
Clinical and Pathological Phenotypes of LRP10 Variant Carriers with Dementia
Abstract
Background:
Rare variants in the low-density lipoprotein receptor related protein 10 gene (LRP10) have recently been implicated in the etiology of Parkinson’s disease (PD) and dementia with Lewy bodies (DLB).
Objective:
We searched for LRP10 variants in a new series of brain donors with dementia and Lewy pathology (LP) at autopsy, or dementia and parkinsonism without LP but with various other neurodegenerative pathologies.
Methods:
Sanger sequencing of LRP10 was performed in 233 donors collected by the Netherlands Brain Bank.
Results:
Rare, possibly pathogenic heterozygous LRP10 variants were present in three patients: p.Gly453Ser in a patient with mixed Alzheimer’s disease (AD)/Lewy body disease (LBD), p.Arg151Cys in a DLB patient, and p.Gly326Asp in an AD patient without LP. All three patients had a positive family history for dementia or PD.
Conclusion:
Rare LRP10 variants are present in some patients with dementia and different brain pathologies including DLB, mixed AD/LBD, and AD. These findings suggest a role for LRP10 across a broad neurodegenerative spectrum.
INTRODUCTION
Genetic factors play an important role in the etiology of both Parkinson’s disease (PD) as well as dementia with Lewy bodies (DLB) [1–3]. Among PD patients, 5–10% are estimated to carry disease-causing rare mutations [3], including variants in the SNCA gene, which are causal for PD as well as DLB. Moreover, common variants in SNCA and GBA are risk factors for both PD and DLB, which further highlights the genetic overlap between these disorders. Although mutations in more than 20 genes are known to cause monogenic forms of PD, additional rare disease-associated genetic variants in familial PD and DLB likely remain to be identified [1, 4].
We recently reported rare variants in the low-density lipoprotein receptor related protein 10 gene (LRP10) in familial forms of PD and DLB, including one large Italian family with genome-wide significant linkage evidence for late-onset, autosomal-dominant inherited PD and DLB. All ten affected relatives and three unaffected relatives (age 45–58 years) carried the same rare LRP10 variant, suggesting an age-related or incomplete penetrance. Several smaller families showed additional, albeit more limited, evidence of segregation of LRP10 variants with disease [5]. In three probands with LRP10 variants, autopsy studies showed diffuse-neocortical Lewy pathology (LP) and concomitant AD-type pathology.
Since our initial report, additional rare LRP10 variants have been reported in patients with PD, DLB, or other neurodegenerative diseases by some but not all studies [6–13]. Three case-control studies searched whole-exome sequencing data from sporadic PD, DLB, and multiple system atrophy (MSA) cases, and detected no evidence of association between rare LRP10 variants and disease [6–8]. However, these studies included mostly sporadic cases and controls with a younger average age, and they might have been underpowered to detect association due to the rarity of pathogenic LRP10 variants, their likely incomplete penetrance, and the etiological heterogeneity of these diseases. A French exome mining study found a p. Tyr307Asn variant that co-segregated with PD in two out of four members of the same family [9]. Although the pathogenicity of this variant may therefore be questioned, phenocopies are known to occur in around 5% of familial PD patients from families with monogenic forms of PD. Lastly, two Chinese and one Dutch cohort studies of sporadic and familial PD and DLB patients provided independent, albeit limited, additional evidence for an association of LRP10 variants with disease [11–13]. In addition, we recently found evidence for enrichment of rare, possibly pathogenic LRP10 variants in a large cohort of patients with progressive supranuclear palsy (PSP), mostly with a pathologically confirmed diagnosis [14]. In conclusion, despite some negative reports, additional evidence has been collected on a role for rare LRP10 variants in independent PD cohorts, and potentially other neurodegenerative diseases such as PSP. However, confirmation in independent large families as well as more extensive functional evidence is needed. Moreover, the findings of possibly pathogenic LRP10 variants in other neurodegenerative diseases need confirmation in pathologically-confirmed series.
Only few studies in the literature deal with the normal function of LRP10 in the human brain. LRP10 encodes transmembrane protein LRP10, which shuttles between the plasma membrane and the Golgi system and co-localizes with VPS35, a retromer component and the product of another known PD-causing gene [15–17]. LRP10 also interacts with GGA proteins, which are reported to affect α-synuclein trafficking and aggregation [18, 19]. Furthermore, LRP10 has been implicated in the metabolism of APOE lipoproteins and direct as well as GGA-mediated regulation of AβPP trafficking and processing, and may therefore also influence amyloid-β homeostasis [16, 20, 21]. Taken together, this suggests a broad role for LRP10 in neurodegeneration.
The aim of this study was to search for LRP10 variants in a series of 233 brain donors from the Netherlands Brain Bank (NBB), who were clinically diagnosed with dementia, and had LP at autopsy or, if LP-negative, had developed parkinsonism in addition to dementia during their lifetime.
MATERIALS AND METHODS
Donor selection
The entire series of brains available from the NBB (n = 3,853) from 1989 to 2017 was considered for inclusion in the study according to the selection criteria. For all donors, a written informed consent for brain autopsy and the use of the material and clinical information for research purposes had been obtained from the donor or the next of kin.
For Group 1, inclusion criteria were: 1) Braak Lewy body stage ≥4 according to the BrainNet Europe (BNE) criteria [22]; 2) presence of dementia; 3) at least low levels of AD type pathology according to National Institute on Aging–Alzheimer’s Association (NIA-AA) guidelines [23]. Out of all brain donors from the NBB, 432 donors showed Braak Lewy body stage ≥4, out of whom 324 showed dementia. Of these donors, 252 showed at least low levels AD type pathology. Some of these donors had already been screened for LRP10 variants in a previous study [5], or did not have frozen tissue available for DNA extraction, and were excluded. Therefore, 126 donors could be included in the current study.
