
Neurotrophic Treatment Initiated During Early Postnatal Development Prevents the Alzheimer-Like Behavior and Synaptic Dysfunction
Abstract
Background:
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by impairments in synaptic plasticity and cognitive performance. Cognitive dysfunction and loss of neuronal plasticity are known to begin decades before the clinical diagnosis of the disease. The important influence of congenital genetic mutations on the early development of AD provides a novel opportunity to initiate treatment during early development to prevent the Alzheimer-like behavior and synaptic dysfunction.
Objective:
To explore strategies for early intervention to prevent Alzheimer’s disease.
Methods:
In the present study, we investigated the effect of treatment during early development with a ciliary neurotrophic factor (CNTF) derived peptidergic compound, P021 (Ac-DGGLAG-NH2) on cognitive function and synaptic plasticity in 3xTg-AD transgenic mouse model of AD. 3xTg-AD and genetic background-matched wild type female mice were treated from birth to postnatal day 120 with P021 in diet or as a control with vehicle diet, and cognitive function and molecular markers of neuroplasticity were evaluated.
Results:
P021 treatment during early development prevented cognitive impairment and increased expressions of pCREB and BDNF that activated downstream various signaling cascades such as PLC/PKC, MEK/ERK and PI3K/Akt, and ameliorated synaptic protein deficit in 4-month-old 3xTg-AD mice.
Conclusion:
These findings indicate that treatment with the neurotrophic peptide mimetic such as P021 during early development can be an effective therapeutic strategy to rescue synaptic deficit and cognitive impairment in familial AD and related tauopathies.
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia, which is characterized by memory loss and other cognitive disabilities serious enough to interfere with daily life [1]. AD accounts for 60–80%of all dementia cases. An estimated 5.7 million Americans were living with AD in 2018. This number includes an estimated 5.5 million people age 65 and older, and approximately 200,000 individuals under age 65 who have mostly the familial younger-onset AD [2]. The behavioral abnormalities in AD result from dysfunction and degeneration of neurons in brain regions involved in cognition and mood such as the hippocampus, entorhinal cortex, basal forebrain, and frontal and parietal lobes [3, 4]. These brain regions suffer from degeneration of synapses and neurons associated with extracellular senile plaques consisting of amyloid-β (Aβ) peptides [5] and intracellular neurofibrillary tangles (NFTs) composed of abnormally hyperphosphorylated tau protein [6]. Although the accumulation of plaques in the brain was reported to cause synaptic dysfunction and degeneration of neurons by inducing disruption of cellular ion homeostasis [7, 8], it does not directly correlate with the severity of cognitive impairment in patients [9]; a large number of people without any cognitive impairment have substantial accumulations of plaques in their brains [10, 11] and the reduction of plaque load in the brain by immunotherapy does not result in cognitive improvement in AD patients [12]. Tangles, on the other hand, do correlate strongly with cognitive decline and with neuronal and synapse loss [7, 13–16]. Of the AD neuropathological features, synapse loss correlates most strongly with dementia, implicating it as important to the disease process [17].
The pathophysiological process of AD is thought to begin decades before the diagnosis [18, 19]. Cognitive impairment may manifest in the preclinical phase of AD substantially [20]. Imaging-based studies suggest an initial deficiency in limbic regions of the brain affecting episodic memory in the early stages of AD [21, 22] that progresses to the cortical regions of the brain followed by other cognitive symptoms, and the dementia syndrome is noticed [23]. The “preclinical AD” is conceptualized to capture this long silence period to facilitate longitudinal clinical research studies [18] and has led to the belief that early preventive efforts may be more effective in slowing preclinical manifestation of AD dementia. Both in Down syndrome and familial AD, despite the disease-causing genetic defects, the AD clinical phenotype does not occur till middle to late middle-old age. Thus, it is rational to investigate if initiation of the drug treatment from birth can have preventive therapeutic effect in a transgenic mouse model of familial AD and tauopathy. It is not impossible that, like familial, a subgroup of sporadic AD could result from a neurotrophic deficit during early development.
Peptide 021 (P021: Ac-DGGLAG-NH2) was generated from a 4-mer ciliary neurotrophic factor (CNTF) peptide by the addition of adamantylated glycine at the C-terminus [24, 25]. Compound P021 was found to ameliorate learning and memory deficits in animal models of aging and AD [26–29]. Oral administration of the compound P021 could exert a neurogenic and neurotrophic effect by inhibiting leukemia inhibitory factor (LIF) signaling pathway and by enhancing transcription and expression of brain derived neurotrophic factor (BDNF) [26, 30, 31]. Chronic treatment with P021 significantly reduced tau pathology both at moderate and severe stages of the pathology via reduction in glycogen synthase kinase-3 beta (GSK3β) activity in 3xTg-AD mice [27, 32, 33]. P021 is a small water-soluble compound that is blood-brain barrier (BBB)-permeable and has suitable pharmacokinetics for oral administration without any adverse effects observed with native CNTF or BDNF [29, 32].
Little information is available on the role of neurotrophins during development in preventing neurobehavioral and synaptic dysfunctions at adult age in AD. Previously, we found that prenatal to early postnatal treatment with P021 can inhibit developmental delay and then at adult age cognitive deficits in TS65Dn trisomic mouse model of Down syndrome [31] and Aβ and tau pathologies and cognitive impairment in 3xTg-AD transgenic mouse model [34]. Chronic treatment with P021 from 3 to 22 months was also found to reverse cognitive impairment and neuroplasticity deficits and prevent Aβ and tau pathologies in 3xTg-AD mice [35, 36]. The present study aimed to address the important practical question whether initiation of the P021 treatment at birth can also prevent cognitive impairment and neuroplasticity deficit. We found that treatment with P021 from birth till postnatal day 120 prevented the memory deficits in 4-month-old 3xTg-AD mice. This therapeutic beneficial effect on cognition was associated with increased pCREB and BDNF expressions which subsequently activated downstream PLC/PKC, MEK/ERK, and PI3K/Akt signaling cascades and decreased neuroplasticity deficit.
MATERIALS AND METHODS
Study outline
3xTg-AD and wild-type (WT) female mice were treated with compound P021 in mouse chow, or as control with vehicle feed from postnatal day 1 to postnatal day 120. Morris water maze and Novel object recognition tests were carried out consecutively from postnatal day 90. The animals were sacrificed at postnatal day 120 for immunohistochemical and biochemical analysis (Fig. 1). There were four study groups as follows: 3xTg-AD mice on vehicle diet (3xTg-Vh, n = 11), 3xTg-AD mice on P021 diet (3xTg-P021, n = 11), WT mice on vehicle diet (WT-Vh, n = 10), and WT mice on P021 diet (WT-P021, n = 10).
Fig. 1
The design of the study. 3×Tg-AD and WT pregnant mice were treated with P021- or vehicle-chow from the day of the delivery of the pups (postnatal day 1 pups) till the weaning of pups on postnatal day 21, at which time point the pups were separated from their mothers and pups of dams treated with P021 were kept on P021 and pups of dams treated with vehicle chow were kept on vehicle chow till they were sacrificed at the age of 120 days. Morris water maze (MWM) and Novel object recognition (NOR) tests were carried out consecutively from postnatal day 90 onwards. The animals were sacrificed at postnatal day 120 for biochemical analysis. P1, postnatal day 1; WT, wild type mice; P021, peptidergic neurotrophic compound P021 diet.
Design and synthesis of P021
The peptidergic compound P021 (Ac-DGGLAG-NH2; mol. wt. of 578.3) corresponds to a biologically active region of human CNTF (amino acid residues 148–151) to which adamantylated glycine was added at the C-terminal to increase its stability and lipophilicity. The peptide was synthesized by solid phase synthesis and purified by reverse-phase high-performance liquid chromatography to 96%purity, and the sequence of the peptide was confirmed by mass spectrometry, as described previously [26, 30]. P021 is quite stable in artificial gastric juice (∼90%during 30 min) and in artificial intestinal juice (∼100%during 120 min). BBB studies on P021, which were carried out through a commercial service (APREDICA, Watertown, MA, USA), demonstrated that a sufficient amount of P021 crossed the BBB to exert its effect in the brain [25].
Animals and housing
The 3xTg-AD mouse is one of the most widely-used animal models described so far as it replicates all histopathological and behavioral hallmarks of AD [37]. The 3xTg-AD mice contain two mutations associated with familial AD (APP Swedish and PSEN1 M146V) and one tau mutation, P301L of frontotemporal dementia. These mice display both plaque and tangle pathology. Cognitive impairments occur several months prior to plaques and tangles [33, 38]. The homozygous 3xTg-AD mice were obtained from Dr. Frank LaFerla (University of California, Irvine) through the Jackson Laboratory (New Harbor, ME, USA). Mice were housed and bred in accordance with approved protocols from our Institutional Animal Care and Use Committee (IACUC), according to the PHS Policy on Human Care and Use of Laboratory animals (revised January 2013). This study was performed on homozygous 3xTg-AD and genetic background matched 129sv WT female mice. Female 3xTg-AD mice were chosen because previous studies demonstrated consistent and higher Aβ burden and worse cognitive performance in female 3xTg-AD than male mice [39–41]. Mice were group-housed (4 animals per cage) with a 12:12 h light/dark cycle and with ad libitum access to food and water.
Treatment of animals with P021
We treated from the day of the delivery of pups till their weaning on postnatal day 21 (PND 21) their 2–3-month-old 3xTg-AD and wild-type (WT) mother mice with compound P021 in mouse chow, or as control with vehicle feed, and the female pups were separated from the dams and kept on the same P021 or vehicle diet till PND120 (Fig. 1). Treatment was administered as 60 nmol peptide/g formulated diet (Research Diets; New Brunswick, NJ). The vehicle-treated control animals received the same diet but without the peptide. On average, each mouse consumed ∼2.7 g diet/day. The body weights of mice were measured every week from the second week.
Behavioral procedures
Morris water maze
The Morris water maze is a behavioral procedure mostly used to study spatial learning and memory, a hippocampal [42]. A total of 42 female mice were subjected to the Morris water maze task after they reached the age of 90 days. The test was performed in a circular white pool (with a diameter of 180 cm and a height of 60 cm) filled with nontoxic white dye-tinted water and maintained at room temperature (20±1°C). The maze was designed with two virtual principal axes, with each line bisecting the maze perpendicular to the other one to divide the maze into four equal quadrants. The end of each line demarcates four cardinal points: north, south, east, and west. A platform was positioned in the middle of one of the quadrants submerged 1 cm below the water surface. Each mouse performed 4 trials for 4 consecutive days from semi-random start positions to find the hidden platform [43]. Each trial was terminated as soon as the mouse climbed onto the hidden platform. If a mouse failed to find the platform within 90 s, it was gently guided to the platform. At the end of each trial, the mouse was left on the platform for 20 s, then removed, dried, and returned to its home cage. A 90 s probe test without platform was performed 24 h after the last trial.
Novel object recognition test
The novel object recognition memory task which mainly requires perirhinal cortex [44] was used to evaluate short term/episodic memory in mice through an evaluation of the difference in the exploration time of novel and familiar objects [45]. The testing apparatus was a classic open field (i.e., a PVC square arena, 50×50 cm, with walls 40 cm high). The open field was placed in a part of the room separated from the investigator and was surmounted by a video camera connected to a computer. The general procedure consisted of three different phases: a habituation phase (4 sessions of 10 min each on four consecutive days), a sample phase (5th day), and a test phase (5th day). On the first four days, the mice were individually submitted each day to a familiarization session of 10 min during which they were introduced in the empty arena in order to become familiar with the apparatus. On the fifth day, each mouse was first submitted to the sample phase where two identical objects were placed in a symmetric position from the center of the arena, and the mouse was allowed to freely explore the objects for 8 min. After a 20 min delay during which the mouse was returned to its home cage, the animal was reintroduced in the arena to perform the test phase. During the test phase, the mouse was exposed to two objects for another 10 min: a familiar object (previously presented during the sample phase) and a new object, placed at the same location as that of other object during the sample phase. Data collection was performed using a video tracking system (ANY-Maze software, version 4.5, Stoelting Co., Wood Dale, IL, USA), that was used to manually score exploratory behavior including time spent exploring the new object and time spent exploring the old object. Each object is assigned a zone and a keyboard button to be identified. The researcher only needs to press the key at the beginning or the end of experiment. The object discrimination was evaluated by the index [DI = (T n –T f)/(T n + T f)] during the test phase.
Tissue processing
After completion of the behavioral tasks at postnatal day 120, Female animals were perfused, and brain tissue was collected for immunohistochemical and biochemical analysis. The animals were anesthetized with avertin (600 mg/kg) and trans cordially perfused with 0.1 M phosphate buffered saline (PBS). After perfusion, the brains were removed from the skull immediately. Hippocampus and cerebral cortex were dissected from the left hemisphere, immediately frozen on dry ice, and then stored at –80°C for biochemical analysis. The complete right hemisphere was immersion fixed in 4%paraformaldehyde in 0.1 M PBS for 24–48 h for immunohistochemical staining.
Western blots
The tissue from left cortex stored at –80°C from each mouse was homogenized in a Teflon-glass homogenizer to generate 10%(w/v) homogenate. The pre-chilled homogenization buffer contained 50 mM Tris–HCl (pH 7.4), 8.5%sucrose, 2 mM EDTA, 2 mM EGTA, 10 mM β-mercaptoethanol plus the following protease and phosphatase inhibitors: 0.5 mM AEBSF, 10μg/mL aprotinin, 10μg/mL leupeptin, 4μg/mL pepstatin, 5 mM benzamidine, 20 mM β-glycerophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, and 100 nM okadaic acid. Each homogenate was boiled in 2×Laemmli sample buffer for 5 min, and protein concentration was measured by Pierce™ 660 nm protein assay (Thermo Scientific, Rockford, IL, USA).
The samples were resolved by 10%SDS-PAGE and electro-transferred onto Immobilon-P membranes (EMD Millipore, Billerica, MA, USA). The blots were then probed with primary antibodies (Table 1) and developed with the corresponding HRP-conjugated secondary antibody and enhanced chemiluminescence kit (Pierce Biotechnology). Densitometric quantification of protein bands in Western blots was analyzed using Multi Gauge version 3.0 software (FUJIFILM North America, Valhalla, NY, USA).
Table 1
Primary antibodies used in this study
| Antibody | Specificity | Species | Type | Dilution | Source |
| CNTF | CNTF | R | Mono- | 1:1000 WB | Abcam (ab46172) |
| BDNF | BDNF | R | Mono- | 1:1000 WB | Santa Cruz Biotechnology |
| TrkB | TrkB | R | Mono- | 1:1000 WB | Abcam (ab33655) |
| GAPDH | GAPDH | R | Poly- | 1:1000 WB | Santa Cruz Biotechnology |
| GluR1 | GluR1 | R | Mono- | 1:1000 WB | EMD Millipore |
| Synaptophysin | Synaptophysin | M | Mono- | 1:3000 WB | Millipore |
| SMI 52 | MAP2 | M | Mono- | 1:4000 WB | Sternberger Monoclonals Inc |
| PSD95 | PSD95 | R | Poly- | 1:1000 WB | Cell Signaling Technology |
| NR1 | NMDAR1 | R | Mono- | 1:500 WB | Abcam |
| GluR 2/3 | GluR2 + 3 | R | Mono- | 1:2000 WB | Abcam |
| Synapsin-1 | Synapsin 1 | R | Poly- | 1:40000 WB | Enzo Life Sciences |
| CREB | CREB | M | Mono- | 1:1000 WB | Cell Signaling Technology |
| p-CREB | Ser133 | R | Mono- | 1:1000 WB | Cell Signaling Technology |
| SMI22 | GFAP | M | Mono- | 1:2000 WB | Sternberger Monoclonals Inc. (Lutherville, MD) |
Mono-, monoclonal; Poly-, polyclonal; p-, phosphorylated; M, mouse; R, rabbit; WB, western blot.
Reverse transcription-polymerase chain reaction analysis (RT-PCR)
The total RNA from left hippocampus was extracted by using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and reversed transcribed using SuperScript III first-strand synthesis system for reverse transcription-polymerase chain reaction (Thermo Fisher Scientific) according to the manufacturer’s instructions. Using the Master-cycler Nexus Thermal Cycler (Eppendorf, Hamburg, Germany), reactions were carried out at 94° for 5 min for initial denaturation and then at 94° for 30 s, 57° for 30 s, and 72° for 45 s. After 38 cycles of amplification, additional extensions were done at 72° for 5 min. Products were resolved and examined by 2%agarose gel electrophoresis. The RT-PCR primers used in this study are shown in Table 2.
Table 2
PCR primers used in this study
| Gene name | Sequence (5’ to 3’) | |
| BDNF | Forward | AGCAGAGTCCATTCAGCACC |
| Reverse | CCGCTAGGAAGCCAACTTCA | |
| CNTF | Forward | GAGAGCCATTATGGGGCCAA |
| Reverse | ACGTGGGTCAACCCTACTTG | |
| NR1 | Forward | ACTCCCAACGACCACTTCAC |
| Reverse | GTAGACGCGCATCATCTCAA | |
| NR2A | Forward | CTCTGATAATCCTTTCCTCCAC |
| Reverse | GACCGAAGATAGCTGTCATTTACT | |
| NR2B | Forward | TCCATCAGCAGAGGTATCTACAG |
| Reverse | CCGTTGACTCCAGACAGGTT | |
| GluR1 | Forward | CAATCACAGGAACATGCGGC |
| Reverse | TCTCTGCGGCTGTATCCAAG | |
| PSD95 | Forward | TCTGTGCGAGAGGTAGCAGA |
| Reverse | AAGCACTCCGTGAACTCCTG | |
| Synapsin | Forward | CAGCACAACATACCCTGTGG |
| Reverse | GGTCTTCCAGTTACCCGACA | |
| Synaptophysin | Forward | TGCCAACAAGACGGAG |
| Reverse | GGCGGATGAGCTAACT | |
| MAP2 | Forward | TTGGTGCCGAGTGAGAAGA |
| Reverse | GTCTGGCAGTGGTTGGTTAA |
Statistical analysis
The statistical analyses were conducted using SPSS version 17.0 (© SPSS Inc., 1989–2007, Chicago, IL, USA), and GraphPad Prism version 8.0 (GraphPad Software Inc., La Jolla, CA, USA). Data are presented as mean±S.E.M. The normality of the data was determined using Kolmogorov–Smirnov test. For analysis involving multiple groups, three-way or two-way ANOVA with post hoc Tukey’s multiple comparisons test (as indicated) was used. Further intergroup comparisons were also performed using unpaired, two-tailed t tests. For all purposes, p < 0.05 was considered as statistically significant.
RESULTS
Treatment with P021 initiated during early development prevents memory deficits during adult age in 3xTg-AD mice
Our previous studies showed that P021 treatment from embryonic day 8 to postnatal day 21 could prevent some of the Down’s syndrome features in Ts65Dn trisomic mice [31] and cognitive deficits and Alzheimer-like pathology in 3xTg-AD mice [34]. In the present study, we investigated whether initiation of P021 treatment at birth can also prevent neuroplasticity deficits and cognitive impairment in 3xTg-AD mouse model of AD. We administered P021 from postnatal day 1 to adult age and studied its effect on the behavior and the neuronal plasticity (Fig. 1). Unlike CNTF which is known to cause aneroxia and severe muscle cramps, P021 treatment did not show any apparent adverse effects, such as motor deficits, nor did it affect body weight (Fig. 2A).
