
Leucine Carboxyl Methyltransferase 1 Overexpression Protects Against Cognitive and Electrophysiological Impairments in Tg2576 APP Transgenic Mice
Abstract
Background:
The serine/threonine protein phosphatase, PP2A, is thought to play a central role in the molecular pathogenesis of Alzheimer’s disease (AD), and the activity and substrate specificity of PP2A is regulated, in part, through methylation and demethylation of its catalytic subunit. Previously, we found that transgenic overexpression of the PP2A methyltransferase, LCMT-1, or the PP2A methylesterase, PME-1, altered the sensitivity of mice to impairments caused by acute exposure to synthetic oligomeric amyloid-β (Aβ).
Objective:
Here we sought to test the possibility that these molecules also controlled sensitivity to impairments caused by chronically elevated levels of Aβ produced in vivo.
Methods:
To do this, we examined the effects of transgenic LCMT-1, or PME-1 overexpression on cognitive and electrophysiological impairments caused by chronic overexpression of mutant human APP in Tg2576 mice.
Results:
We found that LCMT-1 overexpression prevented impairments in short-term spatial memory and synaptic plasticity in Tg2576 mice, without altering APP expression or soluble Aβ levels. While the magnitude of the effects of PME-1 overexpression in Tg2576 mice was small and potentially confounded by the emergence of non-cognitive impairments, Tg2576 mice that overexpressed PME-1 showed a trend toward earlier onset and/or increased severity of cognitive and electrophysiological impairments.
Conclusion:
These data suggest that the PP2A methyltransferase, LCMT-1, and the PP2A methylesterase, PME-1, may participate in the molecular pathogenesis of AD by regulating sensitivity to the pathogenic effects of chronically elevated levels of Aβ.
INTRODUCTION
Protein phosphatase 2A (PP2A) is a serine/threonine phosphatase that has been implicated in the molecular pathogenesis of Alzheimer’s disease (AD) by multiple lines of evidence [1]. PP2A expression and activity is reduced in AD brains [2–5], and PP2A acts as the principal phosphatase for phosphorylated forms of microtubule-associated binding protein, tau, that are linked to AD [6]. In animal models, genetically reducing PP2A activity results in AD-like pathology and cognitive deficits [7–13], while pharmacological activation of PP2A reduces AD-like cognitive impairments and pathology [14–18]. Here we sought to understand the role and therapeutic potential of methylation-dependent PP2A regulation in AD pathogenesis.
PP2A is a heterotrimeric enzyme composed of a catalytic subunit, a structural subunit, and a regulatory subunit [19]. Multiple isoforms exist for each of these subunits, and the combination of different subunit isoforms leads to the formation of different heterotrimeric holoenzymes with different expression patterns and substrate specificities [20]. PP2A activity is regulated through a complex process involving regulated assembly and disassembly, post-translational modifications, and interactions with inhibitory proteins [19, 21, 22]. One of the mechanisms controlling PP2A activity involves methylation of the C-terminal leucine of the catalytic subunit, and in mice, methylation of PP2A is carried out by leucine carboxyl methyltransferase 1 (LCMT-1) and demethylation is carried out by protein phosphatase methylesterase 1 (PME-1). PP2A catalytic subunit methylation promotes the formation of B55α subunit-containing holoenzymes that exhibit the highest tau phosphatase activity [23, 24], and a role for reduced PP2A methylation in AD pathogenesis is suggested by the reduced LCMT-1 expression observed in AD brains [25, 26]. In addition, dysregulated PP2A activity resulting from impaired PP2A methylation is thought to be one of the molecular mechanisms underlying the link between hyperhomocysteinemia and increased risk for AD [27].
We found previously that LCMT-1 and PME-1 overexpression altered sensitivity to cognitive and electrophysiological impairments caused by acute exposure to elevated levels of amyloid-β (Aβ) [28]. To explore further the involvement and therapeutic potential of this pathway in AD, we sought to determine whether LCMT-1 and PME-1 overexpression might alter sensitivity to impairments caused by chronically elevated Aβ levels in APP transgenic mice. To do this, we tested cognitive performance and synaptic plasticity in Tg2576 APP transgenic mice that overexpressed either LCMT-1 or PME-1. We found that LCMT-1 overexpression protected mice from cognitive impairments in a 2-day radial arm water maze task as well as impairments in long-term potentiation (LTP) caused by transgenic expression of mutant APP. The protective effects of LCMT-1 overexpression in Tg2576 mice were associated with trends for reduced tau phosphorylation and restored LCMT-1 expression that suggest possible mechanisms for these effects. Our data also suggest that PME-1 overexpression may increase the sensitivity of mice to impairments caused by expression of the mutant APP transgene in Tg2576 mice. However, the magnitude of the effects in these animals was small and potentially confounded by the emergence of non-cognitive impairments in animals that overexpressed both PME-1 and mutant APP. Together our data support the involvement of LCMT-1 and PME-1 in the molecular pathogenesis of AD as well as their potential as a novel targets for AD prevention or treatment.
MATERIALS AND METHODS
Animals
Subjects in this study were transgenic mice carrying combinations of the CaMKII-tTA [29], tetO-LCMT, tetO-PME [28] and Tg2576 [30] as indicated. Single transgenic animals carrying only either a CaMKII-tTA, tetO-LCMT, or tetO-PME transgene were used as controls. Experimental animals were generated from crosses of either tetO-LCMT; CaMK-tTA or tetO-PME; CaMK-tTA double transgenic mice in a C57BL6/J background to Tg2576 mice in a 129S6 background as outlined in Fig. 1. Equivalent numbers of male and female mice were used for all experiments and animals were tested in groups of 10–12. All procedures involving animals were conducted in strict accordance with protocols approved by the Columbia University Institutional Animal Care and Use Committee (USDA Registration #21-R-0082; AAALAC Accreditation #000687; NYDOH #A141).
Fig. 1
Diagram of the mating scheme in which LCMT-1 or PME-1 overexpressing animals were crossed to Tg2576 transgenic mice to generate offspring in an F1 hybrid background for experiments.
Radial arm water maze
Testing was performed in a 120 cm diameter pool containing a six-arm radial maze insert (San Diego Instruments) and filled with opaque water as described previously [31]. Mice were tested in 15×1 min trials on each of 2 consecutive days. The location of the escape platform was held constant during testing, but the start location was pseudorandomly varied throughout. On the first day, training alternated between visible and hidden platform trials, while on the second day only hidden platform trials were conducted. Water temperature was maintained at approximately 24°C and mice were dried and placed in a clean heated cage between trials to prevent hypothermia. Entries into maze arms that did not contain the escape platform were scored as errors. Data are presented as the average number of errors committed during blocks of 3 training trials.
Visible platform water maze
This task was conducted in a 120 cm diameter pool (San Diego Instruments) filled with opaque water, but without the partitions used for the radial arm water maze task. Training for this task was carried out over 2 days with 3 morning and 3 afternoon trials on each day. Intertrial intervals were 15 to 20 min, and rest periods between morning and afternoon sessions were 2-3 h. Each trial was a maximum of 120 s during which time the animals were required to swim to a marked visible escape platform located just above the water surface. Animals that did not reach the platform within the allotted time were guided to it and allowed to sit there for 15 s before returning to their home cage. The location of the platform was varied among 4 different locations such that it was not present in the same location on any two successive trials. Water temperature was maintained at approximately 24°C, and animals were dried and placed in a clean warmed cage after each trial to prevent hypothermia. Animal movements were recorded using a video-tracking system (Noldus) and time required to reach the platform (latency) and swim speed were determined using Ethovision XT behavioral analysis software (Noldus).
Electrophysiological studies
Extracellular field potential recordings were performed on acute hippocampal slices prepared as described previously [32] from sibling mice with the genotypes and ages indicated in the text. Animals were euthanized by cervical dislocation, a method of euthanasia approved by the Panel on Euthanasia of the American Veterinary Medical Association that yields viable anesthetic-free tissue suitable for electrophysiological recordings. Brains were then rapidly removed and cooled in ice cold ASCF consisting of in mM: 124 NaCl, 4.4 KCl, 1 Na2HPO4, 25 NaHCO3, 2 CaCl2, 2 MgCl2, and 10 glucose. Hippocampi were then dissected and sliced into 400μM sections using a tissue chopper. Slices were incubated at 29°C in an interface chamber under continuous perfusion (2 ml/min) with oxygenated ACSF and allowed to recover for a minimum of 90 min prior to recording responses in the CA1 region to stimulation of Schaffer collateral projections with a bipolar electrode (FHC Inc.). Field potential signals were acquired using a Axoclamp-2A amplifier (Axon Instruments) and pClamp10.6 software (Molecular Devices). Input/output relationships were determined prior to each recording and stimulus intensities that elicited 30%of the maximal response were utilized. Stable baselines were obtained for a minimum of 15 min prior to drug or vehicle application and a theta-burst stimulation protocol consisting of 3 trains separated by 15 s intervals with each train consisting of 10 bursts at 5 Hz and each burst consisting of 5 pulses at 100 Hz was used to elicit LTP. Data analysis was performed using Clampfit 10.6 software (Molecular Devices).
Western blotting
Animals were euthanized by cervical dislocation, an AVMA approved method that allows for rapid removal of non-hypoxic brains. Hippocampi were then rapidly dissected, snap frozen in liquid nitrogen and stored at –80°C prior to homogenization for western blot analysis. Hippocampal homogenates were prepared by sonication at 95°C in aqueous buffer containing 2%lithium dodecyl sulfate and 50 mM Tris pH 7.5. Total protein concentrations were determined by bicinchoninic acid assay according to the manufacturer’s instructions (Micro BCA protein assay kit, Thermo Fisher) and 10–20μg of total protein was loaded per lane on NuPage 4–12%Bis-Tris gels (Invitrogen). Proteins were transferred to PVDF membranes using an i-Blot gel transfer device (Invitrogen). Membranes were blocked with Seablock (Pierce) for 1 h at room temperature, and probed with the indicated primary antibodies (rabbit anti-APP (1:1000) Cell Signaling #2452 RRID:AB_10694227; mouse anti-phospho-tau clone PHF1 (1:500) Peter Davies; mouse anti-phospho-tau clone CP13 (1:500) Peter Davies; rabbit anti-tau clone EP2456Y (1:8000) Abcam #Ab76128 RRID:AB_1524475; rabbit anti-PME (1:1000) Millipore #07-095 RRID:AB_310373; mouse anti-LCMT-1 clone 4A4 (1:1000) Cell Signaling #5691 RRID:AB_10949100; mouse anti-β-actin (1:40,000) Licor #926-42212; rabbit anti-β-actin (1:40,000) Licor #926-42210 RRID:AB_1850027) overnight at 4°C, then washed and incubated with infrared dye-labeled Goat anti-rabbit (IRDye 800CW, LI-COR) and Goat anti-mouse (IRDye 680RD, LI-COR) secondary antibodies at room temperature for 2 h. Immunoreactive bands were detected using an Odyssey 9120 infrared imaging system and analyzed using Image Studio Lite v5.2.5 software (RRID:SCR_014211) (LI-COR).