For Group 2, inclusion criteria were: 1) presence of dementia and parkinsonism; 2) presence of any type of neurodegenerative disease upon autopsy; 3) Braak Lewy body stage 0 or 1 according to the BNE criteria [22]. Parkinsonism was defined as bradykinesia in combination with rigidity and/or tremor. Donors with possible drug-induced parkinsonism were excluded. Out of all brain donors from the NBB, 591 donors showed parkinsonism, of whom 543 had sufficient information on cognitive status. Dementia was present in 394 of these donors, of whom 182 did not show LP, and 148 did not take drugs that could be a sufficient explanation for the presence of parkinsonism. Seven cases had a known causal genetic mutation. Out of the other 141 cases, tissue was available for DNA extraction in 107 cases that had not been screened before for the presence of LRP10 variants
Donors with known disease-causing mutations in genes other than LRP10 were excluded. Fresh-frozen brain tissue from cerebellum, cortical, or subcortical grey matter was available from 126 donors from Group 1 and 107 donors from Group 2.
Clinical information
Clinical information was abstracted from the database from the NBB and included information on sex, age at onset, age at death, disease duration from time at onset of first symptoms, presence and timing of parkinsonism and dementia, a history of parkinsonism or dementia in first-degree or second-degree relatives, the clinical diagnosis by the treating physician, and cause of death.
Neuropathological assessment
For all donors in the study selection, standardized neuropathological examinations had been performed by an experienced neuropathologist (AJMR) (open access: www.brainbank.nl). Neuropathological assessment was done according to NIA-AA and BNE guidelines [22, 23], and standardized evaluation strategies for cerebral amyloid angiopathy (CAA) [24], granulovacuolar degeneration [25], limbic-predominant age-related TDP-43 encephalopathy (LATE) [26], and aging-related tau astrogliopathy (ARTAG) [27].
For the possibly pathogenic LRP10 variant carriers, 17 different brain regions were formalin-fixed and paraffin-embedded, cut into 8μm sections, and stained with hematoxylin and eosin. For selected cortical regions, histological stainings were performed using Congo red and Gallyas silver stains. Immunohistochemical analysis was performed on selected brainstem, limbic, and neocortical regions using primary antibodies against α-synuclein (clone KM51; 1 : 500; Monosan, The Netherlands), hyperphosphorylated tau (clone AT8; 1 : 1000; Innogenetics, Belgium), amyloid-β (clone 6F/3D; 1 : 100; Dako, USA) and phospho-TDP43 (rabbit polyclonal; 1 : 1000 Cosmo Bio, USA). Stages of amyloid-β plaques, neurofibrillary pathology, and neuritic plaques were scored according to the NIA-AA guidelines [23], and stages of LP were scored according to BNE criteria [22]. Standardized staging systems for CAA [24], granulovacuolar degeneration [25], and LATE [26] were applied. Presence of ARTAG was evaluated based on tau immunostaining (clone AT8) of sections of the hippocampus at the level of the lateral geniculate nucleus, amygdala, and temporal pole [27].
Sanger sequencing
Genomic DNA was isolated from brain tissue using standard methods. We performed Sanger sequencing using the reported protocol of Quadri et al. (2018) [5] with minor modifications, for the entire open reading frame and the exon-intron boundaries of LRP10. A total volume of 20μl, containing 2.0μl of 10X Fast Start Taq DNA Polymerase buffer, 1.6μl of 2.5 mM dNTPs, 1.0μl of 10μM forward primer, 1.0μl of 10μM reverse primer, 0.10 or 0.15μl of FastStart Taq. DNA Polymerase (Roche, Basel, Switzerland), and 25 ng of genomic DNA, was used to amplify the fragments. We added 4μl of 1X GC-RICH solution (Roche) for exon 1, 5, and 7. An overview of used primers can be found in Supplementary Table 1. We performed the initial denaturation for 5 min at 94°C followed by 30 cycles of 30 s at 94°C, 30 s at 60°C, 90 s at 72°C, with a final extension step for 5 min at 72°C. We used 5 units of ExoI and 0.5 unit of Fast AP (Thermo Fisher Scientific, Waltham, MA, USA) for 45 min at 37°C and 15 min at 80°C to remove unconsumed dNTPs and primers from the PCR product (3μl). The Big Dye Terminator (version 3.1; Thermo Fisher Scientific) was used according to the manufacturer’s protocol to sequence both DNA strands directly and sephadexG50 (GE Healthcare, Little Chalfont, UK) was used to remove dye terminators. We used an ABI 3730XL Genetic Analyzer (Thermo Fisher Scientific) and Seqscape v3.0 (Thermo Fisher Scientific) for the analysis. The LRP10 NM_0140445.4 transcript was used for sequence variants annotation and the Human Genome Variome Society recommendations [28] was used for variants nomenclature.
We considered variants as possibly pathogenic according to the previously used criteria: 1) heterozygous state; 2) minor allele frequency <0.1% in the genome aggregation database (GnomAD v2.1); 3) exonic location and non-synonymous, or predicted to affect splicing; and 4) predicted as pathogenic by at least five of 11 in-silico programs [5, 13].