Fig. 2
Treatment with P021 started at birth prevents cognitive impairment in 3xTg-AD mice. Treatment with P021 for 120 days had no significant effect on body weight as compared with the Vehicle treatment in 3xTg-AD or WT mice (A). Morris water maze (B-F) and novel object recognition tests (G-H) were carried out from day 90 onwards. B) Escape latency during 4 consecutive days training with hidden platform was higher in 3xTg-AD than WT controls, and treatment with P021 did not reverse it (three-way ANOVA, Days, F (3, 112)=48.93, p = 0.0001; genotype, F (1, 112)=29.99, p = 0.0001; treatment, F (1, 112) = 0.03422, p = 0.8536; two-tailed t tests, WT-Vh versus 3Tg-Vh, p = 0.0405). During the probe trial, while no significant difference was found in first entrance into the target zone (C), number of crossings the target (D) among the four groups of animals, (E) 3×Tg-AD-Vh spent lesser time than the WT-Vh mice and the 3xTg-AD-P021 mice in the target quadrant (Two-way ANOVA, Interaction F (1, 32) = 3.927, p = 0.0562; Treatment F (1, 32) = 10.27, p = 0.0031; genotype F (1, 32) = 4.160, p = 0.0497; Tukey’s multiple comparisons test, WT-Vh versus 3xTg-Vh, p = 0.0060; 3xTg-Vh versus 3xTg-P021, p = 0.0216). G) In the one-trial novel object recognition test, the mice spent equal amounts of time exploring the two identical objects during the 8-min sample phase (Three-way ANOVA, genotypes x treatments x objects). H) One of the two objects was then changed to a novel object during a 5 min test phase. 3×Tg-AD-Vh mice spent less time exploring the novel object than the familiar object in the test phase compared with the WT and 3xTg-AD-P021mice (Two-way ANOVA, Interaction F (1, 28) = 11.02, p = 0.0025, Treatment F (1, 28)=20.93, p = 0.0001, genotype F (1, 28) = 0.9513, p = 0.3377; Tukey’s multiple comparisons test, WT-Vh versus 3xTg-Vh, p = 0.0003; 3xTg-Vh versus 3xTg-P021, p = 0.0002). Data were analyzed by three-way or two-way ANOVA and are shown as mean±S.E.M. n = 10 for WT-Vh; n = 10 for WT-P021; n = 11 for 3xTg-Vh; n = 11 for 3xTg-P021. *p < 0.05, **p < 0.01.
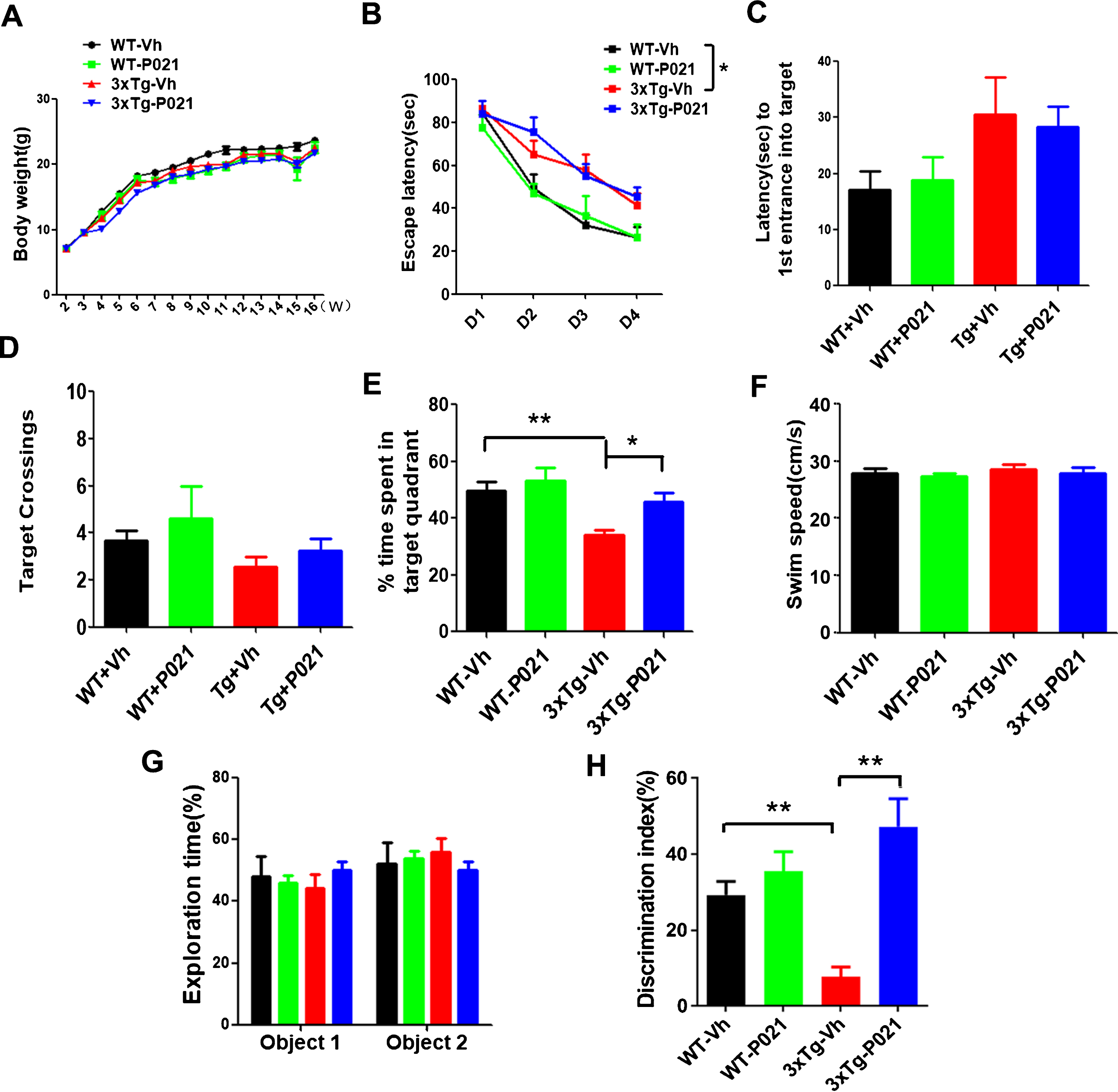
To investigate whether chronic treatment with P021 started from the day of birth can rescue spatial cognitive impairment in 3×Tg-AD mice, Morris water maze test was conducted. While the P021 treatment showed no significant effect on escape latency (Fig. 2B) and no difference in the first entrance into the target platform (Fig. 2C) and number of target crossings (Fig. 2D) in probe test, the P021-treated 3xTg-AD mice significantly spent more time in the target quadrant than the mice treated with vehicle diet (Fig. 2E) and there was no difference in the swim speed between P021- and vehicle-treated mice (Fig. 2F). Altogether, these results suggest a prevention of spatial cognitive impairment by P021 in 3xTg mice.
To examine whether treatment with P021 could rescue the short-term memory impairment in 3×Tg-AD mice, we conducted a one-trial novel object recognition task with a 20 min interval between the sample phase and the test phase. In the one-trial novel object recognition task, mice spent equal amounts of time exploring the two identical objects during the 8 min sample phase (Fig. 2G). After 20 min, one object during sample phase was replaced with a novel object during a 5 min test phase. 3xTg-Vh mice showed impairment by spending lesser time than WT-Vh mice in exploring the novel object in the novel object recognition test, whereas 3×Tg-AD mice treated with P021 spent more time than the mice treated with vehicle only in exploring the novel object than the familiar object in the test phase (Fig. 2H). These data clearly indicate short-term memory impairment in 3×Tg-AD mice was prevented by treatment with P021 started at the time of the birth. Unlike cognitive improvement produced by P021 treatment in wild type middle to old age mice and aged rats in previous studies [24, 26, 46], the 3-4 month old WT mice tested in the present study did not show any significant improvement in cognition. This lack of effect of P021 treatment in 3-4-month-old mice was probably due to optimal cognitive activity at this age.
P021 treatment increases levels of transcription and expression of BDNF in mouse brain
Previously we reported that the beneficial effect of P021 on neurogenesis and cognitive function in aged 3xTg-AD mice and rats occurs through BDNF/TrkB pathway [26, 27]. To learn whether the preventive therapeutic effect of P021 in the present study involved the BDNF/TrkB pathway or CNTF, we investigated changes in levels of transcription and expression of BDNF and CNTF.
To assess the transcription level of BDNF and CNTF, we performed RT-PCR and found that BDNF was dramatically decreased in the hippocampus of 3xTg-Vh mice compared with WT control mice, and 3xTg-P021 mice, while no significant changes were detected in the level of CNTF among the four groups of animals (Fig. 3A, B). Treatment with P021 increased the protein level of BDNF in both WT mice and 3xTg-AD mice; P021 treatment did not show any significant effect on levels of pro-BDNF, TrkB, or CNTF in 3xTg-AD mice (Fig. 3C, D). These results indicate that initiation of P021 treatment at birth can increase the level of BDNF in mice.
