
Increased Insoluble Amyloid-β Induces Negligible Cognitive Deficits in Old AppNL/NL Knock-In Mice
Abstract
Commonly used Alzheimer’s disease mouse models are based on the ectopic overexpression of the human amyloid precursor protein (APP) gene, together with a mutant presenilin gene. Surprisingly, humanized APP knock-in mouse models carrying a single APP Swedish mutation (AppNL), failed to develop amyloid plaque aggregation or cognitive deficits. Here we characterized the effect of this mutation in more advanced ages. We show that 24-month-old AppNL/NL mice, despite presenting an age dependent increase in insoluble amyloid-β oligomers in the prefrontal cortex, they do not develop amyloid plaque deposition, reactive gliosis, or cognitive deficits.
INTRODUCTION
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder characterized by the presence of intracellular neurofibrillary tangles and extracellular amyloid plaques [1]. The latter are mainly composed by aggregation of amyloid-β (Aβ) peptides, produced by a proteolytic cleavage of the amyloid-β protein precursor (AβPP) by β- and γ-secretases [2]. While the etiology of the vast majority of AD cases remains still unknown, there are rare forms of the disease (<1%) that are caused by inherited mutations in AβPP or presenilins (PS), the catalytic subunit of the γ-secretase. These mutations affect AβPP processing by γ-secretase, favoring the release of longer and less soluble Aβ peptides [3, 4].
The discovery of familial AD (FAD) mutations allowed the development of different mouse models that could recapitulate most of the symptoms of AD including the presence of amyloid plaques, increased tau phosphorylation, reactive gliosis and cognitive impairments [5]. Nevertheless, most of these models overexpress AβPP and PS transgenes, which has been associated with side effects and artificial phenotypes [6].
To overcome this problem, Saito et al. developed three new AβPP knock-in mouse models for AD, which express a humanized Aβ sequence carrying either one (Swedish AppNL), two (Swedish and Iberian AppNL - F), or three (Swedish, Iberian, and Artic AppNL - G - F) FAD mutations [7]. Interestingly, only the models with the double (AppNL - F) or the triple (AppNL - G - F) AβPP mutations show signs of AD including amyloid plaques deposits, while mice carrying the single Swedish mutation (AppNL/NL) failed to develop plaques, reactive gliosis, or cognitive deficits [7, 8]. The Swedish mutation is located in the β-cleavage site and it triggers a 2.5 increase in AβPP and Aβ production [7]. In patients, heterozygous Swedish mutation leads to early-onset AD with a mean age of onset of 55 years [9]. The AppNL/NL model has only been characterized at 18 month of age [7, 8], a time when the animals have not undergone the full gamut of aging-related alterations [10]. Therefore, it would be important to determine whether the absence of behavioral defects in these mice is due to an insufficient aging phenotype.
For these reasons, in this study we characterized the effect of the single Swedish mutation in homozygous 24-month-old AppNL/NL male mice, compared to age- and sex-matched C57Bl/6J mice.
MATERIALS AND METHODS
Aβ extraction and ELISA detection
For soluble Aβ extraction, frozen pre-frontal cortices were homogenized in tissue protein extraction reagent (Pierce). Samples were spun down 5 min at 5,000× g and supernatants centrifuged 1 h at 100,000× g. Supernatants were recovered as the soluble fraction. To extract the deposited Aβ, pellets were solubilized with GuHCl (6 M GuHCl/50 mM Tris-HCl), sonicated and centrifuged 20 min at 70,000 rpm. For Aβ quantification, ELISA Meso Scale Discovery (MSD) 96-well plates coated with the human antibody for Aβ40 or Aβ42 (Janssen Pharmaceutica) were used.
Immunohistochemistry
Mice were intracardially perfused with 4% paraformaldehyde (PFA) solution and brains were cut into 50μm-thick slices using a Leica VT1000S vibratome. Before immunohistochemistry, antigen retrieval was performed in boiling 10 mM tri-Sodium Citrate buffer pH 6.0. After blocking, slices were incubated with antibodies against Iba 1 (Wako # 019-19741) and 6E10 (BioLegend #803003) and the corresponding Alexa secondary antibodies. After the washes, slices were incubated with Thioflavin-S and dehydrated using increasing concentrations of ethanol.
Behavioral tests
AppNL/NL and C57/Bl6 male mice were housed in standard mouse cages (3–5 mice per cage) with wood-shaving bedding. Food and water were available ad libitum in temperature- and humidity-controlled rooms with a 12-h light-dark cycle. All experiments were approved by the institutional ethical committee of the KU Leuven for use on experimental animals.
Open field
Locomotor activity was measured in a 50 cm×50 cm arena with bright illumination. Before the test, mice were habituated to the dark for 30 min. Movement was recorded and analyzed using ANY-maze Video Tracking System software (Stoelting Co., IL, USA).
Elevated plus maze
The elevated plus maze had two open arms and two enclosed arms opposite to each other and elevated 30 cm from the surface. Mice were located at the center of the maze and for 10 min the activity was monitored using five infrared beams.
