JP , Rivest S ((2006) ) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49: , 489–502. |
[118] | Venegas C , Kumar S , Franklin BS , Dierkes T , Brinkschulte R , Tejera D , Vieira-Saecker A , Schwartz S , Santarelli F , Kummer MP , Griep A , Gelpi E , Beilharz M , Riedel D , Golenbock DT , Geyer M , Walter J , Latz E , Heneka MT ((2017) ) Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552: , 355–361. |
[119] | Friker LL , Scheiblich H , Hochheiser IV , Brinkschulte R , Riedel D , Latz E , Geyer M , Heneka MT ((2020) ) β-amyloid clustering around ASC fibrils boosts its toxicity in microglia. Cell Rep 30: , 3743–3754.e6. |
[120] | Sarlus H , Heneka MT ((2017) ) Microglia in Alzheimer’s disease. J Clin Invest 127: , 3240–3249. |
[121] | Meyer-Luehmann M , Spires-Jones TL , Prada C , Garcia-Alloza M , de Calignon A , Rozkalne A , Koenigsknecht-Talboo J , Holtzman DM , Bacskai BJ , Hyman BT ((2008) ) Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451: , 720–724. |
[122] | Itagaki S , McGeer PL , Akiyama H , Zhu S , Selkoe D ((1989) ) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24: , 173–182. |
[123] | Maccioni RB , Rojo LE , Fern JA , Kuljis RO ((2009) ) The role of neuroimmunomodulation in Alzheimer’s disease. Neuroimmunomodulation 246: , 240–246. |
[124] | Frackowiak J , Wisniewski HM , Wegiel J , Merz GS , Iqbal K , Wang KC ((1992) ) Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce β-amyloid fibrils. Acta Neuropathol 84: , 225–233. |
[125] | Kaltschmidt B , Uherek M , Volk B , Baeuerle PA , Kaltschmidt C ((1997) ) Transcription factor NF-κB is activated in primary neurons by amyloid β peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci U S A 94: , 2642–2647. |
[126] | Mattson MP , Meffert MK ((2006) ) Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ 13: , 852–860. |
[127] | Hsiao HY , Chen YC , Chen HM , Tu PH , Chern Y ((2013) ) A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington’s disease. Hum Mol Genet 22: , 1826–1842. |
[128] | Mulder SD , Veerhuis R , Blankenstein MA , Nielsen HM ((2012) ) The effect of amyloid associated proteins on the expression of genes involved in amyloid-β clearance by adult human astrocytes. Exp Neurol 233: , 373–379. |
[129] | Ries M , Sastre M ((2016) ) Mechanisms of Aβ clearance and degradation by glial cells. Front Aging Neurosci 8: , 160. |
[130] | González-Reyes RE , Nava-Mesa MO , Vargas-Sáchez K , Ariza-Salamanca D , Mora-Muñoz L ((2017) ) Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci 10: , 427. |
[131] | Zamanian JL , Xu L , Foo LC , Nouri N , Zhou L , Giffard RG , Barres BA ((2012) ) Genomic analysis of reactive astrogliosis. J Neurosci 32: , 6391–6410. |
[132] | Wu T , Dejanovic B , Gandham VD , Gogineni A , Edmonds R , Schauer S , Srinivasan K , Huntley MA , Wang Y , Wang TM , Hedehus M , Barck KH , Stark M , Ngu H , Foreman O , Meilandt WJ , Elstrott J , Chang MC , Hansen DV , Carano RAD , Sheng M , Hanson JE ((2019) ) Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep 28: , 2111–2123.e6. |
[133] | Lian H , Litvinchuk A , Chiang ACA , Aithmitti N , Jankowsky JL , Zheng H ((2016) ) Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J Neurosci 36: , 577–589. |
[134] | Arranz AM , De Strooper B ((2019) ) The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol 18: , 406–414. |
[135] | Zhang X , Lao K , Qiu Z , Rahman MS , Zhang Y , Gou X ((2019) ) Potential astrocytic receptors and transporters in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis 67: , 1109–1122. |