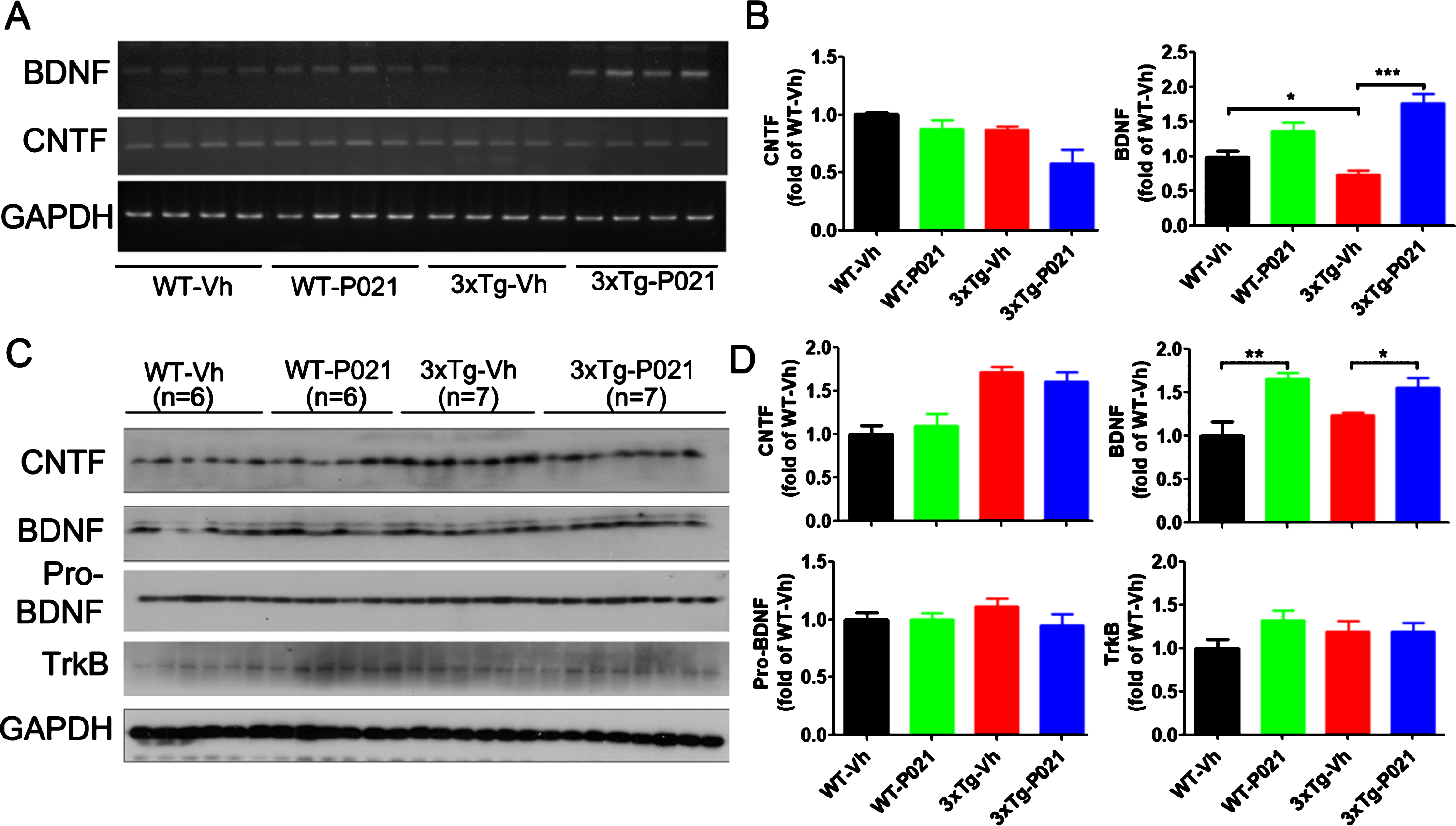
Fig. 3
Treatment with P021 started at birth increases mRNA and protein levels of BDNF but not CNTF. After 120 days treatment with P021- or vehicle- diet, the mice were killed and mRNA level in hippocampus (A, B) and protein level in cortex (C, D) were analyzed. Densitometric quantifications of RT-PCR (B) and western blots (D) normalized with GAPDH. BDNF mRNA level (A, B) significantly decreased in the 3×Tg-AD-Vh compared with the WT-Vh (p = 0.0405) and 3xTg-AD-P021 mice (p = 0.0001). There was no difference in the level of CNTF mRNA among the four groups of animals. P021 treatment increased the protein level of BDNF in WT-P021 mice (p = 0.0016) and 3xTg-AD-P021 mice (p = 0.0172) (C, D); no significant difference was found in CNTF, pro-BDNF and TrkB among the four groups of mice. Data were analyzed by two-way ANOVA followed by a Tukey’s multiple comparisons test and further intergroup comparisons were performed using unpaired, two-tailed t tests. Data are expressed as mean±S.E.M. n = 10 each for WT-Vh and WT-P021; n = 11 each for 3xTg-Vh and 3xTg-P021. *p < 0.05, **p < 0.01, ***p < 0.001.
P021 treatment activates BDNF-TrkB pathway in 3xTg-AD mice
The activation of the BDNF-TrkB pathway is important in the development of memory and the growth of neurons. Here we investigated whether P021 activates the BDNF-TrkB pathway by determining the levels of downstream factors. Western blots results showed that relative levels of phosphorylated-PLC (p-PLC), p-CaMK2, p85 subunit of PI3K, p-Akt, p-GSK3β, p-MEK, and p-ERK were all increased significantly after P021 treatment in the cortex of 3xTg-mice, although only level of p-Akt was notably reduced in 3xTg-Vh mice compared with WT mice (Fig. 4A, B). Altogether, these findings revealed that the initiation of treatment with P021 at birth can activate BDNF-TrkB pathway in 3xTg-AD mice.
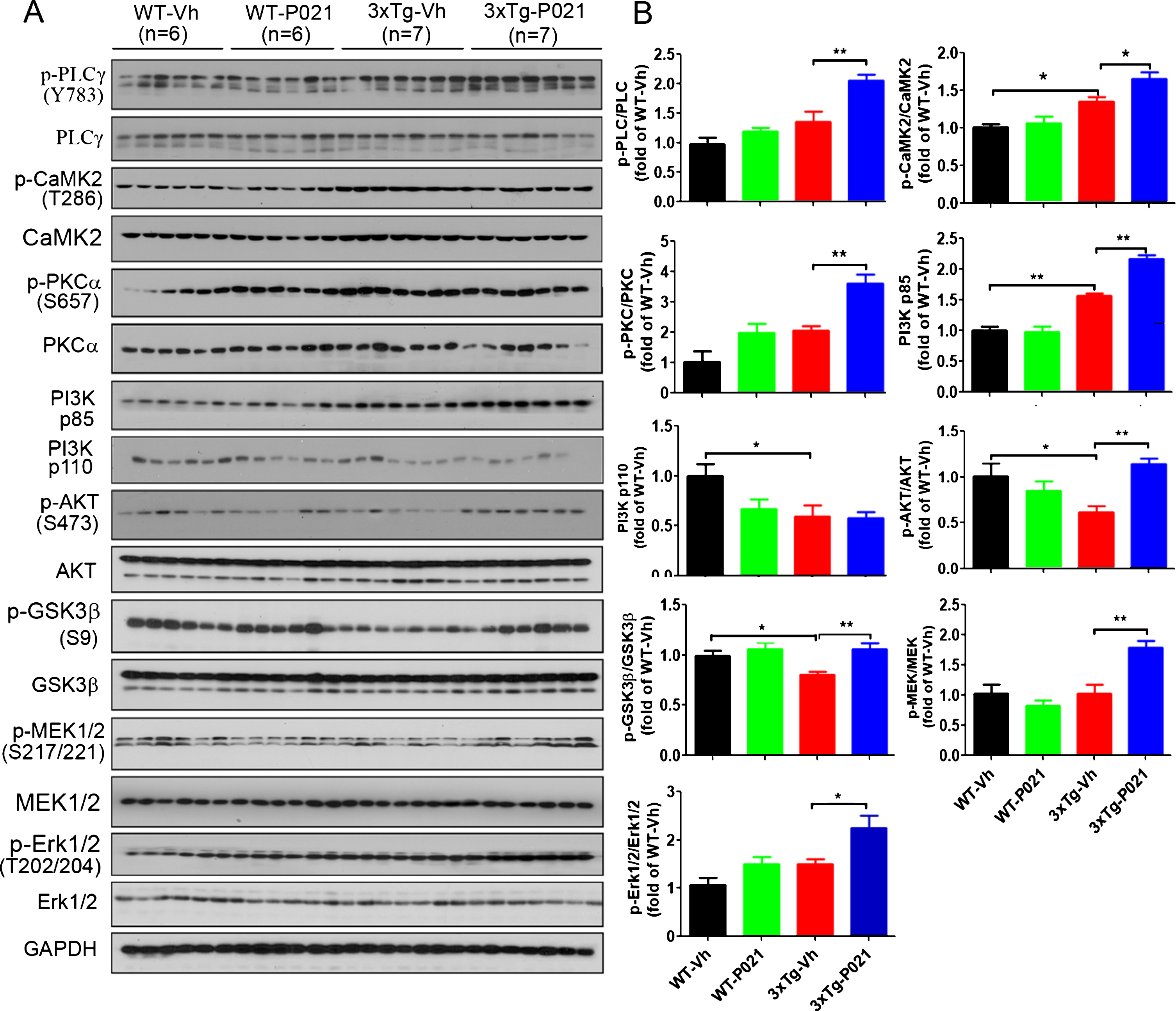
Fig. 4
P021 treatment activates BDNF-TrkB signaling pathway in 3xTg-AD mice. A) Protein levels of PLCγ, CaMK2, PKC, PI3K, AKT, GSK3β, MEK, and ERK in cortex were determined by western blots. B) Densitometric quantifications of blots were normalized to those of GAPDH blots employed as a loading control. P021 treatment increased the ratios of p-PLCγ/PLCγ (p = 0.0016), p-CaMK2/CaMK2 (p = 0.0107), p-PKC/PKC (p = 0.0004), PI3K p85 (p = 0.0001), p-AKT/AKT (p = 0.0001), p-GSK3β/GSK3β (p = 0.0047), p-MEK/MEK (p = 0.0019), and p-ERK/ERK (p = 0.0208) in the 3×Tg-AD mice. Data were analyzed by two-way ANOVA followed by a Tukey’s multiple comparisons test and are expressed as mean±S.E.M. *p < 0.05, **p < 0.01.