Contextual and cued fear conditioning
Mice were first habituated for 5 min in the StartFear cage with a specific context. 24 h later animals were placed in the same cage and exposed two times to a 30 s tone (4 kHz, 80 dB) which co-terminated with an electric shock (2 s, 0.3 mA). The next day, mice were placed again in the same context and freezing was quantified for contextual fear memory. To assess cued memory, the context of the cage was changed (in the visual, tactile, and olfactory dimension) and after 3 min of habituation, animals were exposed to the tone (cue). The percentage of freezing was calculated as reliable readout for fear in rodents.
Social preference/social novelty
The test consisted in three chambers. The left and right chamber contained cylindrical wire cups, and were connected to the central chamber via guillotine doors. In the first trial, mice were habituated for 5 min to the central chamber. Immediately after, a stranger mouse (stranger 1) was placed in one of the wire cups, doors were opened and the test mice was left for free exploration during 10 min (Social preference). Finally, in the last trial (social novelty), a novel mouse (stranger 2) was placed in the second wire cup and test mouse was allowed to explore either stranger 1 or stranger 2 for 10 min. Mice movement was recorded and analyzed with ANY-maze Video Tracking System software (Stoelting Co., IL, USA).
Morris water maze
A circular pool was filled with white-painted water and a scape platform was placed 0.5 cm below the water surface. Mice were trained for 10 days (4 trials/day) to find the hidden platform starting randomly from 4 different positions. Probe trials were performed on day 6 and day 11 by removing the platform and the search pattern of mice was recorded for 100 seconds using EthoVision ® video tracking (Noldus, Wageningen, the Netherlands).
RESULTS
AβPP knock-in mice carrying the double (AppNL - F) and the triple (AppNL - G - F) mutations showed an age-dependent increase of soluble and guanidine hydrochloride (GuHCl)-extracted Aβ42 levels [7]. To analyze whether AppNL/NL aged mice also show accumulation of Aβ peptides, we homogenized prefrontal cortex from AppNL/NL mice at 6, 18, and 24 months of age (n = 4, 6, and 8, respectively) and we measured Aβ levels using mesoscale MSD ELISA plate coated with human-specific antibodies against Aβ40 and Aβ42 peptides. Intriguingly, AppNL/NL mice showed decreased levels of soluble Aβ40 (p = 0.042, 1-way ANOVA Tukey’s post-hoc test, Fig. 1A) and Aβ42 (p = 0.044, 1-way ANOVA Tukey’s post-hoc test, Fig. 1B) between 18 and 24 months of age. However, when we extracted the deposited fractions using GuHCl, we found a significant increase of Aβ40 (6 versus 18 MO: p = 0.0175; 6 versus 24 MO: p = 0.0007, 1-way ANOVA Tukey’s post-hoc test, Fig. 1C) and Aβ42 (6 versus 18 MO: p = 0.008; 6 versus 24 MO: p = 0.0215, 1-way ANOVA Tukey’s post-hoc test, Fig. 1D) in the aged groups (18 and 24 months of age) compared to the young group of 6 months of age.
Fig.1
Increased insoluble amyloid-β oligomers in aged AppNL/NL mice. A, B) ELISA detection of soluble Aβ40 (A) and Aβ42 (B) peptides in the prefrontal cortex from 6 (n = 4), 18 (n = 6), and 24 (n = 8) month-old AppNL/NL mice. C, D) ELISA detection of aggregated GuHCl-extracted fractions of Aβ40 (C) and Aβ42 (D) peptides in the prefrontal cortex from 6 (n = 5), 18 (n = 6), and 24 (n = 8) month-old AppNL/NL mice. Statistical significance (*p < 0.05, **p < 0.005, ***p < 0.001) was evaluated using 1-way ANOVA Turkey’s post-hoc test. E) Representative images from hippocampal and cortical areas from 8-month-old APPswePS1 mice, or 24-month-old WT or AppNL/NL mice stained with Thioflavin (green) and 6E10 (blue) or Iba1 (red)antibody. n = 4 mice per group. Scale bar represents 100μm.