AβELISA
Levels of soluble Aβ1–40 and Aβ1–42 were measured in diethylamine extracted hippocampal homogenates using commercially available human Aβ40 or Aβ42 ELISA kits (Invitrogen) or murine Aβ40 or Aβ42 ELISA kits (FUJIFILM Wako Pure Chemical Corporation) as described previously [33]. Chromogenic signals generated in these assays were quantified by measuring absorbance at 450 nm using an Infinite M200 plate reader (Tecan).
Statistical analysis
Tests for statistical significance between groups were performed using Prism 8 (Graphpad Software, San Diego, CA, USA). 2-way ANOVA comparisons—with or without repeated measures as dictated by the experimental design—were used for analysis of all experiments involving multiple groups with the exception of data shown in Table 1 where comparisons between the control, APP, and APP + LCMT groups was performed using one-way ANOVA. Student’s unpaired, two-tailed t-tests were used for comparisons of data in Table 1 involving two groups. Dunnett’s post-hoc comparisons to controls were performed to test for significant differences among groups in experiments with repeated measures, and Sidak’s post-hoc tests were performed on data from experiments without repeated measures. Tukeys’ comparisons were used to test for significant differences among any of the groups for the data presented in Fig. 2C. The results of these tests together with their associated p values are presented in the legends for their corresponding figure.
Table 1
shows the values obtained for transgenic human Aβ species and endogenous murine Aβ species obtained by ELISAs performed on hippocampal homogenates from 7-8-month-old animals from the indicated genotype groups expressed in pmol of Aβ per gram of total protein±SEM
| Genotype | ||||
| Control (N = 8) | APP (N = 7) | LCMT + APP (N = 7) | T-test | |
| human Aβ40 | n. d. | 491.7 (±47.82) | 484.3 (±45.06) | t = 0.112; p = 0.9126 |
| human Aβ42 | n. d. | 59.37 (±4.58) | 63.21 (±5.95) | t = 0.5112; p = 0.6185 |
| ANOVA | ||||
| murine Aβ40 | 16.18 (±0.276) | 16.01 (±0.634) | 17.23 (±0.688) | F = 1.454; p = 0.2585 |
| murine Aβ42 | 7.882 (±0.496) | 9.112 (±0.619) | 8.707 (±0.578) | F = 1.287; p = 0.2992 |
(n.d., not detected)
Fig. 2
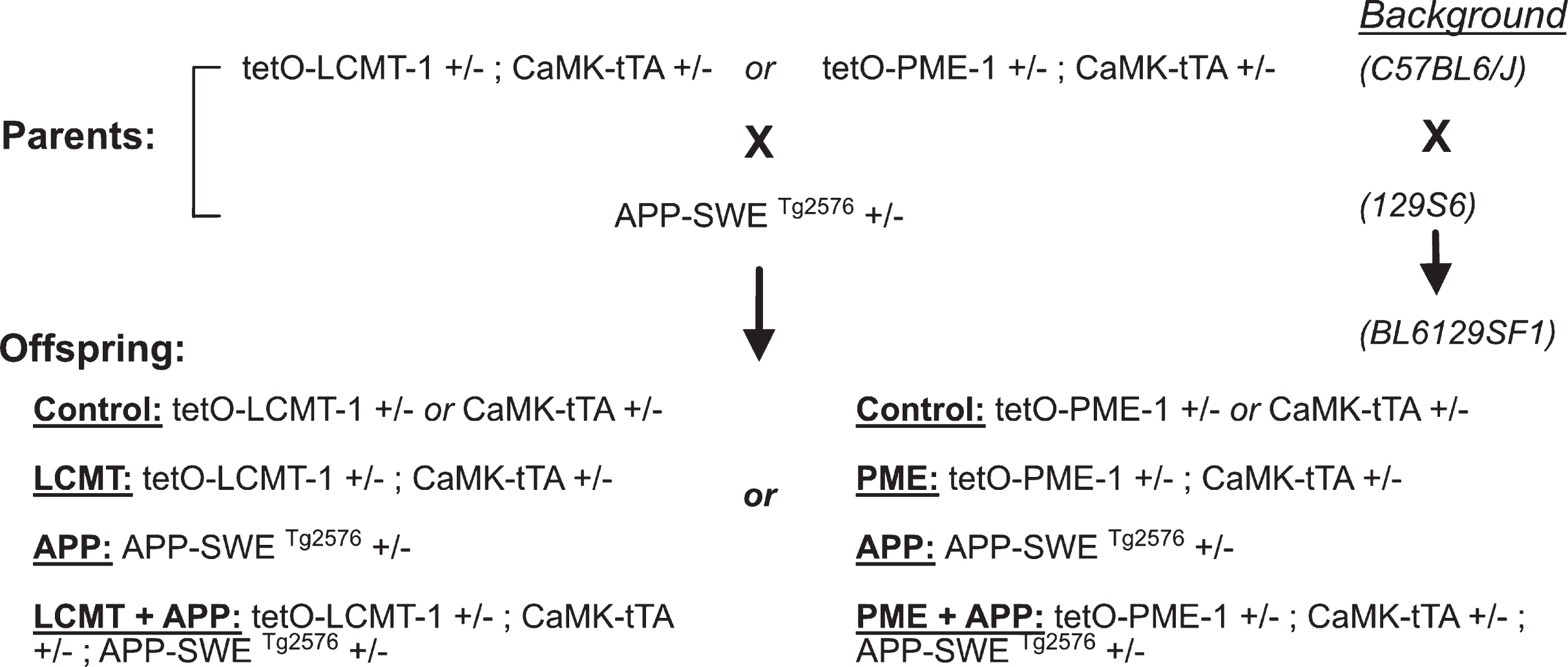
LCMT overexpression protects against cognitive impairments in Tg2576 mice. A) Plot of average number of errors committed (±SEM) during each 3-trial training block of a 2-day radial arm water maze task for 6-7-month-old mice that carried the Tg2576 APP transgene in combination with transgenic LCMT overexpression (LCMT + APP), or 6-7-month-old animals that carried the Tg2576 APP transgene alone (APP), overexpressed LCMT alone (LCMT), or controls. 2-way RM ANOVA for errors on day 2 (blocks 6–10) with group and block as factors shows a significant effect of group (F(3,76) = 9.456, p < 0.0001). Dunnett’s multiple comparisons show that the APP group was significantly different from each of the other three groups (p = 0.0025 for LCMT + APP, p = 0.0003 for LCMT, and p < 0.0001 for control). B) Plot of the average escape latency (±SEM) for 6-7-month-old animals from the indicated groups during training on a visible platform water maze task showed no significant differences between groups (2-way RM ANOVA for effect of group with group and block as factors F(3, 76) = 2.202, p = 0.0947). C) Plot of the average swim speed (±SEM) for the indicated groups during training on the visible platform water maze task described in B showed a significant difference between groups overall (2-way RM ANOVA for effect of group with group and block as factors F(3, 76) = 2.948, p = 0.0381), but no significant differences in pairwise comparisons (Tukey’s comparisons p > 0.05 for all). D) Plot of average number of errors committed (±SEM) during each 3-trial training block of a 2-day radial arm water maze task for 9-10-month-old mice that carried the Tg2576 APP transgene in combination with transgenic LCMT overexpression (LCMT + APP), or 9-10-month-old animals that carried the Tg2576 APP transgene alone (APP), overexpressed LCMT alone (LCMT), or controls. 2-way RM ANOVA for errors on day 2 (blocks 6–10) with group and block as factors shows a significant effect of group (F(3, 65) = 6.135, p = 0.0010). Dunnett’s multiple comparisons show that the APP group was significantly different from each of the other three groups (p = 0.0220 for LCMT + APP, p = 0.0006 for LCMT and p = 0.0029 for control). E) Plot of the average escape latency (±SEM) for 9-10-month-old animals from the indicated groups during training on a visible platform water maze task reveals no significant differences between groups (2-way RM ANOVA for latency with group and block as factors shows no significant effect of group F(3, 64) = 0.961, p = 0.4167). F) Plot of the average swim speed (±SEM) for the indicated groups during training on the visible platform water maze task described in B reveals no significant differences between groups (2-way RM ANOVA for speed with group and block as factors shows no significant effect of group F(3, 64) = 0.5046, p = 0.6805). For experiments on 6-7-month-old animals: N = 27 control, 22 APP, 16 LCMT, 15 LCMT + APP. For experiments on 9-10-month-old animals: N = 17 control, 17 APP, 19 LCMT, 16 LCMT + APP.
RESULTS
Transgenic LCMT-1 overexpression protects against cognitive and electrophysiological impairments in Tg2576 mice
To assess the effects of increased LCMT-1 expression on cognitive impairments resulting from chronically elevated levels of Aβ, we utilized the Tg2576 line of APP transgenic mice [30]. The Tg2576 transgene encodes a 695 amino acid form of human APP carrying the Swedish (K670N, M671L) mutation under the control of the hamster prion protein promoter, and animals that carry this transgene exhibit well-characterized behavioral and electrophysiological impairments [34, 35]. We crossed these animals to mice carrying both the CaMKII-tTA and tetO-LCMT transgenes, which together result in increased expression of murine LCMT-1 in the murine forebrain [28]. We then assessed the cognitive performance of the resulting offspring on a 2-day radial arm water maze task at 6-7 months of age, an early stage in the development of neurodegenerative phenotypes in Tg2576 mice [30, 34, 36, 37], to determine whether LCMT overexpression might prevent or delay the onset of cognitive impairments in these animals. We found that animals that carried the APP transgene alone committed significantly more errors in this task when compared to controls at this age, whereas APP transgenic animals that also expressed the LCMT-1 transgene (LCMT + APP) were indistinguishable in their performance from the control group (Fig. 2A).
We also compared the performance of these same animals on a visible platform water maze task that relies only on an association between a visual cue and the escape platform, rather than spatial memory performance. The performance of all groups in the visible platform water maze task was similar, both with respect to their latency to reach the escape platform, and with respect to their swimming speed (Fig. 2B, C). The absence of impairments in this non-spatial visible platform water maze task suggests that the performance differences we observe in the 2-day radial arm water maze among these groups do not result from non-cognitive differences in visual perception, motivation or swimming ability. Together these data suggest the existence of a cognitive impairment in short-term spatial memory in Tg2576 mice that can be prevented through transgenic overexpression of LCMT-1.
As a further test of the ability of LCMT-1 overexpression to protect against cognitive impairments in Tg2576 mice, we performed the same behavioral tests on a separate group of 9-10-month-old animals, an age when cognitive impairments are reported to be well-established in Tg2576 animals [34]. Again, mice that carried the APP transgene alone committed significantly more errors in this task when compared to controls at this age, whereas the performance APP transgenic animals that also expressed the LCMT-1 transgene (LCMT + APP) was indistinguishable from the control group (Fig. 2D). As was the case at 6-7 months of age, we found that all groups performed similarly in the visible platform water maze task at 9-10 months, further supporting the conclusion that transgenic overexpression of LCMT-1 protects against cognitive impairments in short-term spatial memory in Tg2576 mice (Fig. 2E, F).