Whole exome sequencing, copy number analysis, and APOE genotyping
To exclude (possibly) pathogenic variants in other known genes causing parkinsonism or dementia (Supplementary Table 2) we performed whole exome sequencing (WES) and multiple ligation-dependent probe amplification (MLPA, P051-Parkinson mix 1) in the possibly pathogenic LRP10 variant carriers. WES was performed with the same protocol as previously reported in Jamra et al. (2017) [29]. An average depth of >84× was reached, with 99% of the target region covered >20×.
For copy number analysis, The P051-D1 Parkinson kit (MRC Holland) was used according to the manufacturer’s protocol. An ABI 3730XL Genetic Analyzer (Thermo Fisher Scientific) and Seqscape v3.0 (Thermo Fisher Scientific) were used for analysis. APOE genotyping was performed using TaqMan® SNP Genotyping Assay (Thermo Fisher Scientific).
RESULTS
Clinicopathological characteristics of the study groups
Demographic, clinical, and neuropathological information from the 233 selected donors are summarized in Supplementary Tables 3 and 4. In Group 1, pathological diagnoses included PD with dementia, DLB, AD with LP, mixed AD and Lewy body disease (AD/LBD), frontotemporal dementia (FTD) with LP, and PSP with LP. Distribution of LP was amygdala-predominant (24%), limbic-transitional (38%), or diffuse-neocortical (38%). In Group 2, pathological diagnoses included AD, FTD, vascular dementia, PSP, auto-immune encephalitis, corticobasal degeneration, CRASH syndrome, multiple sclerosis, MSA, neurodegeneration with brain iron accumulation, neuronal intranuclear inclusion disease, and spinocerebellar ataxia. Among donors with a specified clinical diagnosis, 77.5% was confirmed upon autopsy in Group 1 and 61.3% in Group 2.
Genetic findings
Two donors (Patients 1 and 2) from Group 1 carried each a different, possibly pathogenic LRP10 variant: c.1357G>A (p.Gly453Ser) and c.451C>T (p.Arg151Cys) (mean allele frequency (MAF) GnomAD 0.004% and 0.006%, respectively, predicted as pathogenic in 5/11 and 9/11 in-silico programs respectively, Table 1 and Supplementary Table 5). One donor (Patient 3) from Group 2 carried a possibly pathogenic LRP10 variant: c.977G>A (p.Gly326Asp) (MAF GnomAD 0.006%, predicted as pathogenic in 6/11 in-silico programs, Table 1 and Supplementary Table 5). Additional LRP10 variants, which did not fulfill our criteria for possible pathogenicity, are listed in Supplementary Tables 5 and 6. Additional WES and MLPA in possibly pathogenic LRP10 variant carriers showed only one possibly pathogenic variant, namely the ABCA7 p.Gly1741Arg variant in Patient 1 (Supplementary Table 7). APOE genotypes of Patients 1, 2, and 3 were ɛ3/ɛ4, ɛ3/ɛ3, and ɛ2/ɛ4 respectively.
Table 1
Possible pathogenic LRP10 variants
| Genomic position | Nucleotide change | Amino acid change | Exon | Coding effect | dbSNP 142 accession number | MAF GnomAD (alleles) | Functional predictions: pathogenic (total) | Study | Patient number | Clinical diagnosis | Age at onset (y) | Age at death (y) | Pathological diagnosis |
| 14 : 23345514 | c.1357G>A | p.Gly453Ser | 5 | missense | rs146378015 | 0.004% (10) | 5/11 | Current | 1 | DLB | 64 | 72 | Mixed AD/LBD |
| Kia et al., 2019 [6] | n.a. | PD | n.a. | n.a. | n.a. | ||||||||
| 14 : 23344608 | c.451C>T | p.Arg151Cys | 5 | missense | rs774043484 | 0.006% (16) | 9/11 | Current | 2 | AD | 80 | 90 | DLB |
| Vergouw et al., 2019 [13] | Patient II-2 (family 4) | PD with dementia | 66 | 74 | n.a. | ||||||||
| Quadri et al., 2018 [5] | n.a. | control, but neurological status was not available | n.a. | n.a. | n.a. | ||||||||
| 14 : 23345134 | c.977G>A | p.Gly326Asp | 5 | missense | rs547591765 | 0.006% (14) | 6/11 | Current | 3 | AD | 77 | 88 | AD |
| Vergouw et al., 2020 [14] | n.a. | PSP | 55 | 65 | n.a. | ||||||||
| Pihlstrøm et al., 2018 [8] | n.a. | MSA | n.a. | n.a. | MSA |
The Genome Reference Consortium Human Build 37 (hg19) and transcript NM_014045-4 were used. MAF, minor allele frequency; GnomAD, Genome Aggregation Database; n.a., not available; AD, Alzheimer’s disease; PSP, progressive supranuclear palsy; MSA, multiple system atrophy; DLB, dementia with Lewy bodies; PD, Parkinson’s disease; LBD, Lewy body disease.
Clinical phenotype of the LRP10 variant carriers
Patient 1, carrier of the p.Gly453Ser variant, was clinically diagnosed with DLB. Throughout her life, the patient suffered from recurrent depressions, anxiety, and panic attacks. At the age of 64, she developed severe constipation and a tremor of both hands, left more than right, progressing into a hypokinetic-rigid syndrome a year later. Her cognition rapidly deteriorated and neuropsychological examination showed deficits in short term memory, difficulties with calculations, visuoconstruction, and orientation. Brain MRI showed mild general atrophy. Visual hallucinations, personality changes, and apraxia developed during the course of the disease. The patient died at the age of 72 due to pneumonia in an end-stage of dementia. The father of the patient had been diagnosed with PD.