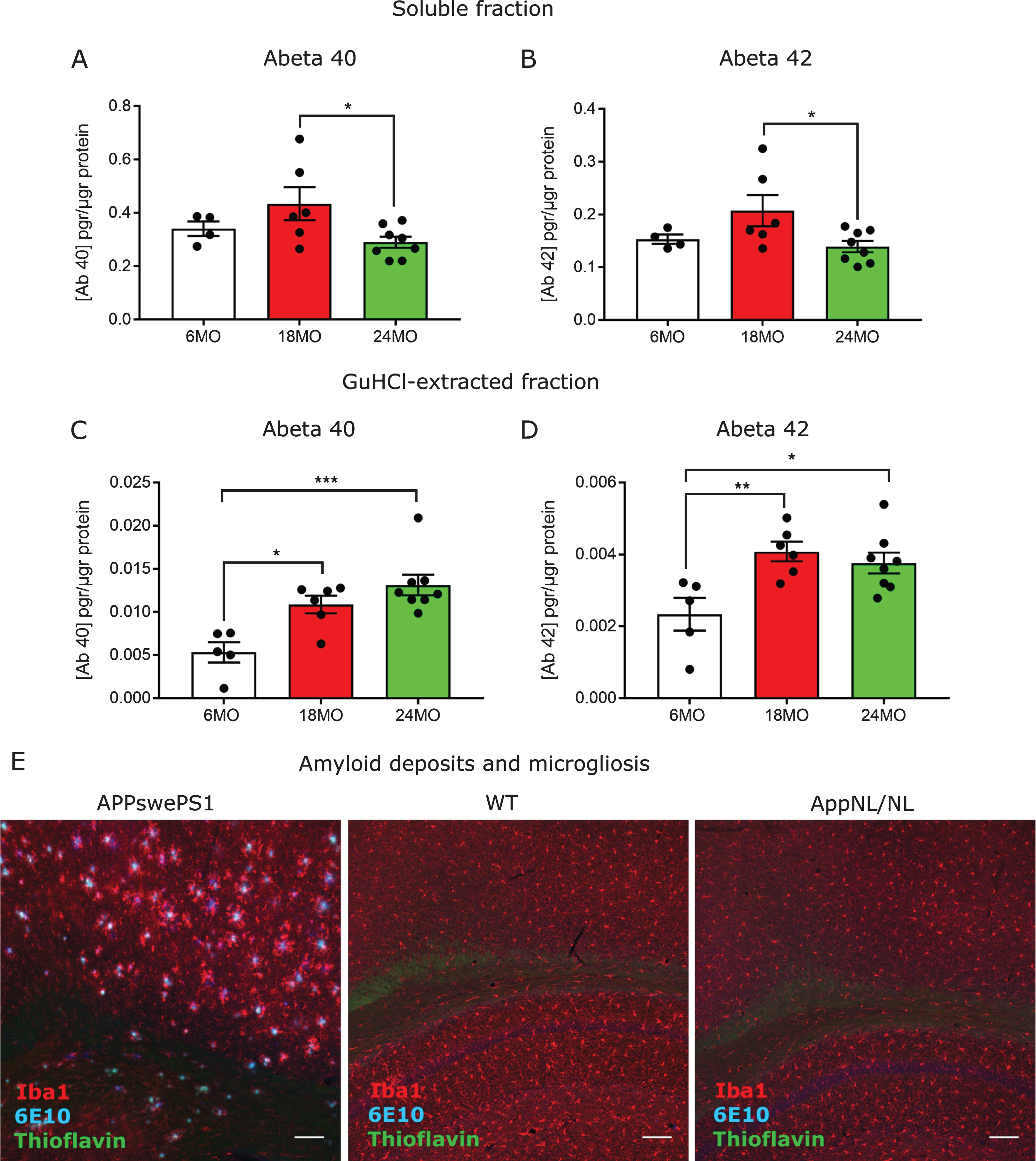
To further elucidate whether the increased levels of non-soluble Aβ peptides resulted in amyloid plaque deposits in the brain, we stained AppNL/NL coronal brain sections with Thioflavin and with the 6E10 antibody against human Aβ. As a positive control, we included sections from 8-month-old APP/PS1 mice, overexpressing AβPP and the PS1 genes with FAD mutations [11], where we observed clear Aβ deposits in the cortex and hippocampus (Fig. 1E). Nonetheless, when we analyzed sections from 24-month-old AppNL/NL mice and age-matched wild-type (WT) controls, no amyloid deposits could be detected in the cortex, hippocampus (Fig. 1E), or prefrontal cortex (Supplementary Figure 1) (n = 4). In addition, we also studied potential signs of gliosis by staining the slices with the microglia-specific marker, Iba1. Accordingly, we found no changes in number and reactivity of microglial cells in old AppNL/NL mice compared to WT (n = 4) (Fig. 1E).
The absence of amyloid plaques in mice with high levels of insoluble forms of Aβ led us to speculate that cognitive defects might have occurred, since there are numerous studies suggesting that an excess of oligomers affects synaptic function [12–14]. Thus, we studied the cognitive functions of 24-month-old AppNL/NL mice compared to WT from the same B57Bl/6J background (n = 11–12 animals per group). Experiments were carried out blinded, and randomized between different genetic groups.
We started assessing locomotor activity using the open field task. During this test, mice were placed in a bright arena for 10 min of free exploration while their trajectories were tracked. Previous models for AD displayed an increased locomotor activity [15]. However, here we show no significant differences in average speed and total distance travelled between AppNL/NL and WT mice (Fig. 2A, B).
Fig.2
No major locomotor and anxiety-related alterations in 24-month-old AppNL/NL mice. A) Average mean speed and distance travelled (B) during the 10 min free exploration in the open field. C) Time spent in the center versus the periphery areas in the open field task n = 12 WT; 11 AppNL/NL mice. D) Explanatory diagram showing the 2 open and 2 closed arms from the elevated plus maze. E) Elevated plus maze shows an increased number of total entries to the open and closed arms in old AppNL/NL (n = 8) mice compared to WT (n = 9). F) No differences in the percentage of entries to the open arms, normalized to the total number of entries between the two genetic groups. Histograms show the mean (±S.E.M). Statistical significance (*p < 0.05, **p < 0.005) was evaluated with an unpaired T-test.