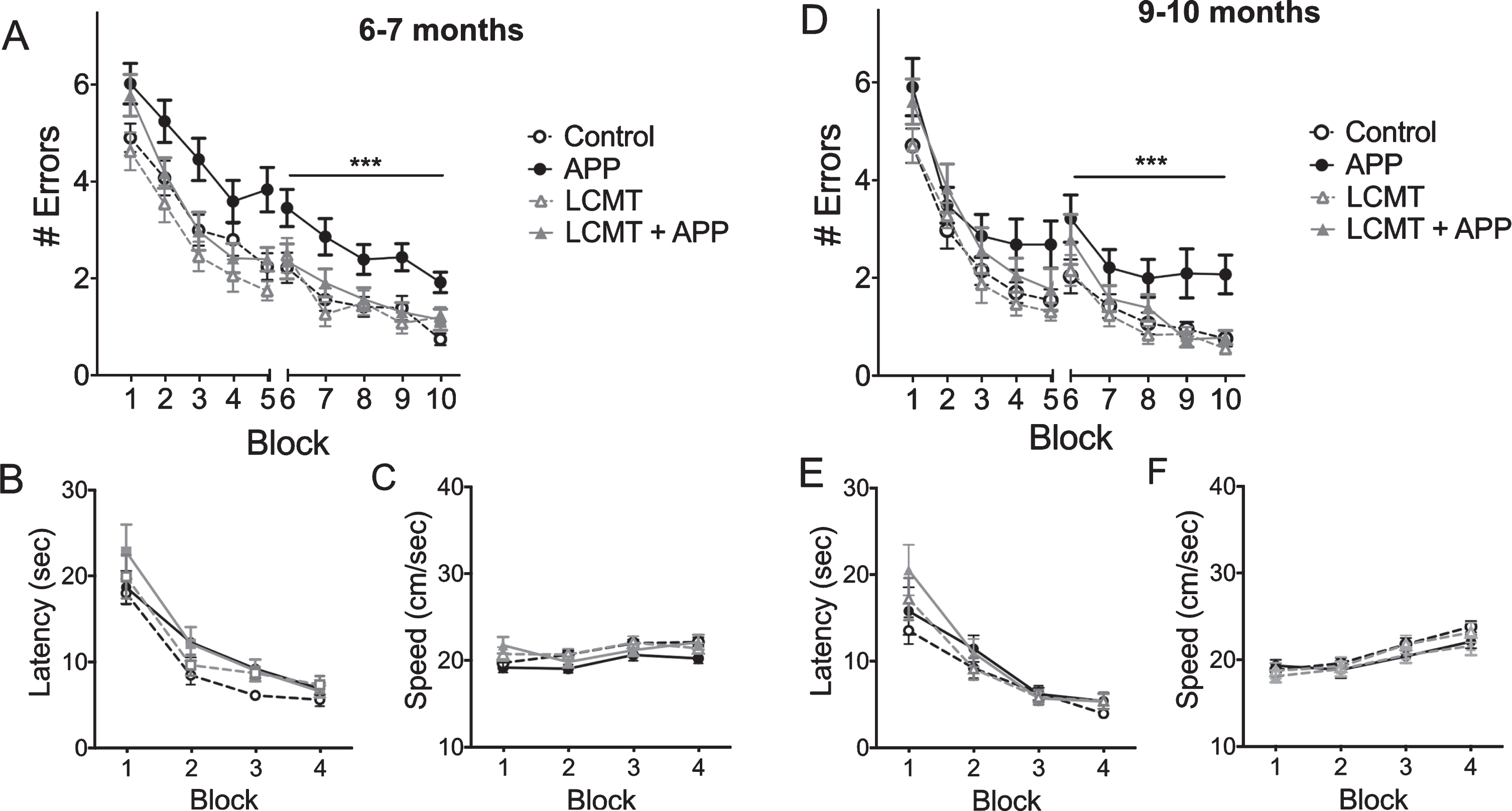
Synaptic plasticity in the hippocampus is thought to be required for normal spatial memory, and impairments in synaptic plasticity have been identified in Tg2576 mice that are thought to contribute to the cognitive impairments exhibited by these animals [36, 37]. To determine whether transgenic LCMT-1 overexpression might also protect against impairments in synaptic plasticity in APP Tg2576 transgenic mice, we compared the magnitude of long-term potentiation (LTP) among APP and LCMT-1 transgenic and control groups at both 7-8 and 10-11 months of age. Acute hippocampal slices were prepared from these animals, and LTP was induced at Schaffer collateral synapses by theta-burst stimulation. Consistent with previous reports [36, 37], we found that the magnitude of LTP in slices from animals that carried the APP transgene alone was significantly decreased relative to controls at both of these ages (Fig. 3A, B). In contrast, the magnitude of LTP in slices prepared from animals expressing both the mutant APP and LCMT-1 transgenes (LCMT + APP) was similar to the control group. These data suggest that, in addition to protecting against cognitive impairments, LCMT-1 overexpression also protects against impairments in synaptic plasticity that result from transgenic expression of mutant APP.
Fig. 3
LCMT overexpression protects against electrophysiological impairments in Tg2576 mice. A) Time course of averaged Schaffer collateral fEPSP responses (±SEM) prior to and following delivery of theta-burst stimulation (arrow) in hippocampal slices prepared from 7-8-month-old mice that carried the Tg2576 APP transgene in combination with transgenic LCMT overexpression (LCMT + APP), or 7-8-month-old animals that carried the Tg2576 APP transgene alone (APP), overexpressed LCMT alone (LCMT), or controls. 2-way RM ANOVA for fEPSP responses over the last 20 min of recording with group and time as factors shows a significant effect of group (horizontal bar; F(3, 60) = 5.297, p = 0.0026). Dunnett’s multiple comparisons show that the APP group was significantly different from each of the other three groups (p = 0.0481 for LCMT + APP, p = 0.0033 for LCMT and p = 0.0019 for control). N = 14 control, 13 APP, 18 LCMT, 19 LCMT + APP. Representative traces of field potential responses prior to TBS, and during the last 20 min of recording are shown above for each of the indicated genotypes. B) Time course of averaged Schaffer collateral fEPSP responses (±SEM) prior to and following delivery of theta-burst stimulation (arrow) in hippocampal slices prepared from 10-11-month-old mice that carried the Tg2576 APP transgene in combination with transgenic LCMT overexpression (LCMT + APP), or 10-11-month-old animals that carried the Tg2576 APP transgene alone (APP), overexpressed LCMT alone (LCMT), or controls. 2-way RM ANOVA for fEPSP responses over the last 20 min of recording with group and time as factors shows a significant effect of group (horizontal bar; F(3, 43) = 7.328, p = 0.0004). Dunnett’s multiple comparisons show that the APP group was significantly different from each of the other three groups (p = 0.0003 for LCMT + APP, p = 0.0183 for LCMT and p = 0.0013 for control). N = 11 control, 12 APP, 12 LCMT, 12 LCMT + APP. Representative traces of field potential responses prior to TBS, and during the last 20 min of recording are shown above for each of the indicated genotypes.
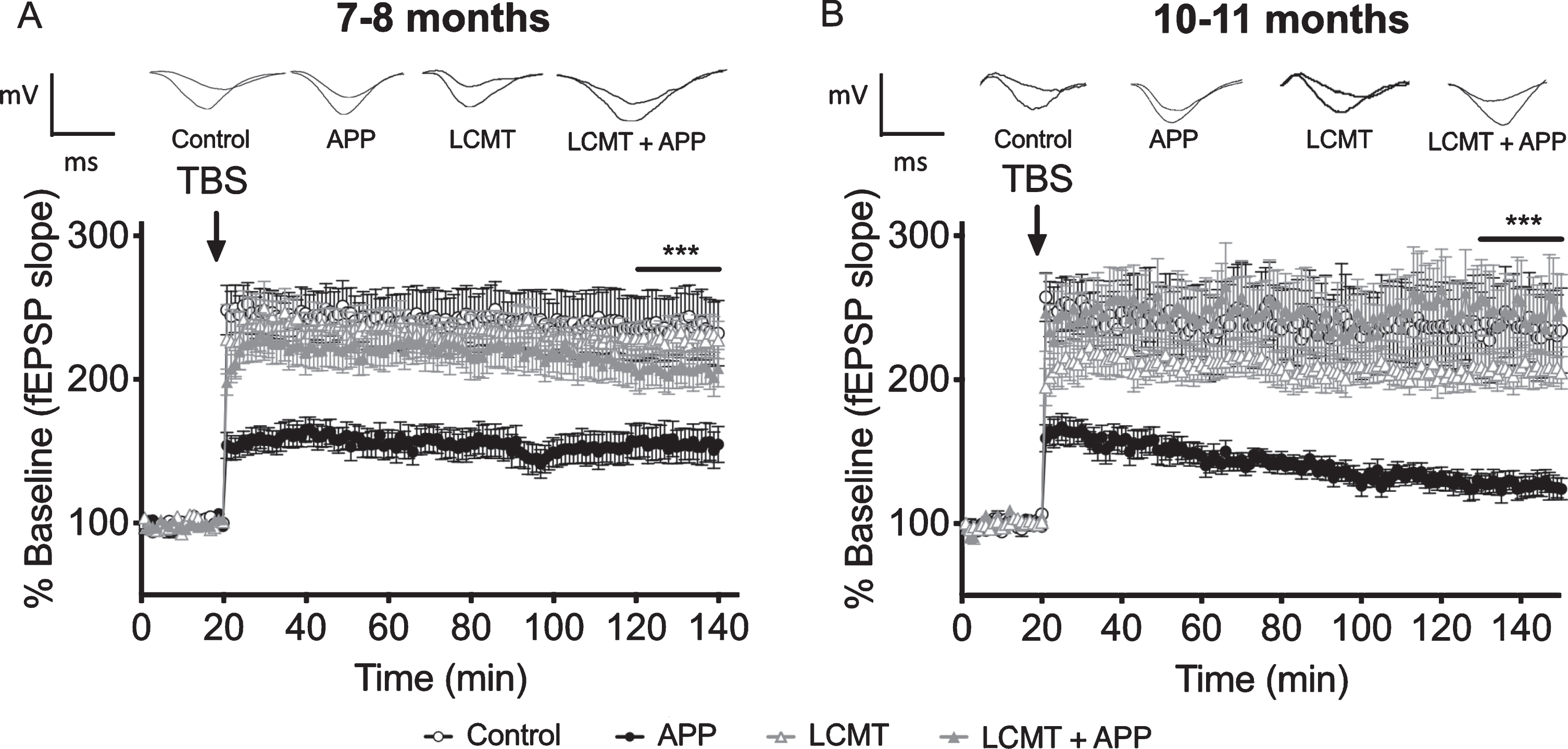
One way in which transgenic LCMT-1 overexpression could protect against cognitive and electrophysiological impairments in Tg2576 mice is by reducing transgenic APP expression or Aβ production in these animals. To test this possibility, we performed western blots for APP on hippocampal homogenates prepared from these groups at 7-8 or 10-11 months of age. As shown in Fig. 4A ,B, and consistent with published reports [30, 38], we found that mice carrying the Tg2576 transgene expressed APP at 4-5 x endogenous levels. However, neither transgenic nor endogenous APP protein levels were significantly affected by transgenic LCMT-1 overexpression. To test the possibility that transgenic LCMT-1 overexpression acted by altering Aβ production in Tg2576 mice, we performed enzyme-linked immunosorbent assays (ELISAs) for human and murine Aβ40 and Aβ42 in hippocampal homogenates prepared from 7-8-month-old mice that expressed the Tg2576 transgene alone or in combination with transgenic LCMT-1 overexpression (Table 1). The results of this analysis revealed no significant differences between groups for either the transgenic human or endogenous murine Aβ40 or Aβ42 species. Together, and consistent with our previous data on the effect of LCMT-1 overexpression on endogenous Aβ levels [28], these data suggest that transgenic LCMT-1 overexpression does not protect against cognitive and electrophysiological impairments in Tg2576 mice by reducing APP expression or Aβ production.