P021 treatment modulates changes in neuroplasticity in the hippocampus in 3xTg-AD mice
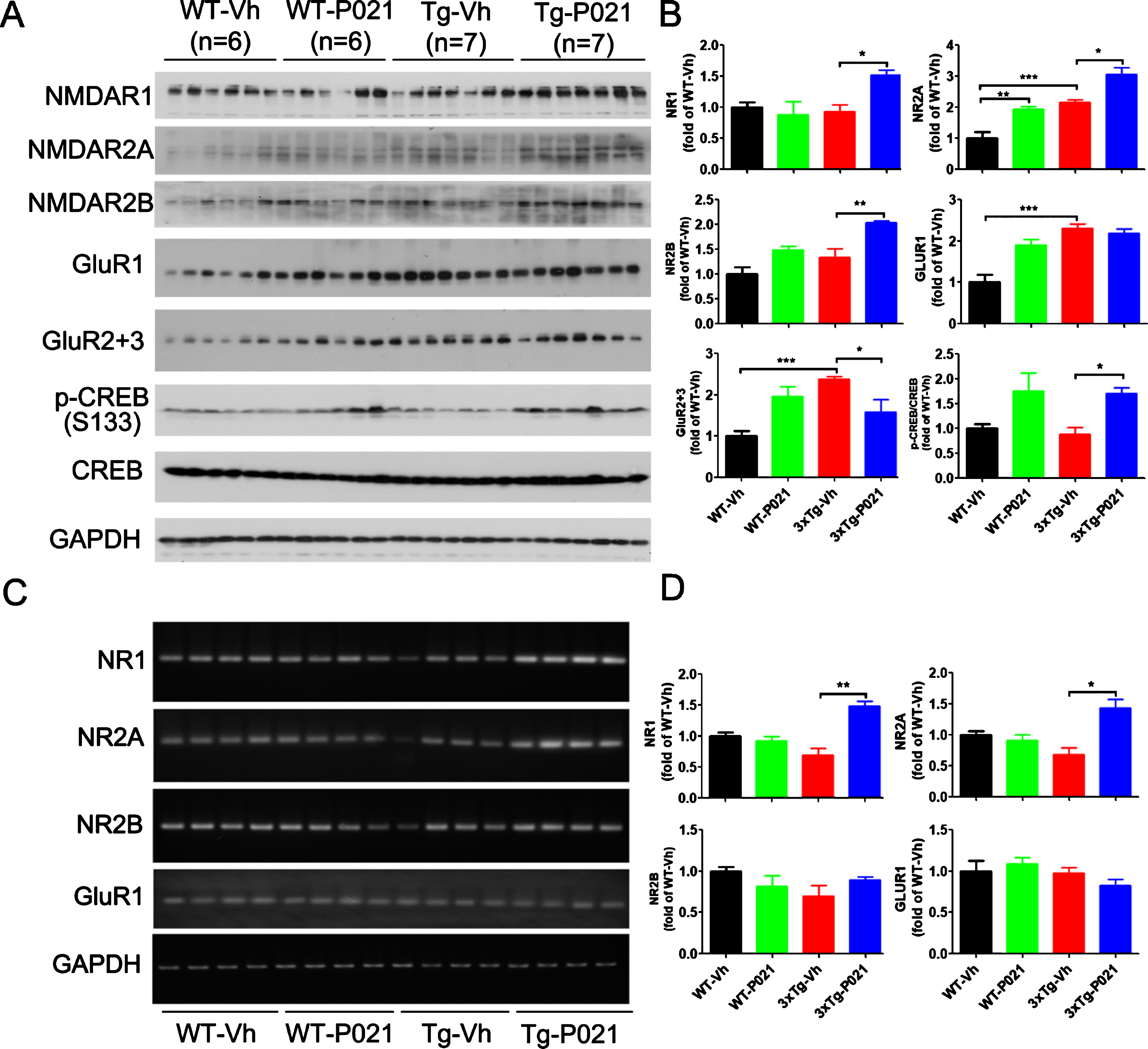
Glutamate is the brain’s major excitatory neurotransmitter, and its release can trigger the depolarization of postsynaptic neurons. AMPA and NMDA receptors are two major ionotropic receptors that are especially suspected of being involved in learning and memory. To learn the effect of P021 on glutamate receptors, we determined their levels in P021- and vehicle-treated control mice by western blots. We found an increase in levels of NR2A, GluR1, GluR2 + 3, and a decrease trend in the level of p-CREB in 3xTg-Vh mice; the level NR2A was further elevated after P021 treatment, while the decrease of p-CREB was reversed in 3xTg-P021 mice. Besides, levels of NR1 and NR2B were significantly increased by P021 treatment, and the level of NR2A was higher in the WT-P021 mice than in WT-Vh mice (Fig. 5A, B). Furthermore, we studied mRNA levels of glutamate receptors using RT-PCR and found that P021 significantly increased the levels of NR1 and NR2A mRNAs and there was a trend of increase in NR2B in the hippocampus, while no statistical difference of NR2B and GluR1 were detected (Fig. 5C, D). Taken together, these results suggest that P021 could promote the synaptic plasticity through modulating their mRNA levels, especially those of NR1 and NR2A, and could also enhance the phosphorylation of CREB in 3xTg mice.
Fig. 5
P021 treatment increases the NMDA receptor-dependent CREB activation in 3×Tg-AD mice. Protein levels in cortex (A) and the mRNA levels in hippocampus (C) were analyzed by Western blots and RT-PCR, respectively. Densitometric quantifications of western blots (B) and RT-PCR (D) were normalized against the loading control GAPDH. The protein expressions of NMDAR1 (p = 0.0107), NMDAR2A (p = 0.0156), NMDAR2B (p = 0.0027) and the ratio of p-CREB/CREB (p = 0.0126) were increased after treatment with P021 (A, B), whereas GluR2 + 3 was decreased (p = 0.0249). The mRNA levels of NMDAR1 (p = 0.0012) and NMDAR2A (p = 0.0155) in hippocampus were increased after treatment with P021 (C, D). Data were analyzed by two-way ANOVA followed by a Tukey’s multiple comparisons test and are expressed as mean±S.E.M. *p < 0.05, **p < 0.01.
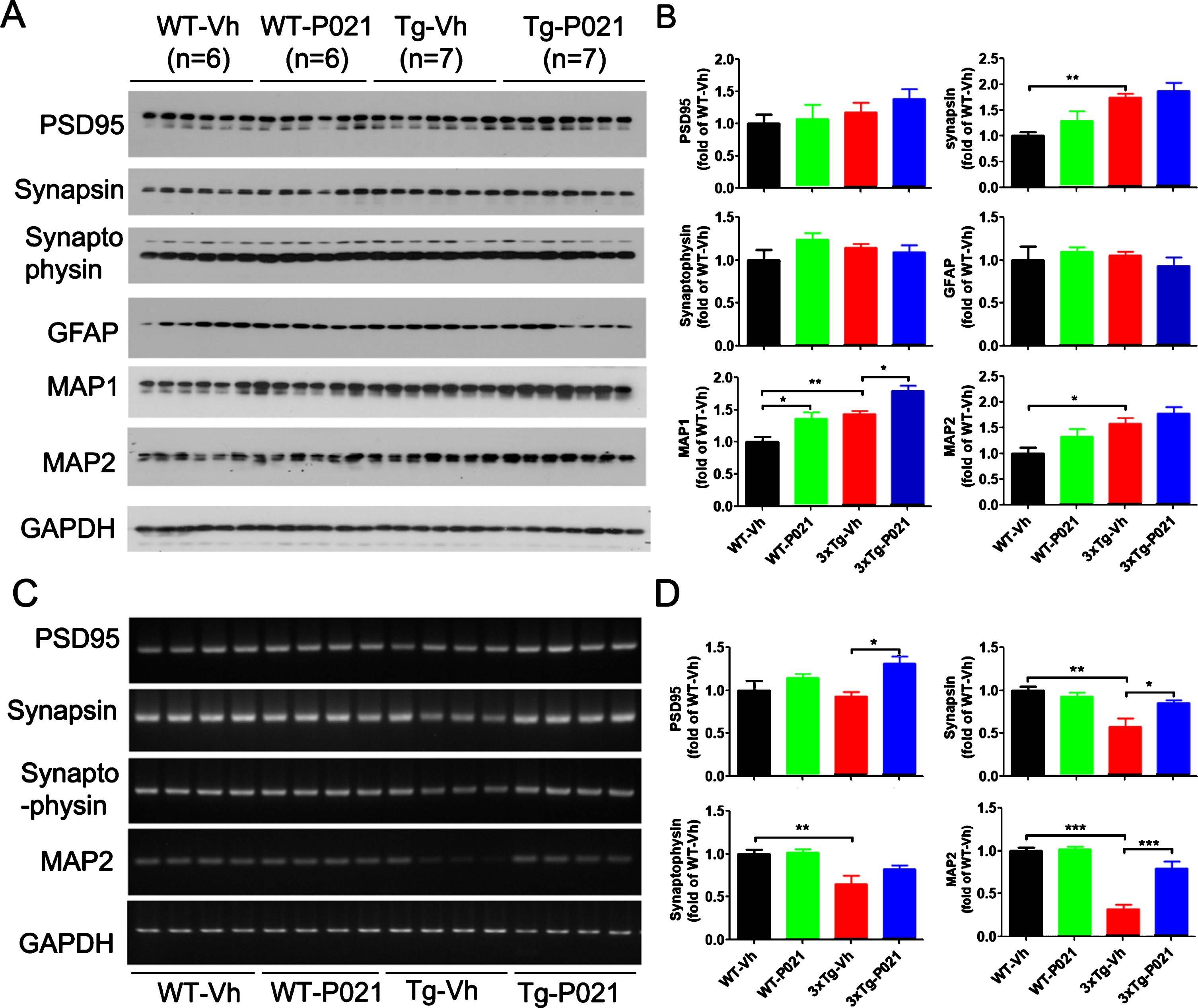
BDNF-TrkB signaling is reported to upregulate translational activity at the synapse. Along with the synaptic markers we also investigated the expression levels of the dendritic marker microtubule-associated protein (MAP) HMW-MAP2, and MAP1b which in adult brain is localized in dendritic spines [47, 48]; both MAPs serve as very useful putative synaptic markers. Western blot results showed that levels of synapsin 1, MAP1b, and MAP2 were increased in the cortex of 3xTg-Vh mice. While 3xTg-P021 mice exhibited an increase in the level of MAP1b, no significant differences in expressions of PSD95 or synaptophysin were found (Fig. 6A, B). We studied astrocytes, a key cell type of neuroinflammation in the brain, with antibody against glial fibrillary acidic protein (GFAP) and found no significant changes in the level of GFAP among the four groups of animals (Fig. 6A, B). In addition, we determined the mRNA levels of the above synaptic markers. We found a trend decrease of PSD95, and robust decrease of synapsin 1, synaptophysin and MAP2 mRNA in the hippocampus in 3xTg-vh mice, and P021 treatment remarkably increased the mRNA levels of synapsin 1, PSD95, and MAP2 (Fig. 6C, D). Altogether, these findings indicated that the beneficial therapeutic effect of P021 on cognitive impairment could be due to the increase in neuronal plasticity.
Fig. 6
P021 treatment rescues dendritic and synaptic deficits in 3×Tg-AD mice. Protein levels in cortex (A) and the mRNA levels in hippocampus (C) were analyzed. Densitometric quantifications of western blots (B) and RT-PCR (D) were normalized against the loading control GAPDH. 3xTg-Vh mice showed increase of protein expressions of synapsin, MAP1, and MAP2, while MAP1 was further increased by P021 treatment (p = 0.0117) (A, B). The mRNA levels (C, D) of synapsin, synaptophysin, MAP1, and MAP2 were decreased, and treatment with P021 rescued the levels of PDS95 (p = 0.0167), synapsin (p = 0.0374), and MAP2 (p = 0.0003) in 3xTg-AD mice. Data were analyzed by two-way ANOVA followed by a Tukey’s multiple comparisons test and are expressed as mean±S.E.M. n = 4. *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
The present study shows for the first time that neurotrophic treatment initiated during early postnatal development can not only prevent the Alzheimer-like cognitive deficit but also ameliorate synaptic dysfunction in adult 3xTg-AD mice. These data suggest that adjusting the brain milieu by providing neurotrophic support at critical period of brain development can be potentially an effective therapeutic strategy for prevention of the disease later in life in genetic cases of AD and related tauopathies.