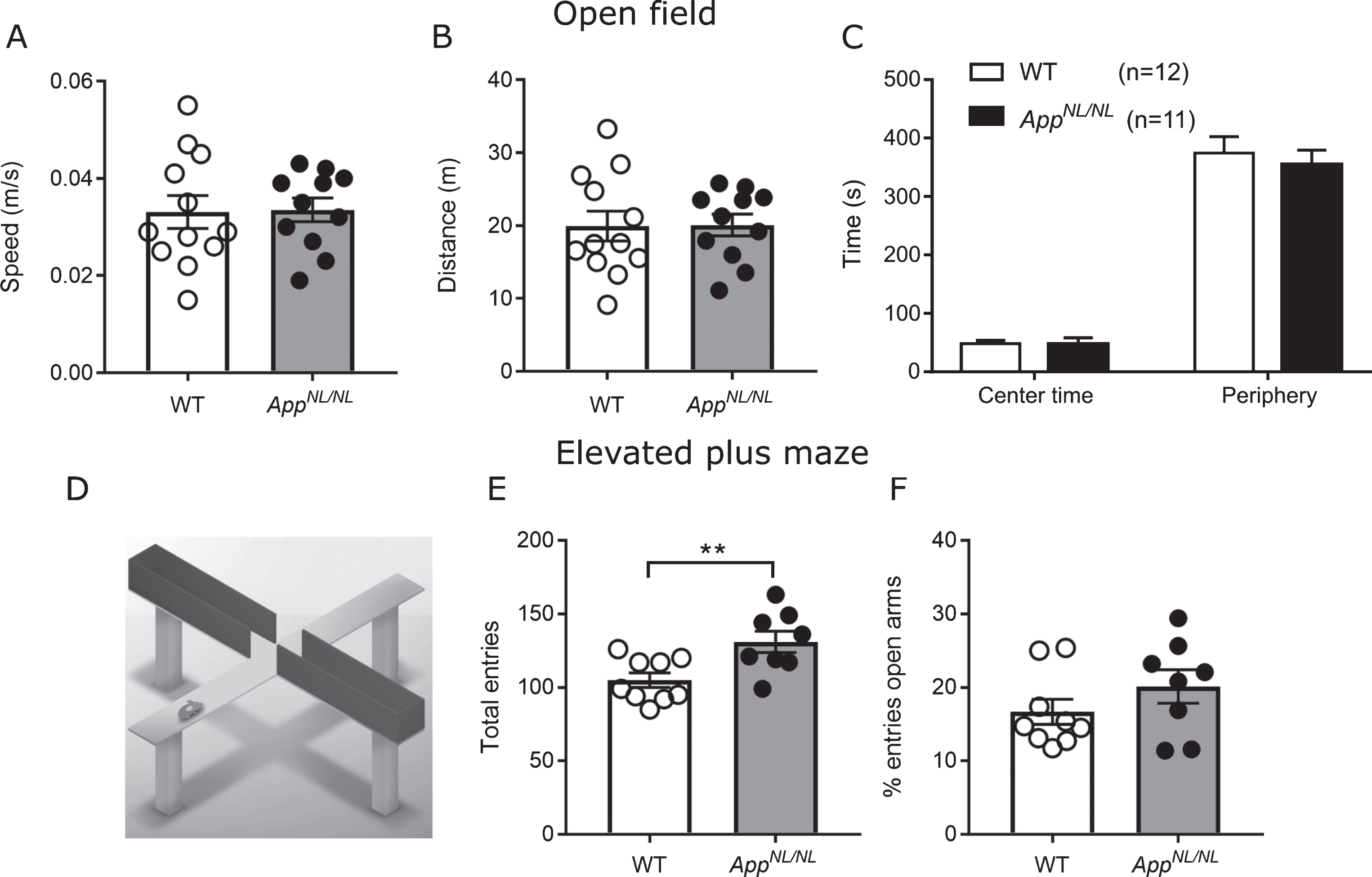
AD has also been associated with changes in anxiety [15, 16]. Time spent or entries to the brightly lit center can reflect anxiety-related behavior: the more anxious the mouse is, the less often it will venture into the open unprotected center. However, we measured no difference in center exploration between the genotypes, with all mice spending most of their time close to the walls (Fig. 2C). In the elevated plus maze (Fig. 2D), AppNL/NL mice were significantly more active than WT (total arm visits WT: 105±4.97; AppNL/NL: 131±7.33 p = 0.0091, unpaired t-test, Fig. 2E), but were not visiting the open arms more frequently (WT: 16.68±1.7; AppNL/NL: 20.14±2.28, p = 0.24, unpaired t-test, Fig. 2F). The increased total number of entries might reflect a general hyperactivity specifically toward closed (dark) arms, since this hyperactivity is not observed in other activity-sensitive readouts, such as speed or distance travelled in the open field. This could indicate that when these two groups of animals are exposed to the brightly lit open field after a period of dark adaptation, all animals would choose the same avoidance behavior and stay close to the wall. However, when presented a choice of open or closed arms, AppNL/NL would be more active in the relative safety of the closed arms.
