
Linking Atrial Fibrillation with Alzheimer’s Disease: Epidemiological, Pathological, and Mechanistic Evidence
Abstract
Many studies have shown a relationship between atrial fibrillation (AF) and vascular dementia. AF is a major risk factor for stroke, and stroke is the greatest risk factor for vascular dementia. However, the relationship between Alzheimer’s disease (AD), the leading cause of dementia, and AF remains unclear. At least four epidemiological studies have reported AF significantly raises the risk of AD 1.5- to 2.5-fold. Chronic cerebral hypoperfusion, resulting from persistent AF, could explain the link as hypoperfusion may mechanistically exacerbate amyloid-β (Aβ) neuropathology, such as senile plaques and amyloid angiopathy, by upregulating Aβ-producing enzymes and lowering Aβ clearance efficiency. In addition, hypoperfusion may exacerbate tau pathology directly through upregulation of tau-phosphorylating enzymes and indirectly via the amyloid cascade. However, most neuropathological studies do not support the direct link between AD pathology and AF but rather suggests vascular neuropathology is related to, or coexistent with, AF and lowers the threshold for clinically-evident AD. Vascular neuropathology may thus mediate the link between AD and AF. From a treatment perspective, an observational study has shown that catheter ablation is associated with less incidence of AD in AF patients, suggesting rhythm-control suppresses hypoperfusion-induced AD neuropathology. In addition, rate-control may lower the rate of cognitive decline in cognitively impaired elderly subjects with AF. Further studies are warranted to clarify the mechanisms underlying the linkage between AF and AD. However, anticoagulation and rhythm-/rate-control against AF may hold promise even for AD patients.
INTRODUCTION
Atrial fibrillation (AF) contributes to the incidence and outcome of ischemic stroke [1]. Estimates of AF prevalence among patients with stroke are variable but recent technical advances have clarified that AF is more common among patients with ischemic stroke than previously reported [2]. Since stroke can cause vascular, or post-stroke, dementia [3], a relationship between AF and vascular dementia is also likely [4]. So is there a relationship between AF and Alzheimer’s disease (AD)? Epidemiological research has offered fairly compelling evidence [5–7]. Why is there such a relationship? Based on the close association between stroke and AD [8], does cardioembolic stroke (CES) induce not only vascular dementia but also AD? Is there a mechanism by which pulse irregularities caused by AF induce functional abnormality in neurons leading to AD? Does cerebral hypoperfusion caused by persistent AF trigger AD? Although it seems premature to draw conclusions about the underlying mechanisms behind the association between AF and AD, we hope this review will provide a guide for future research by focusing on studies differentiating AD from other causes of dementia and speculating on mechanisms linking AF and AD.
EPIDEMIOLOGICAL EVIDENCE
The relationship between AF and AD was first found in the Rotterdam study reported by Ott et al. in 1977 [6]. In this study, 6,584 subjects, aged 55 to 106, registered in 1990–1993, underwent both electrocardiogram examination and cognitive assessment. Dementia was present in 276 people, among whom 206 had AD (75%), 41 vascular dementia, 29 dementia of other causes, and 11 dementia with unexplained etiologies. AF was observed in 195 people with increased frequency with age, and those with AF had significantly higher frequency of dementia with an odds ratio of 2.3 (95% CI, 1.4–3.7). Interestingly, the correlation was particularly strong in young women under 75 years old (odds ratio, 9.2 (2.5–34.3)), and in cases of AD with stroke (AD with cerebrovascular disease (CVD); odds ratio, 4.1 (1.7–9.7)). However, the history of stroke per se was not been related to this correlation.
In addition, a population-based cohort study (Cardiovascular Risk Factors, Aging and Dementia: CAIDE study) that longitudinally followed 1,510 men and women for 7.8 years (62% females; median age, 65 years) showed the hazard ratio for AF was 2.61 (95% CI, 1.05–6.47) for all-cause dementia and 2.54 (95% CI, 1.04–1.61) for AD [5].
Likewise, in another population-based cohort study (Adult Changes in Thought: ACT study) following 3,045 men and women (60% female, average 74 years old) for 6.8 years, the hazard ratio of AF was 1.38 (95% CI, 1.10–1.73) for all-cause dementia, and increased to 1.50 (95% CI, 1.16–1.94) for AD [7]; in this study, cases with past history of stroke were excluded.
