
Dysregulation of Autophagy, Mitophagy, and Apoptosis Genes in the CA3 Region of the Hippocampus in the Ischemic Model of Alzheimer’s Disease in the Rat
Abstract
There is currently no knowledge about the expression profile of the autophagy (BECN1), mitophagy (BNIP3), and apoptosis (CASP3) genes in the CA3 region of the hippocampus after cerebral ischemia. In addition, it is unknown whether genes for BECN1, BNIP3, and CASP3 have any effect on the neuronal death in the CA3 area of the hippocampus due to ischemia. In this study, for the first time, we present, by means of a quantitative PCR protocol with reverse transcriptase, the expression of BECN1 and CASP3 genes in the neuronal CA3 region of the hippocampus with the co-expression of the mitochondrial BNIP3 gene, which genes are associated with Alzheimer’s disease, in the ischemic model of Alzheimer’s disease in the rat. The present study showed that after ischemia, the CASP3 gene was significantly expressed within 7–30 days, the BECN1 gene was significantly overexpressed on the thirtieth day, and the BINP3 gene was lowered below control values during post-ischemic follow-up period. The caspase-dependent neuronal death in the CA3 region of the hippocampus after ischemia is not accompanied by overexpression of the BNIP3 gene. Our data may therefore suggest a new insight into the BNIP3 gene in the regulation of neuronal mitophagy in neurodegeneration in the CA3 region of the hippocampus after ischemia. This indicates no involvement of the BNIP3 gene along with the CASP3 gene in the CA3 region of the hippocampus in delayed neuronal death after brain ischemia.
INTRODUCTION
Transient ischemic brain injury and Alzheimer’s disease (AD) are two known disorders characterized by progressive neurodegeneration, which initially triggers neuronal death in the hippocampus in the CA1 region and then in the CA3 area [1–5], ultimately leading to the development of dementia [6–8]. It has been observed that post-ischemic dementia is associated with both the progressive loss of pyramidal neurons of the CA1 and CA3 hippocampal areas, as well as its gradual atrophy [3, 4, 9, 10], in the same way as in AD [5, 11]. Experimental transient cerebral ischemia in animals and AD cause a sudden loss of neurons in the hippocampus in the CA1 region, whereas in the CA3 region, the death of pyramidal neurons occurs with a significant delay [1, 2, 4, 5]. Previous data have shown that brain ischemia activates neuronal changes and death in the CA1 region of the hippocampus in a mechanism dependent on amyloid and tau protein, thus identifying a new and important phenomenon that regulates survival and/or death after ischemia pyramidal neurons [12, 13]. In addition, we found that autophagy (BECN1) gene expression was not significantly modified at all time points after ischemia, while the expression of the mitophagy (BNIP3) and caspase 3 (CASP3) genes increased on day 2 and decreased to basal values on days 7 and 30 after ischemia [14]. It can be concluded that the mitophagy process is clearly associated with the mechanism of apoptosis during the death of pyramidal neurons in the CA1 region of the hippocampus after brain ischemia. The phenomenon of death of pyramidal neurons occurring at different periods in different areas of the ischemic hippocampus, i.e., CA1 and CA3, was identified on the basis of morphological studies [1–4]; however, molecular processes explaining differences in the response to ischemia of these regions that lead to death of pyramidal neurons in the CA3 region are not currently fully understood. However, to the best of our knowledge, the expression of the BECN1, BNIP3, and CASP3 genes in CA3 hippocampal neurons in the ischemic model of AD in rats has not been previously characterized. We decided to investigate the progress of ischemic disease from the hippocampal region CA1 to CA3 in the ischemic model of AD [3, 4]. In addition, it is unknown whether genes for BECN1, BNIP3, and CASP3 have any effect on the neuronal death in the CA3 area of the hippocampus due to ischemia. The apoptotic and autophagy death of neurons, after brain ischemia, evoke distinctive features of the mechanisms of neuronal cell death, and the boundary between the two types of neuronal cell loss is not completely understood. We will explain the role of the BECN1 gene in the CA3 area of the ischemic hippocampus with a possible crosstalk with the CASP3 and BNIP3 genes if the potential dysfunction of the BECN1 gene is involved in the ischemic etiology of AD. Below, for the first time, we present, by means of a quantitative PCR protocol with reverse transcriptase, the expression of BECN1 and CASP3 genes in the neuronal CA3 region of the hippocampus with the co-expression of the mitochondrial BNIP3 gene, which genes are associated with AD, in the ischemic model of AD in the rat.
MATERIALS AND METHODS
Female Wistar 2-month-old rats (n = 24, body weight 160–180 g) were subjected to transient cerebral ischemia due to cardiac arrest [15]. The rats were kept in pairs per cage at room temperature of 22±1°C, 55±5% humidity, and a 12- h light/dark cycle. All rats had unlimited access to commercial laboratory chow and tap water. All experiments were carried out in the light phase, in identical conditions. Rats used for experiments were treated strictly in accordance with the NIH Guide for the Care and Use of Lab Animals (1985) and Council Directive of the European Communities 142 (86/609/EEC), and with the consent of the local Ethical Committee. Every effort has been made to reduce the number of animals used and to minimize their suffering. The rat brain ischemia model used is clinically representative of reversible cardiac arrest in humans. Global reversible brain ischemia caused by cardiac arrest lasted 10 min [15]. Rats were allowed to survive 2 (n = 8), 7 (n = 8), and 30 days (n = 8) post-ischemia. Sham-operated animals (n = 24) served as controls under the same procedures as ischemic rats but without induced cardiac arrest.