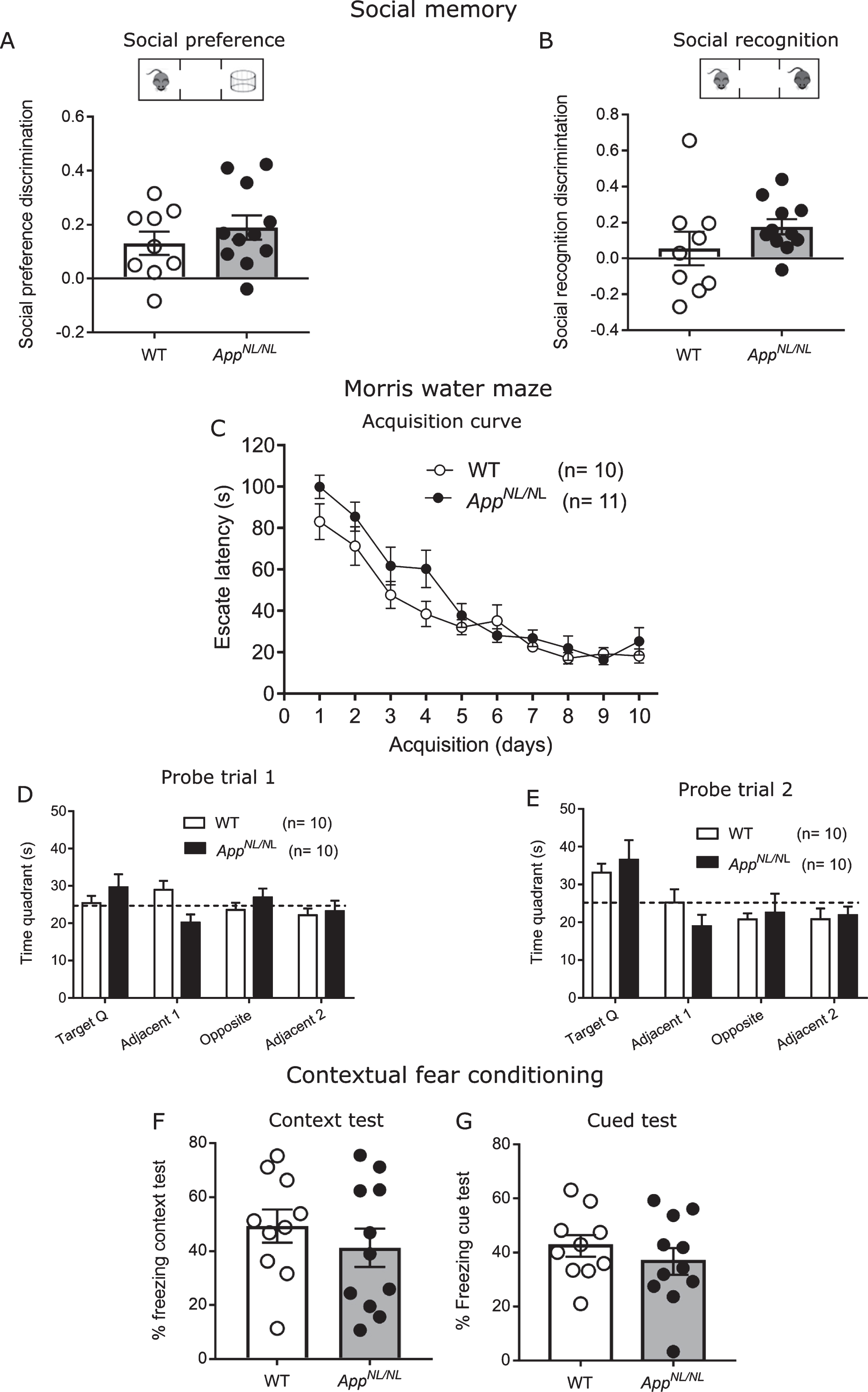
In the clinic, altered social interaction and social recognition have been associated with AD [17], alteration which has been recapitulated in animal models for the disease [18]. Here, we assessed social behavior and recognition memory by presenting either a stranger mouse and an object (social approach) or a familiar and novel stranger mouse (recognition memory). In the social approach phase, preference for exploring a mouse over an object (empty cage) was recorded for 10 min and discrimination index was calculated by [(Time stranger 1-Time object)/ (Time stranger 1 + Time object)]. Our analysis indicated that both genotypes preferred the stranger mouse over the empty cage, and there were no differences between the genotypes (Fig. 3A). When we tested social recognition memory, we observed that AppNL/NL mice spent more time with the novel stranger, while WT did not show this preference (one sample t test comparing discrimination index to chance (=0): AppNL/NL, p = 0.002; WT, p = 0.57; Fig. 3B). This may indicate an impairment in social discrimination in old B57Bl/6J animals, which was not present in the AppNL/NL mice. Discrimination index was calculated as [(Time stranger novel- Time stranger familiar)/ (Time stranger novel + Time stranger familiar)].
Fig.3
24-month-old AppNL/NL mice show no defects in spatial and social learning and memory. A) Social preference test revealed no alterations in the mean discrimination ratio: (Time STR1-Time empty)/ (Time STR1+Time empty). B) Social novelty test showed no differences in the mean discrimination index between 24-month-old WT (n = 8) and AppNL/NL (n = 11) mice: (Time STR1-Time STR2)/ (Time STR1+Time STR2). C) Acquisition curve in the Morris water maze test compares the latency to find the hidden platform during the 10 training days between WT and 24-month-old AppNL/NL mice. D) Probe trial 1 (performed after 5 days of training) and (E) probe trial 2 (performed after 10 days of training) revealed no differences in the time spent swimming in the target quadrant between the two groups. F, G) Contextual and cued-fear conditioning test: Graphs shows mean percentage of freezing during the context test (F); or the cued test (G). n = 10 WT, 11 AppNL/NL mice. Histograms show the mean (±S.E.M). Statistical significance was analyzed with unpaired T-test (A, B, D-G) and with repeated measurements 2-way ANOVA (C).