Patient 2, carrier of the p.Arg151Cys variant, was clinically diagnosed with AD. She died at the age of 90 due to cachexia and decubitus wounds, after a disease duration of 10 years. The dementia was characterized by memory loss, anxiety, visual hallucinations, and delusions. Any other neurological symptoms, in particular parkinsonism, were not mentioned in the clinical records. Her mother was also demented, but the clinical diagnosis of the mother is not known.
Patient 3, carrier of the p.Gly326Asp variant, was clinically diagnosed with AD. The patient had a disease duration of 11 years, starting with mild memory disturbances. Seven years before death, neuropsychological examination only showed mild short-term memory problems, and walking became insecure. In the following years, the patient developed parkinsonism, aphasia, frontal disinhibited behavior, and anxiety. He died at the age of 88 due to pneumonia. The brother and two aunts of the patient also suffered from dementia (Table 2).
Table 2
Clinical and pathological characteristics of possibly pathogenic LRP10 variant carriers
| Patient 1 | Patient 2 | Patient 3 | |
| LRP10 variant | p.Gly453Ser | p.Arg151Cys | p.Gly326Asp |
| Sex | F | F | M |
| Age at death y | 72 | 90 | 88 |
| Clinical diagnosis | DLB | AD | AD |
| Age at onset y | 64 | 80 | 77 |
| Disease duration y | 8 | 10 | 11 |
| Dementia duration y | 6 | 6 | 7 |
| Parkinsonism | yes | not mentioned in clinical records | yes |
| Family history | father with PD | mother with dementia | brother and two aunts with dementia |
| Cause of death | pneumonia at end-stage dementia | cachexia and pressure ulcers at end-stage dementia | pneumonia at end-stage dementia |
| Neuropathological diagnosis | mixed AD/LBD | DLB | AD |
| PMD h | 5.5 | 5.5 | 5.0 |
| APOE genotype | 3/4 | 3/3 | 2/4 |
| Brain weight g | 1043 | 1053 | 1133 |
| Atrophy | moderate frontotemporal and hippocampal atrophy, severe dilation of temporal horn of lateral ventricle | none | mild frontotemporal atrophy, severe diffuse dilation of ventricles |
| Pigmentation substantia nigra | pale | pale | normal |
| Pigmentation locus coeruleus | pale | pale | normal |
| Thal amyloid-β phase [23] | 5 | 3 | 5 |
| Braak neurofibrillary stage [23] | 6 | 4 | 4 |
| CERAD score [23] | C | B | B |
| AD-level [23] | high | intermediate | intermediate |
| Braak Lewy body stage [22] | 6 | 6 | 0 |
| McKeith Lewy body stage [22] | neocortical | neocortical | none |
| Microvascular lesions | yes | no | yes |
| Hippocampal sclerosis | no | no | no |
| Argyrophilic grain disease | no | no | no |
| ARTAG [27] | yes | yes | yes |
| CAA type | 2 | 1 | 1 |
| Thal CAA stage [24] | 1 | 1 | 1 |
| Granulovacuolar degeneration [25] | yes, stage≥4 | yes, stage≥4 | yes, stage≥4 |
| Spongiform changes | severe | no | mild |
| LATE [26] | yes, stage 1 | no | no |
DLB, dementia with Lewy bodies; AD, Alzheimer’s disease; PD, Parkinson’s disease; LBD, Lewy body disease; PMD, postmortem delay; ARTAG, aging-related tau astrogliopathy; CAA, cerebral amyloid angiopathy; LATE, limbic-predominant age-related TDP43 encephalopathy.
Pathological characteristics of the LRP10 variant carriers
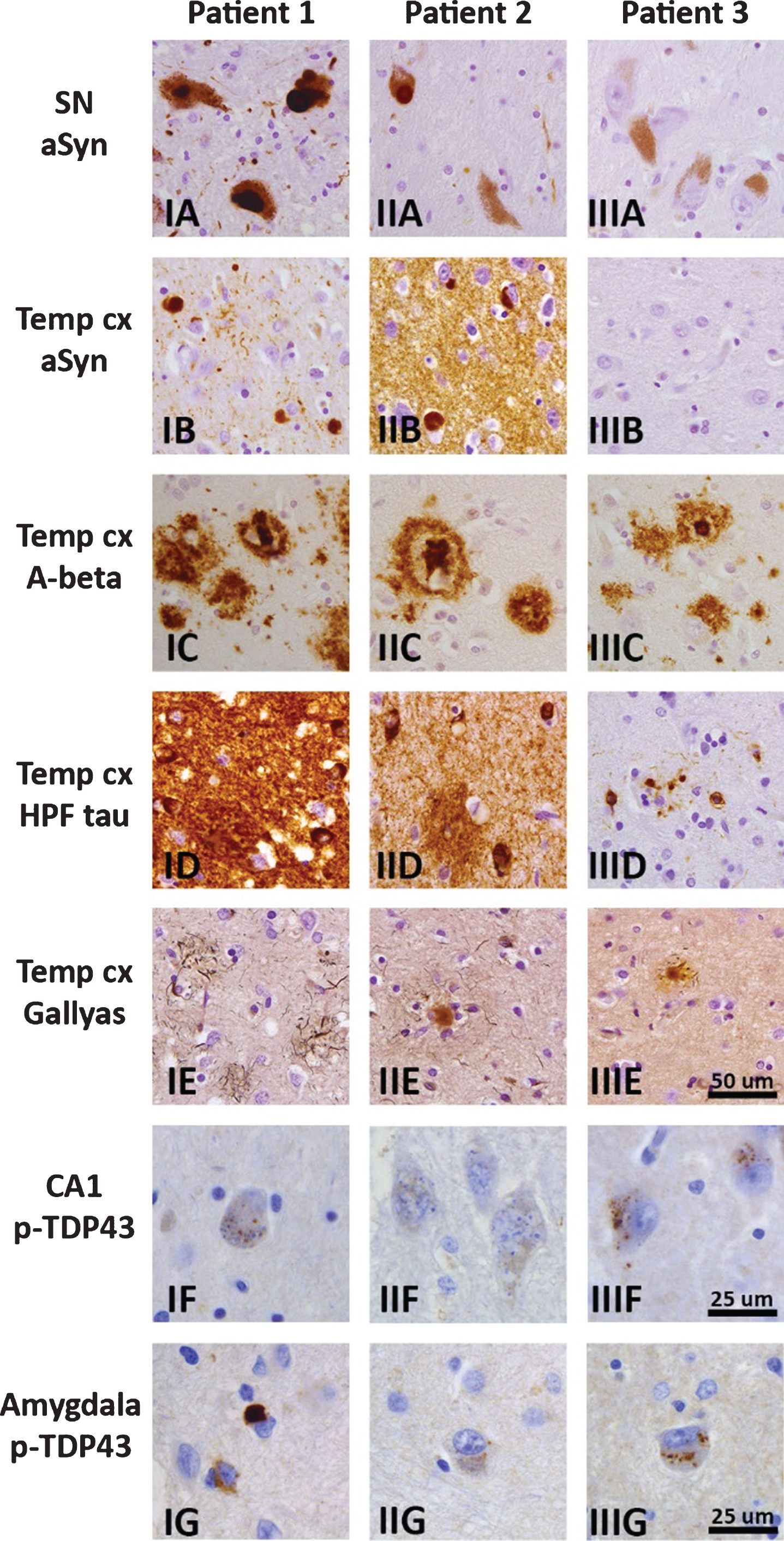