Before samples were taken, the brains were perfused with cold 0.9% NaCl through the left ventricle to rinse the blood vessels from the blood. After removing the brain from the skull, the brain was placed on an ice chilled Petri dish. The brains were cut in the coronal plane by cutting the intersection of the optic nerves. Ischemia and control combined samples of approximately 1 mm3 of the hippocampus volume of the left and right CA3 regions were collected using a narrow scalpel. CA3 samples were immediately placed in the RNALater solution (Life Technologies, USA). Expression of BECN1, BNIP3, and CASP3 genes was assessed by reverse transcription quantitative PCR (RT-qPCR) method. The genes were quantified in each sample taken from the ischemic group and referred to gene expression defined in the respective samples in the control group 1:1.
The isolation of cellular RNA was made according to the method described by Chomczyński and Sacchi [16]. The quality and quantity of RNA was assessed using the Nano Drop 2000 spectrophotometer (Thermo Scientific, USA). The isolated RNA was stored in 80% ethanol at –20°C for further analysis. In later studies, one microgram of total RNA was reverse transcribed into cDNA using a large capacity cDNA kit for reverse transcription according to the manufacturer’s recommendations (Applied Biosystems, USA). The cDNA synthesis was done on Veriti Dx (Applied Biosystems, USA) under the conditions: phase I: 25°C 10 min, phase II: 37°C 2 h, phase III: 85°C 5 min, phase IV: 4°C. The resulting cDNA was amplified by real-time gene expression analysis (qPCR) on the 7900HT Real-Time Fast System (Applied Biosystems, USA) using the Master Mix SYBR-green PCR power mixing reagent using the SDS manufacturer software [12–14]. The amplification protocol consisted of the following cycles: initial denaturation: 95°C 10 min and 40 cycles, each at two different temperatures: 95°C 15 s and 60°C 1 min. The number of DNA copies was assessed by 7900HT Real-Time Fast System (Applied Biosystems, USA) in each amplification cycle. The number of PCR cycles at which the fluorescence level exceeded the specific relative expression of the threshold cycle (CT) was used in the research software (Applied Biosystems, USA) to calculate the number of DNA molecules present in the mixture at the beginning of the reaction. Normalization was evaluated against the endogenous control gene (Rpl13a) [12–14], and the relative amount (RQ) of genes expression was calculated based on the ΔCT method, and the results were presented as