Although the above three cohort studies were population-based, the Intermountain Heart Collaborative Study, which followed up 37,025 hospital cohorts (female 39.9%, average age 60.6 years) showed that during the 5-year observation period, all-cause dementia developed in 1.3% of those without AF versus 3.3% of those with AF, AD in 0.7% versus 1.5%, and vascular dementia in 0.3% versus 0.9% [9]. After multivariate adjustment, the odds ratio of AF for all-cause dementia was 1.73, 1.06 for AD, and 1.73 for vascular dementia but when limited to subjects aged 70 years or less, the odds ratio for AD was even greater at 2.30 with a statistical significance. In this study, senile dementia was distinguished from AD and vascular dementia, which may represent inherent limitation for differential diagnoses of dementia in the elderly and have affected the odds ratios among dementia subtypes. However, in this study, patients with younger-onset AF did show increased prevalence of AD. Furthermore, in a case-control study conducted on 784 subjects at Geriatric Clinic in Italy, AF was associated with 2.0-fold increase in vascular dementia and 1.72-fold increase in AD [10], further supporting the association between AF and AD.
By contrast, in the Swedish Kungsholmen Project (1987–2000), which observed 685 subjects without a history of dementia or stroke, the hazard ratio of dementia associated with AF was 0.9 (95% CI, 0.5–1.7) and that of AD, 0.8 (95% CI, 0.4–1.5), with lack of association of AF with cognitive impairment including AD in the very old during the first follow-up period (1991–1993) of research participants over 75 years of age for 6 years [11]. In a meta-analysis of the correlation between AF and cognitive impairment, the relative risk of dementia increased by 2.7 (95% CI, 1.82–4.00) in AF patients with stroke and by 1.4 (95% CI, 1.19–1.64) with statistical significance even for patients without stroke. When the disease subtypes of dementia were distinguished, the relative risk of vascular dementia was 1.72 (95% CI, 1.27–2.32), while the relative risk of AD was 1.22 (95% CI, 0.96–1.56), suggesting no significant increase in the risk of AD due to AF [12]. However, this meta-analysis did not detail which studies were included for AD risk calculation.
We have listed studies distinguishing AD as a disease subtype of dementia (Table 1). As described above, contradictory results have been reported, but considering the negative study enrolled only elderly people over the age of 75 at the time of baseline [11], the subjects’ age may have impacted the association between AF and AD. In other words, as the subject becomes older, the absolute number of AD cases is irrelevant to increases in AF, making it difficult to detect the positive correlation between AF and AD. Furthermore, in older adults, the proportion of so-called ‘mixed’ vascular/AD cases increase [3, 13], so AD and vascular dementia become difficult to differentiate, which may hamper association studies of dementia subtypes with AF. Consistent with this notion, the Vantaa 85 + Study showed AF was not an independent risk factor for dementia in the very old, and the following neuropathological study showed that the etiology of dementia syndrome was multifactorial, including both AD, and vascular, pathology [14].