Macroscopic examination of the brains of the three possibly pathogenic LRP10 variant carriers showed mild to moderate frontotemporal atrophy in Patients 1 and 3 (Table 2). Microscopy showed LP in brainstem, limbic, and neocortical brain areas in Patients 1 and 2 (Fig. 1. I–IIA, B), but no LP in Patient 3 (Fig. 1. IIIA, B). Nigral degeneration was present in Patients 1 and 2. An intermediate or high level of AD-type pathology was present in Patients 2 and 3, and Patient 1, respectively (Fig. 1. I–IIIC–E). All three patients showed ARTAG, as well as CAA, including capillary CAA in Patients 2 and 3, and granulovacuolar degeneration in hippocampus and amygdala (Fig. 1. I–IIIF, II–IIIG). Patient 1 showed LATE stage 1 (Fig. 1G). The neuropathological diagnoses in the three patients can be summarized as mixed AD/LBD, DLB, and AD, respectively.
Fig. 1
Brain pathology in three patients with possibly pathogenic LRP10 variants, with representative photomicrographs of selected brain regions for Patient 1 (IA–G), Patient 2 (IIA–G), and Patient 3 (IIIA–G). Immunohistochemistry for α-synuclein (clone KM51) showed Lewy bodies and Lewy neurites in brainstem, limbic and neocortical brain areas in Patients 1 and 2 (I–IIA, B), but no α-synuclein immunoreactivity in Patient 3 (IIIA, B). Diffuse and cored amyloid-β plaques (clone 6F/3D) were present in neocortical regions, amygdala, and striatum in all three patients, and extended into the CA4 region, midbrain, and cerebellum in Patients 1 and 3. Amyloid-β positive plaques in the temporal pole are shown in panel I–IIIC. Immunostaining for hyperphosphorylated tau (clone AT8) showed neurofibrillary tangles, threads, and plaques in the hippocampus and temporal pole in all three patients, and extended into the peristriate and striate areas of the occipital cortex in Patient 1. A moderate or high load of neurofibrillary pathology was present in the temporal pole in Patient 3 (IIID), and Patients 1 and 2 (I–IID), respectively. Gallyas silver stain showed a moderate or high load of neuritic plaques in neocortical regions in Patients 2 and 3 (IIE, IIIE), and Patient 1(IE), respectively. Immunostaining for phospho-TDP43 (polyclonal rabbit antibody) showed granulovacuolar degeneration in the hippocampus and amygdala of all three patients (I–IIIF, II–IIIG). Additionally, Patient 1 showed TDP-43-positive neuronal inclusions and threads in the amygdala fitting limbic-predominant age-related TDP-43 encephalopathy (LATE; IG). Scalebar in IIIE represents 50μm and applies to panels A–E. Scalebars in IIIF and IIIG represent 25μm and apply to panels I–IIIF-G. SN, substantia nigra, Temp cx, temporal pole.
DISCUSSION
We found two possibly pathogenic LRP10 variants among 126 donors with dementia and LP, and one among107 donors with dementia and parkinsonism without LP. The first variant, p.Gly453Ser, was carried by a patient with clinically diagnosed DLB and mixed AD/LBD upon autopsy. The second variant, p.Arg151Cys, was carried by a patient clinically diagnosed with AD, and a neuropathological diagnosis of DLB. The third variant, p.Gly326Asp, was carried by a patient with clinically diagnosed AD plus parkinsonism, and pure AD pathology upon autopsy. In the patient with the LRP10 p.Gly453Ser variant, an additional rare variant (p.Gly1741Arg) in ABCA7 was found. Rare disease-associated variants in ABCA7 are considered risk factors for AD and PD [30, 31], and therefore, the ABCA7 p.Gly1741Arg variant may have contributed to the mixed AD/LBD pathology in this patient, together with the LRP10 p.Gly453Ser variant.
