
Aggregates of RNA Binding Proteins and ER Chaperones Linked to Exosomes in Granulovacuolar Degeneration of the Alzheimer’s Disease Brain
Abstract
Granulovacuolar degeneration (GVD) occurs in Alzheimer’s disease (AD) brain due to compromised autophagy. Endoplasmic reticulum (ER) function and RNA binding protein (RBP) homeostasis regulate autophagy. We observed that the ER chaperones Glucose – regulated protein, 78 KDa (GRP78/BiP), Sigma receptor 1 (SigR1), and Vesicle-associated membrane protein associated protein B (VAPB) were elevated in many AD patients’ subicular neurons. However, those neurons which were affected by GVD showed lower chaperone levels, and there was only minor co-localization of chaperones with GVD bodies (GVBs), suggesting that neurons lacking sufficient chaperone-mediated proteostasis enter the GVD pathway. Consistent with this notion, granular, incipient pTau aggregates in human AD and pR5 tau transgenic mouse neurons were regularly co-localized with increased chaperone immunoreactivity, whereas neurons with mature neurofibrillary tangles lacked both the chaperone buildup and significant GVD. On the other hand, APP/PS1 (APPswe/PSEN1dE9) transgenic mouse hippocampal neurons that are devoid of pTau accumulation displayed only few GVBs-like vesicles, which were still accompanied by prominent chaperone buildup. Identifying a potential trigger for GVD, we found cytoplasmic accumulations of RBPs including Matrin 3 and FUS as well as stress granules in GVBs of AD patient and pR5 mouse neurons. Interestingly, we observed that GVBs containing aggregated pTau and pTDP-43 were consistently co-localized with the exosomal marker Flotillin 1 in both AD and pR5 mice. In contrast, intraneuronal 82E1-immunoreactive amyloid-β in human AD and APP/PS1 mice only rarely co-localized with Flotillin 1-positive exosomal vesicles. We conclude that altered chaperone-mediated ER protein homeostasis and impaired autophagy manifesting in GVD are linked to both pTau and RBP accumulation and that some GVBs might be targeted to exocytosis.
INTRODUCTION
In Alzheimer’s disease (AD), hyperphosphorylated tau (pTau) aggregates form neurofibrillary tangles (NFTs), neuropil threads, and the neuritic component of plaques [1] accompanied by extracellular amyloid-β (Aβ) accumulation [2]. Granulovacuolar degeneration (GVD) is a well-documented additional feature of AD pathology with a strong positive correlation between severity of GVD and NFT formation [3]. Numbers of GVD affected neurons in the hippocampus increase with higher Braak NFT stages [4–7]. GVD bodies (GVBs) are rarely found together with NFTs in the same neuron but frequently observed in neurons that were diffusely stained with the antibody AT8 in AD [4, 7–9] and frontotemporal lobar degeneration (FTLD)-tau cases [10] suggesting that these neurons are at the premature-tangle stage.
GVBs were originally observed in hippocampal pyramidal neurons of senile dementia patients [11] and have subsequently been described in further neurodegenerative disorders [12] including tauopathies such as Guam disease [13], Pick’s disease, and progressive supranuclear palsy as well as in the synucleinopathies, sporadic Parkinson’s disease and dementia with Lewy bodies [14]. GVD has recently been reported in C9orf72 mediated familial amyotrophic lateral sclerosis (ALS) [15]. GVD can also be found at low frequency in brains of non-neurologically impaired aged persons [3, 16]. GVBs are composed of 0.5–5μm basophilic cytoplasmic dense-cored granules of various aggregated proteins that accumulate within large, membrane-bound vacuoles which are immunoreactive for late autophagy markers such as Lamp1 and Lamp2, but not with the early autophagy markers LC3 and p62 [17]. Ultrastructural analysis revealed a two-layer membrane morphology of GVBs [18]. These findings suggest that GVBs are late intermediates of the dynamic endo-lysosomal/autophagy pathway accumulating due to autophagy impairment [17].
Neuronal proteostasis is continuously maintained by endoplasmic reticulum (ER)-mediated mechanisms including the unfolded protein response (UPR). Protein aggregates which are formed in spite of this are removed from neurons by autophagy [19, 20]. In AD, ER function and autophagy are compromised due to both NFT buildup and Aβ accumulation [21]. Neuroprotective ER chaperones including Sigma receptor 1 (SigR1) and GRP78/BIP have emerged as critical regulators of ER functions and autophagy [22–24]. Therefore, we set out to explore their relationship with GVD pathology in the framework of the present study.
Searching for factors that trigger GVD, Wiersma et al. recently found that seeding of tau pathology initiates the formation of GVBs in tau P301 L and tau P301 S tg mice in vivo and in primary mouse neurons in vitro. Furthermore, dipeptide repeat (DPR) aggregates, which are mainly processed via autophagy pathway [26] were found within GVBs in neurons of ALS/frontotemporal dementia (FTD) patients with C9orf72 expansion [15]. Similarly, recent studies have emphasized a functional interplay between ER function, autophagy, and RNA binding protein (RBP) homeostasis [27]. Of note, altered autophagy leads to cytoplasmic aggregation of RBPs such as TDP-43, which causes further autophagy impairment. Stress granules (SGs) are RBP-containing organelles which are central in coping with this vicious circle [28]. Interestingly, pTDP-43 is a component of stress granules and has also been shown to accumulate in GVBs [29]. Thus, we investigated the relationship of stress granules and of other RBPs such as FUS and Matrin 3 involved in neurodegeneration with GVBs.
Exocytosis is yet another mechanism that might be activated by neurons to remove harmful protein aggregates. In fact, exocytosis of toxic oligomers including that of TDP-43, Tau, and Aβ has been suggested to underlie the trans-neuronal spread of neurodegenerative pathology [30]. In this context, it appears interesting to speculate that GVBs could be involved in exocytic mechanisms. In the present study, we examined a cohort of AD autopsy brains complemented by studies on pR5 transgenic tauopathy as well as APP/PS1 (APPswe/PSEN1dE9) transgenic mice. Our results suggest that abnormal cytoplasmic RBP aggregation combined with an altered distribution of protective ER (co-) chaperones contributes to GVD pathology and that GVBs containing aggregated pTau and pTDP-43 were found to be associated with the exosomal marker Flotillin 1.
MATERIALS AND METHODS
Antibodies
All primary and secondary antibodies and their dilutions used in this study are listed in Supplementary Table 1. Many of these antibodies have been used by us in previous published studies (see references in Supplementary Table 1). Rabbit polyclonal VAPB antibody was custom made and used by us in previous studies [31–34].
Human postmortem brains
Human postmortem brain samples fixed in buffered formalin (n = 12 AD patients, n = 9 age-matched controls; Table 1) were obtained from the archives of the Institute of Neuropathology, RWTH University Hospital, Aachen (Germany) and from the Department of (Neuro) Pathology, Academic Medical Center (AMC), University of Amsterdam, The Netherlands (see Table 1). Both control and AD cases were selected from a retrospective searchable neuropathologic database, including cases with consent for postmortem brain autopsy and use of their brain material and medical records for research. Controls included in the present study were adult individuals without a history of neurological diseases and without evidence of cognitive decline, based on the last clinical evaluation. We included only cases in which an extensive neuropathological protocol was used (based upon the recommendations of the Brain-Net Europe consortium [35]). The AD cases included in the present study were individuals with a clinical diagnosis of probable mild to moderate AD [36, 37], more recently defined as “Alzheimer’s clinical syndrome” [38]. The selected cases were then re-assessed by neuropathologists using the routine updated protocol for neurodegenerative diseases [39, 40], and included cases with neuropathological verification and AD neuropathological changes and similar load of GVD based on the immunolabelling of GVBs in subicular neurons using the established marker CK1δ [17] (Table 1). We evaluated TDP-43 pathology in both AD and controls cases [41] using routinely used antibody against pTDP-43 (Cosmo Bio Co. LTD, TIP-PTD-M01). None of the control cases displayed TDP-43 pathology. TDP-43 pathology was, however, observed in 5 out of 12 cases, with mild to moderate TDP-43 pathology in the hippocampus (Table 1). We used these cases for co-labelling of pTDP-43 and GVBs. These cases did not show any apparent/significant differences compared to non-TDP-43 positive AD cases in the pattern of immunolabelling of other RBPs and of the proteostasis factors studied. We excluded cases with co-existing other neuropathological lesions (i.e., microvascular infarcts, hippocampal sclerosis, alpha-synuclein inclusions) to avoid confounding factors. The samples were used in compliance with the Declaration of Helsinki. The studies were approved by the Ethical Committees of the Medical Faculty, RWTH Aachen (EK 127/18), and of the Academic Medical Center, Amsterdam (W11_073). The postmortem tissues had been obtained within 6–30 h after death (see Table 1).
Table 1
Patients examined in this study
| Case No. | Age | Gender | PMI (h) | Clinical diagnosis | Thal Phase | Braak stage | CERAD neuritic plaque score | AD neuropathological change | CK1δ IHC | pTDP43 |
| 1 | 71 | M | 12 | AD dementia | 4 | V-VI | 3 | A3, B3, C3 | ++++ | – |
| 2 | 75 | M | 6 | AD dementia | 4 | V-VI | 2 | A3, B3, C2 | ++++ | + |
| 3 | 82 | F | 24 | AD dementia | 4 | V-VI | 3 | A3, B3, C3 | ++++ | + |
| 4 | 79 | M | 12 | AD dementia | 4 | V-VI | 2 | A3, B3, C2 | ++++ | + |
| 5 | 76 | M | 6 | AD dementia | 4 | V-VI | 2 | A3, B3, C2 | ++++ | – |
| 6 | 79 | F | 12 | AD dementia | 4 | V-VI | 2 | A3, B3, C2 | ++++ | + |
| 7 | 76 | F | 6 | AD dementia | 4 | V-VI | 3 | A3, B3, C3 | ++++ | – |
| 8 | 73 | M | 6 | AD dementia | 4 | V-VI | 2 | A3, B3, C2 | ++++ | – |
| 9 | 76 | F | 24 | AD dementia | 4 | V-VI | 2 | A3, B3, C2 | ++++ | – |
| 10 | 68 | M | 24 | AD dementia | 3 | III | 2 | A2, B2, C2 | ++ | – |
| 11 | 76 | F | 24 | AD dementia | 4 | V-VI | 2 | A3, B3, C2 | ++++ | + |
| 12 | 77 | M | 12 | AD dementia | 3 | III | 2 | A2, B2, C2 | ++ | – |
| 1 | 76 | M | 30 | No dementia | 0 | 0 | 0 | 0 | – | – |
| 2 | 76 | M | 7 | No dementia | 1 | 0 | 0 | A1, B0, C0 | + | – |
| 3 | 71 | F | 16 | No dementia | 0 | 0 | 0 | 0 | – | – |
| 4 | 70 | M | 27 | No dementia | 0 | 0 | 0 | 0 | – | – |
| 5 | 81 | F | 24 | No dementia | 0 | 0 | 0 | 0 | – | – |
| 6 | 80 | M | 13 | No dementia | 0 | 0 | 0 | 0 | – | – |
| 7 | 56 | M | 16 | No dementia | 0 | 0 | 0 | 0 | – | – |
| 8 | 54 | M | 12 | No dementia | 0 | 0 | 0 | 0 | – | – |
| 9 | 62 | F | 26 | No dementia | 0 | 0 | 0 | 0 | – | – |
AD, CERAD neuritic plaque score frequent. GVD, granulovacuolar degeneration; PMI, postmortem interval; IHC, DAB immunohistochemistry. Neuropathological assessments: All cases were extensively characterized according to the routine protocol for neurodegenerative diseases. Neuropathological diagnosis of AD was made following the NIA-AA criteria including Thal phasing for Aβ load, Braak-and- Braak-staging for NFTs and CERAD neuritic plaque score to assess the density of neuritic plaques [29, 39, 88–92] (see Material and Methods). CK1δ positive GVD bodies (GVBs): Semiquantitative assessment of GVD was performed by DAB immunohistochemistry using CK1δ antibody as a marker for GVBs. Three hippocampal sections each from the above cases were stained. Abundance of strongly CK1δ-immunoreactive GVBs in subicular neurons was scored as follows: ++++=>20%,+++=10–20%,++=5–10%,+=1–5%, –=<0.1% of subicular neurons stained. This scoring was performed only to select AD cases with a similar load of GVD and to minimize the variability between the cases. It is not meant to confirm the already well-established correlation between GVD stages and histopathological AD stages, which are based on the spread of GVD pathology across various brain regions.