RQ = 2-ΔΔCT [12–14]. Finally, the RQ values were subjected to a logarithmic conversion to RQ logarithm (Log RQ) [12–14]. Log RQ = 0 showed that genes expression in control and ischemic samples did not differ. Log RQ < 0 indicates that we have reduced genes expression in the test sample, while Log RQ > 0 means an increased expression of the genes in the ischemic sample compared to the control.
Statistical evaluation of the results was carried out using the Statistica v. 12 software with the help of non-parametric Kruskal-Wallis test with the “z” test - multiple analyzes of differences between groups. Data are presented as mean±SD. Statistical significance was adopted at p≤0.05.
RESULTS
Expression of the autophagy gene after ischemia
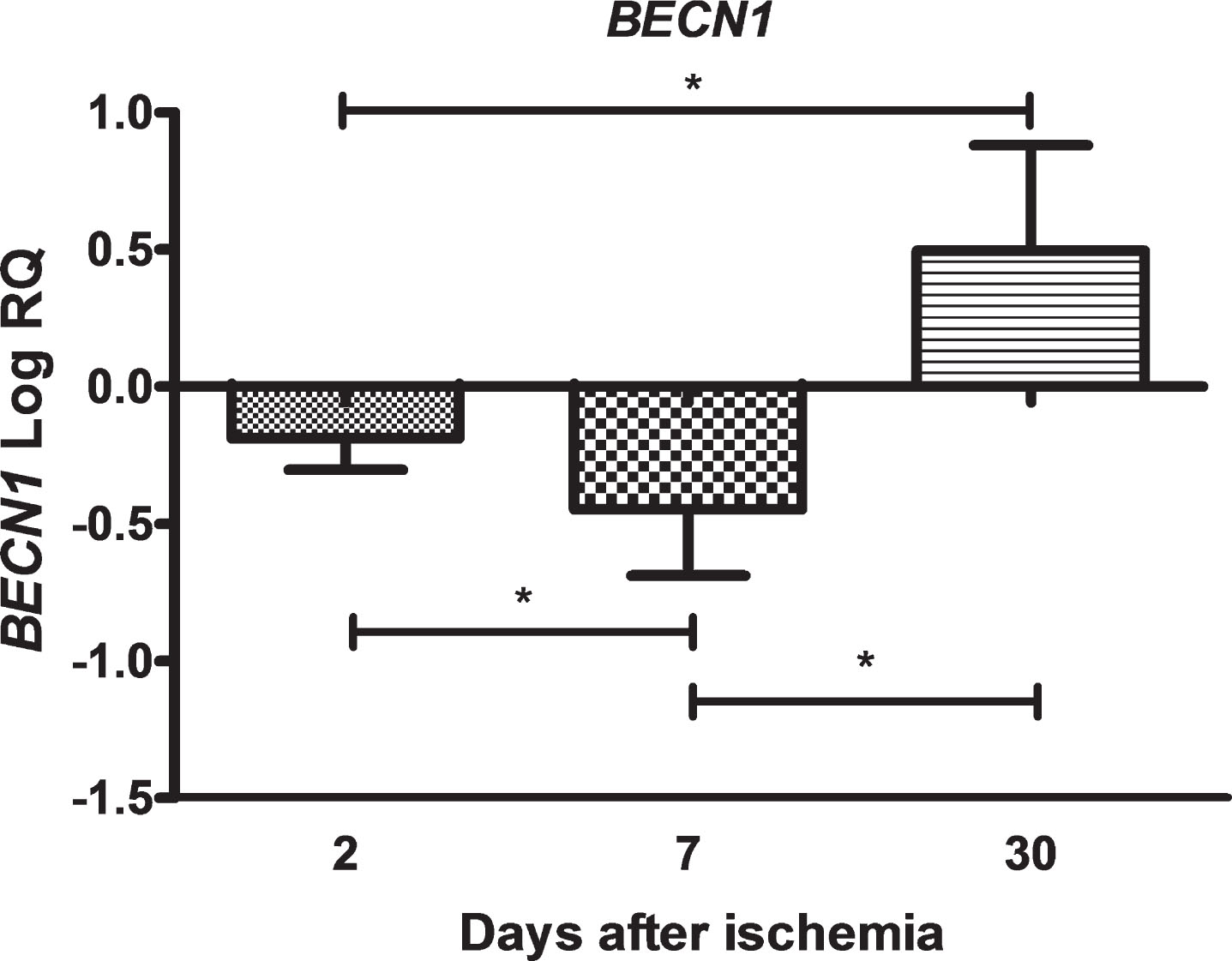
In the CA3 region of the hippocampus, the expression of the BECN1 gene after 10 min brain ischemia with a survival of 2 and 7 days was below the control values and on 30th day increased above control value. On the second day after ischemia, the minimum was –0.467-fold change and maximum –0.007-fold change with median –0.169-fold change. On the seventh day after ischemia, the minimum was –0.867-fold change and maximum –0.095-fold change with median –0.424-fold change. On the thirtieth day after ischemia, the minimum was 0.096-fold change and maximum 1.074-fold change with median 0.408-fold change. Figure 1 illustrates changes in the mean level of expression of the BECN1 gene. The changes were statistically significant at all the times after ischemia (Fig. 1).
Fig.1
The mean expression levels of BECN1 gene in the hippocampus CA3 area in rats 2 (n = 8), 7 (n = 8), and 30 (n = 8) days after brain ischemia. Marked SD, standard deviation. Indicated statistically significant differences in levels of gene expression between 2 and 7 (z = 3.129, p = 0.0052), 2 and 30 (z = 3.845, p = 0.0003), and between 7 and 30 (z = 5.336, p = 0.00001) days after ischemia (Kruskal-Wallis test). *p≤0.01.
Expression of the mitophagy gene after ischemia
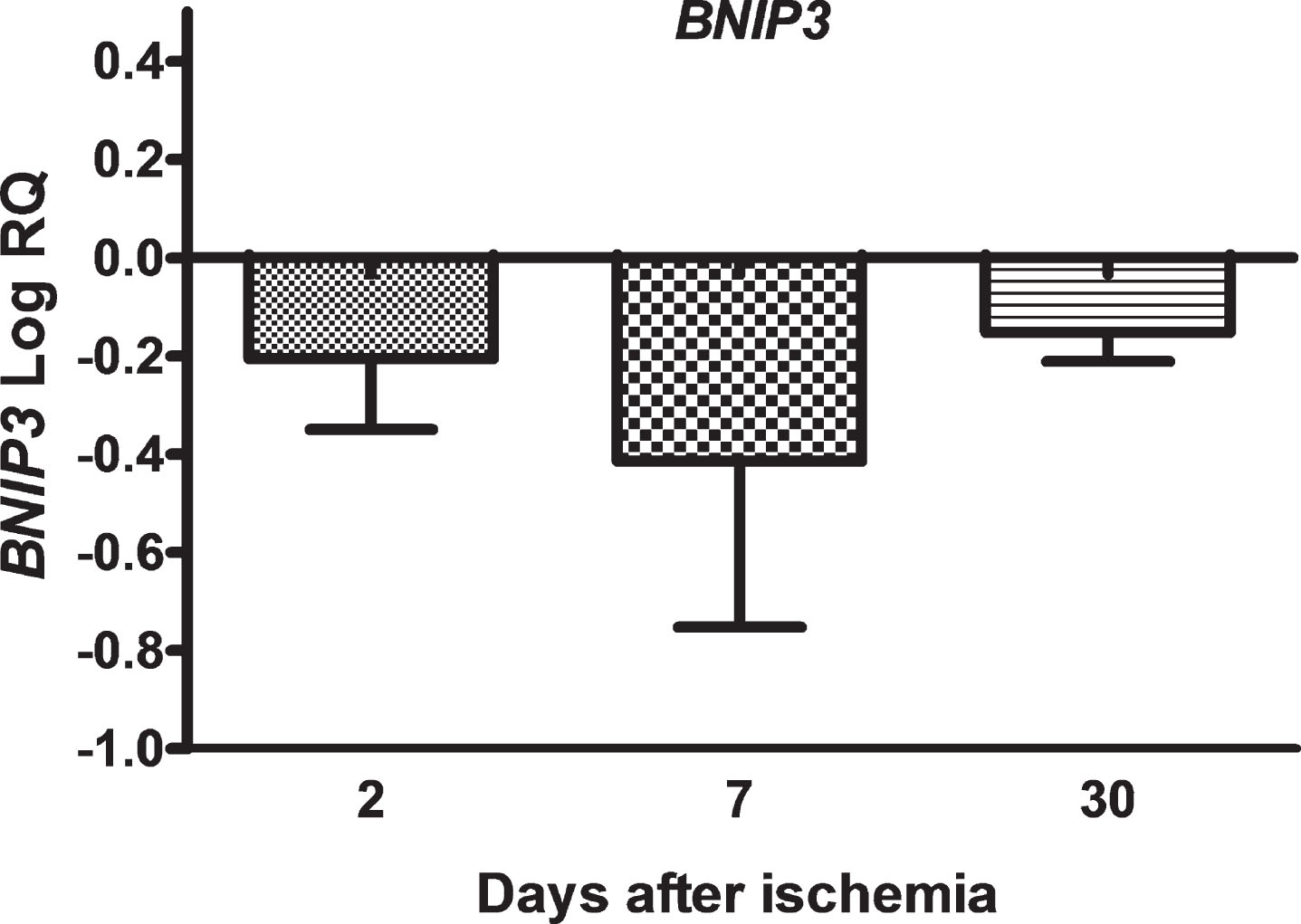
In the CA3 area, the expression of the BNIP3 gene after brain ischemia with a survival of 2, 7, and 30 days was below the control values. On the second day post-ischemia, the minimum was –0.498-fold change and maximum –0.007-fold change with median –0.182-fold change. On the seventh day, the minimum was –0.821-fold change and maximum –0.008-fold change with median –0.550-fold change. On the thirtieth day after ischemia, the minimum was -0.218-fold change and maximum –0.082-fold change with median –0.130-fold change. Figure 2 illustrates alterations in the mean level of expression of the BNIP3 gene. The changes were not statistically significant at all the times after ischemia (Fig. 2).