While the three LRP10 variants are extremely rare in control databases, they have all been reported in independent studies in other patients with neurodegenerative diseases. The p.Gly453Ser variant has previously been found in a patient with clinically diagnosed PD [6]. The p.Arg151Cys variant has previously been identified in a patient with PD with dementia, who had a father with AD and gait problems [13]. This variant was also observed by us in one out of 645 controls who were not neurologically examined [5]. Last, the p.Gly326Asp variant has been previously reported in a pathologically confirmed MSA patient [8], and found by us in a patient with clinically diagnosed PSP [14]. The lack of pathological examination in some of the previously reported patients complicates the overall interpretation of these findings.
We acknowledge that in some of these patients, the LRP10 variant could have been a chance occurrence, with no roles in the disease development. Of note, the identified LRP10 variants are all classified as ‘variants of uncertain significance’ by the American College of Medical Genetics guidelines [32]. However, these guidelines are intended to guide genetic testing in clinical practice, and therefore use strict criteria for pathogenicity. Because of the exploratory nature of the current study and the limited knowledge in the literature on LRP10 mutations, we have used less strict criteria to define pathogenicity. Although more research is needed on the pathogenicity of the described LRP10 variants, our results suggest a pathogenic role for these LRP10 variants in a broad spectrum of neurodegenerative disorders.
LRRK2 is a paradigmatic example of marked pathological pleomorphism associated with pathogenic variants in the same gene, even in different members of the same family [33]. Similarly, LRP10 variants may be related to a variety of neuropathological lesions, including LP, AD pathology, and possibly tauopathies. Especially the association with AD pathology, next to LP, is interesting as all three LRP10 variant carriers reported in this study, as well as the three patients we previously described [5], had an intermediate or high level of AD pathology. Our detection of a possibly pathogenic LRP10 variant in an AD case without LP may suggest a role for LRP10 in the accumulation of amyloid-β or hyperphosphorylated tau. LRP10 is known to be involved in APP trafficking and amyloid-β homeostasis [16, 20], suggesting the relevance of these pathways for the pathogenesis of neurodegeneration in subjects with LRP10 variants. This should be further investigated in future studies.
Of note, all three LRP10 variant carriers in this study had one or more first- or second-degree relatives with parkinsonism or dementia, as expected in an autosomal-dominant inherited disease. Unfortunately, the family of these brain donors could not be contacted. As all patients died several years ago, their house physicians at the time of death have unsuccessfully tried to contact living family members on our request. Therefore, a co-segregation analysis of the LRP10 variants could not be performed, which is a limitation of our study.
In conclusion, we observed possibly pathogenic LRP10 variants in two patients with Lewy body diseases, and in one with pure AD pathology. Further studies of LRP10 in large, well-characterized cohorts of patients with different types of neuropathology are warranted.
ACKNOWLEDGMENTS
We would like to thank all patients who donated their brains to the NBB. This work was supported by a grant of ZonMw [grant number 70-73305-98-102] to FJdJ, and by grants from the Stichting Parkinson Fonds to VB. AJR was supported by NIH grant R01AG061775. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0318r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200318.
REFERENCES
[1] | Vergouw LJM , van Steenoven I , van de Berg WDJ , Teunissen CE , van Swieten JC , Bonifati V , Lemstra AW , de Jong FJ ((2017) ) An update on the genetics of dementia with Lewy bodies. Parkinsonism Relat Disord 43: , 1–8. |
[2] | Keller MF , Saad M , Bras J , Bettella F , Nicolaou N , Simon-Sanchez J , Mittag F , Buchel F , Sharma M , Gibbs JR , Schulte C , Moskvina V , Durr A , Holmans P , Kilarski LL , Guerreiro R , Hernandez DG , Brice A , Ylikotila P , Stefansson H , Majamaa K , Morris HR , Williams N , Gasser T , Heutink P , Wood NW , Hardy J , Martinez M , Singleton AB , Nalls MA , International Parkinson’s Disease Genomics Consortium (IPDGC); Wellcome Trust Case Control Consortium 2 (WTCCC2) ((2012) ) Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’s disease. Hum Mol Genet 21: , 4996–5009. |
[3] | Puschmann A ((2013) ) Monogenic Parkinson’s disease and parkinsonism: Clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord 19: , 407–415. |