Transgenic (tg) mouse brains
APP/PS1
The studies on APP/PS1 mouse tissue were performed in accordance with the guidelines for the care and use of laboratory animals (LANUV approval number 8.87–50.10.37.09.112). Brains of 27- and 50-week-old APP and PSEN1 mutant mice (B6.CgTg (App695)3DboTg (PSEN1dE9) S9Dbo/J) and of their age-matched littermates were used for immunohistochemistry, immunofluorescence (n = 6 for each genotype), and western blotting (n = 3 for each genotype).
pR5 tg mice
Brain sections from 104-week-old pR5 (Tg (Thy1-MAPT)183Gotz) mice and age-matched wildtype mice were used for immunohistochemistry and immunofluorescence (n = 4 for each genotype). Human tau transgenic pR5 mice have been described in detail previously [8, 9]. Experiments were approved by the State Agency for Nature, Environment and Consumer Protection (LANUV) North Rhine-Westphalia, Recklinghausen, Germany (LANUV AZ 84-02.05.40.14.028).
CK1δ positive GVD bodies (GVBs)
Semi-quantitative assessment of GVD was performed in sections stained with DAB immunohistochemistry using CK1δ antibody as a marker for GVBs [17]. Three hippocampal sections each from the above-mentioned cases AD and controls were stained (see Table 1 for details).
Diaminobenzidine (DAB) immunohistochemistry
3–4μm paraffin sections were placed on poly-L-lysine coated slides and allowed to dry in an oven (37°C) overnight and then processed for immunohistochemistry described in detail elsewhere [33]. The sections were deparaffinized in xylene for 20 min and rehydrated in 100%, 96%, and 70% ethanol for 5 min each followed by endogenous peroxidase quenching (0.3% H2O2 in methanol) for 20 min. For antigen retrieval, sections were heated in citrate buffer, pH 6 (DAKO), for 20 min in a pressure cooker. After washing in PBS, sections were incubated with primary antibody (Supplementary Table 1) for 1 h at room temperature or at 4°C overnight. After washing in PBS, sections were incubated with appropriate polymeric HRP-linker secondary antibody (IL Immunologic, Duiven, The Netherlands) for 30 min at room temperature. DAB reagent (DCS Innovative Diagnostic System DAB kit) was used to visualize antibody binding. The sections were then counter-stained with 6% hematoxylin for 3 min. In the case of 82E1 antibody, we used Biotin secondary antibody conjugate (Vector Laboratories, Burlingame, CA, USA) followed by detection using the Vectastain ABC HRP kit (Vector Laboratories). All procedures were performed at room temperature.
Immunofluorescence
Single and double immunofluorescence staining were performed as described by us already elsewhere [33, 42]. In brief, deparaffinized tissue sections were heated in citrate buffer, pH 6 (Dako), for 20 min in a pressure cooker. Sections were then blocked (to avoid non-specific bindings) with ready to use 10% normal goat serum (Life Technologies, MD, USA) for 1 h at room temperature before incubating with primary antibody 4°C overnight. After two times washing in TBS-T for 10 min, the sections were incubated with Alexa conjugated secondary antibody (1 : 500 in PBS) at room temperature for 2 h. Sections were washed in TBS-T (2 x 10 min) and stained for 10 min with 0.1% Sudan Black in 80% ethanol to suppress endogenous lipofuscin auto-fluorescence. Finally, the sections were washed for 5 min in TBST and mounted with Vectashield mounting medium (Vector Laboratories) containing DAPI.
Image acquisition
Images of the DAB-stained sections were taken with a Zeiss Axioplan microscope equipped with a 40x objective and an Axio Cam 506 color camera (Zeiss). Images from immunofluorescence labelled sections were taken with a Zeiss LSM 700 laser scanning confocal microscope using 20x, 40x, and 63x objectives (Zeiss). Images were acquired by averaging 4 scans per area of interest resulting in an image size of 1024x1024 pixels. The laser intensity was kept constant for all the samples examined. Captured confocal images were analyzed using Adobe Photoshop CS5 and ZEN (Blue edition) 2009 software.
Semi-quantitative analysis of co-immunolabelling (human tissue)
Average pixel intensity of the target proteins (SigR1 and VAPB; Fig. 1B, F)/field of view were quantified in three representative sections from each of six AD patients (Braak stages V-VI) and in three representative sections from each of three controls (Braak stage 0) in 8–10 random fields, at low magnification (20x), capturing approx. 30-40 neurons/field of view. Average background pixel intensity was subtracted. Average pixel intensity of the target proteins (SigR1, GRP78, VAPB; Fig. 1C, D, F) in neurons were quantified (E, F) using one representative section each from six AD (Braak stages V-VI) patients and one representative section each from three controls (Braak stage 0). Approx. 20–30 neurons / field of view in 8-10 random 20x fields were found, examining around 100–150 neurons per case by measuring average pixel intensities of the regions of interest drawn around each neuron. We used the unpaired Student’s t-test for comparison between two sample groups. Values represent the mean±standard deviation (SD). Differences between values were regarded as significant when=*p<0.05.
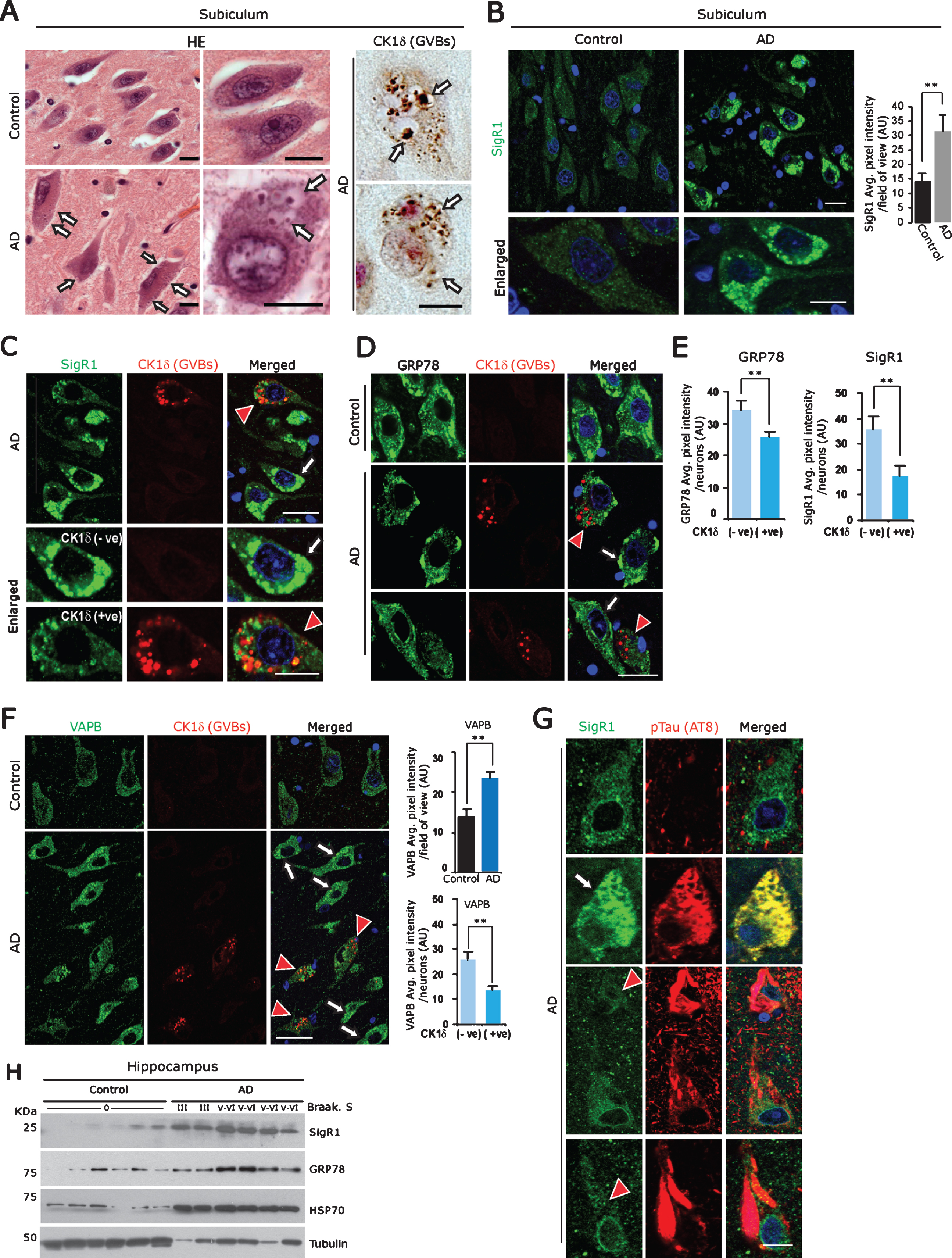
Fig.1
A) Left panel: Representative H&E stained subicular neurons of age-matched control brain compared to an AD case. Arrows: GVBs. Right panel: CK1δ immunoreactive GVBs (arrows) in AD subicular neurons. Scale bars: 20μm. B) Increased levels of the ER chaperone SigR1 in subicular neurons of AD patient compared to the age-matched control. Scale bars: 20 μm. Values represent the mean±SD (**p<0.05). C-F) Double immunofluorescence labeling for CK1δ and for the ER chaperones SigR1 (C) GRP78 (D) and for the UPR co-factor VAPB (F). Note the overall increased levels of SigR1, GRP78 and VAPB in AD subicular neurons (white arrows), but reduced levels of SigR1, GRP78 (C, D, E) and VAPB (F) immunoreactivity in subicular neurons harboring GVBs (red arrowheads, quantification). GVBs harboring neurons are represented as CK1δ (+ve) and neurons without GVBs are denoted as CK1 (–ve) (Scale bars: 20 μm. Values represent the mean±SD (**p<0.05). G) Double immunofluorescence labeling of SigR1 and pTau (AT8). Note the Co-localization of increased SigR1 immunoreactivity with granular pTau (arrow) but decrease of SigR1 immunoreactivity (red arrowheads) in neurons containing mature pTau tangles in AD subicular neurons. Scale bars: 20 μm. H) Immunoblot analysis of hippocampal tissue homogenates from AD cases and their age matched controls showing an increase in ER chaperons SigR1, GRP78 and proteotoxic stress marker HSP70 in AD.
Semi-quantitative analysis of co-immunolabelling (mouse tissue)
Average pixel intensity of the target proteins (SigR1 and VAPB; Supplementery Figure 2D, E) / field of view were quantified in one representative section from each of three APP/PS1 (50-week) transgenic mice and one representative section each from three age-matched littermates. 8–10 random fields in low magnification (20x) were examined, capturing approximately 20–30 neurons/field of view. Average background pixel intensity was subtracted. We used the unpaired Student’s t-test for comparison between two sample groups. Values represent the mean±standard deviation (SD). Differences between values were regarded as significant when *p<0.05.