Table 1
Clinical studies that investigated incidence of Alzheimer’s disease in patients with atrial fibrillation
| Literature | Study | Subjects | Follow-up | Dementia | Results |
| Ott [6] | Rotterdam study | 6,584 subjects, 59.2% female, average 69 years | Cross-sectional study | 206 AD (75%), 41 VaD, 29 other cause, 1 unknown | Age- and sex-adjusted odds ratio: |
| AD with CVD, 4.1 (1.7–9.7) | |||||
| AD without CVD, 1.8 (1.0–3.3) | |||||
| VaD, 1.9 (0.6–5.5) | |||||
| Rusanen [5] | Cardiovascular Risk Factors, Aging and Dementia (CAIDE) study | 1510 subjects, 62% female, mean 65 years | Longitudinal study for 7.8 years | 127 dementia (8.4%), including 102 AD | Hazard ratio: |
| All-cause dementia, 2.61 (1.05–6.47) | |||||
| AD, 2.54 (1.04–6.16) | |||||
| Dublin [7] | Adult Changes in Thought (ACT) study | 3045 subjects, 60% female, mean 74 years | Longitudinal study for 6.8 years | 572 dementia, including 449 AD | Adjusted odds ratio: |
| All-cause dementia, 1.38 (95% CI 1.10–1.73) | |||||
| AD, 1.50 (95% CI 1.16–1.94) | |||||
| Bunch [9] | Intermountain Heart Collaborative study | 37,025 subjects, 39.9% female, average 60.6 years | Longitudinal study 5 years | 1535 dementia (347 AD, 179 VaD) | Odds ratio: |
| All-cause dementia, 1.73 | |||||
| AD, 1.06 | |||||
| AD (70 years or older), 2.30 | |||||
| Di Nisio [10] | Study in Geriatric Clinic (Chieti, Italy) | 784 subjects, 59.2% female, average 77.5 years | Cross-sectional study | 309 dementia (44 VaD, 73 AD, 50 AD with CVD, 81 other causes, 61 non-dementia) | Hazard ratio: |
| AD, 1.72 | |||||
| VaD, 2.0 | |||||
| Marengoni [11] | Kungsholmen project | 685 subjects, 78 years or older at baseline | Longitudinal for 6 years | 170 dementia (143 AD) | Hazard ratio: |
| All-cause dementia, 0.9 (95% CI, 0.5–1.7) | |||||
| AD, 0.8 (95% CI, 0.4–1.5) |
AD, Alzheimer’s disease; CVD, cerebrovascular disease; VaD, vascular dementia.
MECHANISTIC INSIGHTS
The epidemiological studies described above suggest an association between AF and AD, especially in younger individuals or those with stroke. So, what kind of mechanisms underlie the association? Because AF is a strong risk factor for CES [12], it is easy to envisage post-stroke dementia or vascular dementia ensues after the CES. A meta-analysis reported dementia is seen in 20% of the initial stroke patients and 30% of the recurrent stroke patients [15]. Therefore, there seems to be continuity in AF through ischemic stroke leading to post-stroke, or vascular, dementia. AF may also induce cerebral hypoperfusion based on low cardiac output [16], which may cause vascular dementia of hypoperfusive etiology, regardless of whether AF leads to ischemic stroke [16–18]. Indeed, basic research using rodent models has suggested cerebral hypoperfusion induces white matter changes, such as demyelination and axonal damage, and causes working memory impairment [19, 20]. In addition, a close association between hypoperfusion and vascular dementia has been identified [21, 22]. Low cardiac output itself may be a direct risk of stroke, rather than cerebral hypoperfusion. However, only severely reduced ejection fraction (≤20%) can be a specific stroke risk factor [23], which is lower than the level that can be normally induced by AF [24]. This suggests AF can cause vascular dementia through CES and/or cerebral hypoperfusion.
Does continuity exist between AF and AD (Fig. 1)? A hint for the answer is given from a neuropathological analysis of 328 autopsy cases (55% female, average 87.6 years old) in a population-based cohort [25]. In the medical records, 134 subjects (41%) had AF, which was divided into two groups: 1) permanent AF (57 cases) confirmed with an electrocardiogram more than twice, at intervals of 6 to 36 months, and 2) nonpermanent AF (77 cases) confirmed only once, even with repeated electrocardiogram. Intriguingly, when permanent AF was observed, the relative risk of senile plaques, neurofibrillary tangles, and cerebral amyloid angiopathy, which are the three major pathological changes of AD, was 1.47 (95% CI, 0.96–2.28), 1.40 (0.79–2.49), and 1.50 (0.78–2.67) compared to controls without AF, respectively. By contrast, the relative risk of each pathological change associated with nonpermanent AF was reduced at 0.82 (0.49–1.43), 0.42 (0.20–0.87), and 0.74 (0.42–1.30), compared to controls without AF. Although limited number of autopsied cases may underlie the lack of significant differences, the fact that the risk of the three major pathological hallmarks of AD consistently increase with permanent AF, compared to nonpermanent AF, provides insights into the mechanisms linking AF and AD. In addition, the relative risk of neurofibrillary tangles accompanying permanent AF was 1.91 (95% CI 1.01–3.61) when adjusted by the APOE genotypes and presence of chronic heart failure, further suggesting that AF promotes the pathological changes in AD.