[4] | Blauwendraat C , Nalls MA , Singleton AB ((2020) ) The genetic architecture of Parkinson’s disease. Lancet Neurol 19: , 170–178. |
[5] | Quadri M , Mandemakers W , Grochowska MM , Masius R , Geut H , Fabrizio E , Breedveld GJ , Kuipers D , Minneboo M , Vergouw LJM , Carreras Mascaro A , Yonova-Doing E , Simons E , Zhao T , Di Fonzo AB , Chang HC , Parchi P , Melis M , Correia Guedes L , Criscuolo C , Thomas A , Brouwer RWW , Heijsman D , Ingrassia AMT , Calandra Buonaura G , Rood JP , Capellari S , Rozemuller AJ , Sarchioto M , Fen Chien H , Vanacore N , Olgiati S , Wu-Chou YH , Yeh TH , Boon AJW , Hoogers SE , Ghazvini M , IJpma AS , van IJcken WFJ , Onofrj M , Barone P , Nicholl DJ , Puschmann A , De Mari M , Kievit AJ , Barbosa E , De Michele G , Majoor-Krakauer D , van Swieten JC , de Jong FJ , Ferreira JJ , Cossu G , Lu CS , Meco G , Cortelli P , van de Berg WDJ , Bonifati V ; International Parkinsonism Genetics ((2018) ) LRP10 genetic variants in familial Parkinson’s disease and dementia with Lewy bodies: A genome-wide linkage and sequencing study. Lancet Neurol 17: , 597–608. |
[6] | Kia DA , Sabir MS , Ahmed S , Trinh J , Bandres-Ciga S , International Parkinson’s Disease Genomics Consortium ((2018) ) LRP10 in alpha-synucleinopathies. Lancet Neurol 17: , 1032. |
[7] | Guerreiro R , Orme T , Neto JL , Bras J , International DLB Genetics Consortium ((2018) ) LRP10 in alpha-synucleinopathies. Lancet Neurol 17: , 1032–1033. |
[8] | Pihlstrøm L , Schottlaender L , Chelban V , Houlden H , MSA Exome Consortium ((2018) ) LRP10 in alpha-synucleinopathies. Lancet Neurol 17: , 1033–1034. |
[9] | Tesson C , Brefel-Courbon C , Corvol JC , Lesage S , Brice A , French Parkinson’s Disease Genetics Study Group ((2018) ) LRP10 in alpha-synucleinopathies. Lancet Neurol 17: , 1034. |
[10] | Daida K , Nishioka K , Li Y , Yoshino H , Kikuchi A , Hasegawa T , Funayama M , Hattori N ((2019) ) Mutation analysis of LRP10 in Japanese patients with familial Parkinson’s disease, progressive supranuclear palsy, and frontotemporal dementia. Neurobiol Aging 84: , 235.e11–235.e16. |
[11] | Shi CH , Luo HY , Fan Y , Li YS , Xu YM ((2018) ) LRP10 in alpha-synucleinopathies. Lancet Neurol 17: , 1034–1035. |
[12] | Chen Y , Cen Z , Zheng X , Pan Q , Chen X , Zhu L , Chen S , Wu H , Xie F , Wang H , Yang D , Wang L , Zhang B , Luo W ((2019) ) LRP10 in autosomal-dominant Parkinson’s disease. Mov Disord 34: , 912–916. |
[13] | Vergouw LJM , Ruitenberg A , Wong TH , Melhem S , Breedveld GJ , Criscuolo C , De Michele G , de Jong FJ , Bonifati V , van Swieten JC , Quadri M ((2019) ) LRP10 variants in Parkinson’s disease and dementia with Lewy bodies in the South-West of the Netherlands. Parkinsonism Relat Disord 65: , 243–247. |
[14] | Vergouw LJM , Melhem S , Kaat LD , Chiu WZ , Kuipers DJS , Breedveld G , Boon AJW , Wang L-S , Naj AC , Mlynarksi E , Cantwell L , Quadri M , Ross OA , Dickson DW , Schellenberg GD , van Swieten JC , Bonifati V , Jan de Jong F ((2020) ) LRP10 variants in progressive supranuclear palsy. Neurobiol Aging, doi: 10.1016/j.neurobiolaging.2020.04.016. |
[15] | Tang FL , Erion JR , Tian Y , Liu W , Yin DM , Ye J , Tang B , Mei L , Xiong WC ((2015) ) VPS35 in dopamine neurons is required for endosome-to-Golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J Neurosci 35: , 10613–10628. |
[16] | Brodeur J, Theriault C, Lessard-Beaudoain M, Marcil A, Dahan S, Lavoie C ((2012) ) LDLR-related protein 10 (LRP10) regulates amyloid precursor protein (APP) trafficking and processing: Evidence for a role in Alzheimer’s disease, Mol Neurodegener 7: , 31. |
[17] | Zimprich A , Benet-Pages A , Struhal W , Graf E , Eck SH , Offman MN , Haubenberger D , Spielberger S , Schulte EC , Lichtner P , Rossle SC , Klopp N , Wolf E , Seppi K , Pirker W , Presslauer S , Mollenhauer B , Katzenschlager R , Foki T , Hotzy C , Reinthaler E , Harutyunyan A , Kralovics R , Peters A , Zimprich F , Brucke T , Poewe W , Auff E , Trenkwalder C , Rost B , Ransmayr G , Winkelmann J , Meitinger T , Strom TM ((2011) ) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89: , 168–175. |
[18] | von Einem B , Eschbach J , Kiechle M , Wahler A , Thal DR , McLean PJ , Weishaupt JH , Ludolph AC , von Arnim CAF , Danzer KM ((2017) ) The Golgi-localized, gamma ear-containing, ARF-binding (GGA) protein family alters alpha synuclein (alpha-syn) oligomerization and secretion. Aging (Albany NY) 9: , 1677–1697. |
[19] | Chen YC , Farzadfard F , Gharaei N , Chen WCW , Cao J , Lu TK ((2017) ) Randomized CRISPR-Cas transcriptional perturbation screening reveals protective genes against alpha-synuclein toxicity. Mol Cell 68: , 247&257 e245. |
[20] | Sugiyama T , Kumagai H , Morikawa Y , Wada Y , Sugiyama A , Yasuda K , Yokoi N , Tamura S , Kojima T , Nosaka T , Senba E , Kimura S , Kadowaki T , Kodama T , Kitamura T ((2000) ) A novel low-density lipoprotein receptor-related protein mediating cellular uptake of apolipoprotein E-enriched beta-VLDL in vivo. Biochemistry 39: , 15817–15825. |