Then, spatial learning and memory was evaluated in the Morris water maze. Mice were trained for 10 consecutive days (4 trials/day; 15–30 min inter-trial interval) to find a hidden platform (15 cm) in a circular pool (150 cm). To assess spatial reference memory, we performed 2 probe trials after 5 (probe trial 1) and 10 (probe trial 2) days. During these trials the platform was removed and the swimming trajectories were tracked to quantify the amount of time mice swam in the target quadrant where the platform was located.
AppNL/NL mice displayed delayed learning during the first 5 days of the training (F (1,19) = 10.19; p = 0.005, RM-ANOVA, group factor, Fig. 3C). But the performance equalized in the second week of training, indicating that AppNL/NL mice did learn to find the hidden platform but needed more time to reach the same performance as the WT controls (Fig. 3C, Supplementary Figure 2A, B).
Probe trials revealed accuracy to remember a certain spatial location. Both groups needed 2 weeks of training to demonstrate a robust preference for the target quadrant. This preference was not above chance (=25%) in probe trial 1, but in probe 2, the time spent in the target quadrant was significantly above 25% in both groups, without genotype differences (Fig. 3D, E, Supplementary Figure 2C, D). This allows for the conclusion that AppNL/NL learned slower than WT, but both groups needed 2 weeks of training to show robust spatial reference memory.
Finally, we assessed the contextual fear memory in old AppNL/NL mice using contextual and cued fear conditioning test. In this test, mice received an electric shock paired with a tone in a specific context. 24 h later, animals were placed in the same context and the amount of freezing was quantified as a read-out for fear memory. Figure 3E revealed comparable freezing between old AppNL/NL and WT animals during the context test. To analyze whether the mice were able to associate the aversive electric shock with the conditioned stimulus, animals were placed in a new context and exposed to the same acoustic stimulus that was previously paired with shock. Similarly, to the context test, percentage of freezing in the cued test was not affected in AppNL/NL mice compared to WT controls (Fig. 3F). In conclusion, we did not observe any genotype effect in contextual fear conditioning. However, we observed a rather high variability in freezing between the animals. Therefore, we analyzed whether freezing (readout for fear memory) correlated with the amount of Aβ in the brain. As shown in Supplementary Table 1, we did not find a significant correlation with any of the different Aβ peptides.
In conclusion, we find very subtle, but genotype-specific differences in Morris water maze learning but not in reference memory, in social recognition but not social approach, and in activity in safe spaces but not in open field.
DISCUSSION
To conclude, in this work we provide for the first time a complete behavioral and biochemical characterization of the aged AppNL/NL mice, which in our view, is the closest model mimicking molecularly one genetic human AD condition (Swedish mutation). The results presented here revealed that the Swedish mutation alone, despite inducing an age-dependent increase of insoluble Aβ levels, was not sufficient to trigger amyloid plaques deposition, reactive gliosis, or major cognitive deficits, even when combined with aging in 24-month-old AppNL/NL mice.
Intriguingly, the increased levels of insoluble Aβ with age were accompanied by decreased levels of Aβ in the soluble fraction. This is similar to what happens in AD brains, where it has been shown that the percentages of soluble Aβ40 and Aβ42 were also decreased compared to healthy brains, as the insoluble percentages increased [19]. One possible explanation for this phenotype would lie in the general impairment of the “protein quality control” machinery that associates with age [20, 21].
We also found it surprising that in this model, the increased levels of insoluble Aβ oligomers in old AppNL/NL mice did not result in amyloid plaques deposition in the brain. This might suggest that Aβ oligomers only start aggregating above certain threshold that was not reached in the brains from old AppNL/NL mice. Alternatively, it could also suggest the activation of protective proteolytic or survival pathways in this model, which prevent the development of an AD phenotype [22].
Interestingly, the APP23 mouse model overexpressing human AβPP Swedish under the Thy 1.2 promoter shows amyloid plaques and massive glia reaction in hippocampus and cortex at 6 months of age [23]. In addition, APPswe mice expressing human AβPP Swedish under the neuron-specific enolase promoter (NSE), exhibit cognitive alterations in the Morris water maze test at the age of 12 months [24]. These studies, compared to the data shown here, suggest that the ectopic expression of the AβPP transgene, might be partially responsible for the observed AD-related alterations, instead of the Swedish mutation itself. In fact, overexpression of human WT AβPP gene alone has been reported to cause memory impairments [25]. Supporting the notion that AD-like phenotype can be due to ectopic expression, the general behavioral phenotype from the AβPP knock-in models generated by Saito, is surprisingly mild compared to other overexpression models. For example, the AppNL - G - F model carrying three AβPP mutations, shows an accumulation of amyloid plaques already at 6 months of age [7], but does not exhibit defects in spatial learning, assessed with the place preference task. Regarding behavioral alterations, while Masuda et al. described an impaired spatial reversal learning in 13–14-month-old AppNL - G - F mice [8]; Latif-Hernández et al., reported no defects in spatial reversal reference memory in the Morris water maze test at 12 months of age [26]. In this work, we observed a mild delay in spatial learning during the first 5 days of acquisition in the Morris water maze test in 24-month-old AppNL/NL mice. However, this delayed learning did not affect the memory for the platform location during the probe trials.