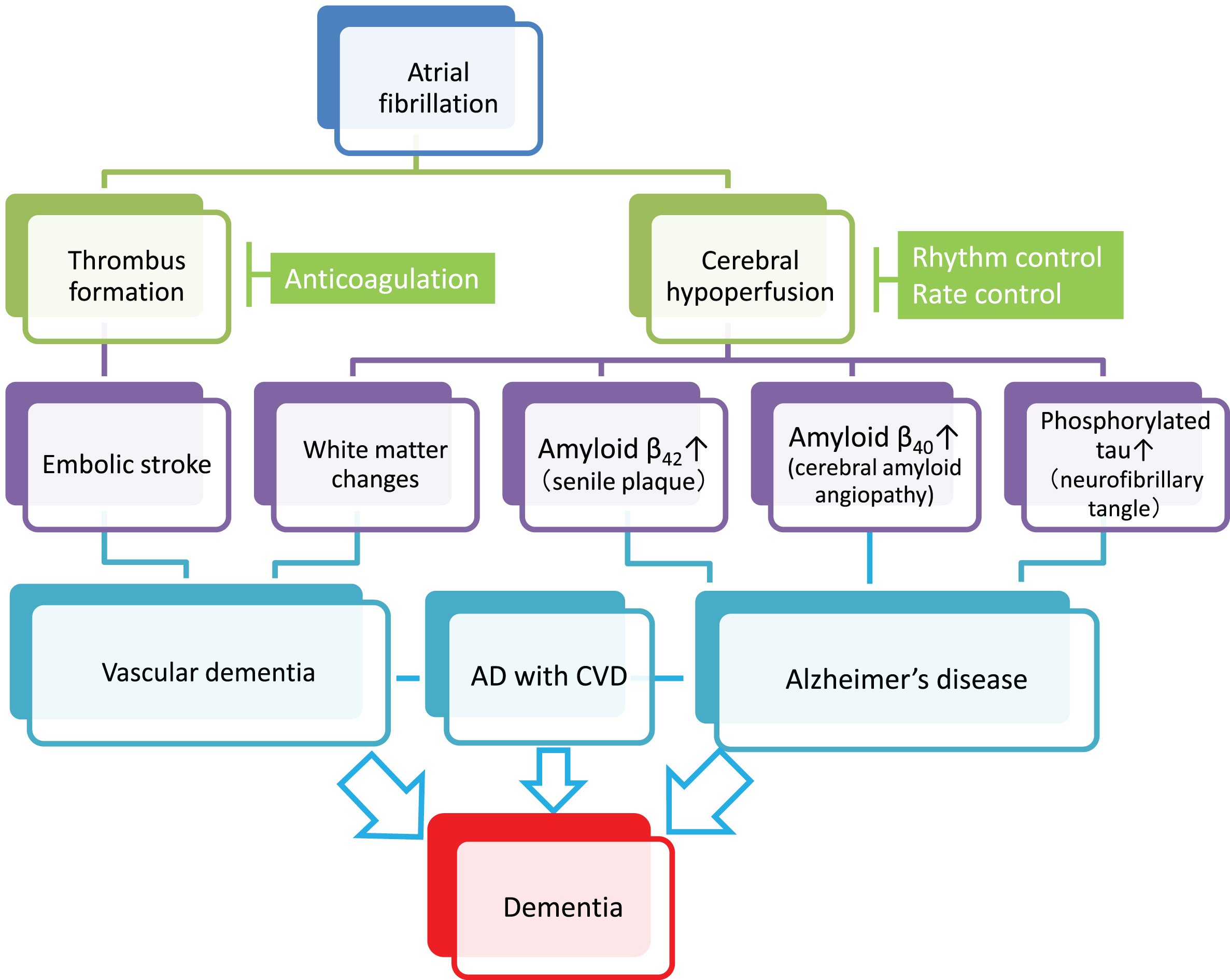
Fig.1
Plausible mechanisms by which atrial fibrillation (AF) induces vascular dementia and Alzheimer’s disease (AD). AF causes cerebral infarction due to the embolic mechanism through thrombus formation within the heart chamber and can be a risk of vascular dementia (left cascade). Meanwhile, persistent AF may accelerate the three major pathological hallmarks of AD, namely senile plaques, amyloid angiopathy, and neurofibrillary tangles, through AF-associated cerebral hypoperfusion (right cascade). In addition, cerebral hypoperfusion can cause hypoperfusive vascular dementia. Although further research should be conducted to reach definitive conclusions, anticoagulation therapy may prevent vascular dementia, while rhythm/rate control may prevent AD. Since AD frequently coexists with cerebrovascular diseases (AD with CVD) in the elderly, and both dementing disorders may be induced by AF, optimal management of AF should be considered for prevention of the two major subtypes of dementia. Possible confounding pathologies that contribute to dementia in AF patients are not included in the figure for simplification (see text for details).