[21] | von Einem B , Wahler A , Schips T , Serrano-Pozo A , Proepper C , Boeckers TM , Rueck A , Wirth T , Hyman BT , Danzer KM , Thal DR , von Arnim CA ((2015) ) The Golgi-localized gamma-ear-containing ARF-binding (GGA) proteins alter amyloid-beta precursor protein (APP) processing through interaction of their GAE domain with the beta-site APP cleaving enzyme 1 (BACE1). PLoS One 10: , e0129047. |
[22] | Alafuzoff I , Ince PG , Arzberger T , Al-Sarraj S , Bell J , Bodi I , Bogdanovic N , Bugiani O , Ferrer I , Gelpi E , Gentleman S , Giaccone G , Ironside JW , Kavantzas N , King A , Korkolopoulou P , Kovacs GG , Meyronet D , Monoranu C , Parchi P , Parkkinen L , Patsouris E , Roggendorf W , Rozemuller A , Stadelmann-Nessler C , Streichenberger N , Thal DR , Kretzschmar H ((2009) ) Staging/typing of Lewy body related alpha-synuclein pathology: A study of the BrainNet Europe Consortium. Acta Neuropathol 117: , 635–652. |
[23] | Montine TJ , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Trojanowski JQ , Vinters HV , Hyman BT , National Institute on Aging, Alzheimer’s Association ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol 123: , 1–11. |
[24] | Thal DR , Griffin WS , de Vos RA , Ghebremedhin E ((2008) ) Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol 115: , 599–609. |
[25] | Thal DR , Del Tredici K , Ludolph AC , Hoozemans JJ , Rozemuller AJ , Braak H , Knippschild U ((2011) ) Stages of granulovacuolar degeneration: Their relation to Alzheimer’s disease and chronic stress response. Acta Neuropathol 122: , 577–589. |
[26] | Nelson PT , Dickson DW , Trojanowski JQ , Jack CR , Boyle PA , Arfanakis K , Rademakers R , Alafuzoff I , Attems J , Brayne C , Coyle-Gilchrist ITS , Chui HC , Fardo DW , Flanagan ME , Halliday G , Hokkanen SRK , Hunter S , Jicha GA , Katsumata Y , Kawas CH , Keene CD , Kovacs GG , Kukull WA , Levey AI , Makkinejad N , Montine TJ , Murayama S , Murray ME , Nag S , Rissman RA , Seeley WW , Sperling RA , White Iii CL , Yu L , Schneider JA ((2019) ) Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 142: , 1503–1527. |
[27] | Kovacs GG , Ferrer I , Grinberg LT , Alafuzoff I , Attems J , Budka H , Cairns NJ , Crary JF , Duyckaerts C , Ghetti B , Halliday GM , Ironside JW , Love S , Mackenzie IR , Munoz DG , Murray ME , Nelson PT , Takahashi H , Trojanowski JQ , Ansorge O , Arzberger T , Baborie A , Beach TG , Bieniek KF , Bigio EH , Bodi I , Dugger BN , Feany M , Gelpi E , Gentleman SM , Giaccone G , Hatanpaa KJ , Heale R , Hof PR , Hofer M , Hortobagyi T , Jellinger K , Jicha GA , Ince P , Kofler J , Kovari E , Kril JJ , Mann DM , Matej R , McKee AC , McLean C , Milenkovic I , Montine TJ , Murayama S , Lee EB , Rahimi J , Rodriguez RD , Rozemuller A , Schneider JA , Schultz C , Seeley W , Seilhean D , Smith C , Tagliavini F , Takao M , Thal DR , Toledo JB , Tolnay M , Troncoso JC , Vinters HV , Weis S , Wharton SB , White CL 3rd , Wisniewski T , Woulfe JM , Yamada M , Dickson DW ((2016) ) Aging-related tau astrogliopathy (ARTAG): Harmonized evaluation strategy. Acta Neuropathol 131: , 87–102. |
[28] | den Dunnen JT , Dalgleish R , Maglott DR , Hart RK , Greenblatt MS , McGowan-Jordan J , Roux AF , Smith T , Antonarakis SE , Taschner PE ((2016) ) HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat 37: , 564–569. |
[29] | Trujillano D , Bertoli-Avella AM , Kumar Kandaswamy K , Weiss ME , Koster J , Marais A , Paknia O , Schroder R , Garcia-Aznar JM , Werber M , Brandau O , Calvo Del Castillo M , Baldi C , Wessel K , Kishore S , Nahavandi N , Eyaid W , Al Rifai MT , Al-Rumayyan A , Al-Twaijri W , Alothaim A , Alhashem A , Al-Sannaa N , Al-Balwi M , Alfadhel M , Rolfs A , Abou Jamra R ((2017) ) Clinical exome sequencing: Results from 2819 samples reflecting 1000 families. Eur J Hum Genet 25: , 176–182. |
[30] | De Roeck A , Van Broeckhoven C , Sleegers K ((2019) ) The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol 138: , 201–220. |
[31] | Nuytemans K , Maldonado L , Ali A , John-Williams K , Beecham GW , Martin E , Scott WK , Vance JM ((2016) ) Overlap between Parkinson disease and Alzheimer disease in ABCA7 functional variants. Neurol Genet 2: , e44. |
[32] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , Grody WW , Hegde M , Lyon E , Spector E , Voelkerding K , Rehm HL , ACMG Laboratory Quality Assurance Committee ((2015) ) Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: , 405–424. |
[33] | Zimprich A , Biskup S , Leitner P , Lichtner P , Farrer M , Lincoln S , Kachergus J , Hulihan M , Uitti RJ , Calne DB , Stoessl AJ , Pfeiffer RF , Patenge N , Carbajal IC , Vieregge P , Asmus F , Muller-Myhsok B , Dickson DW , Meitinger T , Strom TM , Wszolek ZK , Gasser T ((2004) ) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44: , 601–607. |