To date, there are no effective interventions that can cure, halt or prevent the progression of AD. Both in human AD cases and in various transgenic mouse models of this disease including 3xTg-AD mice used in the present study, cognitive dysfunction and loss of neuronal plasticity are known to precede Aβ and tau pathologies [49–51]. Thus, an ideal drug would be one that can prevent and rescue neurodegeneration and cognitive impairment.
AD is characterized by abnormalities in synaptic plasticity associated with deficits in BDNF function [52–54]. Studies have shown reduction (21–30%) in pro-BDNF in patients with mild cognitive impairment and major reduction (40%) in terminal patients [53]. The blood level of BDNF is decreased in both AD and mild cognitive impairment patients [55]. Decreased BDNF protein levels were also reported in 3xTg-AD transgenic mouse line [56]. BDNF mRNA and protein levels were found to be reduced in postmortem brain samples of AD patients [57–59]. Although neurotrophic factors have been suggested as essential contributors to the etiology of AD, BDNF itself does not have pharmacokinetics suitable for systemic administrations due to its short plasma half-life and poor BBB penetration [60].
P021 is a small water-soluble compound that was administered successfully orally in diet in the present study. The compound has plasma half-life > 3 h and stability of ∼90%in gastric and ∼100%in intestinal juice and is BBB permeable [27, 33]. Thus, this small molecule mimetic overcomes the main limitation associated with therapeutic usage of neurotrophic factors such as CNTF and BDNF as peripherally administered neurotrophic factors poorly reach the central nervous system. In previous studies we showed that P021 can enhance dentate gyrus neurogenesis and memory processes via inhibiting leukemia inhibitory factor (LIF) signaling pathway [26, 30, 31]. In the present study we found that initiation of P021 treatment at birth can increase the transcription and the translation of BDNF in the brain in 3xTg-AD mice. The present study provides a novel evidence that treatment with P021 initiated during early development can prevent the Alzheimer-like cognitive impairment and synaptic dysfunction, probably via increased BDNF expression which activates the BDNF-TrkB pathway.
BDNF protein is widely distributed in the neuronal cell body, axons, and dendrites in the CNS [61], and is also widely involved in neural plasticity important for learning and memory [62, 63]. BDNF signaling is elicited when it binds to TrkB which modifies its downstream targets leading to various neuronal processes. BDNF activates the signaling cascades such as PLC/PKC, MEK/ERK, and PI3K/Akt [64]. Activation of PI3K/Akt pathway through BDNF/TrkB interaction inhibits cell apoptosis and leads to protein synthesis. The PI3K/Akt pathway also modulates NMDAR-dependent synaptic plasticity [65–67] and regulates adult neural hippocampal progenitor proliferation and differentiation through activation of CREB [68, 69]. The MEK/ERK signaling cascade regulates protein synthesis during neuronal differentiation [70]. It also actives CREB that is critical for early response gene expression [71, 72], for cytoskeleton protein synthesis [66], and dendritic growth and branching in hippocampal neurons [73, 74]. BDNF through PLCγ-dependent pathway evokes activation of CAMK2 and PKC, which subsequently increase Ca2 + ion concentration [75, 76], while the PKC dependent pathway is reported to enhance synaptic plasticity [77, 78].
The transcription of BDNF gene is regulated by CREB phosphorylation [79]. The phosphorylation of CREB is known to play a critical role in long-term synaptic plasticity and memory formation [80, 81]. The neuroprotective effect can also be achieved due to synaptic NMDAR stimulation and subsequent increase of the nuclear Ca2 + influx, which also results in activation of CREB and increase in expression of genes coding proteins involved in neuroprotection [82, 83]. In the present study, we found that treatment with P021 during early development can increase the expression of NMDAR and significantly enhance the phosphorylation of CREB in 3xTg-AD mice.
Synaptic plasticity, the property of synapses to undergo long-term changes in synaptic strength, is believed to be the cellular substrate of learning and memory [84, 85]. Cognitive deficits in AD are strongly associated with failure of hippocampal LTP [86], aberrant expression of synaptic proteins [87] in the hippocampus. In the present study, we found a remarkable increase in the mRNA levels of synapsin, PSD95, and MAP2 by treatment with P021 in 3xTg-AD mice. These data provide strong evidence that neurotrophic treatment during early development can exert long-lasting effect on synaptic plasticity and can enhance cognitive performance.
BDNF expression depends on various forms of cellular and synaptic activity, initiated by stimuli of different modalities [88]. In the present study, the P021 treatment during early development was found to increase BDNF gene expression in the hippocampus in 3xTg-AD mice. The relationship between BDNF expression level and stimulus-evoked cellular activity indicates reciprocal effect. In the present study, on the one hand, increased expression of BDNF by P021-induced inhibition of the binding of LIF to LIF/CNTF/gp130 tripartite receptor strengthened synaptic potentiation, improved performance in cognitive function, on the other hand, further increase in BDNF expression found was probably a consequence of glutamatergic NMDARs with subsequent activation of CREB and its binding to BDNF promoter and initiation of its transcription.
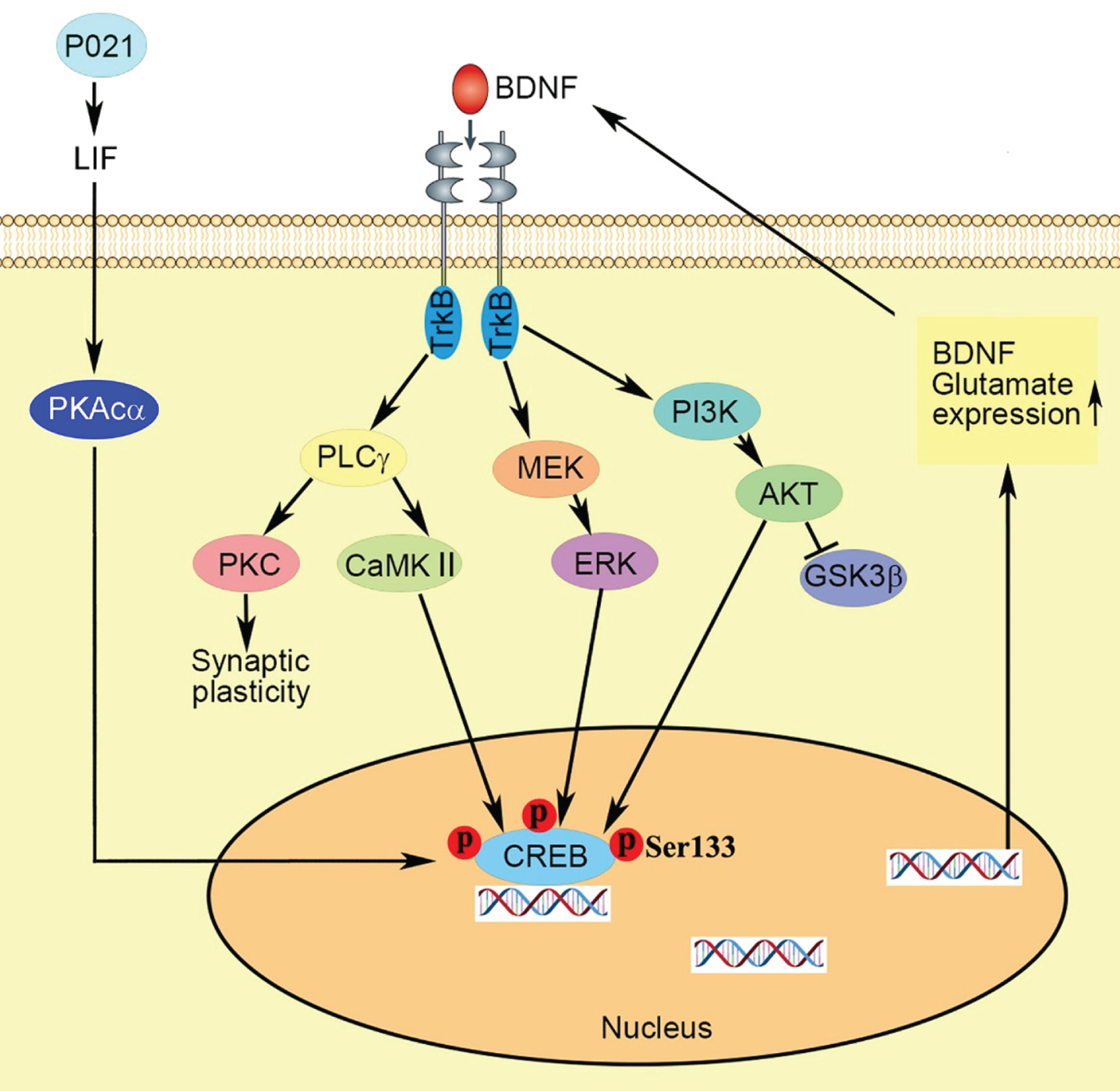
In summary (Fig. 7), we found neurotrophic P021 treatment initiated during early development can prevent the behavioral impairment and synaptic plasticity dysfunction in 3xTg-AD mice, probably via increased BDNF expression that modifies its downstream targets leading to various neuronal processes, increasing p-CREB and consequently expression of genes coding proteins and amelioration of synaptic protein deficit. Thus, adjusting the brain milieu by providing neurotrophic support at critical period of brain development can be potentially an effective therapeutic strategy for the prevention of AD-like neuronal plasticity deficit and cognitive impairment.
Fig. 7
Proposed mechanism of the therapeutic beneficial effect of treatment with P021 started from the day of the birth. Chronic treatment with P021 increases BDNF expression through LIF signaling/ increase in PKAcα and activation of CREB which activates the BDNF-TrkB pathways. The interaction between the receptor TrkB and neurotrophin activates three main intracellular signaling pathways. The MAPK/ERK kinase (MEK) -extracellular signal-regulated kinase (ERK) pathway promotes neuronal differentiation and growth, and the phosphatidylinositol 3-kinase (PI3K)–Akt pathway promotes survival and growth of neurons, and the PLCγ–Ca2 + pathway which stimulates protein kinase C (PKC) and Ca2 +-dependent protein kinase (CaMKII) enhances synaptic plasticity. Activation of CaMKII, ERK and Akt mediates the activation of transcription factor cyclic AMP-responsive element-binding protein (CREB) which enhances expressions of BDNF and glutamate receptors.