So, what is the underlying mechanism by which this distinct difference arises between permanent and nonpermanent AF? In the same study, the relative risk of cerebral infarction was 1.84 (95% CI 1.12–3.01) in permanent, and 1.81 (95% CI 1.13–2.84) in nonpermanent, AF with no apparent difference, making it unlikely that CES caused by AF mediates the difference. However, the crucial difference between permanent and nonpermanent AF is the reduction in cardiac output in permanent AF, which increases the likelihood of cerebral hypoperfusion [16]. In general, in vital organs, such as the brain, the mechanism of autoregulation maintains circulation, even if cardiac output decreases [26]. However, long-term AF eventually leads to a failure of this compensatory mechanism and a decrease in cerebral circulation, as demonstrated in a study that measured cerebral blood flow by the 133Xenon inhalation method [27]. In this study, younger subjects aged 35 to 50 years had a higher degree of cerebral blood flow reduction by 17.5% due to permanent AF compared to age-matched controls.
In support of such in vivo observations, simulation of AF using two coupled lumped-parameter models (systemic and cerebrovascular circulations, respectively) showed variability of the cerebral hemodynamic variables led to transient hypoperfusion, even if the cerebral autoregulation system is able to maintain a normal mean flow during AF [28]. This study also showed that the major difference between AF and sinus rhythm is the larger standard deviations of each cerebrovascular variable, suggesting AF induces fluctuations within the parameters of cerebral hemodynamics. This simulation explains the close linkage between AF and cerebral hypoperfusion, even if it is transient. These two lines of evidence from in vivo and simulation studies may, at least partially, explain the correlation between AF and AD, even in younger patients with presumed intact cerebral autoregulation systems.
Additional evidence for the association between hypoperfusion and AD can be found from a prospective assessment of 1,039 participants enrolled in the Framingham Offspring Study without prevalent stroke, cognitive decline, and dementia [29]. The study reported an independent association of cardiac output with incident dementia (adjusted HR 1.66) and with AD (adjusted HR 1.65) at median 7.7 years of follow-up. This study, although not restricted to AF patients, suggests an important association between AF and AD in a population-based cohort. Furthermore, lower cardiac index was associated with an increased risk for the development of dementia (HR = 2.92) and AD (HR = 2.87), suggesting cerebral hypoperfusion has a role in the acceleration of cognitive decline. In addition, a pathological study has reported amyloid-β (Aβ) itself affected cardiac function in patients with AD. Echocardiographic examination suggested patients with AD presented with diastolic dysfunction and histopathological examination revealed that Aβ40 and Aβ42 were both present in the myocardium. Aβ40 and Aβ42 expression was increased in heart tissue of AD patients, as in the brain. Aβ cleared from the brain into the circulation may accumulate in the heart of AD patients, suggesting Aβ plays a pathogenic role in diastolic dysfunction of the heart [30]. As diastolic dysfunction is a risk for AF [31], AD may induce AF through Aβ accumulation in the heart, suggesting the relationship between AD and AF is bidirectional.
One of the mechanisms by which senile plaques and amyloid angiopathy are induced by long-term cerebral hypoperfusion is through expression of β- (BACE 1) and γ-secretase, whose activity centers on presenilin (PSEN) are enhanced thus increasing the amount of Aβ spliced from amyloid-β protein precursor (AβPP) in the BACE 1 - PSEN - AβPP regulatory system [32–34] (Fig. 2). Aging itself also increases the expression of BACE 1[35] and Aβ production. Aβ42 with higher aggregation ability accumulates in the brain parenchyma, while Aβ40 with relatively lower aggregation ability accumulates in cerebral blood vessels, forming senile plaques and cerebral amyloid angiopathy, respectively. Cerebral amyloid angiopathy leads to circulatory insufficiency in the brain due to dysfunction of vascular smooth muscle [36], resulting in an increase in Aβ production. Among the several subtypes of vascular dementia, the positive rate of AD pathology is highest (87%) in hypoperfusive dementia, which supports the existence of such a mechanism [37].