Fig.2
The mean expression levels of BNIP3 gene in the hippocampus CA3 area 2 (n = 8), 7 (n = 8) and 30 (n = 8) days after brain ischemia. Marked SD, standard deviation. No statistical significance at all times after ischemia (Kruskal-Wallis test).
Expression of the apoptosis gene after ischemia
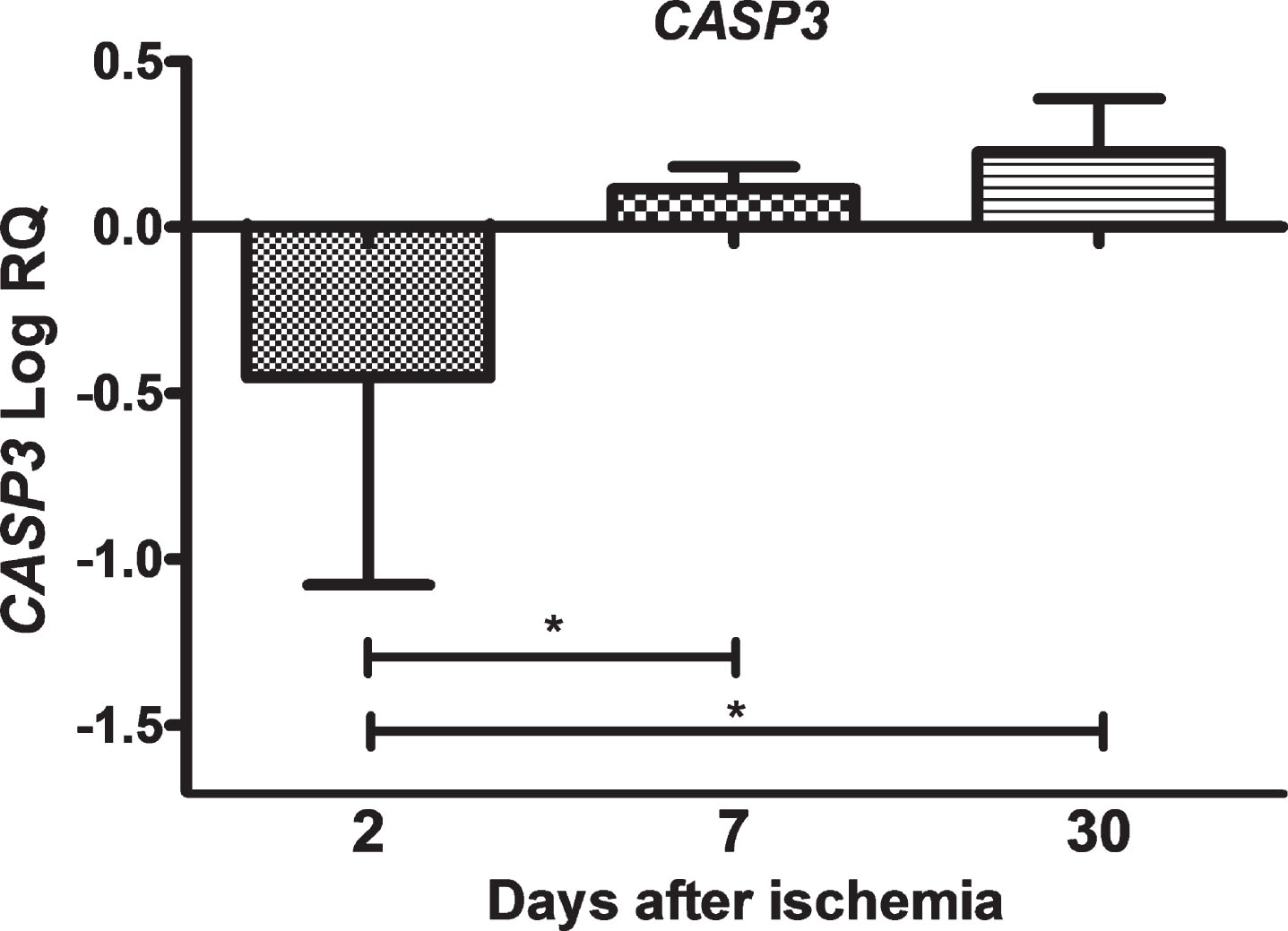
In the CA3 area of the hippocampus, the expression of the CASP3 gene after ischemic injury with a survival of 2 days was below the control values and higher than the control values on days 7–30. On the second day after ischemia, the minimum was –1.699-fold change and maximum –0.010-fold change with median –0.174-fold change. On the seventh day post-ischemia, the minimum was 0.056-fold change and maximum 0.215-fold change with median 0.089-fold change. On the thirtieth day, the minimum was 0.018-fold change and maximum 0.424-fold change with median 0.224-fold change. Figure 3 illustrates changes in the mean expression of the CASP3 gene. The changes were statistically significant between 2 and 7 days and between 2 and 30 days post-ischemia (Fig. 3).
Fig.3
The mean expression levels of CASP3 gene in the hippocampus CA3 area in rats 2 (n = 8), 7 (n = 8), and 30 (n = 8) days after brain ischemia. Marked SD, standard deviation. Indicated statistically significant differences in levels of gene expression between 2 and 7 (z = 3.705, p = 0.0006) and between 2 and 30 days (z = 3.807, p = 0.0004) after ischemia (Kruskal-Wallis test). *p≤0.001.
DISCUSSION
We provide evidence of significant downstream expression of the BECN1 gene within 2–7 days after ischemia with significant overexpression on day 30 in the neurons of the CA3 hippocampal area. However, the expression of the BNIP3 gene during the entire post-ischemic follow-up period was below the control values. On the other hand, the expression of the CASP3 gene responsible for the apoptosis phenomenon after cerebral ischemia on the second day was below the control values in the hippocampal CA3 region to achieve overexpression slightly above control values in 7–30 days, which was statistically significant. Our data may therefore suggest a new insight into the BNIP3 gene in the regulation of neuronal mitophagy in neurodegeneration in the CA3 region of the hippocampus after ischemia. These studies show that the BNIP3 gene is not upregulated in the ischemic CA3 region of the hippocampus, and certainly does not play a key role in activating the unique neuronal death program, resulting in less pronounced delayed apoptosis. This suggests the lack of involvement of the BNIP3 gene along with the CASP3 gene in the CA3 region of the hippocampus in delayed neuronal death after brain ischemia.