Fig. 4
The effect of LCMT overexpression on APP expression, tau phosphorylation, and LCMT protein levels in Tg2576 mice. A, B) Representative western blots showing anti-APP, phospho-tau serine 396/404 and serine 202, total tau, and LCMT immunoreactivity in hippocampal homogenates prepared from 7-8 (A) or 9-10-month-old (B) control, LCMT overexpressing, Tg2576 APP transgenic, or LCMT overexpressing/APP transgenic mice together with corresponding histograms showing average normalized immunoreactivity (±SEM) in each group. APP genotype significantly affected APP expression (2-way ANOVA with APP and LCMT genotype as factors: F(1, 20) = 169.7, p < 0.0001 for 7-8-month-old mice and F(1, 20) = 1157, p < 0.0001 for 10-11-month-old mice). Sidak’s multiple comparisons showed no significant differences in APP expression between control and LCMT groups (p = 0.9873 for 7-8-month-old mice and p = 0.9804 for 10-11-month-old mice) or between APP and LCMT + APP groups (p = 0.9815 for 7-8-month-old and, p = 0.9843 for 10-11-month-old mice). Animals carrying an APP transgene alone showed a trend for increased tau phosphorylation at serine residues 396/404 (APP:107.3±10.46%versus control: 100±11.53, LCMT: 98.3±10.01, APP:107.3±10.46, LCMT + APP: 97.5±14.22 at 7-8 months, and APP:118.0±9.92%versus control: 100±5.15, LCMT: 107.2±7.28, LCMT + APP: 107.5±8.99 at 10-11 months) that was not statistically significant (2-way ANOVA test for interaction with APP and LCMT genotype as factors: F(1,20) = 0.174, p = 0.6811 for 7-8-month-old mice and F(1,20) = 1.212, p = 0.2840 for 10-11-month-old mice). Animals carrying an APP transgene alone showed a similar trend for increased tau phosphorylation at serine residue 202 (APP:106.9±9.37%versus control: 100±11.68, LCMT: 100.5±7.64, LCMT + APP: 97.0±9.36 at 7-8 months, and APP:114.0±18.47%versus control: 100±13.92, LCMT: 97.0±9.701, LCMT + APP: 102.6±16.65 at 10-11 months) that was not statistically significant (2-way ANOVA test for interaction with APP and LCMT genotype as factors: F(1,20) = 0.2877, p = 0.5976 for 7-8-month-old mice and F(1,20) = 0.07876, p = 0.7819 for 10-11-month-old mice). Neither LCMT overexpression nor APP transgene expression significantly affected endogenous tau levels (2-way ANOVA for effect of LCMT transgene with APP and LCMT genotype as factors: F(1,44) = 0.0111, p = 0.9166 for 7-8-month-old mice, and F(1,44) = 0.06945, p = 0.7934 for 10-11-month-old mice);(2-way ANOVA for effect of APP transgene with APP and LCMT genotype as factors: F(1,44) = 0.0223, p = 0.8819 for 7-8-month-old mice and F(1,44) = 4.196, p = 0.0465 for 10-11-month-old mice). As expected, transgenic LCMT expression significantly affected LCMT protein levels (2-way ANOVA with LCMT and APP genotype as factors: F(1,20) = 14.1, p = 0.0012 for 7-8-month-old mice and F(1,20) = 46.97, p < 0.0001 for 10-11-month-old mice. In addition, for 10-11-month-old mice, Sidak’s multiple comparisons show a significant decrease in total LCMT expression in the APP transgenic group relative to controls (p = 0.0204), and in the LCMT + APP group relative to LCMT alone group (p = 0.0134). Transgenic LCMT overexpression did not significantly affect the amount of PP2A catalytic subunit that was demethylated at Leucine 309 (2-way ANOVA with LCMT and APP genotype as factors: F(1,20) = 0.5316, p = 0.4744 and F(1,20) = 0.3604, p = 0.5550 respectively for 7-8-month-old mice, and F(1,20) = 0.7365, p = 0.4010 and F(1,20) = 0.0877, p = 0.7702 respectively for 10-11-month-old mice). Transgenic LCMT overexpression also did not significantly affect the total amount of PP2A catalytic subunit (2-way ANOVA with LCMT and APP genotype as factors: F(1,20) = 0.4440, p = 0.5128 and F(1,20) = 0.1909, p = 0.6669 respectively for 7-8-month-old mice, and F(1,20) = 0.1213, p = 0.7313 and F(1,20) = 0.2888, p = 0.5969 respectively for 10-11-month-old mice). For all blots band intensities in each lane were normalized to corresponding actin or total tau immunoreactivity in that lane and values were expressed as a percentage of the mean of the control group (N = 6 per group).
Aβ has been suggested to exert its pathogenic effects, at least in part, through the microtubule binding protein, tau, and consistent with this model, genetically eliminating tau has been found to protect against impairments in APP transgenic mice [39, 40]. Elevated levels of Aβ have also been found to increase tau phosphorylation at sites linked to human neurodegeneration [41–46]. Since protein phosphatase 2A plays a central role in regulating tau phosphorylation [1], one way LCMT-1 overexpression might exert its protective effects is by increasing PP2A-mediated tau phosphatase activity, thereby reducing pathogenic, Aβ-induced increases in tau phosphorylation. To explore this possibility, we performed western blots for two AD-related phospho-tau epitopes that were shown previously to respond to both PP2A activity and Aβ levels [10, 28, 41, 43–45, 47–49]. Our analysis revealed no significant differences in tau phosphorylation among any of the groups tested at either 7-8 or 10-11 months of age for either the serine 396/404 or serine 202 phospho-tau epitopes (Fig. 4A, B). However, we did observe a trend for increased tau phosphorylation at these sites in APP transgenic animals that was absent in animals expressing both the APP and LCMT-1 transgenes, raising the possibility that increased tau dephosphorylation downstream of LCMT-1 may contribute to its protective effects.
LCMT-1 levels have been reported to be reduced in AD brains, suggesting that reduced LCMT-1 activity may contribute to the molecular pathogenesis of AD [25, 26]. To further explore the relationship between LCMT-1 expression and AD-related pathology, we compared the levels of LCMT-1 expression in hippocampal homogenates prepared from controls and mice that expressed the APP and LCMT-1 transgenes alone or together. Consistent with a potential role for reduced LCMT-1 expression in AD pathogenesis, we found a significant reduction in LCMT-1 protein levels in 10-11-month-old APP transgenic mice that was compensated for by transgenic LCMT-1 overexpression (Fig. 4A, B). If reduced LCMT-1 protein levels contribute to the cognitive and electrophysiological impairments in Tg2576 animals, then compensation for this decrease could underlie the protective effects of transgenic LCMT-1 overexpression that we observe. Moreover, if the APP-related decrease in LCMT-1 levels also contributes to the trend for increased tau phosphorylation in these animals, then transgenic LCMT-1 overexpression may similarly counteract this effect. However, we were unable to detect a significant effect of LCMT overexpression on demethyl-PP2A levels in either control or APP transgenic animals, suggesting that either subtle changes in the levels of PP2A methylation underlie the effects of LCMT overexpression, or LCMT acts via mechanisms other than increased PP2A methylation to protect mice from impairments caused by Tg2576 expression (Fig 4A, B).
Transgenic PME-1 overexpression produces a trend for increased behavioral and electrophysiological impairments in Tg2576 mice
PP2A methylation by LCMT-1 is reversed by the demethylating activity of PME-1 [50, 51]. Consistent with the opposing actions of these enzymes, we found previously that transgenic overexpression of LCMT-1 and PME-1 also exerted opposite effects on sensitivity to impairments caused by acute exposure to exogenously applied Aβ [28]. To test whether PME-1 overexpression might also increase the sensitivity of mice to impairments caused by chronically elevated levels of Aβ, we generated animals that expressed the PME-1 and Tg2576 transgenes (PME + APP), the Tg2576 (APP) or PME-1 (PME) transgenes alone, and non-expressing controls. To determine whether PME-1 overexpression might advance the onset of cognitive impairments in AβPP transgenic mice, we compared their performance in a 2-day radial arm water maze task at 3-4 months of age, when cognitive impairments are not yet apparent in Tg2576 animals. We found that there was a clear trend for increased errors in the group that expressed both the APP and PME transgenes. However, the difference between this group and the others did not reach statistical significance (Fig. 5A). In addition, the PME + APP group also exhibited a significant increase in escape latency in the initial training blocks of a visible platform water maze task (Fig. 5B). Since this task requires only a simple association between the visible platform and escape, their impaired performance in this task suggests that the combination of mutant APP and PME-1 overexpression adversely affects a non-cognitive aspect behavior required for normal water maze performance that may also adversely impact performance the performance of this group in the 2-day radial arm water maze task.
Fig. 5
The effect of PME overexpression on cognitive impairments in Tg2576 mice. A) Plot of average number of errors committed (±SEM) during each 3-trial training block of a 2-day radial arm water maze task for 3-4-month-old mice that carried the Tg2576 APP transgene in combination with transgenic PME overexpression (PME + APP), carried the Tg2576 APP transgene alone (APP), overexpressed PME alone (PME), or controls. The PME + APP showed a trend for increased errors compared to all other groups that was not statistically significant (2-way RM ANOVA for errors on day 2 (blocks 6–10) with group and block as factors, F(3,51) = 1.783, p = 0.1621 for group). N = 12 control, 12 APP, 16 PME, 15 PME + APP. B) Plot of the average escape latency (±SEM) for 3-4-month-old animals from the indicated groups during training on a visible platform water maze task reveals significant differences between groups (2-way RM ANOVA for latency with group and block as factors shows no significant effect of group F(3,50) = 7.865, p = 0.0002). Dunnett’s multiple comparisons show that the PME + APP group was significantly different from each of the other three groups (p = 0.0002 for control, p = 0.0018 for APP, and p = 0.0026 for PME). N = 13 control, 10 APP, 16 PME, 15 PME + APP. C) Plot of the average swim speed (±SEM) for the indicated groups during training on the visible platform water maze task described in B reveals no significant differences between groups (2-way RM ANOVA for speed with group and block as factors shows no significant effect of group F(3,50) = 0.6083, p = 0.6127). D) Plot of average number of errors committed (±SEM) during each 3-trial training block of a 2-day radial arm water maze task for 6-7-month-old mice that carried the Tg2576 APP transgene in combination with transgenic PME overexpression (PME + APP), carried the Tg2576 APP transgene alone (APP), overexpressed PME alone (PME), or controls. 2-way RM ANOVA for errors on day 2 (blocks 6–10) with group and block as factors shows a significant effect of group (F(3,72) = 15.46, p < 0.0001). Dunnett’s multiple comparisons show that the both the PME + APP and APP groups were significantly different from both control and PME alone groups (p < 0.001 for all comparisons) but were not significantly different from one another (p = 0.2956). E) Plot of the average escape latency (±SEM) for 6-7-month-old animals from the indicated groups during training on a visible platform water maze task reveals significant differences between groups (2-way RM ANOVA for effect of group with group and block as factors F(3,72) = 4.954, p = 0.0035). Dunnett’s multiple comparisons show that the PME + APP group was significantly different from the control (p = 0.0012) and PME (p = 0.0167) groups, but not the APP group (p = 0.1064). F) Plot of the average swim speed (±SEM) for the indicated groups during training on the visible platform water maze task described in B reveals significant differences between groups (2-way RM ANOVA for effect of group with group and block as factors F(3,72) = 12.15, p < 0.0001). Dunnett’s multiple comparisons show that the PME + APP group was significantly different from each of the other groups (p < 0.0001 for control, p = 0.0034 for PME alone, p = 0.0005 for APP alone). For experiments on 6-7-month-old animals: N = 27 control, 22 APP, 13 PME, 14 PME + APP.