Fig.2
Vascular changes may influence Alzheimer’s disease lesion burden. A) Scheme and arrows indicate how cerebrovascular factors affect amyloid-β (Aβ) processing and clearance. (1) Hypoperfusion, as a result of arteriolosclerosis or blood–brain barrier disruption, elevates BACE1 levels and activity, leading to Aβ overproduction. (2) Aβ overproduction leads to amyloid plaque formation and cerebral amyloid angiopathy (CAA); CAA impairs Aβ clearance efficiency, thus forming a cycle of Aβ clearance failure. (3) CAA further potentiates deficiencies of blood flow by aggravating preexisting arteriosclerosis and BBB disruption, thus forming a cycle of Aβ overproduction. AF can modify and drive such vicious cycles through concomitant hypoperfusion. B) AF may accelerate Alzheimer’s disease indirectly through cerebrovascular disease such as cardioembolic stroke and white matter changes. Note that arteriolosclerosis is known to be associated with ischemic stroke while CAA with white matter changes, which may further drive the vicious cycle of dementia.
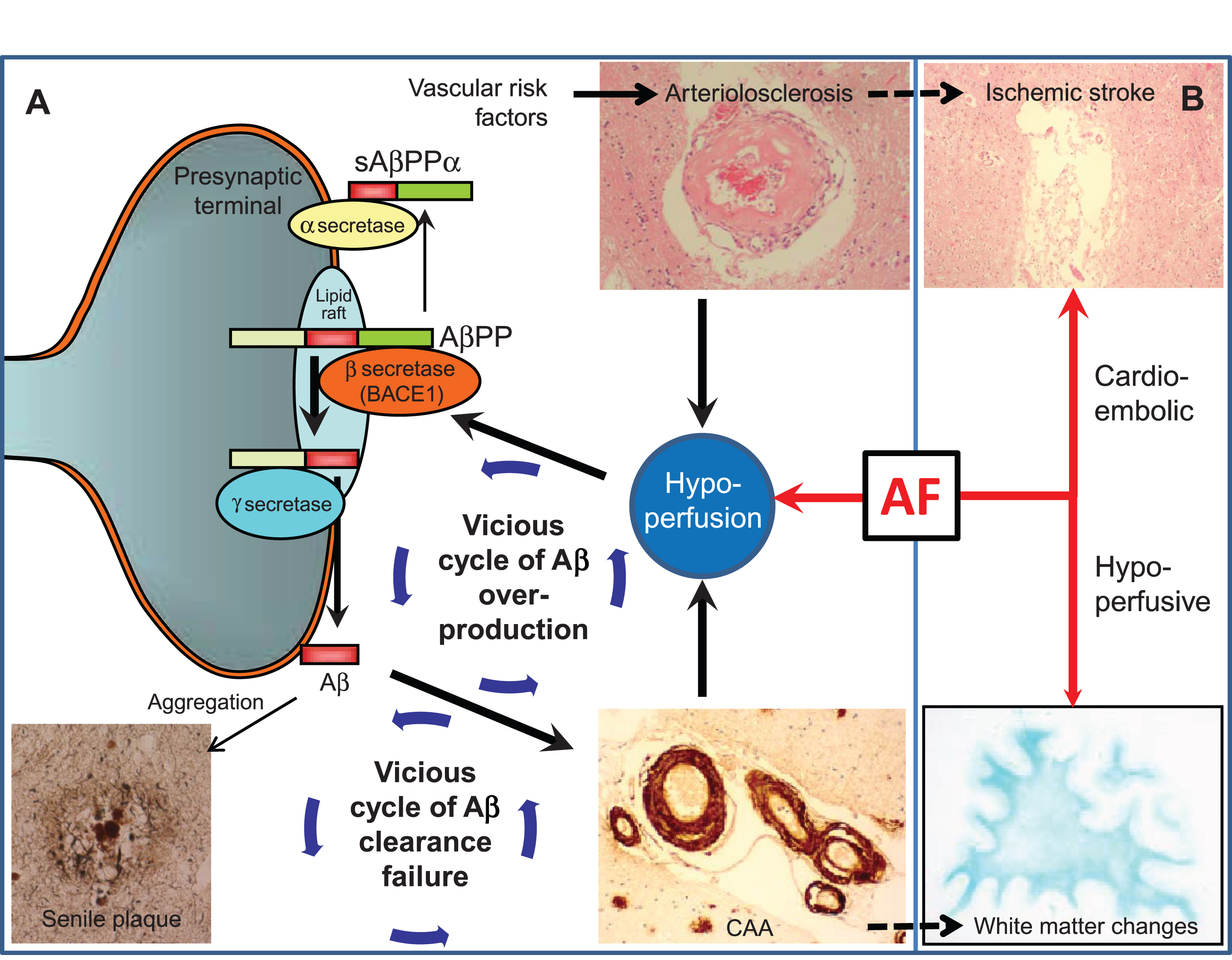
Another important mechanism by which hypoperfusion induces Aβ accumulation is clearance failure [8] (Fig. 2). Cerebral amyloid angiopathy has also been shown to interfere with the Aβ clearance pathway, inducing the accumulation of Aβ and leading to a cycle of Aβ accumulation [38]. Cerebral perfusion can be a driving force of Aβ clearance, as carotid stenosis impairs the perivascular drainage pathway [39], with aortic or brachiocephalis stenosis impairing the paravascular counterpart [40]. Such mechanisms have been shown mechanistically using two-photon imaging [41, 42], identifying cerebral perfusion as a driving force.
According to the amyloid hypothesis, accumulation of Aβ induces neurofibrillary tangles, mainly composed of phosphorylated tau, ultimately leading to neuronal cell death [43]. Chronic hypoperfusion is known to activate the tau phosphorylation enzymes glycogen synthase kinase-3 beta and cyclin-dependent kinase-5 [44], suggesting chronic hypoperfusion is directly involved in the phosphorylation of tau, independent of Aβ pathology.