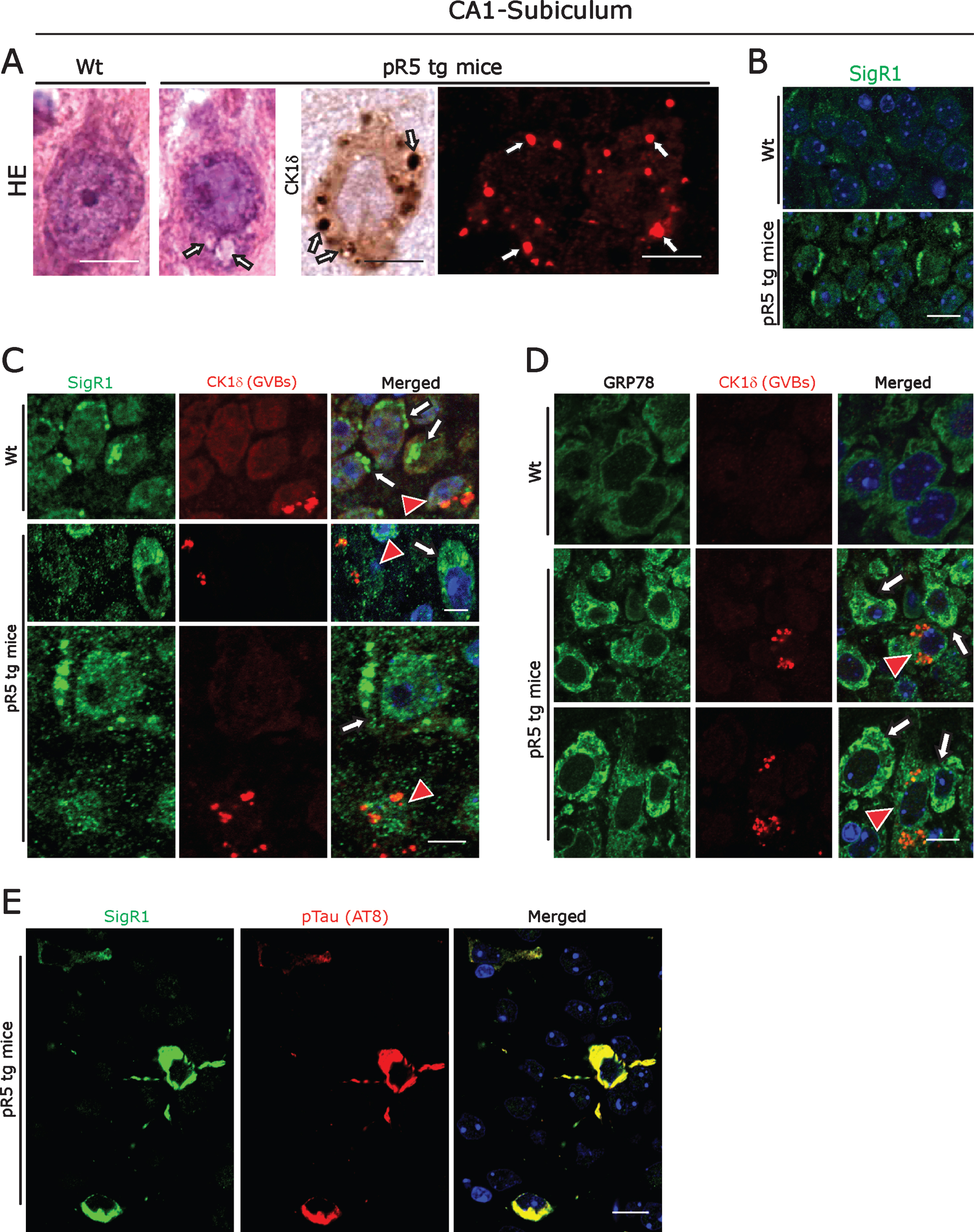
Fig.2
A) Left panel: Representative H&E stained CA1- subicular neurons of 104-week-old-pR5 tg mice compared to an age-matched wt mouse. Arrows: GVBs. Right panel: CK1δ immunoreactive GVBs (arrows) in pR5 tg mice CA1- subicular neurons. Scale bars: 15μm. B) Increased levels of the ER chaperone SigR1 in CA1- subicular neurons compared to the wt mice. Scale bars: 15μm. C, D) Double immunofluorescence labeling for CK1δ and for the ER chaperones SigR1 (C) and GRP78 (D). Note the reduced levels of SigR1, GRP78 immunoreactivity in neurons harboring GVBs (red arrowheads). White arrows: Increased levels of SigR1 and GRP78 in neurons without GVBs. Scale bars: 15μm. E) Double immunofluorescence labeling of SigR1 and pTau (AT8). Note the Co-localization of increased SigR1 immunoreactivity with pTau: Scale bar: 15μm.
Western blot analysis
Western blots of brain tissue were performed as described previously [33]. Briefly, human and mouse brain tissues were homogenized in ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer, with protease inhibitor cocktail (Roche Life Science, Penzberg, Germany). The crude lysates were briefly centrifuged and then processed for the bicinchoninic acid protein assay (BCA; Thermo Scientific) according to the manufacturer’s protocol. Equal amounts of protein were boiled in Laemmli sample buffer for 5-10 min and processed for SDS-PAGE. The protein gels were transferred onto polyvinylidene fluoride (PVDF) membranes. The blots were then blocked with 4% skimmed milk in TBS-T for 30 min and incubated with specific primary antibody (Supplementary Table 1) overnight at 4°C under agitation. Thereafter, the blots were washed three times in TBS-T for 10 min each and incubated for 1 h with appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (Thermo Scientific). Immunoreactive protein bands were visualized by exposing the blots on an X-ray film (Fuji Films). Quantification of the band intensities was performed after normalizing to tubulin levels using Adobe Photoshop CS5. Values represent the mean±SD (**p<0.05).
RESULTS
ER stress and aberrant association of ER chaperones with GVBs
GVD is predominantly present in brain regions that are most affected by AD pathology such as hippocampus (CA1, CA2, CA4, and subiculum) [29]. GVBs can be detected as membrane bound vesicular structures by routine hematoxylin-eosin (Fig. 1A) or by silver staining. We used the established GVB marker, Casein kinase 1 isoform delta (CK1δ) [29] to label GVBs in subicular neurons (Fig. 1A; Supplementary Figure 1A).
ER chaperones are critical regulators in maintaining proteotoxic effects and/or autophagy overload due to Aβ and pTau accumulations in AD brain [43]. Consistent with this, we observed significantly elevated overall immunoreactivity for the ER chaperone SigR1 in subicular neurons of the AD brains (Fig. 1B). However, those neurons which were affected by GVD, which comprise about 25% of the total neuronal population in the cases examined by us, showed lower SigR1 levels, and there was only minor co-localization of SigR1 with GVBs (Fig. 1C). Similarly, immunoreactivity for GRP78, a prime initiator of the UPR, was reduced in subicular neurons harboring GVBs (Fig. 1D, E), and VAPB, a UPR co-factor [44], was found to be generally increased in subicular neurons of AD brains. Those neurons which harbored GVBs showed a less pronounced VAPB buildup (Fig. 1F). GVBs showed only minor, if any colocalization with any of these ER chaperones (Fig. 1C, D, F). Similarly, human AD neurons harboring small granular pTau deposits showed elevated SigR1 immunoreactivity, and these pTau aggregates were regularly co-localized with SigR1 (Fig. 1G; Supplementary Figure 1B), suggesting that SigR1 chaperone is involved in clearing such small granular pTau accumulations. Neurons with mature NFTs lacked both the chaperone buildup and significant GVBs (Fig. 1G; Supplementary Figure 1C). On the other hand, moderately affected dentate gyrus neurons [45] which carried only a minor load of pTau and GVBs (Supplementary Figure 1G) and showed particularly high levels of SigR1 (Supplementary Figure 1E) and GRP78 (not shown). The overall increased levels of ER chaperones were confirmed by western blot analyses of AD patient and age-matched control hippocampi (Fig. 1H).
ER chaperones and GVBs in pR5 Tau tg mice
The pR5 mouse strain is a well-established tauopathy model that develops NFTs due to overexpression of the longest human tau isoform harboring the P301 L mutation [46, 47]. We confirmed that 104-week-old pR5 mice displayed CK1δ immunoreactive GVBs coinciding with NFT pathology [8, 9] in hippocampus (Fig. 2A; Supplementary Figure 1A). These GVBs were morphologically similar to the GVBs present in human AD; they have already been shown to be immunoreactive for several UPR markers [9]. We found that, similar to the situation in human AD brain, CA1-subicular neurons of pR5 mice consistently showed overall increased levels of the ER chaperones SigR1, GRP78, and VAPB (Fig. 2B-D; Supplementary Figure 1F). Again, those neurons that harbored GVBs showed decreased SigR1 and GRP78 levels, and there was only minor, if any co-localization of SigR1 with GVBs (Fig. 2C, D). Also consistent with the changes observed in human AD brain, granular, pre-tangle pTau aggregates co-localized with SigR1 immunoreactivity (Fig. 2E) and some of them were also found to be co-localized with GVBs (Supplementary Figure 1H).
CK1δ-positive incipient GVBs and ER chaperones in APP/PS1 transgenic mice
APP/PS1 tg mouse brains showed prominent extracellular Aβ plaque pathology (Supplementary Figure 2A), but their hippocampal pyramidal neurons, in contrast to the pR5 mice, harbored only rather small, probably incipient accumulations of GVB-like, CK1δ-immunoreactive granular structures (Supplementary Figures 1A, 2C). They were distinct from mature human GVBs in many respects, such as smaller size, frequent presence of granular cores without outer layer morphology, and recurrent appearance in distinct groups, but not scattered over the neuronal cytoplasm (Supplementary Figures 1A, 2B, C). Like human AD and pR5 mouse neurons, SigR1 and VAPB were found at overall elevated levels in APP/PS1 mouse neurons; however, in contrast to the human AD patient and pR5 mouse GVBs, the CK1δ-immunoreactive granules in APP/PS1 mouse neurons were SigR1- and VAPB-positive (Supplementary Figure 2D, E). Similarly, these structures were strongly ubiquitin immunoreactive (Supplementary Figure 2F). Consistent with the immunohistochemical results, immunoblot analysis of total APP/PS1 mouse brain homogenates revealed a significant age-dependent increase in the proteotoxic stress markers SigR1, GRP78, Hsp70, and Hsp27 as well as ubiquitin conjugates (Supplementary Figure 2G).
RNA binding protein homeostasis and GVBs
pTau can alter autophagy at multiple steps [48] and can trigger the formation of GVBs in vivo as well as in vitro [25]. Similarly, DPR aggregates, which alter autophagy pathways [26], were found within GVBs in neurons of ALS/FTD patients with C9orf72 expansion [15]. RBPs such as TDP-43 form aggregates in AD patient neurons and quite often granular pTDP-43 aggregates co-localize with GVBs in AD patient hippocampal neurons [29]. Furthermore, RBPs associated with SGs progressively accumulate together with tau in mouse models of tauopathy, as well as in human AD and FTLD-Tau brain tissue [49]. Therefore, in an attempt to identify the trigger for GVD, we next examined RBPs in subicular neurons affected by GVD. Subicular neurons from AD patients contained several NFTs, resembling pTDP-43-immunoreactive aggregates and many granular or vesicular aggregates (20–30%), some of which were co-localized with GVBs as described previously by others [29, 41, 50] (Fig. 3A, lower panel). Consistent with this, pR5 mouse hippocampal neurons also frequently showed globular and vesicular pTDP43-immunoreactive structures (Fig. 3B) which were co-localized with the GVB marker CK1δ (Fig. 3B, lower panel). Even though pR5 mouse neurons displayed abundant pTau-positive NFTs, larger tangle-like pTDP-43 aggregates were absent (Fig. 3B). To decipher whether the co-localization of pTDP-43 with GVBs is indicative of a more general buildup of RBPs in AD neurons, we examined AD brains with antibodies against Matrin 3 and FUS as well as SG proteins used by us previously when studying ALS pathomechanisms [42, 51, 52]. Matrin 3 is of prime importance because it regulates the transcription and stability of RBPs including TDP-43 and FUS [53]. Moreover, MATR3 mutations lead to ALS and distal vacuolar myopathy in patients [54–56]. We found nuclear accumulation and cytoplasmic mis-localization of Matrin 3 in those neurons which contained GVBs. These globular or vesicular cytoplasmic Matrin 3-immunoreactive structures co-localized with GVBs (Fig. 3C). In contrast, neurons in control cases showed basal levels of nuclear Matrin 3-immunoreactivity and no cytoplasmic Matrin 3 (Fig. 3C, upper panel). Of note, Matrin 3 levels were unchanged and there was no obvious cytoplasmic mis-localization of Matrin 3-immunoreactivity present in pR5 mouse hippocampal neurons (Fig. 3D). Consistent with the pattern of Matrin 3-immunoreactivity, cytoplasmic mis-localization and co-localization of FUS together with GVBs (Fig. 3E) was observed in human AD neurons. However, there was no clear-cut link between GVD pathology and reduced or increased intensity of nuclear FUS staining. Interestingly, even though pR5 hippocampal neurons showed significant granular cytoplasmic FUS labelling, FUS immunoreactivity was not co-localized with GVBs (Fig. 3F; lower panel).