Overall, the altered cognitive abilities described in any of the mentioned studies, including ours, were minimal compared to the phenotypes described in the more aggressive overexpression models such as APP/PS1 [11] or 5xFAD [27].
Another possible explanation for the absence of major behavioral phenotype in these animals is the lack of tau pathology, which is needed in combination with amyloidosis to trigger cognitive deficits [28, 29]. In this work, we have not characterized the levels of tau phosphorylation in the aged AppNL/NL mice. However, in the original paper, Saito et al. reported no changes in tau phosphorylation in the knock-in models [7].
One possible confounder in the current study is that Aβ measurements were done in the same animals that previously underwent the battery of behavioral tests. Thus, an effect of the behavioral activities on amyloid production cannot be excluded, i.e., physical exercise and environmental enrichment has been suggested to reduce Aβ levels and amyloid deposition [30, 31].
In addition, it is important to note that the old ages of the animals may impair their acoustic and visual capacities which can interfere with the behavioral test [32, 33]. However, all animals were able to find the platform, and show a significant increase of freezing in the cued test, suggesting their capability to detect the external visual and auditory cues was not affected.
Altogether, in this paper we have shown that in mouse models, a single AβPP mutation expressed under physiological levels is not sufficient to trigger amyloid plaques or major cognitive deficits despite increased levels of insoluble Aβ oligomers. Further work is needed to determine the murine-specific factors preventing amyloid plaque accumulation in this model or human specific factors enhancing these effects.
ACKNOWLEDGMENTS
We thank Véronique Hendrickx and Jonas Verwaeren for the assistance with breeding the mouse, and Jonny Draffin for the critical reading of the manuscript. The work is supported by the Fonds voor Wetenschappelijk Onderzoek (FWO), the KU Leuven and VIB, a Methusalem grant of the KU Leuven/Flemish Government, and grants from stichting Alzheimer Onderzoek (SAO-Belgium) and Vlaams Initiatief voor Netwerken voor Dementie Onderzoek (VIND, Strategic Basic Research Grant 135043). BDS is supported by the Bax-Vanluffelen Chair for Alzheimer’s Disease and “Opening the Future”, and by funds from the UK-DRI, which is supported by MRC, Alzheimer society UK and Alzheimer Research UK. CGD is supported by an Innovation Ingenio-Consolider grant CSD2010-00045 and by Spanish Ministry of Science and Spanish Ministry use colonies. Annerieke Sierksma for the help with the statistical analysis, and Jonn of Economy and Competitiveness grants SAF2013-45392 and SAF2016-76722.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0410r2).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: 10.3233/JAD-180410.
REFERENCES
[1] | Holtzman DM , Morris JC , Goate AM ((2011) ) Alzheimer’s disease: The challenge of the second century. Sci Transl Med 3: , 77sr1. |
[2] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[3] | Veugelen S , Saito T , Saido TC , Chávez-Gutiérrez L , De Strooper B ((2016) ) Familial Alzheimer’s disease mutations in presenilin generate amyloidogenic Aβ peptide seeds. Neuron 90: , 410–416. |
[4] | Szaruga M , Munteanu B , Lismont S , Veugelen S , Horré K , Mercken M , Saido TC , Ryan NS , De Vos T , Savvides SN , Gallardo R , Schymkowitz J , Rousseau F , Fox NC , Hopf C , De Strooper B , Chávez-Gutiérrez L ((2017) ) Alzheimer’s-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell 170: , 443–456.e14. |
[5] | LaFerla FM , Green KN ((2012) ) Animal models of Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006320. |
[6] | Saito T , Matsuba Y , Yamazaki N , Hashimoto S , Saido TC ((2016) ) Calpain activation in Alzheimer’s model mice is an artifact of APP and presenilin overexpression. J Neurosci 36: , 9933–9936. |
[7] | Saito T , Matsuba Y , Mihira N , Takano J , Nilsson P , Itohara S , Iwata N , Saido TC ((2014) ) Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci 17: , 661–663. |
[8] | Masuda A , Kobayashi Y , Kogo N , Saito T , Saido TC , Itohara S ((2016) ) Cognitive deficits in single App knock-in mouse models. Neurobiol Learn Mem 135: , 73–82. |
[9] | Mullan M , Crawford F , Axelman K , Houlden H , Lilius L , Winblad B , Lannfelt L ((1992) ) A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N–terminus of β–amyloid. Nat Genet 1: , 345–347. |
[10] | Fox JG ((2007) ), The Mouse in biomedical research Academic Press. |
[11] | Radde R , Bolmont T , Kaeser SA , Coomaraswamy J , Lindau D , Stoltze L , Calhoun ME , Jäggi F , Wolburg H , Gengler S , Haass C , Ghetti B , Czech C , Hölscher C , Mathews PM , Jucker M ((2006) ) Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7: , 940–946. |