Statistically significant downstream expression of the BECN1 gene was observed within 2–7 days after brain ischemia in the hippocampal CA3 area. The subsequent expression of the BECN1 gene was significantly increased on day 30 after ischemia. In contrast to the CA3 region, there were no statistically significant changes in BECN1 gene expression within 2–30 days after cerebral ischemia in the CA1 region of the hippocampus [14]. These data suggest the possible ineffective role of the BECN1 gene in autophagy in the presumed delayed neuronal death in the CA1 region of the hippocampus caused by cerebral ischemia [14]. But data from CA3 area suggest a possible effective protective and slowing role of the BECN1 gene against the progressive death of neurons 30 days after ischemic brain injury. Beclin 1, a key factor in the formation of autophagosomes, has been shown to be suppressed both at the level of mRNA and protein in the brains cortex of patients with early AD [17]. We partially confirmed the above decrease in BECN1 gene expression in brains from AD patients in the ischemic CA3 region of the hippocampus within 2–7 days after ischemia, but we observed increased gene expression on day 30 after ischemia and this difference may result from inability to study gene expression at specific time points and brain structures during the development of AD in patients. Autophagy dysfunction is referred as a secondary pathologic mechanism for the development of neurodegenerative diseases, such as AD and probably ischemic neurodegeneration [14, 17, 18]. Finally, some studies indicate that BECN1 gene plays an important role in the processing of the amyloid-β protein precursor (AβPP), as well as in the autophagy removal of proteins susceptible to aggregation from the brains of patients with AD and probably also in ischemic neurodegeneration [17, 18]. Regulation of the expression of beclin 1 in the brain tissue of patients with AD at the transcriptional level has not been fully elucidated. Probably our data seem to partially explain this problem as a rather complicated and complex reaction of autophagy to ischemia, which can make neurons more susceptible to death after ischemia.