Fig.3
A) DAB immunohistochemistry (upper panel) for pTDP-43 showing immunostaining associated with NFT-like structures and double immunofluorescence labeling (lower panel) demonstrating co-localization of pTDP-43 immunoreactivity with GVBs in human AD subicular neurons. Scale bars: 15μm. B) DAB immunohistochemistry for pTDP-43 showing diffuse nuclear immunoreactivity in wt mice CA1-subicular neurons (upper panel) and cytoplasmic granular accumulations (middle panel, white arrows) and rare globular accumulations (middle panel, black arrow) in pR5 tg mice. Double immunofluorescence labeling (lower panel) demonstrating co-localization of pTDP-43 immunoreactivity with GVBs in pR5 tg mice CA1- subicular neurons. Scale bars: 15μm. C) Matrin 3 and CK1δ double immunofluorescence: Increased levels of Matrin 3 in the nucleus and vesicular immunoreactive structures co-localizing with GVBs in human AD subicular neurons (middle and lower panels). Note the normal nuclear Matrin 3 levels in a neuron from the control case (upper panel). Scale bar: 20μm. D) Matrin 3 and CK1δ double immunofluorescence showing no cytoplasmic mis-localization of Matrin 3 as well as no co-localization with GVBs in pR5 tg mice CA1-subicular neurons. Scale bar: 15μm. E, F) FUS and CK1δ double immunofluorescence: Strong co-localization of cytoplasmic FUS accumulations with GVBs in human AD patient subicular neurons (E), while there is no co-localization of cytoplasmic FUS with GVBs in pR5 tg mice CA1-subicular neurons (F). Scale bars: 15μm. G) CK1δ-immunoreactive GVBs often co-stained with G3BP. Note the loss of nuclear G3BP immunoreactivity (white arrowheads) in the neurons containing GVBs. Scale bars: 15μm. (H) Loss of S6-kinase nuclear immunoreactivity (white arrowhead) and increased cytoplasmic accumulation in subicular AD neurons, which consistently showed CK1δ labeling (White arrow). Note the S6-Kinase-immunoreactive NFT-like structure (red arrowhead). Scale bars: 20μm. I) Increased cytoplasmic accumulations of S6-kinase in CA1-subicular neurons of pR5 tg mice, which were consistently co-localized with CK1δ labeling (arrow). Scale bar: 15μm.
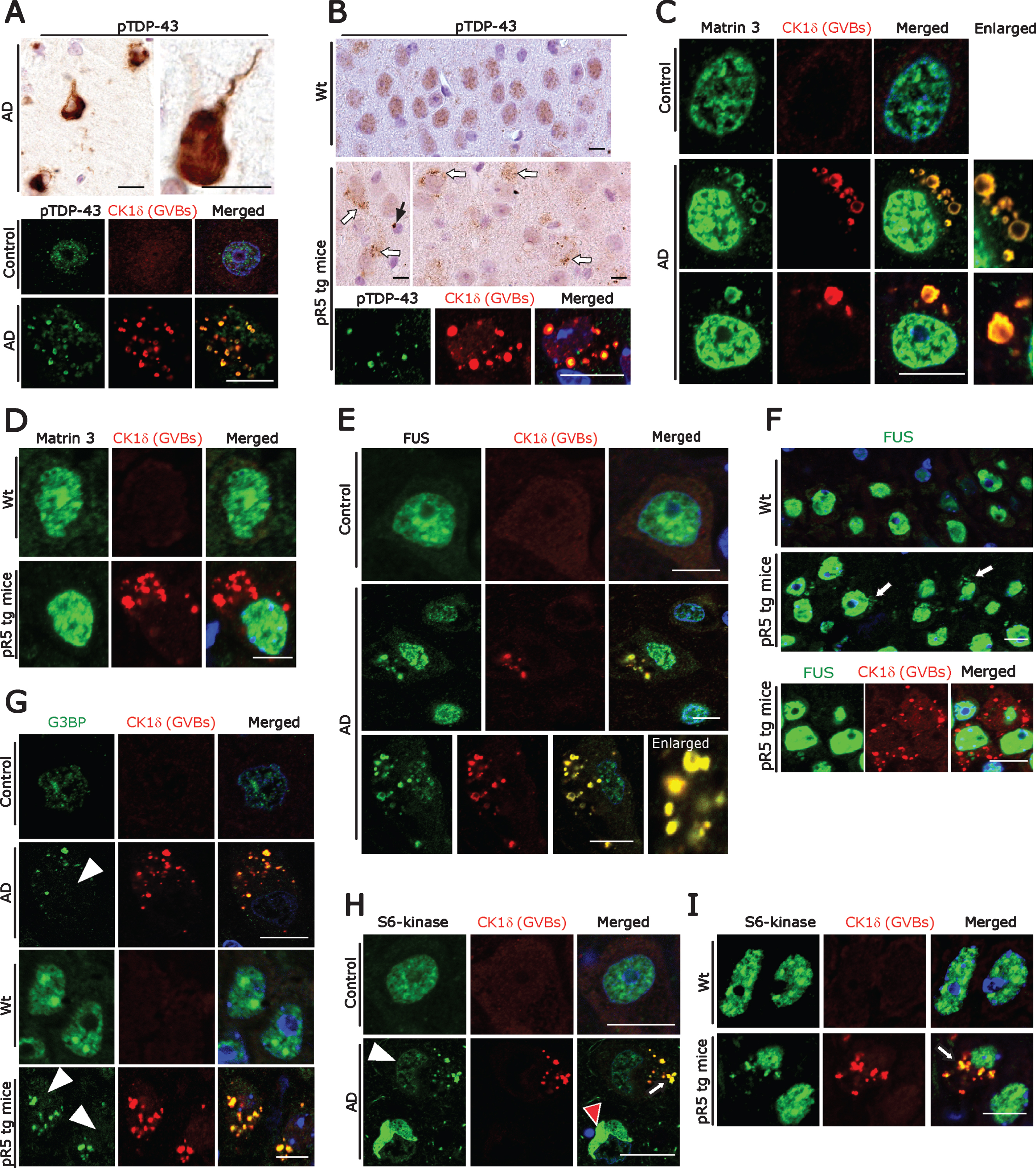
Because aggregation of RBPs proceeds through the SG pathway, we stained SGs together with GVBs. Using antibodies against the SG marker G3BP as previously described by us and others [42, 57], we found that many human AD as well as the pR5 mouse neurons develop cytoplasmic accumulations of G3BP, along with reduced nuclear G3BP immunoreactivity (Fig. 3G). These cytoplasmic accumulations were often CK1δ-positive (Fig. 3G). SGs including processing bodies (P-bodies), which are also cytoplasmic accumulations of non-translating messenger ribonucleoprotein complexes (mRNPs), are cleared by autophagy [58]. Compared to the controls we found increased granular cytoplasmic immunoreactivity and reduced nuclear immunoreactivity for the P-body marker S6 kinase in AD brain subicular neurons (Fig. 3H; Supplementary Figure 3A); some of the granular S6 kinase staining co-localized with CK1δ-positive GVBs (Fig. 3H, lower panel). Similarly, increased granular cytoplasmic immunoreactivity of S6 kinase co-localized with CK1δ-positive GVBs in pR5 hippocampal neurons (Fig. 3I). Taken together, abnormal cytoplasmic RBP aggregates co-localize with GVBs, suggesting that RBP aggregates possibly trigger GVB formation and that GVBs might actually be involved in the removal of such aggregates. They are in line with a recent study showing that DPR aggregates contribute to GVD pathology in C9orf72-related FTLD/ALS [15].
Endosomes, exosomes, and GVD
Recent studies suggested that exosomes from AD brain contain Aβ oligomers and that exosomes may act as vehicles for the neuron-to-neuron transfer of toxic Aβ oligomers in cell culture models [59, 60]. Blocking the formation, secretion or uptake of exosomes was found to reduce both the spread of oligomers and their toxicity in these cell culture systems [59, 60]. Considering that most exosomal vesicles originate from endosomes [61], we therefore examined endosomes in neurons affected by GVD. Staining with antibodies against Rab11 and Mannnose-6-phosphate confirmed that these late endosomal markers co-localize with GVBs in AD patient subicular neurons (Supplementary Figure 3D), as shown already by others [17]. Additional immunohistochemistry revealed that GVBs also co-localize with the early and intermediate endosomal markers EEA1 and Rab7 (Supplementary Figure 3D) and with the Golgi marker GM130 (Supplementary Figure 3C). Of note, in a recent study we showed that the Golgi protein GolginA4, that was found to be elevated in cerebrospinal fluid of AD patients, is a major constituent of GVBs [62] (Fig. 4A). Thus, we hypothesized that GVBs might actually undergo exocytosis.
Fig.4
A) Co-immunofluorescence labeling of CK1δ-immunoreactive GVBs together with Golgin A4 in AD patient subicular and pR5 tg mice CA1-subicular neurons. Scale bars: 15μm. B) Increased size and density of Flotillin-1-immunoreactive vesicles in AD patient subicular neurons and in APP/PS1 tg mice and pR5 tg mice CA1-subicular neurons compared to the control and to wt mice. Upper right panel: DAB immunohistochemistry showing increased Flotillin-1-immunoreactive vesicles associated with GBVs (white arrows). Scale bars: 15μm. C, D) Co-immunofluorescence labeling of CK1δ-immunoreactive GVBs, together with Flotillin-1 in AD brain subicular neurons (C) and in pR5 tg mice CA1-subicular neurons (D). Note that while many GVBs are co-localized with Flotillin-1 (white arrows); few actually appear to be completely enclosed by Flotillin-1 labeled vesicles (yellow arrows). Scale bars: 10μm. E, F) Co-immunofluorescence of GolginA4 together with Flotillin-1, in AD brain subicular neurons (E) and in pR5 tg mice CA1-subicular neurons (F). Note that Golgin A4-labeled granules often co-localize with Flotillin-1 labeled vesicles (arrows). Scale bars: 10μm.