In our present study, we did not assay the level of P021 that reached the brain in mice which will have to wait future studies. The present study employed only female animals. Thus, the gender difference in response to the P021 treatment remains to be studied. In the present study we did not directly investigate whether the therapeutic beneficial effect of P021 in 3xTg-AD mice involved rescue of any neurotrophic deficit and/or was a result of increased neurotrophic activity. The fact that we also found increased in the level of BDNF and beneficial effect of P021 treatment on several markers of synaptic plasticity in the WT animals in the present study indicates that the treatment could be potentially beneficial due an increase in neurotrophic activity over and beyond normal level. While 3xTg-AD is an artificial over-expression transgenic mouse model of familial AD and frontolobar dementia, in previous studies we found therapeutic beneficial effect of P021 treatment in aged WT mice and rats, Ts 65Dn trisomic mouse model of Down’s syndrome, mouse model of traumatic brain injury, rat models of sporadic AD and autism and macular degeneration-like changes in aged mice and rats [24, 26, 29–31, 89, 90]. Because of the ability of P021 to promote neuro-regeneration and synaptic plasticity it has the potential to treat not only AD but also other neurodegenerative diseases and conditions. P021 treatment is apparently very safe. We did not find any negative effects of the compound either when the treatment was carried out during early development or at adult age and up to 18 months in mice. However, a double-blind placebo control human clinical trial remains to be carried out to learn the actual therapeutic potential of P021.
ACKNOWLEDGMENTS
We are thankful to Maureen Marlow for copyediting the manuscript. This work was supported in part by funds from the New York State Office for People with Developmental Disabilities, Jinan University, and Guangzhou Medical University.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1599r1).
REFERENCES
[1] | Iqbal K , Grundke-Iqbal I ((2011) ) Opportunities and challenges in developing Alzheimer disease therapeutics. Acta Neuropathol 122: , 543–549. |
[2] | ((2020) ) 2020 Alzheimer’s disease facts and figures. Alzheimers Dement 16: , 391–460. |
[3] | Wenk GL ((2003) ) Neuropathologic changes in Alzheimer’s disease. J Clin Psychiatry 64 Suppl 9: , 7–10. |
[4] | Ballinger EC , Ananth M , Talmage DA , Role LW ((2016) ) Basal forebrain cholinergic circuits and signaling in cognition and cognitive decline. Neuron 91: , 1199–1218. |
[5] | Glenner GG , Wong CW ((1984) ) Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: , 885–890. |
[6] | Grundke-Iqbal I , Iqbal K , Tung YC , Quinlan M , Wisniewski HM , Binder LI ((1986) ) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83: , 4913–4917. |
[7] | Ingelsson M , Fukumoto H , Newell KL , Growdon JH , Hedley-Whyte ET , Frosch MP , Albert MS , Hyman BT , Irizarry MC ((2004) ) Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62: , 925–931. |
[8] | Spires-Jones TL , Hyman BT ((2014) ) The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82: , 756–771. |
[9] | Tomlinson BE , Blessed G , Roth M ((1970) ) Observations on the brains of demented old people. J Neurol Sci 11: , 205–242. |
[10] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[11] | Perez-Nievas BG , Stein TD , Tai HC , Dols-Icardo O , Scotton TC , Barroeta-Espar I , Fernandez-Carballo L , de Munain EL , Perez J , Marquie M , Serrano-Pozo A , Frosch MP , Lowe V , Parisi JE , Petersen RC , Ikonomovic MD , Lopez OL , Klunk W , Hyman BT , Gomez-Isla T ((2013) ) Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136: , 2510–2526. |
[12] | Holmes C , Boche D , Wilkinson D , Yadegarfar G , Hopkins V , Bayer A , Jones RW , Bullock R , Love S , Neal JW , Zotova E , Nicoll JA ((2008) ) Long-term effects of Abeta42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 372: , 216–223. |
[13] | Tapiola T , Overmyer M , Lehtovirta M , Helisalmi S , Ramberg J , Alafuzoff I , Riekkinen P Sr. , Soininen H ((1997) ) The level of cerebrospinal fluid tau correlates with neurofibrillary tangles in Alzheimer’s disease. Neuroreport 8: , 3961–3963. |
[14] | Arriagada PV , Growdon JH , Hedley-Whyte ET , Hyman BT ((1992) ) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42: , 631–639. |
[15] | Giannakopoulos P , Herrmann FR , Bussiere T , Bouras C , Kovari E , Perl DP , Morrison JH , Gold G , Hof PR ((2003) ) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60: , 1495–1500. |
[16] | Hong S , Beja-Glasser VF , Nfonoyim BM , Frouin A , Li S , Ramakrishnan S , Merry KM , Shi Q , Rosenthal A , Barres BA , Lemere CA , Selkoe DJ , Stevens B ((2016) ) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: , 712–716. |
[17] | Koffie RM , Hyman BT , Spires-Jones TL ((2011) ) Alzheimer’s disease: Synapses gone cold. Mol Neurodegener 6: , 63. |
[18] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR Jr. , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[19] | Mehta D , Jackson R , Paul G , Shi J , Sabbagh M ((2017) ) Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs 26: , 735–739. |
[20] | Rajan KB , Wilson RS , Weuve J , Barnes LL , Evans DA ((2015) ) Cognitive impairment 18 years before clinical diagnosis of Alzheimer disease dementia. Neurology 85: , 898–904. |
[21] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[22] | Jack CR Jr. , Petersen RC , Xu YC , Waring SC , O’Brien PC , Tangalos EG , Smith GE , Ivnik RJ , Kokmen E ((1997) ) Medial temporal atrophy on MRI in normal aging and very mild Alzheimer’s disease. Neurology 49: , 786–794. |
[23] | Braak H , Braak E ((1996) ) Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol 92: , 197–201. |
[24] | Chohan MO , Li B , Blanchard J , Tung YC , Heaney AT , Rabe A , Iqbal K , Grundke-Iqbal I ((2011) ) Enhancement of dentate gyrus neurogenesis, dendritic and synaptic plasticity and memory by a neurotrophic peptide. Neurobiol Aging 32: , 1420–1434. |
[25] | Li B , Wanka L , Blanchard J , Liu F , Chohan MO , Iqbal K , Grundke-Iqbal I ((2010) ) Neurotrophic peptides incorporating adamantane improve learning and memory, promote neurogenesis and synaptic plasticity in mice. FEBS Lett 584: , 3359–3365. |
[26] | Bolognin S , Buffelli M , Puolivali J , Iqbal K ((2014) ) Rescue of cognitive-aging by administration of a neurogenic and/or neurotrophic compound. Neurobiol Aging 35: , 2134–2146. |
[27] | Kazim SF , Blanchard J , Dai CL , Tung YC , LaFerla FM , Iqbal IG , Iqbal K ((2014) ) Disease modifying effect of chronic oral treatment with a neurotrophic peptidergic compound in a triple transgenic mouse model of Alzheimer’s disease. Neurobiol Dis 71: , 110–130. |
[28] | Khatoon S , Chalbot S , Bolognin S , Puolivali J , Iqbal K ((2015) ) Elevated tau level in aged rat cerebrospinal fluid reduced by treatment with a neurotrophic compound. J Alzheimers Dis 47: , 557–564. |
[29] | Kazim SF , Iqbal K ((2016) ) Neurotrophic factor small-molecule mimetics mediated neuroregeneration and synaptic repair: Emerging therapeutic modality for Alzheimer’s disease. Mol Neurodegener 11: , 50. |
[30] | Chohan MO , Bragina O , Kazim SF , Statom G , Baazaoui N , Bragin D , Iqbal K , Nemoto E , Yonas H ((2015) ) Enhancement of neurogenesis and memory by a neurotrophic peptide in mild to moderate traumatic brain injury. Neurosurgery 76: , 201–214; discussion 214-205. |
[31] | Kazim SF , Blanchard J , Bianchi R , Iqbal K ((2017) ) Early neurotrophic pharmacotherapy rescues developmental delay and Alzheimer’s-like memory deficits in the Ts65Dn mouse model of Down syndrome. Sci Rep 7: , 45561. |
[32] | Iqbal K , Liu F , Gong CX ((2016) ) Tau and neurodegenerative disease: The story so far. Nat Rev Neurol 12: , 15–27. |
[33] | Baazaoui N , Iqbal K ((2018) ) A novel therapeutic approach to treat Alzheimer’s disease by neurotrophic support during the period of synaptic compensation. J Alzheimers Dis 62: , 1211–1218. |
[34] | Wei W , Wang Y , Liu Y , Dai CL , Tung YC , Liu F , Iqbal K ((2020) ) Prenatal to early postnatal neurotrophic treatment prevents Alzheimer-like behavior and pathology in mice. Alzheimers Res Ther 12: , 102. |
[35] | Baazaoui N , Iqbal K ((2017) ) Prevention of dendritic and synaptic deficits and cognitive impairment with a neurotrophic compound. Alzheimers Res Ther 9: , 45. |
[36] | Baazaoui N , Iqbal K ((2017) ) Prevention of amyloid-beta and tau pathologies, associated neurodegeneration, and cognitive deficit by early treatment with a neurotrophic compound. J Alzheimers Dis 58: , 215–230. |
[37] | Oddo S , Caccamo A , Kitazawa M , Tseng BP , LaFerla FM ((2003) ) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging 24: , 1063–1070. |
[38] | Billings LM , Oddo S , Green KN , McGaugh JL , LaFerla FM ((2005) ) Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45: , 675–688. |
[39] | Clinton LK , Billings LM , Green KN , Caccamo A , Ngo J , Oddo S , McGaugh JL , LaFerla FM ((2007) ) Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis 28: , 76–82. |