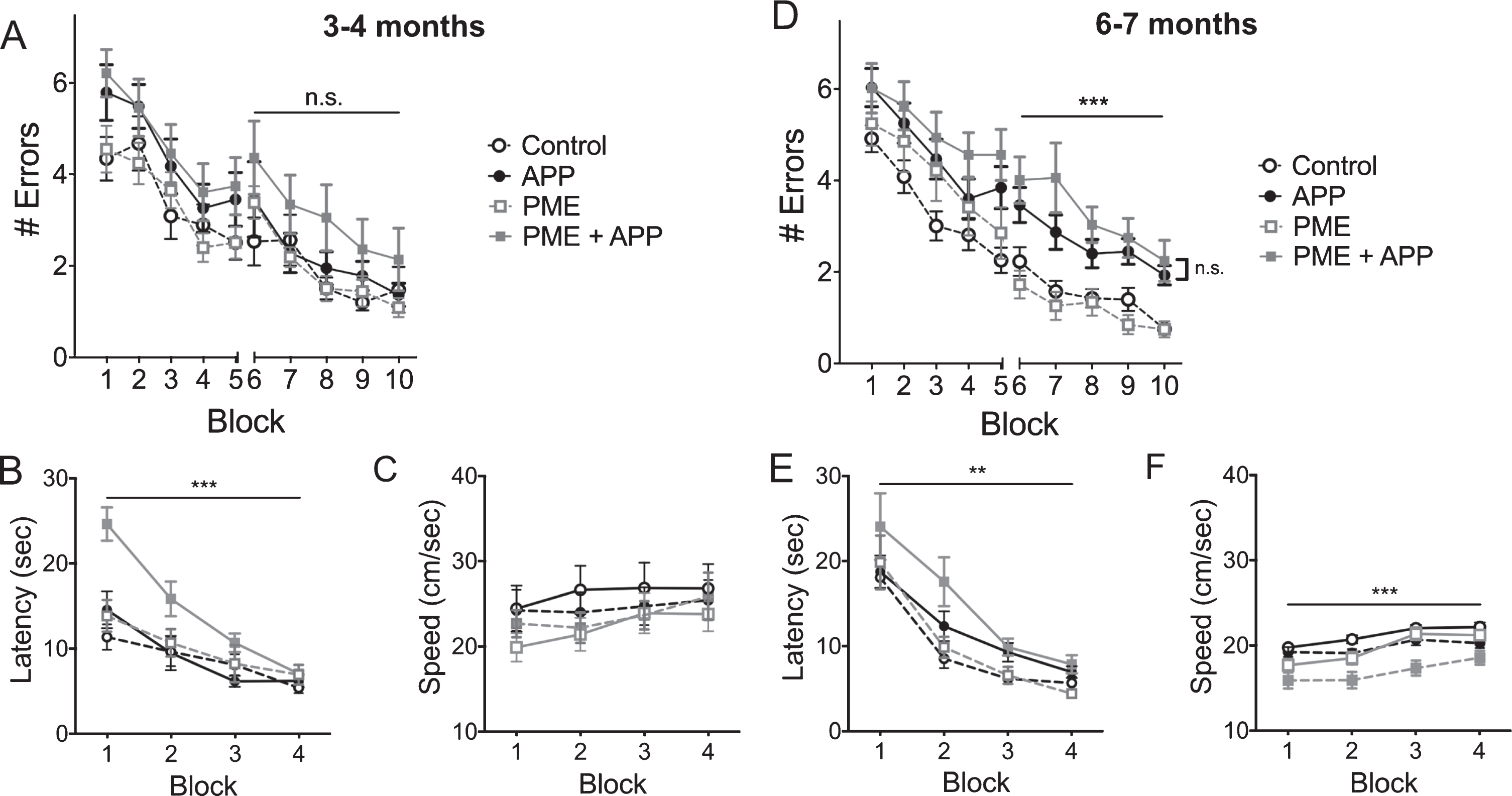
To test whether PME-1 overexpression may increase the severity of cognitive impairments in mutant APP transgenic mice, we performed these same behavioral tests on a separate group of animals at 6-7 months of age. Here, we found that animals that expressed APP alone, or in combination with the PME-1 transgene were comparably impaired in their performance on a 2-day radial arm water maze task (Fig. 5D). As was the case for these animals at 3-4 months, we found that the APP + PME-1 group showed a significant reduction in escape latency on a visible platform water maze task at this age (Fig. 5E). These animals also showed a significant reduction in swimming speed at that was not evident at 3-4 months, and together this reduced swim speed, and the increased escape latency of this group further support the conclusion that the combination of mutant APP expression and PME-1 overexpression impairs non-cognitive aspects of behavior required for normal water maze performance (Fig. 5F).
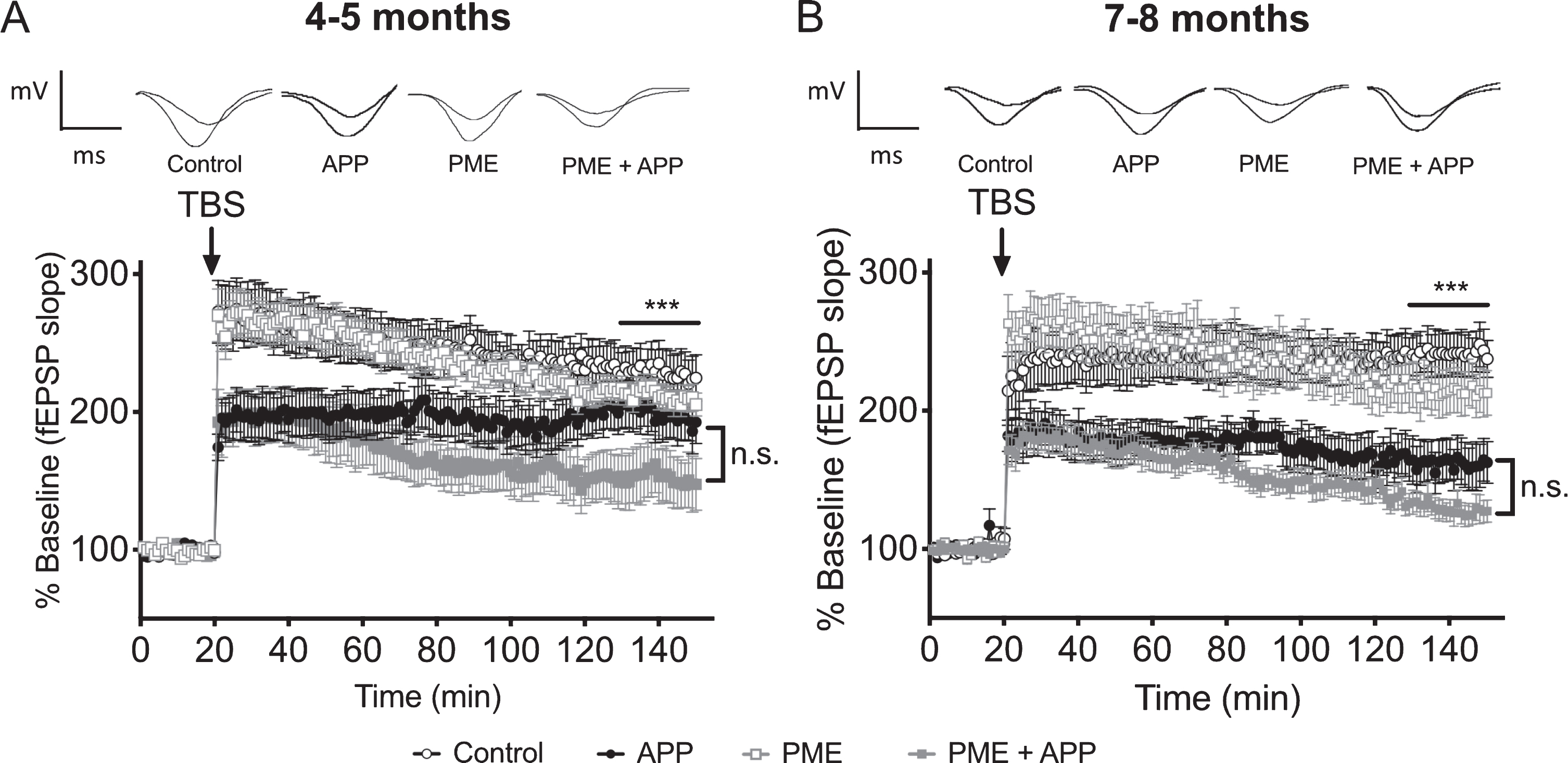
To determine whether transgenic PME-1 overexpression might advance the onset or increase the severity of synaptic plasticity impairments in Tg2576 mice, we compared the magnitude of LTP among groups of mice that expressed both the APP and PME-1 transgenes, the APP transgene alone, the PME-1 transgene alone, or neither transgene. We found that at both 4-5 months and 7-8 months of age, mice that co-expressed the APP and PME-1 transgenes exhibited a clear trend for reduced LTP relative to animals that carried the APP transgene alone, and the fact that the apparent magnitude of the difference between these two groups was greater at 4-5 months compared to 7-8 months is consistent with PME-1 overexpression promoting an earlier onset of LTP impairments in APP transgenic animals (Fig. 6A, B). However, in neither case did the difference between these groups reach statistical significance in post-hoc pairwise comparisons.
Fig. 6
The effect of PME overexpression on LTP impairments in Tg2576 mice. A) Time course of averaged Schaffer collateral fEPSP responses (±SEM) prior to and following delivery of theta-burst stimulation (arrow) in hippocampal slices prepared from 4-5-month-old mice that carried the Tg2576 APP transgene in combination with transgenic PME overexpression (PME + APP), or 4-5-month-old animals that carried the Tg2576 APP transgene alone (APP), overexpressed PME alone (PME), or controls. 2-way RM ANOVA for fEPSP responses over the last 20 min of recording with group and time as factors shows a significant effect of group (horizontal bar; F(3,65) = 4.717, p = 0.0049), however, the trend for increased impairment in the PME + APP group relative to the APP alone group was not statistically significant (Dunnett’s multiple comparisons p = 0.1165). N = 17 control, 13 APP, 24 PME, 15 PME + APP. Representative traces of field potential responses prior to TBS, and during the last 20 min of recording are shown above for each of the indicated genotypes. B) Time course of averaged Schaffer collateral fEPSP responses (±SEM) prior to and following delivery of theta-burst stimulation (arrow) in hippocampal slices prepared from 7-8-month-old mice that carried the Tg2576 APP transgene in combination with transgenic PME overexpression (PME + APP), or 7-8-month-old animals that carried the Tg2576 APP transgene alone (APP), overexpressed PME alone (PME), or controls. 2-way RM ANOVA for fEPSP responses over the last 20 min of recording with group and time as factors shows a significant effect of group (horizontal bar; F(3,45) = 15.17, p < 0.0001); however, the trend for increased impairment in the PME + APP group relative to the APP alone group was not statistically significant (Dunnett’s multiple comparisons p = 0.2791). N = 11 control, 8 APP, 14 PME, 16 PME + APP. Representative traces of field potential responses prior to TBS, and during the last 20 min of recording are shown above for each of the indicated genotypes.