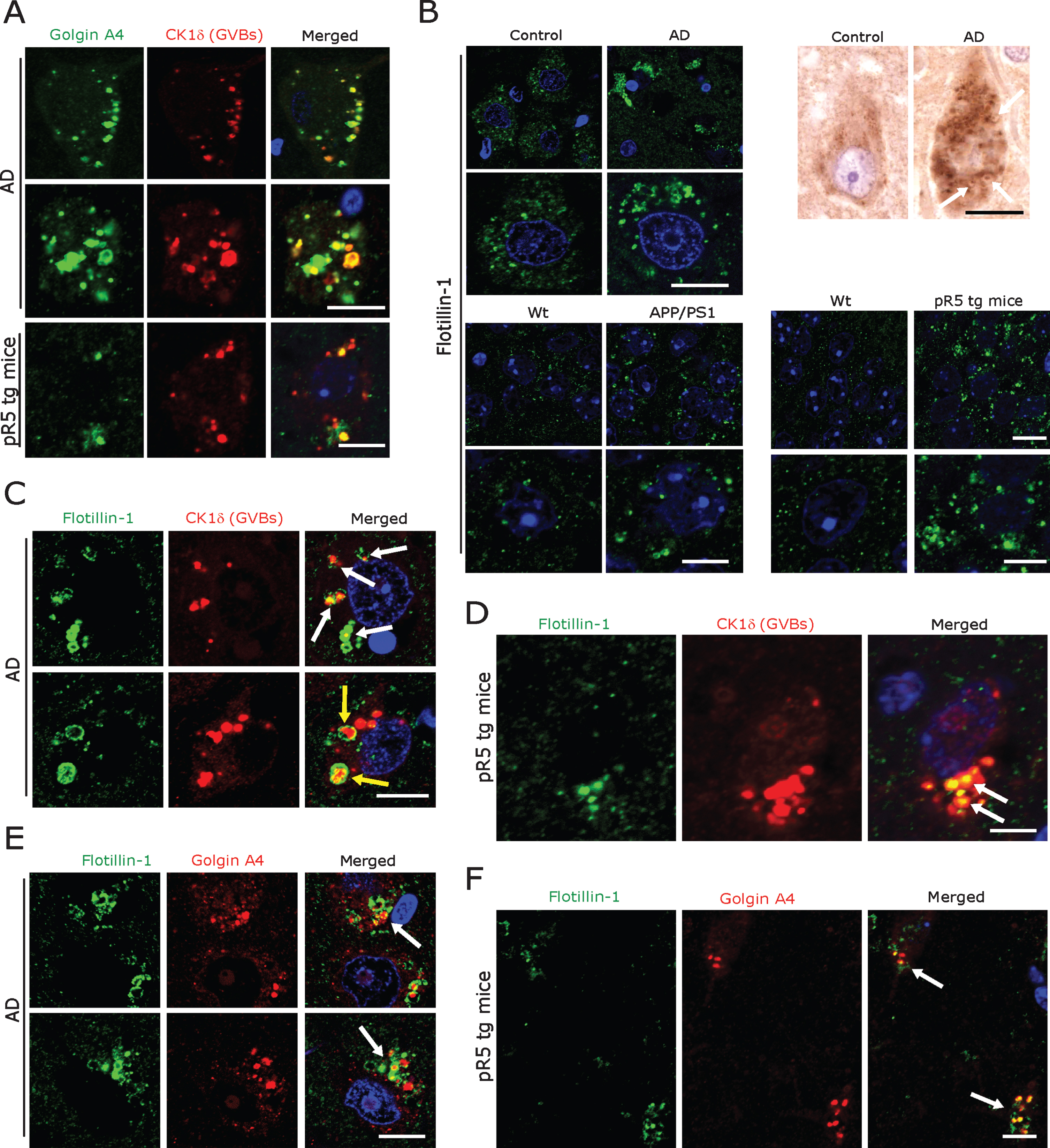
Even though not specific, universally applicable exosomal markers have evolved so far. Tetraspanins such as CD9, CD63, and CD81 and components of the endosomal sorting complex required for transport (ESCRT) including TSG101, Flotillin 1, and Alix [59, 60, 63] are currently being used to label exosomal vesicles. We found a consistent pattern of immunolabelling of vesicles at low density in subicular neurons of human control cases and of control mice using both a monoclonal (BD Biosciences) and a polyclonal antibody (Cell Signaling) against Flotillin 1 (Fig. 4B). In contrast, incubation with the monoclonal antibody against CD9 (antibody online, Germany) yielded abundant cytoplasmic immunoreactivity in many other cells in both human AD and control and mouse brains (Supplementary Figure 3B). Based on these observations, we decided to focus on the monoclonal Flotillin 1 antibody (BD Biosciences) for our subsequent analyses. The same monoclonal antibody has also been used for the staining of human autopsy specimens in the recent study by Sardar Sinha et al. on exosomal mechanisms in AD [60]. We found that Flotillin 1-positive vesicles were markedly increased in number and size in subicular neurons of AD patients and in CA1-subicular neurons of pR5 and APP/PS1 tg mice; in many of these pyramidal neurons, Flotillin-1 immunoreactivity was co-localized with CK1δ-positive (Fig. 4C, D) and Golgin A4-positive GVBs (Fig. 4E, F).
Prompted by these observations, we next examined whether these Flotillin-1-positive vesicles also contain pTau and RBP aggregates, as detected by us in GVBs (see above and Fig. 3, Supplementary Figure 1). We observed frequent and prominent co-localization of Flotillin-1-immunoreactive vesicles with granular pTau in human AD subicular neurons and in pR5 mouse hippocampal neurons (Fig. 5A). Some of these Flotillin-1-immunoreactive vesicular structures actually appeared to enclose smaller granular pTau immunoreactive species (Fig. 5A; arrows). Similarly, pTDP-43-immunoreactive aggregates were found to be co-localized with or even enclosed by Flotillin-1-positive vesicles (Fig. 5B) in both human AD and pR5 mouse neurons. In line with a recent report showing co-localization of 82E1-immunoreactive intracellular Aβ oligomers with Flotillin-1-positive granular structures in human AD brain [60], we found (albeit minor) co-localization of Flotillin-1-positive vesicles with 82E1-positive intracellular deposits in human AD subicular neurons (Fig. 5C) and in hippocampal APP/PS1 mouse neurons (Fig. 5D). However, only very minor, if any, co-localization of Aβ-(81E1-) immunoreactive intraneuronal material with CK1δ-positive GVBs was observed by us in AD patient subicular neurons (not shown). There was no detectable co-labelling of extracellular 82E1-positive Aβ in AD patient and APP/PS1 mouse hippocampi with our Flotillin-1 antibody (not shown).
Fig.5
A, B) Double immunofluorescence for Flotillin-1 together with pTau- (A) and pTDP-43 (B), demonstrating that these aggregates are often co-localized with and/or are enclosed by Flotillin-1 labeled vesicles (arrows) in AD patient subicular and in pR5 tg mice CA1-subicular (lower panels), neurons, maximum intensity projections (below) to show the co-localization. Scale bars: 15μm. C) Double immunofluorescence of Aβ oligomer (82E1 antibody) and Flotillin-1. There was a rather rare, somewhat arbitrary coincidence of Flotillin-1-labeled structures with 82E1 immunoreactivity within human AD subicular neurons (arrows) in the vicinity of 82E1-labeled Aβ plaques (P). Scale bars: 15μm. D) In APP/PS1 tg mice 82E1 immunoreactive intracellular material is often co-localized with Flotillin-1-labeled vesicles (arrows) within subicular neurons in the vicinity of Aβ plaques (P). Scale bars: 10μm.
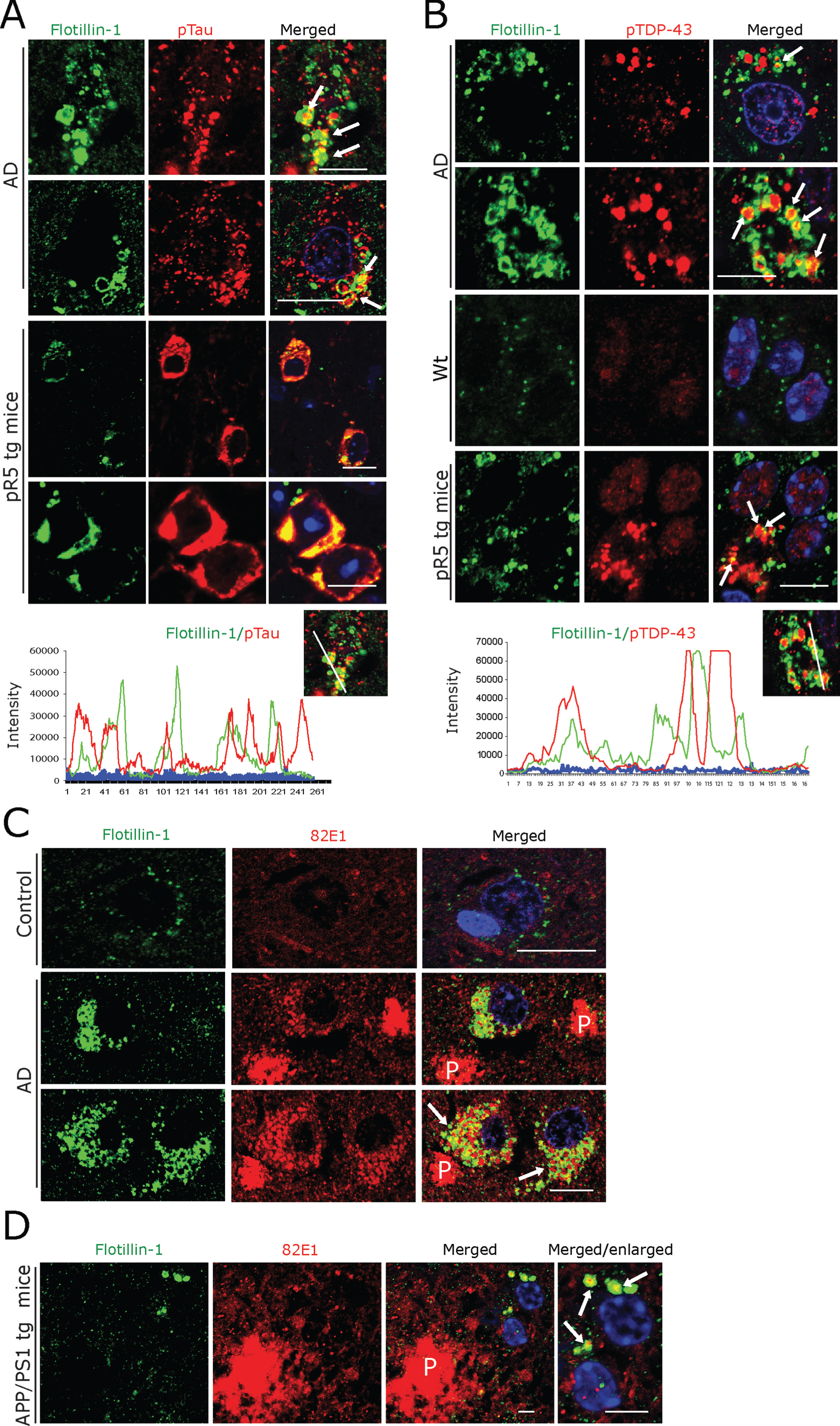
Fig.6
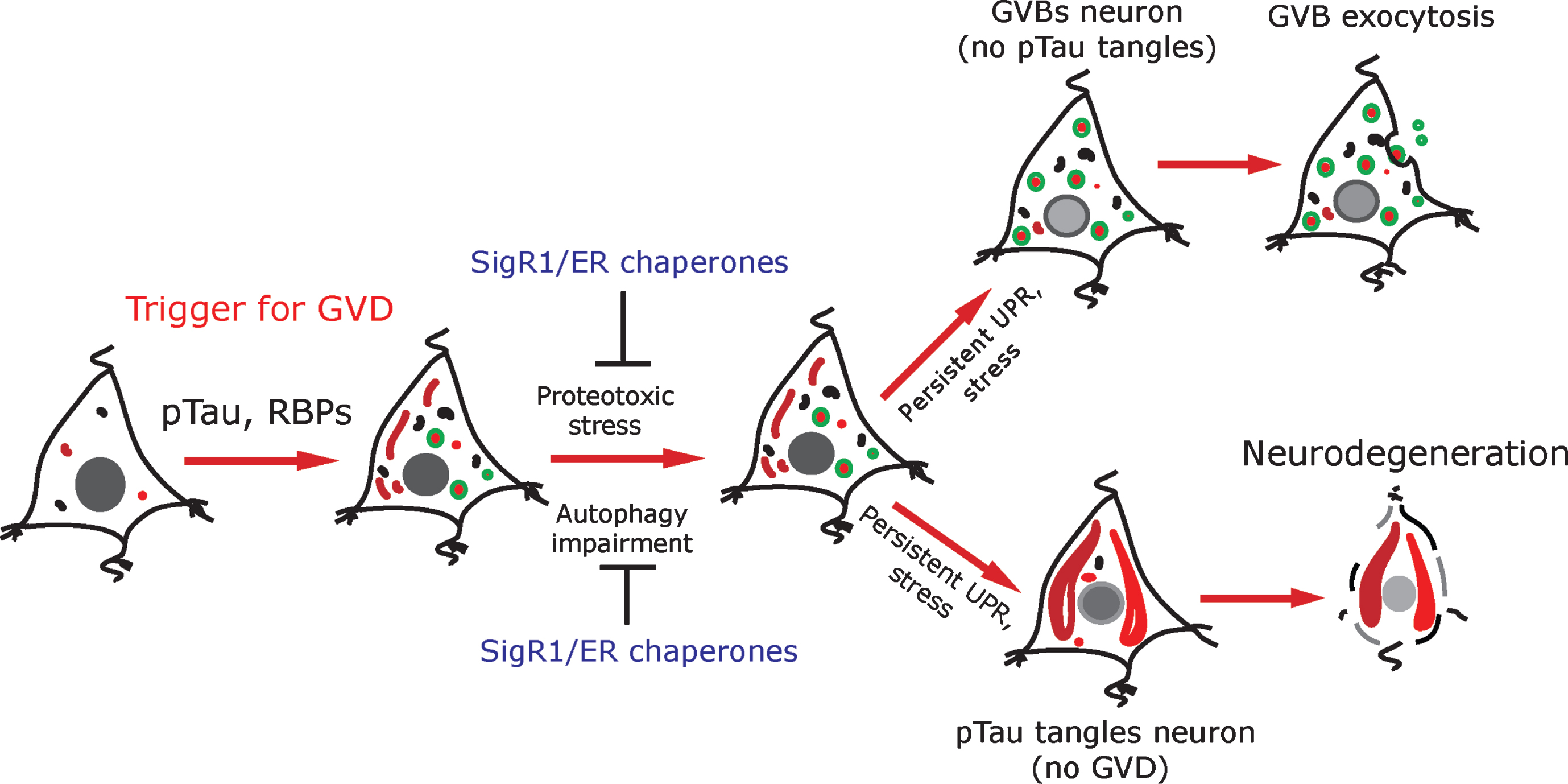
Schematic representation: pTau and RBPs can potentially trigger GVD in AD neurons. GVD affected neurons seldom contain pre-tangle pTau and show an activated UPR. SigR1 and other ER chaperones are elevated to minimize the misfolded protein stress as a protective mechanism. Persistent UPR could potentiate further tau phosphorylation and leads to mature tangle formation and promotes neurodegeneration. On the other hand, neurons with higher load of GVBs do not contain mature tangles. Some GVBs might undergo exocytosis.