There were no statistically significant changes in the expression of the BNIP3 gene, which was below control values within 2–30 days after brain ischemia in the CA3 region of the hippocampus. In the CA1 region, it was noticed that the expression of the BNIP3 gene was significantly elevated on the 2nd day after ischemia [14], when the delayed death of neurons is triggered by ischemia [1–4], which is consistent with the data on translocation of BNIP3 protein into the nucleus [19]. Other available studies have shown an increase in protein in the cerebral cortex at the peak of 48 h after ischemia [20], which is consistent with the maximum expression of the BNIP3 gene in the CA1 region [14], but the expression of the gene in CA1 was significantly reduced within 7–30 days of observation [14]. In the CA3 region of the hippocampus, data suggest a lack of BNIP3 gene activity in the regulation of mitophagy leading to mitochondrial dysfunction, the consequence of which is the deregulation of the ability to remove dysfunctional mitochondria and toxic proteins after brain ischemia. Continuous removal of proteins and mitochondrial remnants is important for the proper functioning of neurons. Abnormalities in the expression of mitochondrial genes lead to mitochondria dysfunctions and are important in the progression and pathogenesis of AD [18]. Our data may therefore suggest a new insight into the BNIP3 gene in regulation neuronal of mitophagy in the CA3 region of the hippocampus and neurodegeneration after ischemia. This present study shows that the BNIP3 gene in neurons is downregulated in the ischemic CA3 region of the hippocampus, but still plays a key role in activating a unique neuronal protection program or death without removing altered proteins or mitochondria. Research conducted by Radenovic et al. [21] showed that the number of empty synaptic terminals and terminals with one mitochondrion increased more significantly in the CA1 region of the hippocampus during recirculation. In contrast, the number of terminals with two or more mitochondria increased at the same time in the CA3 region of the hippocampus. In addition, the average number of mitochondria per terminal was significantly higher in the CA3 area compared to CA1. The authors suggested that this quantitative difference could be considered a sign of neuroprotection and that the increased number of mitochondria in the hippocampal CA3 region may defend terminals against ischemic damage [21]. The suggestion that the amyloid-β (Aβ) peptide and hyperphosphorylated tau protein trigger defective mitophagy in AD [18] is strongly supported by our observations in the ischemic model of AD.
Caspases are thought to be key factors in the development of neuronal apoptosis in post-ischemic brain [22]. CASP3 gene expression was increased in the hippocampal CA3 region after transient cerebral ischemia within 7–30 days. With regard to the increased expression of this gene in the CA1 region of the hippocampus after ischemia [14], we observed 7–30 days delay in the CA3 region. The caspase-activated DNAse/inhibitor of caspase-activated DNAse complex was identified as a caspase substrate. When caspase cleaves the above complex, the caspase-activated DNAse is transferred to the neuron nucleus, causing DNA fragmentation. Seven to 30 days after ischemia, overexpression of the CASP3 gene appears to be a key trigger for neuronal death in the CA3 region of the hippocampus. There is a high probability that the above process may also occur in CA3 hippocampal neurons in the brains of patients with AD and may partially explain the mechanism of neuronal loss in the hippocampus in AD [5]. Importantly, since the AβPP was shown to be a caspase substrate and was elevated for a long time after ischemic injury [3, 4], this effect may also affect the development of long-term deficits [6, 7]. It should be emphasized that the Aβ peptide intensifies the phosphorylation of tau protein, it causes inhibition of microtubule development leading to apoptosis [23]. In addition, caspase cleaves the GGA3 protein, and thus increases β-secretase activity [24] leading to the vicious cycle. Activated caspase 3 protein cleaves nuclear DNA repair enzymes, such as PARP 1, which causes damage to nuclear DNA and subsequent apoptosis of neurons [14, 21, 25]. On the other hand, an excessive activation of PARP 1 causes depletion of nicotinamide adenine dinucleotide and ATP, which ultimately leads to cellular energy failure and necrotic cell death [14, 22].
The present study showed that after ischemia, the CASP3 gene was significantly increased within 7–30 days, the BECN1 gene was significantly overexpressed on the thirtieth day, and the BINP3 gene was below control values during post-ischemic follow-up period. As discussed above, the caspase-dependent neuronal death in the CA3 region of the hippocampus after ischemia is not accompanied by overexpression of the BNIP3 gene. It is clear that one ischemic injury can multiply more than one mode of neuronal damage and death in the same population of neurons and different structures (see CA1 and CA3 areas) [21], and in many cases one neuron can show signs of more than one neuron death mode. Activated CASP3 gene precedes the activation of the autophagy gene (on 30th day) to inhibit or reduce the survival of neurons provided by autophagy, and therefore the caspase may enhance apoptosis [26, 27]. On the other hand, it interferes with the processing of the AβPP and increases the production of Aβ peptide [3, 4, 12, 28]. Above supports the view that CASP3 gene overexpression increases β-secretase activity [12, 24]. The currently observed reduced expression of the BNIP3 gene may indirectly cause disorders in the processing of the AβPP and the clearance of dysfunctional mitochondria and toxic proteins such as amyloid and altered tau protein in neurons of the CA3 region. We need more research on the mechanisms of autophagy and mitophagy and therapeutic aspects that will use animal models, cell cultures and clinical trials of AD.