To explore the effects of PME-1 overexpression on the molecular pathogenesis of cognitive and electrophysiological impairments in Tg2576 mice, we performed a series of western blots analogous to those described above for LCMT-1. Hippocampal homogenates were prepared from 4-5- and 7-8-month-old mice that expressed both the APP and PME-1 transgenes, the APP transgene alone, the PME-1 transgene alone, or neither transgene. PME-1 transgene expression resulted in a 14–25-fold increase in PME-1 immunoreactivity over endogenous levels with a high degree of inter-individual variability (minimum of 2.3-fold/maximum of 58-fold) that was not significantly affected by expression of the mutant APP transgene (Fig. 7A, B). In addition to increased PME-1 expression, animals expressing the PME-1 transgene exhibited significant increases in the amount of demethylated (L309) PP2A catalytic subunit, as well as a small but significant increase in the total amount of PP2A catalytic subunit in the PME alone group relative to controls, but not the PME + APP group relative APP alone (Fig. 7A, B).
Fig. 7
The effect of PME overexpression on APP expression, tau phosphorylation and PME protein levels in Tg2576 mice. A, B) Representative western blots showing anti-APP, phospho-tau serine 396/404 and serine 202, total tau, PME, demethylated PP2A/C and total PP2A/C immunoreactivity in hippocampal homogenates prepared from 4-5 (A) or 7-8-month-old (B) control, PME overexpressing, Tg2576 APP transgenic, or PME overexpressing/APP transgenic mice together with corresponding histograms showing average normalized immunoreactivity (±SEM) in each group. APP genotype significantly affected APP expression (2-way ANOVA with APP and PME genotype as factors: F(1,20) = 564.7, p < 0.0001 for 4-5-month-old mice and F(1,20) = 251.3, p < 0.0001 for 7-8-month-old mice). Sidak’s multiple comparisons showed no significant differences in APP expression between control and PME groups (p = 0.9504 for 4-5 and p = 0.9542 for 7-8-month-old mice) or between APP and PME + APP groups (p = 0.5961 for 4-5-month-old and, p = 0.9313 for 7-8-month-old mice). Animals carrying an APP transgene showed a trend for increased tau phosphorylation at serine residues 396/404 (APP:113.3±3.82, PME + APP: 111.3±6.05 versus control: 100±8.58, PME: 102.1±4.08 at 4-5 months, and APP:119.5±8.42, PME + APP: 115.9±7.41 versus control: 100±4.98, PME: 99.0±14.06 at 7-8 months) that did not reach statistical significance (2-way ANOVA with APP and PME genotype as factors: F(1,20) = 3.725, p = 0.0679 for 4-5-month-old mice and F(1,20) = 3.806, p = 0.0652 for 7-8-month-old mice), and was not affected by PME overexpression (Sidak’s multiple comparisons between APP and PME + APP groups: p = 0.9499 for 4-5-month-old and, p = 0.9554 for 7-8-month-old mice). Similarly, animals carrying an APP transgene showed a trend for increased tau phosphorylation at serine residue 202 (APP:108.9±4.78, PME + APP: 117.9±8.82 versus control: 100±6.13, PME: 101.6±6.87 at 4-5 months, and APP: 128.7±11.25, PME + APP: 124.3±16.02 versus control: 100±11.51, PME: 105.2±10.04), that did not reach statistical significance (2-way ANOVA with APP and PME genotype as factors: F(1,20) = 3.423, p = 0.0791 for 4-5-month-old mice and F(1,20) = 3.721, p = 0.0680 for 7-8-month-old mice), and was not affected by PME overexpression (Sidak’s multiple comparisons between APP and PME + APP groups (p = 0.5895 for 4-5-month-old and, p = 0.9750 for 7-8-month-old mice). Neither PME nor APP transgenes significantly affected endogenous tau levels (2-way ANOVA for effect of PME transgene with APP and PME genotype as factors: F(1,44) = 1.098, p = 0.3005 for 4-5-month-old mice and F(1,44) = 0.6706, p = 0.4173 for 7-8-month-old mice; 2-way ANOVA for effect of APP transgene with APP and PME genotype as factors: F(1,44) = 0.6987, p = 0.4077 for 4-5-month-old mice and F(1,44)<0.0001, p = 0.9985 for 7-8-month-old mice). Total PME expression (endogenous + transgenic) showed a significant effect of PME genotype (2-way ANOVA with PME and APP genotype as factors: F(1,20) = 20.39, p = 0.0002 for 4-5-month-old mice and F(1,20) = 7.611, p = 0.0121 for 7-8-month-old mice). Sidak’s multiple comparisons showed no significant differences in PME expression between control and APP groups (p > 0.9999 for 4-5 and 7-8-month-old mice) or between PME and PME + APP groups (p = 0.6028 for 4-5-month-old and, p = 0.5471 for 7-8-month-old mice). Transgenic PME overexpression significantly increased the amount of PP2A catalytic subunit that was demethylated at Leucine 309 (2-way ANOVA with PME and APP genotype as factors: F(1,20) = 59.54, p < 0.0001 for 4-5-month-old mice and F(1,20) = 24.55, p < 0.0001 for 7-8-month-old mice). Sidak’s multiple comparisons showed significant differences in demethyl-PP2A/C immunoreactivity between control and PME groups (p < 0.0001 for 4-5 and p = 0.0017 for 7-8-month-old mice) or between APP and PME + APP groups (p = 0.0003 for 4-5-month-old and, p = 0.0119 for 7-8-month-old mice). Transgenic PME overexpression also led to a small but significant increase in the total amount of PP2A catalytic subunit in the PME relative to control group but not the PME + APP relative to APP alone group (2-way ANOVA for effect of PME with PME and APP genotype as factors: F(1,20) = 6.077, p = 0.0229 for 4-5-month-old mice and F(1,20) = 10.73, p = 0.0048 for 7-8-month-old mice). Sidak’s multiple comparisons showed significant differences in total PP2A/C immunoreactivity between control and PME groups (p = 0.0230 for 4-5 and p = 0.0313 for 7-8-month-old mice) but not APP and PME + APP groups (p = 0.7376 for 4-5-month-old and p = 0.1374 for 7-8-month-old mice). For all blots band intensities in each lane were normalized to corresponding actin or total tau immunoreactivity in that lane, and values were expressed as a percentage of the mean of the control group (N = 6 per group).
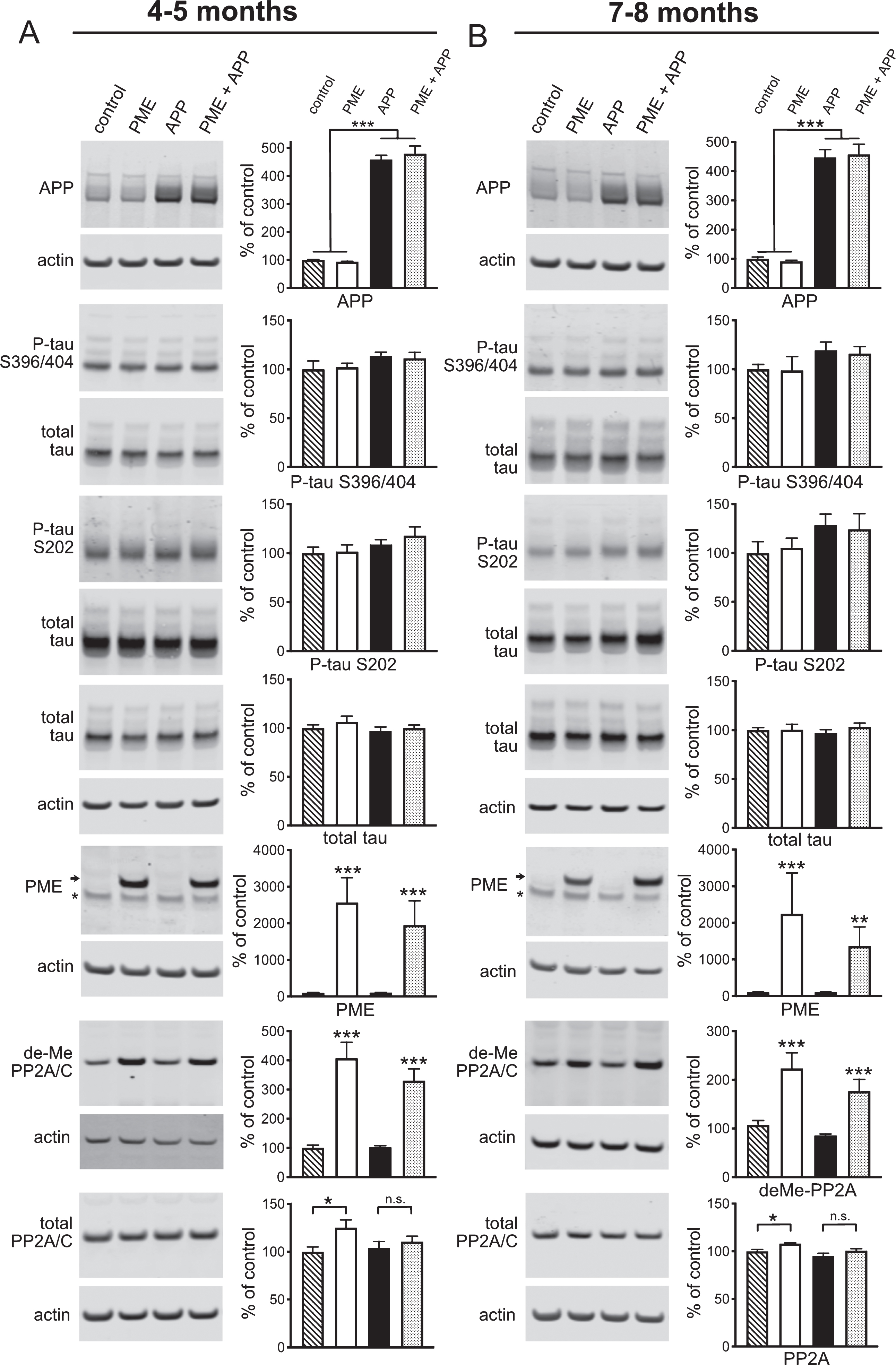
A comparison of APP immunoreactivity in these homogenates showed the same 4-5-fold increase in APP protein levels in animals that carried the mutant APP transgene that we observed in Fig. 4, with no significant effect of transgenic PME-1 expression on either transgenic or endogenous APP levels (Fig. 7A, B). Western blots on these homogenates for total and phosphorylated forms of tau showed a trend for increased tau phosphorylation at the serine 202 and 396/404 residues in APP transgenic mice that was similar to what we observed in Fig. 4 (Fig. 7A, B). However, unlike the mutant APP + LMCT-1 transgene expressing animals, this trend was also apparent in the mutant APP + PME-1 transgene expressing animals. This apparent difference between the effects of LCMT-1 and PME-1 overexpression on APP-related increases in tau phosphorylation is consistent with their opposing actions on PP2A-dependent tau dephosphorylation as well as the possibility that changes in PP2A-dependent tau dephosphorylation may contribute to their effects on Aβ sensitivity. However, the magnitude of these apparent changes in tau phosphorylation in both the PME-1 and LCMT-1 overexpressing mice, and the absence of significant changes in PP2A methylation in the LCMT-1 overexpressing mice suggest that alternate or additional, tau or PP2A-independent mechanisms may underlie or contribute to the effects LCMT-1 and PME-1 overexpression on Aβ sensitivity (see Discussion).