DISCUSSION
ER stress and UPR activation are early phenomena in neurodegenerative diseases [24] including AD [4, 64, 65]. Thus, in AD pathogenesis, ER alterations leading to inadequate UPR should cause autophagy impairment and subsequent accumulation of GVBs. ER chaperones like GRP78, SigR1, and SIL1 and ER tethering proteins such as VAPB are determinants of ER functions including protein quality control/UPR and autophagy [66–69]. Mutations in such ER proteins can lead to familial neurodegenerative disorders [70–73]. In addition, they have been shown to be abnormally modified in AD [74–77], Parkinson’s disease [75], Huntington’s disease [78], and ALS [33, 34, 67], disorders that feature distinct ultrastructural ER alterations and defective protein degradation pathways [79]. Consistent with these studies, we found increased immunoreactivity for SigR1, GRP78, and VAPB in AD patient subicular neurons. SigR1 was often co-localized with small granular pTau aggregates (Fig. 1G), suggesting that SigR1 is involved in maintaining protein homeostasis in these neurons. In contrast, this buildup of GRP78, SigR1, and VAPB was lacking in those subicular neurons which showed GVD (Fig. 1C–F), suggesting that those neurons that can no longer maintain protein homeostasis by chaperone activation engage the GVD pathway. These results are in line with the findings that UPR is activated in pre-tangle neurons and that GVBs display immunoreactivity for UPR markers such as PKR-like ER kinase (pPERK), phosphorylated inositol-requiring enzyme 1α (pIRE1), and phosphorylated eukaryotic translation initiation factor 2α (peIF2α) in both human AD and pR5 mice brain, suggesting that the UPR is activated in these neurons even before mature NFTs are formed [55, 57]. Thus, activated UPR in these neurons could be neuroprotective initially; however, persistent UPR might promote further tau hyperphosphorylation [10, 80].
Studies on human AD brains complemented by recent in vitro and in vivo experiments have shown that the extent of tau pathology strongly correlates with GVB load, suggesting that tau buildup can trigger the formation of GVBs [5, 7, 25]. Other recent studies have demonstrated that DPR aggregates contribute to GVD in neurons of ALS/FTD patients with C9orf72 expansion [15]. Consistent with this we recently reported GVD pathology in cases of C9orf72-related FTLD [62]. Furthermore, RBPs such as TDP-43 form aggregates in AD patient neurons and quite often such granular pTDP-43 aggregates co-localize with GVBs [29, 81]. Finally, RBPs associated with SGs progressively accumulate together with tau in neurons of tauopathy mice as well as in human AD and FTLD-tau brain tissue [49]. Thus, it appears reasonable to speculate that not only pTau, but also RBPs could trigger GVD. Consistent with this notion we observed cytoplasmic mis-localization of Matrin 3 and FUS together with pTDP-43 within CK1δ-positive GVBs. Similarly, the SG protein G3BP together with the P-body marker S6 kinase showed increased granular and NFT-like cytoplasmic immunoreactivity co-localizing with pTau and with GVBs in human AD subicular and pR5 mouse hippocampal neurons (Figs. 3 and 5). Thus, our results support the concept that GVBs function as an early, reversible, and protective structure due to their appearance parallel to the buildup of SGs and other RBP accumulations [81]. Further, autophagy impairment could potentiate GVD, supporting a vicious circle, similar to the recently described pathomechanisms related to the buildup and propagation of mutated TDP-43, C9orf72, FUS [51, 82], SOD1 [83], and VAPB [84, 85].
The evidence discussed thus far suggests that GVBs form a neuronal intracellular compartment designed to accumulate heterogeneous materials awaiting intracellular degradation. Another potential mechanism for dealing with such material is the exocytotic release. Consistent with this hypothesis, exocytotic vesicles or exosomes from blood, CSF and cultured neuronal cells have been shown to contain tau, Aβ, and α-synuclein [30, 59, 86, 87]. In line with these findings, we recently showed that CSF obtained from AD patients contains elevated levels of the Golgi protein GolginA4, which we also found to accumulate extensively in GVBs [62]. We now observed that CK1δ-positive GVBs frequently co-localize with Flotillin-1-positive vesicles; moreover, aggregated pTDP-43 and pTau that were present in GVBs were also found to be co-localized with Flotillin-1-positive vesicles. Together, these results are compatible with the notion that GVBs might actually be exocytosed. However, these descriptive observations have to be complemented by functional/experimental work to validate this hypothesis.
Finally, the recently published observation of intracellular Aβ-positive granular structures co-localizing with Flotillin-1-immunoreactive vesicles in AD brain neurons [60] prompted us to ask whether intraneuronal Aβ-(82E1-) immunoreactivity co-localizes with Flotillin-1-positive vesicles in our series of AD cases. We found that such co-localizations were also present in our cases, albeit at a rather low frequency. However, there was virtually no detectable co-localization of Aβ with CK1δ-positive GVBs. Moreover, the small CK1δ-positive granular structures found in hippocampal neurons of APP/PS-1 mice (Supplementary Figures 1 and 2) were clearly different from the mature GVBs detected by us using the same antibody in human AD subicular and pR5 mouse hippocampal neurons, consistent with the absence of tau pathology in APP/PS-1 mice. Apparently the elevated SigR1, GRP78 and VAPB levels in APP/PS-1 mouse neurons (Supplementary Figure 2) are part of the stress response which keeps proteotoxic stress at manageable levels.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-190722.
ACKNOWLEDGMENTS
This work was supported by the Confocal Microscopy Facility, a Core Facility of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University. We thank A. Knischewski, H. Wiederholt, E. Pascual, C. Krude and H. Mader (Institute of Neuropathology, RWTH Aachen University) for technical support and S. Gründer (Institute of Physiology, RWTH Aachen University) and his lab members for confocal microscopy. We also thank lab members of the Department of Neuro (Pathology), Academic Medical Centre, Amsterdam, The Netherlands, for their generous help. We thank the team that established the Dutch ALS Tissue Bank including Prof. M. de Visser and also acknowledge the contribution of the Dutch ALS Foundation to this tissue collection.
This work was supported by German Motor Neuron Disease Network (BMBF-MND-Net; Funds 360644) and by the EU Joint Program Neurodegenerative Diseases (JPND Fly SMALS) to JW and by the Interdisciplinary Centre for Clinical Research (IZKF Aachen, N7-4), the German Myopathy Society (DGM MND-Net) and the Initiative Therapieforschung ALS e.V. to AG and JW.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0722r2).