These pathological and experimental studies suggest that persistent AF leads to cerebral hypoperfusion (circulation failure), even without cerebral infarction, and over a long period of time, causing an increase in Aβ production and failure in Aβ clearance, which directly or indirectly induces tau phosphorylation. Further elucidation of the mechanisms involved in this process is required but it is apparent rhythm or rate disorders may be a factor in cerebral circulation failure, as well as embolic mechanisms due to AF.
PATHOLOGICAL EVIDENCE
Many studies that included autopsies have not found a link between AF and AD-type pathologies, i.e., plaques and tangles, though several large clinical studies have shown a link between AF and AD, as shown above. A correlation between diabetes mellitus and AD may provide an illustrative parallel: many clinical studies have shown the link between diabetes and AD [45], though studies that added autopsy have found no link [46]. Indeed, participants in the Rush longitudinal cohort study of aging underwent annual clinical evaluations and a brain autopsy at death, and found no relationship between diabetes and global or regional AD pathology, including in the mesial temporal lobe [47]; rather, diabetes was associated with gross subcortical infarcts.
This association may be extrapolated to the apparent AF-AD link. In other words, ‘mixed’ vascular/AD cases may be clinically labelled as AD and the contribution of vascular factors may be underestimated in patients with AF. There is a possibility vascular neuropathology, such as small vessel pathology, may mediate the true AF-dementia link, and AD pathology may be an epiphenomenon or remain subclinical in the elderly. In addition, in clinical studies, a multitude of confounders may be omitted from analysis, such as co-morbidities of AF, including atherosclerosis and arteriosclerosis, and other co-existing neuropathologies such as α-synuclein, or even TDP-43, pathologies. These ‘clinically invisible’ pathologies may thus be under-evaluated or underdiagnosed in epidemiological studies, though they are thoroughly examined in details in neuropathological studies.