Age is a major non-modifiable risk factor for brain ischemia and AD [29–32]. The incidence of cerebral ischemia and AD increases significantly with age in both men and women, covering almost the same number of cases occurring in persons over 65 years of age [29]. Although the entire older population is at risk of ischemic stroke and AD, there are significant gender differences in the occurrence of brain ischemia and AD [29, 32]. In general, the incidence of ischemic stroke is higher in men than in women, but at the same age, the number of cases of AD is higher in women than in men [29]. The above evidence was the main justification for conducting the current experiments on relatively young female rats. As we already know, changes in AD develop slowly and clinical symptoms appear at least a decade from the beginning of the disease. Therefore, in our model, we need adequate time for the development of lesions characteristic of AD, which, as we know, depends on the survival time, and in this case, the time after the episode of cerebral ischemia. Experiments on adults and/or old rats would be at risk of high mortality [31] and no or limited development of AD-related changes. The influence of age and sex on the incidence of ischemic stroke, functional neurological recovery of health and mortality after stroke have been demonstrated in both human and animal models [29]. Although local cerebral ischemia is technically difficult to perform in older rodents, not to mention complete cerebral ischemia, focal cerebral ischemia in older animals is considered to be a clinically more appropriate and mimicking stroke model in humans [29, 30]. It should be noted that this does not apply to ischemic models of AD.
Conclusion
Loss of neurons in the hippocampus is an early morphological symptom of AD, and we have found that the death of pyramidal neurons in the hippocampal region of CA3 in an ischemic model of AD develops in a manner dependent on overexpression of BECN1 and CASP3 genes at a very early stages after cerebral ischemia. Our observations indicate the key role of brain damage caused by ischemia-reperfusion in the development of AD through primary neurodegeneration of the hippocampus, including the CA3 region, which is associated with progressive deterioration of cognitive function together with prolonged survival. Thus, damage to the above-mentioned area causes the deterioration of short-term memory, which leads to the inability to create new memories. Alterations in the CA3 area in the post-ischemic brain are particularly important in memory disorders, including AD. However, further research is needed to determine whether injury and loss of pyramidal neurons in the CA3 area are causative events or independent consequences of an ischemic episode occurring in parallel and leading to the development of dementia following an ischemia with the AD phenotype. Apparently, the prevention of cerebral ischemia-reperfusion injury and the early start of stroke treatment in humans can reduce the development of AD. Finally, the rodent model used in this study appears to be a useful approach to determining the role of genes directly related to AD. In-depth research into the common genetic mechanisms associated with brain ischemia-reperfusion injury and AD, accelerate the understanding of new pathological pathways in both diseases, and conduct future research into brain ischemia or AD in new directions.
ACKNOWLEDGMENTS
The authors acknowledge the financial support from the following institutions: the Mossakowski Medical Research Centre, Polish Academy of Sciences, Warsaw, Poland (T3-RP) and the Medical University of Lublin, Lublin, Poland (DS 475/19-SJC).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0966r1).
REFERENCES
[1] | Krajewski S , Mai JK , Krajewska M , Sikorska M , Mossakowski MJ , Reed JC ((1995) ) Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci 15: , 6364–6376. |
[2] | Sadowski M , Wisniewski HM , Jakubowska-Sadowska K , Tarnawski M , Lazarewicz JW , Mossakowski MJ ((1999) ) Pattern of neuronal loss in the rat hippocampus following experimental cardiac arrest-induced ischemia. J Neurol Sci 168: , 13–20. |
[3] | Pluta R ((2000) ) The role of apolipoprotein E in the deposition of β-amyloid peptide during ischemia-reperfusion brain injury. A model of early Alzheimer’s disease. Ann N Y Acad Sci 903: , 324–334. |
[4] | Pluta R , Ułamek M , Jabłoński M ((2009) ) Alzheimer’s mechanisms in ischemic brain degeneration. Anat Rec 292: , 1863–1881. |
[5] | Padurariu M , Ciobica A , Mavroudis I , Fotiou D , Baloyannis S ((2012) ) Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer’s disease patients. Psychiatr Danub 24: , 152–158. |
[6] | Kiryk A , Pluta R , Figiel I , Mikosz M , Ułamek M , Niewiadomska G , Jabłoński M , Kaczmarek L ((2011) ) Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav Brain Res 219: , 1–7. |
[7] | Cohan CH , Neumann JT , Dave KR , Alekseyenko A , Binkert M , Stransky K , Lin HW , Barnes CA , Wright CB , Perez-Pinzon MA ((2015) ) Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS One 10: , e0124918. |
[8] | Kim JH , Lee Y ((2018) ) Dementia and death after stroke in older adults during a 10-year follow-up: Results from a competing risk model. J Nutr Health Aging 22: , 297–301. |
[9] | Jabłoński M , Maciejewski R , Januszewski S , Ułamek M , Pluta R ((2011) ) One year follow up in ischemic brain injury and the role of Alzheimer factors. Physiol Res 60: (Suppl 1), S113–S119. |