[136] | Batarseh YS , Duong QV , Mousa YM , Al Rihani SB , Elfakhri K , Kaddoumi A ((2016) ) Amyloid-β and astrocytes interplay in amyloid-β related disorders. Int J Mol Sci 17: , 338. |
[137] | Jones RS , Minogue AM , Connor TJ , Lynch MA ((2013) ) Amyloid-β-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J Neuroimmune Pharmacol 8: , 301–311. |
[138] | Carrero I , Gonzalo MR , Martin B , Sanz-Anquela JM , Arévalo-Serrano J , Gonzalo-Ruiz A ((2012) ) Oligomers of beta-amyloid protein (Aβ1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp Neurol 236: , 215–227. |
[139] | Tarassishin L , Casper D , Lee SC ((2014) ) Aberrant expression of interleukin-1β and inflammasome activation in human malignant gliomas. PLoS One 9: , e103432. |
[140] | Apelt J , Schliebs R ((2001) ) β-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res 894: , 21–30. |
[141] | Kawana N , Yamamoto Y , Ishida T , Saito Y , Konno H , Arima K , Satoh JI ((2013) ) Reactive astrocytes and perivascular macrophages express NLRP3 inflammasome in active demyelinating lesions of multiple sclerosis and necrotic lesions of neuromyelitis optica and cerebral infarction. Clin Exp Neuroimmunol 4: , 296–304. |
[142] | Liu HD , Li W , Chen ZR , Hu YC , Zhang DD , Shen W , Zhou ML , Zhu L , Hang CH ((2013) ) Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res 38: , 2072–2083. |
[143] | Johann S , Heitzer M , Kanagaratnam M , Goswami A , Rizo T , Weis J , Troost D , Beyer C ((2015) ) NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63: , 2260–2273. |
[144] | Bertheloot D , Latz E , Franklin BS ((2021) ) Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol Immunol 18: , 1106–1121. |
[145] | Chi H , Chang HY , Sang TK ((2018) ) Neuronal cell death mechanisms in major neurodegenerative diseases. Int J Mol Sci 19: , 3082. |
[146] | Andreone BJ , Larhammar M , Lewcock JW ((2019) ) Cell death and neurodegeneration. Cold Spring Harb Perspect Biol 12: , a036434. |
[147] | Fink SL , Cookson BT ((2005) ) Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect Immun 73: , 1907–1916. |
[148] | Dhuriya YK , Sharma D ((2018) ) Necroptosis: A regulated inflammatory mode of cell death. J Neuroinflammation 15: , 199. |
[149] | Shi J , Gao W , Shao F ((2016) ) Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci 42: , 245–254. |
[150] | Galluzzi L , Vitale I , Aaronson SA , Abrams JM , Adam D , Agostinis P , Alnemri ES , Altucci L , Amelio I , Andrews DW , Annicchiarico-Petruzzelli M , Antonov A V. , Arama E , Baehrecke EH , Barlev NA , Bazan NG , Bernassola F , Bertrand MJM , Bianchi K , Blagosklonny M V. , Blomgren K , Borner C , Boya P , Brenner C , Campanella M , Candi E , Carmona-Gutierrez D , Cecconi F , Chan FKM , Chandel NS , Cheng EH , Chipuk JE , Cidlowski JA , Ciechanover A , Cohen GM , Conrad M , Cubillos-Ruiz JR , Czabotar PE , D’Angiolella V , Dawson TM , Dawson VL , De Laurenzi V , De Maria R , Debatin KM , Deberardinis RJ , Deshmukh M , Di Daniele N , Di Virgilio F , Dixit VM , Dixon SJ , Duckett CS , Dynlacht BD , El-Deiry WS , Elrod JW , Fimia GM , Fulda S , García-Sáez AJ , Garg AD , Garrido C , Gavathiotis E , Golstein P , Gottlieb E , Green DR , Greene LA , Gronemeyer H , Gross A , Hajnoczky G , Hardwick JM , Harris IS , Hengartner MO , Hetz C , Ichijo H , Jäättelä M , Joseph B , Jost PJ , Juin PP , Kaiser WJ , Karin M , Kaufmann T , Kepp O , Kimchi A , Kitsis RN , Klionsky DJ , Knight RA , Kumar S , Lee SW , Lemasters JJ , Levine B , Linkermann A , Lipton SA , Lockshin RA , López-Otín C , Lowe SW , Luedde T , Lugli E , MacFarlane M , Madeo F , Malewicz M , Malorni W , Manic G , Marine JC , Martin SJ , Martinou JC , Medema JP , Mehlen P , Meier P , Melino S , Miao EA , Molkentin JD , Moll UM , Muñoz-Pinedo C , Nagata S , Nuñez G , Oberst A , Oren M , Overholtzer M , Pagano M , Panaretakis T , Pasparakis M , Penninger JM , Pereira DM , Pervaiz S , Peter ME , Piacentini M , Pinton P , Prehn JHM , Puthalakath H , Rabinovich GA , Rehm M , Rizzuto R , Rodrigues CMP , Rubinsztein DC , Rudel T , Ryan KM , Sayan E , Scorrano L , Shao F , Shi Y , Silke J , Simon HU , Sistigu A , Stockwell BR , Strasser A , Szabadkai G , Tait SWG , Tang D , Tavernarakis N , Thorburn A , Tsujimoto Y , Turk B , Vanden Berghe T , Vandenabeele P , Vander Heiden MG , Villunger A , Virgin HW , Vousden KH , Vucic D , Wagner EF , Walczak H , Wallach D , Wang Y , Wells JA , Wood W , Yuan J , Zakeri Z , Zhivotovsky B , Zitvogel L , Melino G , Kroemer G ((2018) ) Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25: , 486–541. |
[151] | Ma Y , Jiang J , Gao Y , Shi T , Zhu X , Zhang K , Lu K , Xue B ((2018) ) Research progress of the relationship between pyroptosis and disease. Am J Transl Res 10: , 2213–2219. |
[152] | McKenzie BA , Dixit VM , Power C ((2020) ) Fiery cell death: Pyroptosis in the central nervous system. Trends Neurosci 43: , 55–73. |
[153] | Bergsbaken T , Fink SL , Cookson BT ((2009) ) Pyroptosis: Host cell death. Nat Rev Microbiol 7: , 99–109. |
[154] | Shi J , Zhao Y , Wang K , Shi X , Wang Y , Huang H , Zhuang Y , Cai T , Wang F , Shao F ((2015) ) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: , 660–665. |
[155] | Panicker N , Kam T-I , Neifert S , Hinkle J , Mao X , Karuppagounder S , Wang H , Kumar M , Pirooznia S , Pletnikova O , Troncoso J , Dawson VL , Dawson TM ((2020) ) NLRP3 inflammasome activation in dopamine neurons contributes to neurodegeneration in Parkinson’s Disease. J Fed Am Soc Exp Biol 34: (S1), 1. |
[156] | Kametani F , Hasegawa M ((2018) ) Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front Neurosci 12: , 25. |
[157] | Takeda S ((2019) ) Tau propagation as a diagnostic and therapeutic target for dementia: Potentials and unanswered questions. Front Neurosci 13: , 1274. |
[158] | Pooler AM , Phillips EC , Lau DHW , Noble W , Hanger DP ((2013) ) Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep 14: , 389–394. |
[159] | Asai H , Ikezu S , Tsunoda S , Medalla M , Luebke J , Wolozin B , Butovsky O , Ikezu T , Therapeutics E ((2015) ) Depletion of microglia and inhibition of exosome synthe. Nat Neurosci 18: , 1584–1593. |
[160] | Wang Y , Balaji V , Kaniyappan S , Krüger L , Irsen S , Tepper K , Chandupatla R , Maetzler W , Schneider A , Mandelkow E , Mandelkow EM ((2017) ) The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 12: , 5. |
[161] | Stancu IC , Cremers N , Vanrusselt H , Couturier J , Vanoosthuyse A , Kessels S , Lodder C , Brône B , Huaux F , Octave JN , Terwel D , Dewachter I ((2019) ) Aggregated tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol 137: , 599–617. |
[162] | Ising C , Venegas C , Zhang S , Scheiblich H , Schmidt S V. , Vieira-Saecker A , Schwartz S , Albasset S , McManus RM , Tejera D , Griep A , Santarelli F , Brosseron F , Opitz S , Stunden J , Merten M , Kayed R , Golenbock DT , Blum D , Latz E , Buée L , Heneka MT ((2019) ) NLRP3 inflammasome activation drives tau pathology. Nature 575: , 669–673. |