REFERENCES
[1] | Trojanowski JQ , Schmidt ML , Shin RW , Bramblett GT , Rao D , Lee VM ((1993) ) Altered tau and neurofilament proteins in neuro-degenerative diseases: diagnostic implications for Alzheimer’s disease and Lewy body dementias. Brain Pathol 3: , 45–54. |
[2] | Masters CL , Simms G , Weinman NA , Multhaup G , McDonald BL , Beyreuther K ((1985) ) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82: , 4245–4249. |
[3] | Ball MJ , Lo P ((1977) ) Granulovacuolar degeneration in the ageing brain and in dementia. J Neuropathol Exp Neurol 36: , 474–487. |
[4] | Hoozemans JJ , van Haastert ES , Nijholt DA , Rozemuller AJ , Eikelenboom P , Scheper W ((2009) ) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 174: , 1241–1251. |
[5] | Ghoshal N , Smiley JF , DeMaggio AJ , Hoekstra MF , Cochran EJ , Binder LI , Kuret J ((1999) ) A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer’s disease. Am J Pathol 155: , 1163–1172. |
[6] | Ghoshal N , Garcia-Sierra F , Wuu J , Leurgans S , Bennett DA , Berry RW , Binder LI ((2002) ) Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp Neurol 177: , 475–493. |
[7] | Yamazaki Y , Matsubara T , Takahashi T , Kurashige T , Dohi E , Hiji M , Nagano Y , Yamawaki T , Matsumoto M ((2011) ) Granulovacuolar degenerations appear in relation to hippocampal phosphorylated tau accumulation in various neurodegenerative disorders. PloS One 6: , e26996. |
[8] | Kohler C ((2016) ) Granulovacuolar degeneration: a neurodegenerative change that accompanies tau pathology. Acta Neuropathol 132: , 339–359. |
[9] | Kohler C , Dinekov M , Gotz J ((2014) ) Granulovacuolar degeneration and unfolded protein response in mouse models of tauopathy and Abeta amyloidosis. Neurobiol Dis 71: , 169–179. |
[10] | Nijholt DA , van Haastert ES , Rozemuller AJ , Scheper W , Hoozemans JJ ((2012) ) The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol 226: , 693–702. |
[11] | Simchowicz T ((1911) ) Histologische und histopathologische Arbeiten über die Grosshirnrinde mit besonderer Berücksichtigung der pathologischen Anatomie der Geisteskrankheiten. In Histologische Studien über die senile Demenz, NisslF, AlzheimerA, eds. Gustav Fischer Verlag, Jena, Germany pp. 267–444. |
[12] | Kahn J , Anderton BH , Probst A , Ulrich J , Esiri MM ((1985) ) Immunohistological study of granulovacuolar degeneration using monoclonal antibodies to neurofilaments. J Neurol Neurosurg Psychiatry 48: , 924–926. |
[13] | Hirano A , Dembitzer HM , Kurland LT , Zimmerman HM ((1968) ) The fine structure of some intraganglionic alterations. Neurofibrillary tangles, granulovacuolar bodies and “rod-like” structures as seen in Guam amyotrophic lateral sclerosis and parkinsonism-dementia complex. J Neuropathol Exp Neurol 27: , 167–182. |
[14] | Schwab C , DeMaggio AJ , Ghoshal N , Binder LI , Kuret J , McGeer PL ((2000) ) Casein kinase 1 delta is associated with pathological accumulation of tau in several neurodegenerative diseases. Neurobiol Aging 21: , 503–510. |
[15] | Riku Y , Duyckaerts C , Boluda S , Plu I , Le Ber I , Millecamps S , Salachas F , Brainbank NeuroCEB Neuropathology Network, Yoshida M , Ando T , Katsuno M , Sobue G , Seilhean D ((2019) ) Increased prevalence of granulovacuolar degeneration in C9orf72 mutation. Acta Neuropathol 138: , 783–793. |
[16] | Xu M , Shibayama H , Kobayashi H , Yamada K , Ishihara R , Zhao P , Takeuchi T , Yoshida K , Inagaki T , Nokura K ((1992) ) Granulovacuolar degeneration in the hippocampal cortex of aging and demented patients–a quantitative study. Acta Neuropathol 85: , 1–9. |
[17] | Funk KE , Mrak RE , Kuret J ((2011) ) Granulovacuolar degeneration (GVD) bodies of Alzheimer’s disease (AD) resemble late-stage autophagic organelles. Neuropathol Appl Neurobiol 37: , 295–306. |
[18] | Okamoto K , Hirai S , Iizuka T , Yanagisawa T , Watanabe M ((1991) ) Reexamination of granulovacuolar degeneration. Acta Neuropathol 82: , 340–345. |
[19] | Boland B , Yu WH , Corti O , Mollereau B , Henriques A , Bezard E , Pastores GM , Rubinsztein DC , Nixon RA , Duchen MR , Mallucci GR , Kroemer G , Levine B , Eskelinen EL , Mochel F , Spedding M , Louis C , Martin OR , Millan MJ ((2018) ) Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov 17: , 660–688. |
[20] | Williams A , Jahreiss L , Sarkar S , Saiki S , Menzies FM , Ravikumar B , Rubinsztein DC ((2006) ) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76: , 89–101. |
[21] | Nixon RA , Yang DS ((2011) ) Autophagy failure in Alzheimer’s disease–locating the primary defect. Neurobiol Dis 43: , 38–45. |
[22] | Vollrath JT , Sechi A , Dreser A , Katona I , Wiemuth D , Vervoorts J , Dohmen M , Chandrasekar A , Prause J , Brauers E , Jesse CM , Weis J , Goswami A ((2014) ) Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances. Cell Death Dis 12: , 243. |
[23] | Li J , Ni M , Lee B , Barron E , Hinton DR , Lee AS ((2008) ) The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ 15: , 1460–1471. |
[24] | Hetz C , Saxena S ((2017) ) ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13: , 477–491. |
[25] | Wiersma VI , van Ziel AM , Vazquez-Sanchez S , Nolle A , Berenjeno-Correa E , Bonaterra-Pastra A , Clavaguera F , Tolnay M , Musters RJP , van Weering JRT , Verhage M , Hoozemans JJM , Scheper W ((2019) ) Granulovacuolar degeneration bodies are neuron-selective lysosomal structures induced by intracellular tau pathology. Acta Neuropathol 138: , 943–970. |
[26] | Cristofani R , Crippa V , Vezzoli G , Rusmini P , Galbiati M , Cicardi ME , Meroni M , Ferrari V , Tedesco B , Piccolella M , Messi E , Carra S , Poletti A ((2018) ) The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72-related neurodegenerative diseases. Cell Stress Chaperones 23: , 1–12. |
[27] | Ling SC , Polymenidou M , Cleveland DW ((2013) ) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79: , 416–438. |
[28] | Wolozin B ((2012) ) Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener 7: , 56. |
[29] | Thal DR , Del Tredici K , Ludolph AC , Hoozemans JJ , Rozemuller AJ , Braak H , Knippschild U ((2011) ) Stages of granulovacuolar degeneration: their relation to Alzheimer’s disease and chronic stress response. Acta Neuropathol 122: , 577–589. |
[30] | Howitt J , Hill AF ((2016) ) Exosomes in the pathology of neurodegenerative diseases. J Biol Chem 291: , 26589–26597. |
[31] | Nachreiner T , Esser M , Tenten V , Troost D , Weis J , Kruttgen A ((2010) ) Novel splice variants of the amyotrophic lateral sclerosis-associated gene VAPB expressed in human tissues. Biochem Biophys Res Commun 394: , 703–708. |
[32] | Prause J , Goswami A , Katona I , Roos A , Schnizler M , Bushuven E , Dreier A , Buchkremer S , Johann S , Beyer C , Deschauer M , Troost D , Weis J ((2013) ) Altered localization, abnormal modification and loss of function of Sigma receptor-1 in amyotrophic lateral sclerosis. Hum Mol Genet 22: , 1581–1600. |
[33] | Jesse CM , Bushuven E , Tripathi P , Chandrasekar A , Simon CM , Drepper C , Yamoah A , Dreser A , Katona I , Johann S , Beyer C , Wagner S , Grond M , Nikolin S , Anink J , Troost D , Sendtner M , Goswami A , Weis J ((2017) ) ALS-associated endoplasmic reticulum proteins in denervated skeletal muscle: implications for motor neuron disease pathology. Brain Pathol 27: , 781–794. |
[34] | Goswami A , Jesse CM , Chandrasekar A , Bushuven E , Vollrath JT , Dreser A , Katona I , Beyer C , Johann S , Feller AC , Grond M , Wagner S , Nikolin S , Troost D , Weis J ((2015) ) Accumulation of STIM1 is associated with the degenerative muscle fibre phenotype in ALS and other neurogenic atrophies. Neuropathol Appl Neurobiol 41: , 304–318. |
[35] | Bell JE , Alafuzoff I , Al-Sarraj S , Arzberger T , Bogdanovic N , Budka H , Dexter DT , Falkai P , Ferrer I , Gelpi E , Gentleman SM , Giaccone G , Huitinga I , Ironside JW , Klioueva N , Kovacs GG , Meyronet D , Palkovits M , Parchi P , Patsouris E , Reynolds R , Riederer P , Roggendorf W , Seilhean D , Schmitt A , Schmitz P , Streichenberger N , Schwalber A , Kretzschmar H ((2008) ) Management of a twenty-first century brain bank: experience in the BrainNet Europe consortium. Acta Neuropathol 115: , 497–507. |
[36] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr. , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[37] | Albert MS , DeKosky ST , Dickson D , Dubois B , Feldman HH , Fox NC , Gamst A , Holtzman DM , Jagust WJ , Petersen RC , Snyder PJ , Carrillo MC , Thies B , Phelps CH ((2011) ) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 270–279. |
[38] | Jack CR Jr. , Bennett DA , Blennow K , Carrillo MC , Dunn B , Haeberlein SB , Holtzman DM , Jagust W , Jessen F , Karlawish J , Liu E , Molinuevo JL , Montine T , Phelps C , Rankin KP , Rowe CC , Scheltens P , Siemers E , Snyder HM , Sperling R , Contributors ((2018) ) NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14: , 535–562. |
[39] | Kovacs GG ((2019) ) Molecular pathology of neurodegenerative diseases: principles and practice. J Clin Pathol 72: , 725–735. |
[40] | Kovacs G ((2014) ) Neuropathology of Neurodegenerative Diseases: A Practical Guide, Cambridge University Press, Cambridge. |
[41] | McAleese KE , Walker L , Erskine D , Thomas AJ , McKeith IG , Attems J ((2017) ) TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol 27: , 472–479. |
[42] | Dreser A , Vollrath JT , Sechi A , Johann S , Roos A , Yamoah A , Katona I , Bohlega S , Wiemuth D , Tian Y , Schmidt A , Vervoorts J , Dohmen M , Beyer C , Anink J , Aronica E , Troost D , Weis J , Goswami A ((2017) ) The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ 24: , 1655–1671. |
[43] | Yang Y , Turner RS , Gaut JR ((1998) ) The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem 273: , 25552–25555. |
[44] | Chen HJ , Anagnostou G , Chai A , Withers J , Morris A , Adhikaree J , Pennetta G , de Belleroche JS ((2010) ) Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J Biol Chem 285: , 40266–40281. |
[45] | Llorens-Martin M , Blazquez-Llorca L , Benavides-Piccione R , Rabano A , Hernandez F , Avila J , DeFelipe J ((2014) ) Selective alterations of neurons and circuits related to early memory loss in Alzheimer’s disease. Front Neuroanat 8: , 38. |
[46] | Gotz J , Chen F , Barmettler R , Nitsch RM ((2001) ) Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem 276: , 529–534. |
[47] | Gotz J , Nitsch RM ((2001) ) Compartmentalized tau hyperphosphorylation and increased levels of kinases in transgenic mice. Neuroreport 12: , 2007–2016. |
[48] | Nixon RA , Yang DS ((2011) ) Autophagy failure in Alzheimer’s disease – locating the primary defect. Neurobiol Dis 43: , 38–45. |
[49] | Maziuk BF , Apicco DJ , Cruz AL , Jiang L , Ash PEA , da Rocha EL , Zhang C , Yu WH , Leszyk J , Abisambra JF , Li H , Wolozin B ((2018) ) RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol Commun 6: , 71. |
[50] | Kadokura A , Yamazaki T , Kakuda S , Makioka K , Lemere CA , Fujita Y , Takatama M , Okamoto K ((2009) ) Phosphorylation-dependent TDP-43 antibody detects intraneuronal dot-like structures showing morphological characters of granulovacuolar degeneration. Neurosci Lett 463: , 87–92. |