[10] | Bivard A , Lillicrap T , Maréchal B , Garcia-Esperon C , Holliday E , Krishnamurthy V , Levi CR , Parsons M ((2018) ) Transient ischemic attack results in delayed brain atrophy and cognitive decline. Stroke 49: , 384–390. |
[11] | Bloom GS ((2014) ) Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71: , 505–508. |
[12] | Kocki J , Ułamek-Kozioł M , Bogucka-Kocka A , Januszewski S , Jabłoński M , Gil-Kulik P , Brzozowska J , Petniak A , Furmaga-Jabłońska W , Bogucki J , Czuczwar SJ , Pluta R ((2015) ) Dysregulation of amyloid precursor protein, β-secretase, presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J Alzheimers Dis 47: , 1047–1056. |
[13] | Pluta R , Bogucka-Kocka A , Ułamek-Kozioł M , Bogucki J , Kocki J , Czuczwar SJ ((2018) ) Ischemic tau protein gene induction as an additional key factor driving development of Alzheimer’s phenotype changes in CA1 area of hippocampus in an ischemic model of Alzheimer’s disease. Pharmacol Rep 70: , 881–884. |
[14] | Ułamek-Kozioł M , Kocki J , Bogucka-Kocka A , Januszewski S , Bogucki J , Czuczwar SJ , Pluta R ((2017) ) Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol Rep 69: , 1289–1294. |
[15] | Pluta R , Lossinsky AS , Mossakowski MJ , Faso L , Wiśniewski HM ((1991) ) Reassessment of new model of complete cerebral ischemia in rats. Method of induction of clinical death, pathophysiology and cerebrovascular pathology. Acta Neuropathol 83: , 1–11. |
[16] | Chomczynski P , Sacchi N ((1987) ) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: , 156–159. |
[17] | Meng T , Lin S , Zhuang H , Huang H , He Z , Hu Y , Gong Q , Feng D ((2019) ) Recent progress in the role of autophagy in neurological diseases. Cell Stress 3: , 141–161. |
[18] | Reddy PH , Oliver DM ((2019) ) Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 8: , E488. |
[19] | Schmidt-Kastner R , Aguirre Chen C , Kietzmann T , Saul I , Busto R , Ginsberg MD ((2004) ) Nuclear localization of the hypoxia-regulated pro-apoptotic protein BNIP3 after global brain ischemia in the rat hippocampus. Brain Res 1001: , 133–142. |
[20] | Althaus J , Bernaudin M , Petit E , Toutain J , Touzani O , Rami A ((2006) ) Expression of the gene encoding the pro-apoptotic BNIP3 protein and stimulation of hypoxia-inducible factor-1alpha (HIF-1a) protein following focal cerebral ischemia in rats. Neurochem Int 48: , 687–695. |
[21] | Radenovic L , Korenic A , Maleeva G , Osadchenko I , Kovalenko T , Skibo G ((2011) ) Comparative ultrastructural analysis of mitochondria in the CA1 and CA3 hippocampal pyramidal cells following global ischemia in Mongolian gerbils. Anat Rec 294: , 1057–1065. |
[22] | Radak D , Katsiki N , Resanovic I , Jovanovic A , Sudar-Milovanovic E , Zafirovic S , Mousad SA , Isenovic ER ((2017) ) Apoptosis and acute brain ischemia in ischemic stroke. Curr Vasc Pharmacol 15: , 115–122. |
[23] | Wen Y , Yang SH , Liu R , Perez EJ , Brun-Ziukemagel AM , Koulen P , Simpkins JW ((2007) ) Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta 1772: , 473–483. |
[24] | Sarajarvi T , Haapasalo A , Viswanathan J , Mäkinen P , Laitinen M , Soininen H , Hiltunen M ((2009) ) Down-regulation of seladin-1 increases BACE1 levels and activity through enhanced GGA3 depletion during apoptosis. J Biol Chem 284: , 34433–34443. |
[25] | Obulesu M , Lakshmi MJ ((2014) ) Apoptosis in Alzheimer’s disease: An understanding of the physiology, pathology and therapeutic avenues. Neurochem Res 39: , 2301–2312. |
[26] | Wirawan E , Vande Walle L , Kersse K , Cornelis S , Claerhout S , Vanoverberghe I , Roelandt R , De Rycke R , Verspurten J , Declercq W , Agostinis P , Vanden Berghe T , Lippens S , Vandenabeele P ((2010) ) Caspase-mediated cleavage of beclin-1 inactivates beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis 1: , e18. |
[27] | Siddiqui MA , Mukherjee S , Manivannan P , Malathi K ((2015) ) RNase L cleavage products promote switch from autophagy to apoptosis by caspase-mediated cleavage of beclin-1. Int J Mol Sci 16: , 17611–17636. |
[28] | Pluta R , Kida E , Lossinsky AS , Golabek AA , Mossakowski MJ , Wisniewski HM ((1994) ) Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest: I. Extracellular accumulation of Alzheimer’s β-amyloid protein precursor in the brain. Brain Res 649: , 323–328. |
[29] | Popa-Wagner A , Buga AM , Doeppner TR , Hermann DM ((2014) ) Stem cell therapies in preclinical models of stroke associated with aging. Front Cell Neurosci 8: , 347. |
[30] | Tatarishvili J , Oki K , Monni E , Koch P , Memanishvili T , Buga AM , Verma V , Popa- Wagner A , Brüstle O , Lindvall O , Kokaia Z ((2014) ) Human induced pluripotent stem cells improve recovery in stroke-injured aged rats. Restor Neurol Neurosci 32: , 547–558. |
[31] | Sandu RE , Buga AM , Uzoni A , Petcu EB , Popa-Wagner A ((2015) ) Neuroinflammation and comorbidities are frequently ignored factors in CNS pathology. Neural Regen Res 10: , 1349–1355. |
[32] | Slevin M , Matou S , Zeinolabediny Y , Corpas R , Weston R , Liu D , Boras E , Di Napoli M , Petcu E , Sarroca S , Popa-Wagner A , Love S , Font MA , Potempa LA , Al-Baradie R , Sanfeliu C , Revilla S , Badimon L , Krupinski J ((2015) ) Monomeric C-reactive protein-a key molecule driving development of Alzheimer’s disease associated with brain ischaemia? Sci Rep 5: , 13281. |