[163] | Doyle L , Wang M ((2019) ) Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 8: , 727. |
[164] | Iraci N , Gaude E , Leonardi T , Costa ASH , Cossetti C , Peruzzotti-Jametti L , Bernstock JD , Saini HK , Gelati M , Vescovi AL , Bastos C , Faria N , Occhipinti LG , Enright AJ , Frezza C , Pluchino S ((2017) ) Extracellular vesicles are independent metabolic units with asparaginase activity. Nat Chem Biol 13: , 951–955. |
[165] | Abal M ((2017) ) Characterizing the contribution of inflammasome-derived exosomes in the activation of the immune response. Ann Transl Med 5: , 7–9. |
[166] | Cypryk W , Nyman TA , Matikainen S ((2018) ) From inflammasome to exosome - Does extracellular vesicle secretion constitute an inflammasome-dependent immune response? Front Immunol 9: , 2188. |
[167] | Qu Y , Franchi L , Nunez G , Dubyak GR ((2007) ) Nonclassical IL-1β secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol 179: , 1913–1925. |
[168] | De Rivero Vaccari JP , Brand F , Adamczak S , Lee SW , Perez-Barcena J , Wang MY , Bullock MR , Dietrich WD , Keane RW ((2016) ) Exosome-mediated inflammasome signaling after central nervous system injury. J Neurochem 136: , 39–48. |
[169] | Sarkar S , Rokad D , Malovic E , Luo J , Harischandra DS , Jin H , Anantharam V , Huang X , Lewis M , Kanthasamy A , Kanthasamy AG ((2019) ) Manganese activates NLRP3 inflammasome signaling and propagates exosomal release of ASC in microglial cells.eaat. Sci Signal 12: , 9900. |
[170] | Prada I , Gabrielli M , Turola E , Iorio A , D’Arrigo G , Parolisi R , De Luca M , Pacifici M , Bastoni M , Lombardi M , Legname G , Cojoc D , Buffo A , Furlan R , Peruzzi F , Verderio C ((2018) ) Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol 135: , 529–550. |
[171] | Zahid A , Li B , Kombe AJK , Jin T , Tao J ((2019) ) Pharmacological inhibitors of the nlrp3 inflammasome. Front Immunol 10: , 2538. |
[172] | Mullard A ((2019) ) NLRP3 inhibitors stoke anti-inflammatory ambitions. Nat Rev Drug Discov 18: , 405–407. |
[173] | Coll RC , Robertson AAB , Chae JJ , Higgins SC , Muñoz-Planillo R , Inserra MC , Vetter I , Dungan LS , Monks BG , Stutz A , Croker DE , Butler MS , Haneklaus M , Sutton CE , Núñez G , Latz E , Kastner DL , Mills KHG , Masters SL , Schroder K , Cooper MA , O’Neill LAJ ((2015) ) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: , 248–257. |
[174] | Coll RC , Hill JR , Day CJ , Zamoshnikova A , Boucher D , Massey NL , Chitty JL , Fraser JA , Jennings MP , Robertson AAB , Schroder K ((2019) ) MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol 15: , 556–559. |
[175] | Dempsey C , Rubio Araiz A , Bryson KJ , Finucane O , Larkin C , Mills EL , Robertson AAB , Cooper MA , O’Neill LAJ , Lynch MA ((2017) ) Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav Immun 61: , 306–316. |
[176] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[177] | Walsh DM , Selkoe DJ ((2007) ) Aβ oligomers - A decade of discovery. J Neurochem 101: , 1172–1184. |
[178] | Hayden EY , Teplow DB ((2013) ) Amyloid β -protein oligomers and Alzheimer’s disease. Alzheimers Res Ther 5: , 60. |
[179] | Ricciarelli R , Fedele E ((2017) ) The amyloid cascade hypothesis in Alzheimer’s disease: It’s time to change our mind. Curr Neuropharmacol 15: , 926–935. |
[180] | Zhang Y , Dong Z , Song W ((2020) ) NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct Target Ther 5: , 37. |