DISCUSSION
Here, we sought to determine whether increased LCMT-1 activity would protect against cognitive impairments caused by chronically elevated Aβ levels. To do this, we utilized the Tg2576 line of transgenic mice maintained in a congenic 129S6 background that are reported to be free of visual impairments caused by the rd1 mutation and to exhibit more consistent behavioral performance than animals maintained in the original mixed B6;SJL background [52, 53]. We crossed these animals to a previously described line of LCMT-1 overexpressing transgenic mice maintained in a C57BL6/J background to generate animals in a defined mixed F1 genetic background [28]. Tg2576 transgene expression in the progeny of these crosses resulted in significantly elevated levels of both Aβ40 and Aβ42 (Table 1), as well as characteristic age-dependent cognitive and electrophysiological impairments [34, 35].
We found that transgenic LCMT-1 overexpression protected against cognitive and electrophysiological impairments in animals that carried the Tg2576 transgene. Moreover, these protective affects were evident at both 6-7 and 9-10 months of age and were without effect on either endogenously or transgenically produced APP, Aβ40, or Aβ42. These results are consistent with our previous observations of the protective effects of transgenic LCMT-1 overexpression on cognitive and electrophysiological impairments caused by acutely elevated Aβ levels [28], and with the hypothesis that LCMT-1 overexpression blocks the pathological response to these proteins, rather than their production. Since Tg2576 transgenic mice produce increased levels of full-length APP, these data also suggest that LCMT-1 overexpression may protect against the pathological actions of other APP proteolytic products, in addition to Aβ40 and Aβ42.
PME-1 opposes the activity of LCMT-1 by demethylating PP2A [50, 51, 54], and we found previously that PME-1 overexpression increased sensitivity to impairments caused by acutely elevated Aβ levels [28]. We therefore sought to determine whether increased PME-1 activity would also increase the severity or advance the onset of cognitive or electrophysiological impairments in Tg2576 animals. While the results of these experiments were consistent with PME overexpression leading to an earlier onset and/or increased severity of these impairments, the magnitude of these effects were small and potentially confounded by the emergence of non-cognitive impairments in PME overexpressing Tg2576 animals.
One of the ways in which LCMT-1 overexpression could impact pathogenic responses to Aβ, is through increased tau dephosphorylation. Increased LCMT-1 expression could lead to an increase in PP2A methylation and PP2A-mediated tau dephosphorylation that antagonizes pathogenic increases in tau phosphorylation caused by Aβ exposure. Our data revealed trends for changes in tau phosphorylation that were consistent with this model, as well as a possible role for restored LCMT-1 expression in LCMT-1 overexpressing Tg2576 mice that parallels a proposed role for the decreased LCMT-1 expression observed in AD brains.
However, the modest changes in tau phosphorylation that we observed in these animals suggest that additional, or alternate downstream targets may account for, or contribute to the protective effects of LCMT-1 overexpression. PP2A has been found to interact with multiple kinases including extracellular signal-related kinase (ERK), c-Jun amino terminal kinase (JNK), cyclin-dependent kinase-5 (CDK5), and glycogen synthase kinase-3β (GSK3β), as well as metabotropic glutamate receptor-5 (mGluR5) and NMDA receptors, each of which have been implicated in AD [12, 49, 55–59]. If LCMT-1 overexpression protects against Aβ-induced impairments by altering PP2A activity, then alterations in PP2A’s interactions with one or more of these proteins may underlie its protective effects. Moreover, the net effect of these alterations may also be impacted by the complexity of the regulatory relationships among PP2A and these targets. For example, while PP2A dephosphorylates tau residues that are direct targets of GSK3β, it also activates GSK3β through dephosphorylation at serine 9, and evidence suggests that GSK3β may, in turn, positively or negatively regulate PP2A [59–64].
Similarly, our inability to detect a change in demethyated PP2A levels in the LCMT-1 overexpressing mice raises the possibility that LCMT-1 transgene expression may protect against cognitive and electrophysiological impairments in Tg2576 mice via mechanisms that are independent of PP2A methylation. One possibility is that PME-1 or LCMT-1 with may affect PP2A biogenesis or subcellular distribution through physical association PP2A or its subunits independent of their effect on methylation, and this possibility is consistent with differences in the subcellular distribution of PME-1 and LCMT-1 that have been described [65, 66]. Another possibility is that the effects of LCMT-1 or PME-1 overexpression on Aβ-related impairments may result from their actions on targets other than PP2A. In this regard, the phosphatases, PP4 and PP6 share a high degree of sequence similarity to PP2A including a conserved C-terminal leucine on the catalytic subunit that is subject to methylation by LCMT-1, and, in the case of PP4, demethylation by PME-1 [67, 68]. If transgenic overexpression of LCMT-1 or PME-1 alters the methylation levels of PP4 and/or PP6, then the resulting changes in the activity of these phosphatases could also contribute the effects of LCMT-1 or PME-1 overexpression on Aβ-related impairments. Identifying the possible contributions of different PP2A substrates and LCMT-1 targets to the protective effects of LCMT-1 overexpression will both further our understanding of the molecular mechanisms underlying Aβ-induced impairments, and reveal additional targets in this pathway that may be therapeutically relevant for AD prevention or treatment.
ACKNOWLEDGMENTS
This work was supported by a National Institutes of Health grant 1RO1NS092045-01. The authors thank Dr. Peter Davies of the Albert Einstein Medical College for the generous gifts of the PHF1 and CP13 antibodies, and Dr. Karen Hsiao-Ashe of the University of Minnesota for the generous gift of the Tg2576 mouse line.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0462r2)
REFERENCES
[1] | Sontag JM , Sontag E ((2014) ) Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front Mol Neurosci 7: , 16. |
[2] | Sontag E , Luangpirom A , Hladik C , Mudrak I , Ogris E , Speciale S , White CL , 3rd ((2004) ) Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol 63: , 287–301. |
[3] | Vogelsberg-Ragaglia V , Schuck T , Trojanowski JQ , Lee VM ((2001) ) PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp Neurol 168: , 402–412. |
[4] | Gong CX , Shaikh S , Wang JZ , Zaidi T , Grundke-Iqbal I , Iqbal K ((1995) ) Phosphatase activity toward abnormally phosphorylated tau: Decrease in Alzheimer disease brain. J Neurochem 65: , 732–738. |
[5] | Gong CX , Singh TJ , Grundke-Iqbal I , Iqbal K ((1993) ) Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem 61: , 921–927. |
[6] | Martin L , Latypova X , Wilson CM , Magnaudeix A , Perrin ML , Terro F ((2013) ) Tau protein phosphatases in Alzheimer’s disease: The leading role of PP2A. Ageing Res Rev 12: , 39–49. |
[7] | Sun L , Liu SY , Zhou XW , Wang XC , Liu R , Wang Q , Wang JZ ((2003) ) Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience 118: , 1175–1182. |
[8] | Wang X , Blanchard J , Kohlbrenner E , Clement N , Linden RM , Radu A , Grundke-Iqbal I , Iqbal K ((2010) ) The carboxy-terminal fragment of inhibitor-2 of protein phosphatase-2A induces Alzheimer disease pathology and cognitive impairment. FASEB J 24: , 4420–4432. |
[9] | Yin YY , Liu H , Cong XB , Liu Z , Wang Q , Wang JZ , Zhu LQ ((2010) ) Acetyl-L-carnitine attenuates okadaic acid induced tau hyperphosphorylation and spatial memory impairment in rats. J Alzheimers Dis 19: , 735–746. |
[10] | Kins S , Crameri A , Evans DR , Hemmings BA , Nitsch RM , Gotz J ((2001) ) Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J Biol Chem 276: , 38193–38200. |
[11] | Schild A , Ittner LM , Gotz J ((2006) ) Altered phosphorylation of cytoskeletal proteins in mutant protein phosphatase 2A transgenic mice. Biochem Biophys Res Commun 343: , 1171–1178. |
[12] | Louis JV , Martens E , Borghgraef P , Lambrecht C , Sents W , Longin S , Zwaenepoel K , Pijnenborg R , Landrieu I , Lippens G , Ledermann B , Gotz J , Van Leuven F , Goris J , Janssens V ((2011) ) Mice lacking phosphatase PP2A subunit PR61/B’delta (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3beta. Proc Natl Acad Sci U S A 108: , 6957–6962. |
[13] | Bolognin S , Blanchard J , Wang X , Basurto-Islas G , Tung YC , Kohlbrenner E , Grundke-Iqbal I , Iqbal K ((2012) ) An experimental rat model of sporadic Alzheimer’s disease and rescue of cognitive impairment with a neurotrophic peptide. Acta Neuropathol 123: , 133–151. |
[14] | Basurto-Islas G , Blanchard J , Tung YC , Fernandez JR , Voronkov M , Stock M , Zhang S , Stock JB , Iqbal K ((2014) ) Therapeutic benefits of a component of coffee in a rat model of Alzheimer’s disease. Neurobiol Aging 35: , 2701–2712. |
[15] | Corcoran NM , Martin D , Hutter-Paier B , Windisch M , Nguyen T , Nheu L , Sundstrom LE , Costello AJ , Hovens CM ((2010) ) Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J Clin Neurosci 17: , 1025–1033. |
[16] | van Eersel J , Ke YD , Liu X , Delerue F , Kril JJ , Gotz J , Ittner LM ((2010) ) Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci U S A 107: , 13888–13893. |
[17] | Vitek MP , Christensen DJ , Wilcock D , Davis J , Van Nostrand WE , Li FQ , Colton CA ((2012) ) APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer’s disease transgenics. Neurodegener Dis 10: , 122–126. |
[18] | Asam K , Staniszewski A , Zhang H , Melideo SL , Mazzeo A , Voronkov M , Huber KL , Perez E , Stock M , Stock JB , Arancio O , Nicholls RE ((2017) ) Eicosanoyl-5-hydroxytryptamide (EHT) prevents Alzheimer’s disease-related cognitive and electrophysiological impairments in mice exposed to elevated concentrations of oligomeric beta-amyloid. PLoS One 12: , e0189413. |