[12] | Mucke L , Selkoe DJ ((2012) ) Neurotoxicity of amyloid β-protein: Synatic and network dysfunction. Cold Spring Harb Perspect Med 2: , a006338. |
[13] | Parihar MS , Brewer GJ ((2010) ) Amyloid-β as a modulator of synaptic plasticity. J Alzheimers Dis 22: , 741–763. |
[14] | Ferreira ST , Klein WL ((2011) ) The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol Learn Mem 96: , 529–543. |
[15] | Webster SJ , Bachstetter AD , Nelson PT , Schmitt FA , Van Eldik LJ ((2014) ) Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet 5: , 88. |
[16] | Gimenez-Llort L , Ramirez-Boix P , García Y , LaFerla FM , Sanfeliu C ((2010) ) Modeling neuropsychiatric symptoms of Alzheimer’s disease in 3xTgAD mice: Neophobia, anxiety and aggressiveness at advanced stages of the disease. Alzheimers Dement 6: , 227. |
[17] | Torralva T , Dorrego F , Sabe L , Chemerinski E , Starkstein SE ((2000) ) Impairments of social cognition and decision making in Alzheimer’s disease. Int Psychogeriatr 12: , 359–368. |
[18] | Faizi M , Bader PL , Saw N , Nguyen T-V V. , Beraki S , Wyss-Coray T , Longo FM , Shamloo M ((2012) ) Thy1-hAPP Lond/Swe + mouse model of Alzheimer’s disease displays broad behavioral deficits in sensorimotor, cognitive and social function. Brain Behav 2: , 142–154. |
[19] | Wang J , Dickson DW , Trojanowski JQ , Lee VM-Y ((1999) ) The levels of soluble versus insoluble brain Aβ distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol 158: , 328–337. |
[20] | Cuervo AM ((2008) ) Autophagy and aging: Keeping that old broom working. Trends Genet 24: , 604–612. |
[21] | Carmona-Gutierrez D , Hughes AL , Madeo F , Ruckenstuhl C ((2016) ) The crucial impact of lysosomes in aging and longevity. Ageing Res Rev 32: , 2–12. |
[22] | Murphy MP , LeVine III H ((2010) ) Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis 19: , 311–323. |
[23] | Sturchler-Pierrat C , Abramowski D , Duke M , Wiederhold KH , Mistl C , Rothacher S , Ledermann B , Bürki K , Frey P , Paganetti PA , Waridel C , Calhoun ME , Jucker M , Probst A , Staufenbiel M , Sommer B ((1997) ) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A 94: , 13287–13292. |
[24] | Hwang DY , Cho JS , Lee SH , Chae KR , Lim HJ , Min SH , Seo SJ , Song YS , Song CW , Paik SG , Sheen YY , Kim YK ((2004) ) Aberrant expressions of pathogenic phenotype in Alzheimer’s diseased transgenic mice carrying NSE-controlled APPsw. Exp Neurol 186: , 20–32. |
[25] | Simón A-M , Schiapparelli L , Salazar-Colocho P , Cuadrado-Tejedor M , Escribano L , López de Maturana R , Del Río J , Pérez-Mediavilla A , Frechilla D ((2009) ) Overexpression of wild-type human APP in mice causes cognitive deficits and pathological features unrelated to Aβ levels. Neurobiol Dis 33: , 369–378. |
[26] | Latif-Hernandez A , Shah D , Craessaerts K , Saido T , Saito T , De Strooper B , Van der Linden A , D’Hooge R ((2017) ) Subtle behavioral changes and increased prefrontal-hippocampal network synchronicity in APP NL–G–F mice before prominent plaque deposition. Behav Brain Res. doi: 10.1016/j.bbr.2017.11.017. |
[27] | Schneider F , Baldauf K , Wetzel W , Reymann KG ((2014) ) Behavioral and EEG changes in male 5xFAD mice. Physiol Behav 135: , 25–33. |
[28] | Marciniak E , Leboucher A , Caron E , Ahmed T , Tailleux A , Dumont J , Issad T , Gerhardt E , Pagesy P , Vileno M , Bournonville C , Hamdane M , Bantubungi K , Lancel S , Demeyer D , Eddarkaoui S , Vallez E , Vieau D , Humez S , Faivre E , Grenier-Boley B , Outeiro TF , Staels B , Amouyel P , Balschun D , Buee L , Blum D ((2017) ) Tau deletion promotes brain insulin resistance. J Exp Med 214: , 2257–2269. |
[29] | Underwood E ((2016) ) Tau protein— not amyloid— may be key driver of Alzheimer’s symptoms. http://www.sciencemag.org/news/2016/05/tau-protein-not-amyloid-may-be-key-driver-alzheimer-s-symptoms. |
[30] | Lazarov O , Robinson J , Tang Y-P , Hairston IS , Korade-Mirnics Z , Lee VM-Y , Hersh LB , Sapolsky RM , Mirnics K , Sisodia SS ((2005) ) Environmental enrichment reduces Aβ levels and amyloid deposition in transgenic mice. Cell 120: , 701–713. |
[31] | He X , Liu D , Zhang Q , Liang F , Dai G , Zeng J , Pei Z , Xu G , Lan Y ((2017) ) Voluntary exercise promotes glymphatic clearance of amyloid beta and reduces the activation of astrocytes and microglia in aged mice. Front Mol Neurosci 10: , 144. |
[32] | Kolesnikov AV , Fan J , Crouch RK , Kefalov VJ ((2010) ) Age-related deterioration of rod vision in mice. J Neurosci 30: , 11222–11231. |
[33] | Yang C-H , Schrepfer T , Schacht J ((2015) ) Age-related hearing impairment and the triad of acquired hearing loss. Front Cell Neurosci 9: , 276. |