[40] | Carroll JC , Rosario ER , Kreimer S , Villamagna A , Gentzschein E , Stanczyk FZ , Pike CJ ((2010) ) Sex differences in beta-amyloid accumulation in 3xTg-AD mice: Role of neonatal sex steroid hormone exposure. Brain Res 1366: , 233–245. |
[41] | Hirata-Fukae C , Li HF , Hoe HS , Gray AJ , Minami SS , Hamada K , Niikura T , Hua F , Tsukagoshi-Nagai H , Horikoshi-Sakuraba Y , Mughal M , Rebeck GW , LaFerla FM , Mattson MP , Iwata N , Saido TC , Klein WL , Duff KE , Aisen PS , Matsuoka Y ((2008) ) Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res 1216: , 92–103. |
[42] | Morris RG , Garrud P , Rawlins JN , O’Keefe J ((1982) ) Place navigation impaired in rats with hippocampal lesions. Nature 297: , 681–683. |
[43] | Vorhees CV , Williams MT ((2006) ) Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1: , 848–858. |
[44] | Olarte-Sanchez CM , Amin E , Warburton EC , Aggleton JP ((2015) ) Perirhinal cortex lesions impair tests of object recognition memory but spare novelty detection. Eur J Neurosci 42: , 3117–3127. |
[45] | Bevins RA , Besheer J ((2006) ) Object recognition in rats and mice: A one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc 1: , 1306–1311. |
[46] | Swaminath G , Jaeckel P , Guo Q , Cardozo M , Weiszmann J , Lindberg R , Wang Y , Schwandner R , Li Y ((2010) ) Allosteric rescuing of loss-of-function FFAR2 mutations. FEBS Lett 584: , 4208–4214. |
[47] | Diaz-Nido J , Serrano L , Hernandez MA , Avila J ((1990) ) Phosphorylation of microtubule proteins in rat brain at different developmental stages: Comparison with that found in neuronal cultures. J Neurochem 54: , 211–222. |
[48] | Tortosa E , Montenegro-Venegas C , Benoist M , Hartel S , Gonzalez-Billault C , Esteban JA , Avila J ((2011) ) Microtubule-associated protein 1B (MAP1B) is required for dendritic spine development and synaptic maturation. J Biol Chem 286: , 40638–40648. |
[49] | Jack CR Jr. , Knopman DS , Jagust WJ , Petersen RC , Weiner MW , Aisen PS , Shaw LM , Vemuri P , Wiste HJ , Weigand SD , Lesnick TG , Pankratz VS , Donohue MC , Trojanowski JQ ((2013) ) Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol 12: , 207–216. |
[50] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 39: , 409–421. |
[51] | Iqbal K , Kazim SF , Bolognin S , Blanchard J ((2014) ) Shifting balance from neurodegeneration to regeneration of the brain: A novel therapeutic approach to Alzheimer’s disease and related neurodegenerative conditions. Neural Regen Res 9: , 1518–1519. |
[52] | Fumagalli F , Racagni G , Riva MA ((2006) ) The expanding role of BDNF: A therapeutic target for Alzheimer’s disease? Pharmacogenomics J 6: , 8–15. |
[53] | Budni J , Bellettini-Santos T , Mina F , Garcez ML , Zugno AI ((2015) ) The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis 6: , 331–341. |
[54] | Mattson MP ((2008) ) Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci 1144: , 97–112. |
[55] | Lee JG , Shin BS , You YS , Kim JE , Yoon SW , Jeon DW , Baek JH , Park SW , Kim YH ((2009) ) Decreased serum brain-derived neurotrophic factor levels in elderly korean with dementia. Psychiatry Investig 6: , 299–305. |
[56] | Castello NA , Green KN , LaFerla FM ((2012) ) Genetic knockdown of brain-derived neurotrophic factor in 3xTg-AD mice does not alter Abeta or tau pathology. PLoS One 7: , e39566. |
[57] | Fahnestock M , Garzon D , Holsinger RM , Michalski B (2002) Neurotrophic factors and Alzheimer’s disease: Are wefocusing on the wrong molecule? J Neural Transm Suppl, 241-252. |
[58] | Allen SJ , Dawbarn D ((2006) ) Clinical relevance of the neurotrophins and their receptors. Clin Sci (Lond) 110: , 175–191. |
[59] | Song JH , Yu JT , Tan L ((2015) ) Brain-derived neurotrophic factor in Alzheimer’s disease: Risk, mechanisms, and therapy. Mol Neurobiol 52: , 1477–1493. |
[60] | Poduslo JF , Curran GL ((1996) ) Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res Mol Brain Res 36: , 280–286. |
[61] | Conner JM , Lauterborn JC , Yan Q , Gall CM , Varon S ((1997) ) Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: Evidence for anterograde axonal transport. J Neurosci 17: , 2295–2313. |
[62] | Fritsch B , Reis J , Martinowich K , Schambra HM , Ji Y , Cohen LG , Lu B ((2010) ) Direct current stimulation promotes BDNF-dependent synaptic plasticity: Potential implications for motor learning. Neuron 66: , 198–204. |
[63] | Leal G , Afonso PM , Salazar IL , Duarte CB ((2015) ) Regulation of hippocampal synaptic plasticity by BDNF. Brain Res 1621: , 82–101. |
[64] | Numakawa T , Suzuki S , Kumamaru E , Adachi N , Richards M , Kunugi H ((2010) ) BDNF function and intracellular signaling in neurons. Histol Histopathol 25: , 237–258. |
[65] | Baydyuk M , Xu B ((2014) ) BDNF signaling and survival of striatal neurons. Front Cell Neurosci 8: , 254. |
[66] | Gonzalez A , Moya-Alvarado G , Gonzalez-Billaut C , Bronfman FC ((2016) ) Cellular and molecular mechanisms regulating neuronal growth by brain-derived neurotrophic factor. Cytoskeleton (Hoboken) 73: , 612–628. |
[67] | Kowianski P , Lietzau G , Czuba E , Waskow M , Steliga A , Morys J ((2018) ) BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol 38: , 579–593. |
[68] | Finkbeiner S ((2000) ) CREB couples neurotrophin signals to survival messages. Neuron 25: , 11–14. |
[69] | Aguiar AS Jr. , Castro AA , Moreira EL , Glaser V , Santos AR , Tasca CI , Latini A , Prediger RD ((2011) ) Short bouts of mild-intensity physical exercise improve spatial learning and memory in aging rats: Involvement of hippocampal plasticity via AKT, CREB and BDNF signaling. Mech Ageing Dev 132: , 560–567. |
[70] | Chen BY , Wang X , Wang ZY , Wang YZ , Chen LW , Luo ZJ ((2013) ) Brain-derived neurotrophic factor stimulates proliferation and differentiation of neural stem cells, possibly by triggering the Wnt/beta-catenin signaling pathway. J Neurosci Res 91: , 30–41. |
[71] | Finkbeiner S , Tavazoie SF , Maloratsky A , Jacobs KM , Harris KM , Greenberg ME ((1997) ) CREB: A major mediator of neuronal neurotrophin responses. Neuron 19: , 1031–1047. |
[72] | Ying SW , Futter M , Rosenblum K , Webber MJ , Hunt SP , Bliss TV , Bramham CR ((2002) ) Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: Requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci 22: , 1532–1540. |
[73] | McAllister AK , Katz LC , Lo DC ((1997) ) Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron 18: , 767–778. |
[74] | Kwon M , Fernandez JR , Zegarek GF , Lo SB , Firestein BL ((2011) ) BDNF-promoted increases in proximal dendrites occur via CREB-dependent transcriptional regulation of cypin. J Neurosci 31: , 9735–9745. |
[75] | Amaral MD , Pozzo-Miller L ((2007) ) BDNF induces calcium elevations associated with IBDNF, a nonselective cationic current mediated by TRPC channels. J Neurophysiol 98: , 2476–2482. |
[76] | Yoshii A , Constantine-Paton M ((2010) ) Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol 70: , 304–322. |
[77] | Mattson MP , Maudsley S , Martin B ((2004) ) BDNF and 5-HT: A dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci 27: , 589–594. |
[78] | Melemedjian OK , Tillu DV , Asiedu MN , Mandell EK , Moy JK , Blute VM , Taylor CJ , Ghosh S , Price TJ ((2013) ) BDNF regulates atypical PKC at spinal synapses to initiate and maintain a centralized chronic pain state. Mol Pain 9: , 12. |
[79] | Tao X , Finkbeiner S , Arnold DB , Shaywitz AJ , Greenberg ME ((1998) ) Ca2+influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20: , 709–726. |
[80] | Benito E , Barco A ((2010) ) CREB’s control of intrinsic and synaptic plasticity: Implications for CREB-dependent memory models. Trends Neurosci 33: , 230–240. |
[81] | Kandel ER ((2012) ) The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5: , 14. |
[82] | Konur S , Ghosh A ((2005) ) Calcium signaling and the control of dendritic development. Neuron 46: , 401–405. |
[83] | Wang H , Peng RY ((2016) ) Basic roles of key molecules connected with NMDAR signaling pathway on regulating learning and memory and synaptic plasticity. Mil Med Res 3: , 26. |
[84] | Neves G , Cooke SF , Bliss TV ((2008) ) Synaptic plasticity, memory and the hippocampus: A neural network approach to causality. Nat Rev Neurosci 9: , 65–75. |
[85] | Martin SJ , Grimwood PD , Morris RG ((2000) ) Synaptic plasticity and memory: An evaluation of the hypothesis. Annu Rev Neurosci 23: , 649–711. |
[86] | Selkoe DJ ((2002) ) Alzheimer’s disease is a synaptic failure. Science 298: , 789–791. |
[87] | Masliah E , Mallory M , Alford M , DeTeresa R , Hansen LA , McKeel DW Jr. , Morris JC ((2001) ) Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 56: , 127–129. |
[88] | Kuczewski N , Porcher C , Lessmann V , Medina I , Gaiarsa JL ((2009) ) Activity-dependent dendritic release of BDNF and biological consequences. Mol Neurobiol 39: , 37–49. |
[89] | Liu Y , Wei W , Baazaoui N , Liu F , Iqbal K ((2019) ) Inhibition of AMD-like pathology with a neurotrophic compound in aged rats and 3xTg-AD mice. Front Aging Neurosci 11: , 309. |
[90] | Bolognin S , Blanchard J , Wang X , Basurto-Islas G , Tung YC , Kohlbrenner E , Grundke-Iqbal I , Iqbal K ((2012) ) An experimental rat model of sporadic Alzheimer’s disease and rescue of cognitive impairment with a neurotrophic peptide. Acta Neuropathol 123: , 133–151. |