[19] | Lambrecht C , Haesen D , Sents W , Ivanova E , Janssens V ((2013) ) Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol Biol 1053: , 283–305. |
[20] | Reynhout S , Janssens V ((2019) ) Physiologic functions of PP2A: Lessons from genetically modified mice. Biochim Biophys Acta Mol Cell Res 1866: , 31–50. |
[21] | Sents W , Ivanova E , Lambrecht C , Haesen D , Janssens V ((2013) ) The biogenesis of active protein phosphatase 2A holoenzymes: A tightly regulated process creating phosphatase specificity. FEBS J 280: , 644–661. |
[22] | Wu CG , Zheng A , Jiang L , Rowse M , Stanevich V , Chen H , Li Y , Satyshur KA , Johnson B , Gu TJ , Liu Z , Xing Y ((2017) ) Methylation-regulated decommissioning of multimeric PP2A complexes. Nat Commun 8: , 2272. |
[23] | Longin S , Zwaenepoel K , Louis JV , Dilworth S , Goris J , Janssens V ((2007) ) Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic Subunit. J Biol Chem 282: , 26971–26980. |
[24] | Xu Y , Chen Y , Zhang P , Jeffrey PD , Shi Y ((2008) ) Structure of a protein phosphatase 2A holoenzyme: Insights into B55-mediated Tau dephosphorylation. Mol Cell 31: , 873–885. |
[25] | Sontag E , Hladik C , Montgomery L , Luangpirom A , Mudrak I , Ogris E , White CL , 3rd ((2004) ) Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J Neuropathol Exp Neurol 63: , 1080–1091. |
[26] | Park HJ , Lee KW , Oh S , Yan R , Zhang J , Beach TG , Adler CH , Voronkov M , Braithwaite SP , Stock JB , Mouradian MM ((2018) ) Protein phosphatase 2A and its methylation modulating enzymes LCMT-1 and PME-1 are dysregulated in tauopathies of progressive supranuclear palsy and Alzheimer disease. J Neuropathol Exp Neurol 77: , 139–148. |
[27] | Zhuo JM , Wang H , Pratico D ((2011) ) Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol Sci 32: , 562–571. |
[28] | Nicholls RE , Sontag JM , Zhang H , Staniszewski A , Yan S , Kim CY , Yim M , Woodruff CM , Arning E , Wasek B , Yin D , Bottiglieri T , Sontag E , Kandel ER , Arancio O ((2016) ) PP2A methylation controls sensitivity and resistance to beta-amyloid-induced cognitive and electrophysiological impairments. Proc Natl Acad Sci U S A 113: , 3347–3352. |
[29] | Mayford M , Bach ME , Huang YY , Wang L , Hawkins RD , Kandel ER ((1996) ) Control of memory formation through regulated expression of a CaMKII transgene. Science 274: , 1678–1683. |
[30] | Hsiao K , Chapman P , Nilsen S , Eckman C , Harigaya Y , Younkin S , Yang F , Cole G ((1996) ) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: , 99–102. |
[31] | Alamed J , Wilcock DM , Diamond DM , Gordon MN , Morgan D ((2006) ) Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat Protoc 1: , 1671–1679. |
[32] | Fa M , Orozco IJ , Francis YI , Saeed F , Gong Y , Arancio O (2010) Preparation of oligomeric beta-amyloid 1-42 and induction of synaptic plasticity impairment on hippocampal slices. J Vis Exp 1884. |
[33] | Casali BT , Landreth GE ((2016) ) Abeta extraction from murine brain homogenates. Bio Protoc 6: , e1787. |
[34] | Stewart S , Cacucci F , Lever C ((2011) ) Which memory task for my mouse? A systematic review of spatial memory performance in the Tg2576 Alzheimer’s mouse model. J Alzheimers Dis 26: , 105–126. |
[35] | Webster SJ , Bachstetter AD , Nelson PT , Schmitt FA , Van Eldik LJ ((2014) ) Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet 5: , 88. |
[36] | Chapman PF , White GL , Jones MW , Cooper-Blacketer D , Marshall VJ , Irizarry M , Younkin L , Good MA , Bliss TV , Hyman BT , Younkin SG , Hsiao KK ((1999) ) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2: , 271–276. |
[37] | Jacobsen JS , Wu CC , Redwine JM , Comery TA , Arias R , Bowlby M , Martone R , Morrison JH , Pangalos MN , Reinhart PH , Bloom FE ((2006) ) Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 103: , 5161–5166. |
[38] | Kawarabayashi T , Younkin LH , Saido TC , Shoji M , Ashe KH , Younkin SG ((2001) ) Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 21: , 372–381. |
[39] | Bloom GS ((2014) ) Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71: , 505–508. |
[40] | Spires-Jones TL , Hyman BT ((2014) ) The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82: , 756–771. |
[41] | Shipton OA , Leitz JR , Dworzak J , Acton CE , Tunbridge EM , Denk F , Dawson HN , Vitek MP , Wade-Martins R , Paulsen O , Vargas-Caballero M ((2011) ) Tau protein is required for amyloid beta-induced impairment of hippocampal long-term potentiation. J Neurosci 31: , 1688–1692. |
[42] | Ma QL , Yang F , Rosario ER , Ubeda OJ , Beech W , Gant DJ , Chen PP , Hudspeth B , Chen C , Zhao Y , Vinters HV , Frautschy SA , Cole GM ((2009) ) Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J Neurosci 29: , 9078–9089. |
[43] | Jin M , Shepardson N , Yang T , Chen G , Walsh D , Selkoe DJ ((2011) ) Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A 108: , 5819–5824. |
[44] | De Felice FG , Wu D , Lambert MP , Fernandez SJ , Velasco PT , Lacor PN , Bigio EH , Jerecic J , Acton PJ , Shughrue PJ , Chen-Dodson E , Kinney GG , Klein WL ((2008) ) Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging 29: , 1334–1347. |
[45] | Zempel H , Thies E , Mandelkow E , Mandelkow EM ((2010) ) Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci 30: , 11938–11950. |
[46] | Song MS , Rauw G , Baker GB , Kar S ((2008) ) Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur J Neurosci 28: , 1989–2002. |
[47] | Sontag E , Nunbhakdi-Craig V , Lee G , Bloom GS , Mumby MC ((1996) ) Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron 17: , 1201–1207. |
[48] | Sontag E , Nunbhakdi-Craig V , Sontag JM , Diaz-Arrastia R , Ogris E , Dayal S , Lentz SR , Arning E , Bottiglieri T ((2007) ) Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J Neurosci 27: , 2751–2759. |
[49] | Arif M , Kazim SF , Grundke-Iqbal I , Garruto RM , Iqbal K ((2014) ) Tau pathology involves protein phosphatase 2A in parkinsonism-dementia of Guam. Proc Natl Acad Sci U S A 111: , 1144–1149. |
[50] | Lee J , Chen Y , Tolstykh T , Stock J ((1996) ) A specific protein carboxyl methylesterase that demethylates phosphoprotein phosphatase 2A in bovine brain. Proc Natl Acad Sci U S A 93: , 6043–6047. |
[51] | Ogris E , Du X , Nelson KC , Mak EK , Yu XX , Lane WS , Pallas DC ((1999) ) A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. J Biol Chem 274: , 14382–14391. |
[52] | Rustay NR , Cronin EA , Curzon P , Markosyan S , Bitner RS , Ellis TA , Waring JF , Decker MW , Rueter LE , Browman KE ((2010) ) Mice expressing the Swedish APP mutation on a 129 genetic background demonstrate consistent behavioral deficits and pathological markers of Alzheimer’s disease. Brain Res 1311: , 136–147. |
[53] | Wolf A , Bauer B , Abner EL , Ashkenazy-Frolinger T , Hartz AM ((2016) ) A comprehensive behavioral test battery to assess learning and memory in 129S6/Tg2576 mice. PLoS One 11: , e0147733. |
[54] | Xing Y , Li Z , Chen Y , Stock JB , Jeffrey PD , Shi Y ((2008) ) Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell 133: , 154–163. |
[55] | Kins S , Kurosinski P , Nitsch RM , Gotz J ((2003) ) Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol 163: , 833–843. |
[56] | Plattner F , Angelo M , Giese KP ((2006) ) The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem 281: , 25457–25465. |
[57] | Mao L , Yang L , Arora A , Choe ES , Zhang G , Liu Z , Fibuch EE , Wang JQ ((2005) ) Role of protein phosphatase 2A in mGluR5-regulated MEK/ERK phosphorylation in neurons. J Biol Chem 280: , 12602–12610. |
[58] | Chan SF , Sucher NJ ((2001) ) An NMDA receptor signaling complex with protein phosphatase 2A. J Neurosci 21: , 7985–7992. |
[59] | Qian W , Shi J , Yin X , Iqbal K , Grundke-Iqbal I , Gong CX , Liu F ((2010) ) PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J Alzheimers Dis 19: , 1221–1229. |
[60] | Zhou XW , Winblad B , Guan Z , Pei JJ ((2009) ) Interactions between glycogen synthase kinase 3beta, protein kinase B, and protein phosphatase 2A in tau phosphorylation in mouse N2a neuroblastoma cells. J Alzheimers Dis 17: , 929–937. |
[61] | Yao XQ , Li XC , Zhang XX , Yin YY , Liu B , Luo DJ , Wang Q , Wang JZ , Liu GP ((2012) ) Glycogen synthase kinase-3beta regulates leucine-309 demethylation of protein phosphatase-2A via PPMT1 and PME-1. FEBS Lett 586: , 2522–2528. |
[62] | Wang Y , Yang R , Gu J , Yin X , Jin N , Xie S , Wang Y , Chang H , Qian W , Shi J , Iqbal K , Gong CX , Cheng C , Liu F ((2015) ) Cross talk between PI3K-AKT-GSK-3beta and PP2A pathways determines tau hyperphosphorylation. Neurobiol Aging 36: , 188–200. |
[63] | Chu D , Tan J , Xie S , Jin N , Yin X , Gong CX , Iqbal K , Liu F ((2016) ) GSK-3beta is dephosphorylated by PP2A in a Leu309 methylation-independent manner. J Alzheimers Dis 49: , 365–375. |
[64] | Jin N , Shi R , Jiang Y , Chu D , Gong CX , Iqbal K , Liu F ((2019) ) Glycogen synthase kinase-3beta suppresses the expression of protein phosphatase methylesterase-1 through beta-catenin. Aging (Albany NY) 11: , 9672–9688. |
[65] | Sontag JM , Nunbhakdi-Craig V , Sontag E ((2013) ) Leucine carboxyl methyltransferase 1 (LCMT1)-dependent methylation regulates the association of protein phosphatase 2A and tau protein with plasma membrane microdomains in neuroblastoma cells. J Biol Chem 288: , 27396–27405. |
[66] | Longin S , Zwaenepoel K , Martens E , Louis JV , Rondelez E , Goris J , Janssens V ((2008) ) Spatial control of protein phosphatase 2A (de)methylation. Exp Cell Res 314: , 68–81. |
[67] | Hwang J , Lee JA , Pallas DC ((2016) ) Leucine carboxyl methyltransferase 1 (LCMT-1) methylates protein phosphatase 4 (PP4) and protein phosphatase 6 (PP6) and differentially regulates the stable formation of different PP4 holoenzymes. J Biol Chem 291: , 21008–21019. |
[68] | Wandzioch E , Pusey M , Werda A , Bail S , Bhaskar A , Nestor M , Yang JJ , Rice LM ((2014) ) PME-1 modulates protein phosphatase 2A activity to promote the malignant phenotype of endometrial cancer cells. Cancer Res 74: , 4295–4305. |