[51] | Naumann M , Pal A , Goswami A , Lojewski X , Japtok J , Vehlow A , Naujock M , Gunther R , Jin M , Stanslowsky N , Reinhardt P , Sterneckert J , Frickenhaus M , Pan-Montojo F , Storkebaum E , Poser I , Freischmidt A , Weishaupt JH , Holzmann K , Troost D , Ludolph AC , Boeckers TM , Liebau S , Petri S , Cordes N , Hyman AA , Wegner F , Grill SW , Weis J , Storch A , Hermann A ((2018) ) Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun 9: , 335. |
[52] | Marrone L , Drexler HCA , Wang J , Tripathi P , Distler T , Heisterkamp P , Anderson EN , Kour S , Moraiti A , Maharana S , Bhatnagar R , Belgard TG , Tripathy V , Kalmbach N , Hosseinzadeh Z , Crippa V , Abo-Rady M , Wegner F , Poletti A , Troost D , Aronica E , Busskamp V , Weis J , Pandey UB , Hyman AA , Alberti S , Goswami A , Sterneckert J ((2019) ) FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol 138: , 67–84. |
[53] | Salton M , Elkon R , Borodina T , Davydov A , Yaspo ML , Halperin E , Shiloh Y ((2011) ) Matrin 3 binds and stabilizes mRNA. PloS One 6: , e23882. |
[54] | Muller TJ , Kraya T , Stoltenburg-Didinger G , Hanisch F , Kornhuber M , Stoevesandt D , Senderek J , Weis J , Baum P , Deschauer M , Zierz S ((2014) ) Phenotype of matrin-3-related distal myopathy in 16 German patients. Ann Neurol 76: , 669–680. |
[55] | Senderek J , Garvey SM , Krieger M , Guergueltcheva V , Urtizberea A , Roos A , Elbracht M , Stendel C , Tournev I , Mihailova V , Feit H , Tramonte J , Hedera P , Crooks K , Bergmann C , Rudnik-Schoneborn S , Zerres K , Lochmuller H , Seboun E , Weis J , Beckmann JS , Hauser MA , Jackson CE ((2009) ) Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet 84: , 511–518. |
[56] | Johnson JO , Pioro EP , Boehringer A , Chia R , Feit H , Renton AE , Pliner HA , Abramzon Y , Marangi G , Winborn BJ , Gibbs JR , Nalls MA , Morgan S , Shoai M , Hardy J , Pittman A , Orrell RW , Malaspina A , Sidle KC , Fratta P , Harms MB , Baloh RH , Pestronk A , Weihl CC , Rogaeva E , Zinman L , Drory VE , Borghero G , Mora G , Calvo A , Rothstein JD , Italsgen , Drepper C , Sendtner M , Singleton AB , Taylor JP , Cookson MR , Restagno G , Sabatelli M , Bowser R , Chio A , Traynor BJ ((2014) ) Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17: , 664–666. |
[57] | Vanderweyde T , Yu H , Varnum M , Liu-Yesucevitz L , Citro A , Ikezu T , Duff K , Wolozin B ((2012) ) Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci 32: , 8270–8283. |
[58] | Buchan JR , Kolaitis RM , Taylor JP , Parker R ((2013) ) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153: , 1461–1474. |
[59] | Rajendran L , Honsho M , Zahn TR , Keller P , Geiger KD , Verkade P , Simons K ((2006) ) Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A 103: , 11172–11177. |
[60] | Sardar Sinha M , Ansell-Schultz A , Civitelli L , Hildesjo C , Larsson M , Lannfelt L , Ingelsson M , Hallbeck M ((2018) ) Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol 136: , 41–56. |
[61] | Babst M , Katzmann DJ , Estepa-Sabal EJ , Meerloo T , Emr SD ((2002) ) Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell 3: , 271–282. |
[62] | Kork F , Jankowski J , Goswami A , Weis J , Brook G , Yamoah A , Anink J , Aronica E , Fritz S , Huck C , Schipke C , Peters O , Tepel M , Noels H , Jankowski V ((2018) ) Golgin A4 in CSF and granulovacuolar degenerations of patients with Alzheimer disease. Neurology 91: , e1799–e1808. |
[63] | Yanez-Mo M , Siljander PR , Andreu Z , Zavec AB , Borras FE , Buzas EI , Buzas K , Casal E , Cappello F , Carvalho J , Colas E , Cordeiro-da Silva A , Fais S , Falcon-Perez JM , Ghobrial IM , Giebel B , Gimona M , Graner M , Gursel I , Gursel M , Heegaard NH , Hendrix A , Kierulf P , Kokubun K , Kosanovic M , Kralj-Iglic V , Kramer-Albers EM , Laitinen S , Lasser C , Lener T , Ligeti E , Line A , Lipps G , Llorente A , Lotvall J , Mancek-Keber M , Marcilla A , Mittelbrunn M , Nazarenko I , Nolte-’t Hoen EN , Nyman TA , O’Driscoll L , Olivan M , Oliveira C , Pallinger E , Del Portillo HA , Reventos J , Rigau M , Rohde E , Sammar M , Sanchez-Madrid F , Santarem N , Schallmoser K , Ostenfeld MS , Stoorvogel W , Stukelj R , Van der Grein SG , Vasconcelos MH , Wauben MH , De Wever O ((2015) ) Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 4: , 27066. |
[64] | Ho YS , Yang X , Lau JC , Hung CH , Wuwongse S , Zhang Q , Wang J , Baum L , So KF , Chang RC ((2012) ) Endoplasmic reticulum stress induces tau pathology and forms a vicious cycle: implication in Alzheimer’s disease pathogenesis. J Alzheimers Dis 28: , 839–854. |
[65] | Lindholm D , Wootz H , Korhonen L ((2006) ) ER stress and neurodegenerative diseases. Cell Death Differ 13: , 385–392. |
[66] | Hendershot LM ((2004) ) The ER function BiP is a master regulator of ER function. Mt Sinai J Med 71: , 289–297. |
[67] | Filezac de L’Etang A , Maharjan N , Cordeiro Brana M , Ruegsegger C , Rehmann R , Goswami A , Roos A , Troost D , Schneider BL , Weis J , Saxena S ((2015) ) Marinesco-Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat Neurosci 18: , 227–238. |
[68] | Hayashi T , Su TP ((2007) ) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131: , 596–610. |
[69] | Moustaqim-Barrette A , Lin YQ , Pradhan S , Neely GG , Bellen HJ , Tsuda H ((2014) ) The amyotrophic lateral sclerosis 8 protein, VAP, is required for ER protein quality control. Hum Mol Genet 23: , 1975–1989. |
[70] | Funke AD , Esser M , Kruttgen A , Weis J , Mitne-Neto M , Lazar M , Nishimura AL , Sperfeld AD , Trillenberg P , Senderek J , Krasnianski M , Zatz M , Zierz S , Deschauer M ((2010) ) The p.P56S mutation in the VAPB gene is not due to a single founder: the first European case. Clin Genet 77: , 302–303. |
[71] | Senderek J , Krieger M , Stendel C , Bergmann C , Moser M , Breitbach-Faller N , Rudnik-Schoneborn S , Blaschek A , Wolf NI , Harting I , North K , Smith J , Muntoni F , Brockington M , Quijano-Roy S , Renault F , Herrmann R , Hendershot LM , Schroder JM , Lochmuller H , Topaloglu H , Voit T , Weis J , Ebinger F , Zerres K ((2005) ) Mutations in SIL1 cause Marinesco-Sjogren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet 37: , 1312–1314. |
[72] | Nishimura AL , Al-Chalabi A , Zatz M ((2005) ) A common founder for amyotrophic lateral sclerosis type 8 (ALS8) in the Brazilian population. Hum Genet 118: , 499–500. |
[73] | Krieger M , Roos A , Stendel C , Claeys KG , Sonmez FM , Baudis M , Bauer P , Bornemann A , de Goede C , Dufke A , Finkel RS , Goebel HH , Haussler M , Kingston H , Kirschner J , Medne L , Muschke P , Rivier F , Rudnik-Schoneborn S , Spengler S , Inzana F , Stanzial F , Benedicenti F , Synofzik M , Lia Taratuto A , Pirra L , Tay SK , Topaloglu H , Uyanik G , Wand D , Williams D , Zerres K , Weis J , Senderek J ((2013) ) SIL1 mutations and clinical spectrum in patients with Marinesco-Sjogren syndrome. Brain 136: , 3634–3644. |
[74] | Uchida N , Ujike H , Tanaka Y , Sakai A , Yamamoto M , Fujisawa Y , Kanzaki A , Kuroda S ((2005) ) A variant of the sigma receptor type-1 gene is a protective factor for Alzheimer disease. Am J Geriatr Psychiatry 13: , 1062–1066. |
[75] | Mishina M , Ohyama M , Ishii K , Kitamura S , Kimura Y , Oda K , Kawamura K , Sasaki T , Kobayashi S , Katayama Y , Ishiwata K ((2008) ) Low density of sigma1 receptors in early Alzheimer’s disease. Ann Nucl Med 22: , 151–156. |
[76] | Feher A , Juhasz A , Laszlo A , Kalman J Jr. , Pakaski M , Kalman J , Janka Z ((2012) ) Association between a variant of the sigma-1 receptor gene and Alzheimer’s disease. Neurosci Lett 517: , 136–139. |
[77] | Huang Y , Zheng L , Halliday G , Dobson-Stone C , Wang Y , Tang HD , Cao L , Deng YL , Wang G , Zhang YM , Wang JH , Hallupp M , Kwok J , Chen SD ((2011) ) Genetic polymorphisms in sigma-1 receptor and apolipoprotein E interact to influence the severity of Alzheimer’s disease. Curr Alzheimer Res 8: , 765–770. |
[78] | Miki Y , Mori F , Kon T , Tanji K , Toyoshima Y , Yoshida M , Sasaki H , Kakita A , Takahashi H , Wakabayashi K ((2014) ) Accumulation of the sigma-1 receptor is common to neuronal nuclear inclusions in various neurodegenerative diseases. Neuropathology 34: , 148–158. |
[79] | Kuijpers M , van Dis V , Haasdijk ED , Harterink M , Vocking K , Post JA , Scheper W , Hoogenraad CC , Jaarsma D ((2013) ) Amyotrophic lateral sclerosis (ALS)-associated VAPB-P56S inclusions represent an ER quality control compartment. Acta Neuropathol Commun 1: , 24. |
[80] | Hoozemans JJM , van Haastert ES , Nijholt DAT , Rozemuller AJM , Eikelenboom P , Scheper W ((2009) ) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 174: , 1241–1251. |
[81] | Castellani RJ , Gupta Y , Sheng B , Siedlak SL , Harris PL , Coller JM , Perry G , Lee HG , Tabaton M , Smith MA , Wang X , Zhu X ((2011) ) A novel origin for granulovacuolar degeneration in aging and Alzheimer’s disease: parallels to stress granules. Lab Invest 91: , 1777–1786. |
[82] | Halliday G , Bigio EH , Cairns NJ , Neumann M , Mackenzie IR , Mann DM ((2012) ) Mechanisms of disease in frontotemporal lobar degeneration: gain of function versus loss of function effects. Acta Neuropathol 124: , 373–382. |
[83] | Saccon RA , Bunton-Stasyshyn RK , Fisher EM , Fratta P ((2013) ) Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 136: , 2342–2358. |
[84] | Tran D , Chalhoub A , Schooley A , Zhang W , Ngsee JK ((2012) ) A mutation in VAPB that causes amyotrophic lateral sclerosis also causes a nuclear envelope defect. J Cell Sci 125: , 2831–2836. |
[85] | Suzuki H , Kanekura K , Levine TP , Kohno K , Olkkonen VM , Aiso S , Matsuoka M ((2009) ) ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J Neurochem 108: , 973–985. |
[86] | Asai H , Ikezu S , Tsunoda S , Medalla M , Luebke J , Haydar T , Wolozin B , Butovsky O , Kugler S , Ikezu T ((2015) ) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18: , 1584–1593. |
[87] | Danzer KM , Kranich LR , Ruf WP , Cagsal-Getkin O , Winslow AR , Zhu L , Vanderburg CR , McLean PJ ((2012) ) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 7: , 42. |
[88] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[89] | Hyman BT , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Carrillo MC , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Thies B , Trojanowski JQ , Vinters HV , Montine TJ ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8: , 1–13. |
[90] | Thal DR , Rub U , Orantes M , Braak H ((2002) ) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[91] | Montine TJ , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Trojanowski JQ , Vinters HV , Hyman BT , National Institute on Aging, Alzheimer’s Association ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123: , 1–11. |
[92] | KovacsGG, GelpiE ((2012) ) Clinical neuropathology practice news 3-2012: the “ABC” in AD-revised and updated guideline for the neuropathologic assessment of Alzheimer’s disease. Clin Neuropathol 31: , 116–118. |