For instance, a neuropathological study using a large autopsy cohort of 334 elderly persons (mean age, 84.0 years old at death) modeled the association between 43 different clinical and pathological variables and the severity of cognitive impairment [48]. There were associations between cognitive impairment and several neuropathological findings but not with heart disease, including AF, although demographic information was only available in 15% of the cohort. In addition, among 1593 patients with primary neuropathological diagnosis of AD, of which 250 had AF, patients with AF showed (paradoxically) milder AD neuropathology with higher frequency of vascular neuropathology [49], suggesting AF-induced or co-incident vascular neuropathology contributed in lowering the threshold for clinically-evident dementia. The mean age at death was 88.9 years old in those with heart failure and 84.4 years old in those without heart failure. This notion was further confirmed by another neuropathological study using 84 autopsy-confirmed AD cases, in which presence of cerebrovascular changes was associated with lower Braak and Braak stage [50]. The mean age at death was 79.1 years old and the AF prevalence was 12% in those with cerebrovascular changes and 75.1 years old and 2.9% in those without cerebrovascular changes. The most common forms of cerebrovascular changes were circle of Willis atherosclerosis followed by arteriosclerosis, lacunes, and microinfarcts. Such cerebrovascular changes were associated with aggregate vascular burden but not with individual vascular risk factors, including AF. Taken together, these results suggest the higher frequency of AD diagnosis among AF patients found in prior studies may be explained by symptomatic and asymptomatic vascular neuropathology uncovering mild forms of AD or by other concomitant pathological processes only evident in neuropathological studies. In clinical settings, one cannot entirely exclude the influence of possible confounders underlying the apparent link between AF and AD.
Why is there such discrepancy about the AF-AD link between epidemiological studies and neuropathological studies (Table 2)? As indicated above, epidemiological studies suggest an association between AF and AD, especially in younger individuals. The contribution of AF to AD pathogenesis may be obscured in the elderly because of the age-dependent, absolute increase of AD prevalence independent of AF (compare Tables 1 and 2 for age). Another issue may be the method of AF evaluation in the studies. For example, AF may not be properly diagnosed due to lack of adequate monitoring [51], and, with the exception of the study by Dublin et al. [25], neuropathological studies do not typically distinguish persistent AF from non-persistent AF when taking medical history and examination. Non-persistent or paroxysmal AF detected at one time point does not always mean patients have low cardiac output or cerebral hypoperfusion. The lack of distinction between persistent and non-persistent AF in neuropathological studies may, at least partially, explain the discrepancy between epidemiological and neuropathological findings.
Table 2
Key pathological studies that evaluated association between AF and AD
| Literature | Study | Subjects on neuropathology | AF | Results |
| Rastas [14] | Vantaa 85 + study | 290 population-based subjects; average 88.4 years (age data from the 553 study subjects) | 55 AF patients; 235 non-AF subjects | Plaque and tangle pathology was not different between AF and non-AF subjects |
| Dublin [25] | Adult Changes in Thought (ACT) study | 328 population-based subjects; average 87.6 years | 194 never AF patients; 77 non-permanent AF; 57 permanent AF | Patients with permanent AF were more likely to have AD changes than those without AF or those with nonpermanent AF |
| Sposato [49] | National Alzheimer’s Coordinating Center database | 1,593 cases of primary neuropathological diagnosis of AD; average 84.4 years (AF+, HF–) and 88.9 years (AF+, HF+) | 250 AF subjects | AF was associated with milder AD neuropathology |
| Bangen [50] | University of California San Diego Alzheimer’s Disease Research Center | 84 AD cases on neuropathology; average 79.1 years in AD with cerebrovascular changes and 75.1 years in AD without cerebrovascular changes | 6 (12%) AF in AD with cerebrovascular changes and 1 (3%) AF in AD without cerebrovascular changes | Presence of cerebrovascular changes was associated with lower Braak and Braak stage |
AF, atrial fibrillation; HF, heart failure.
Generally, the findings derived from population-based, longitudinal studies may not always reconcile with the cross-sectional neuropathological observations targeting only specific diseases (AD in this case). In a large observational study, patients with mild dementia may be underdiagnosed and patients may be incorrectly dichotomized into vascular dementia or AD due to lack of neuroimaging. In reality, most patients may have mixed pathology [13, 52]. Lack of neuroimaging may lead to underdiagnoses of silent stroke common in AF patients [12], including cortical microinfarcts [53], which have been associated with cerebral hypoperfusion [54]. However, a strong link between AF and AD with CVD in an epidemiological study [6] may not be inconsistent with an association between AF and milder AD neuropathology with higher frequency of vascular neuropathology [49]. Although the former study is population-based, and the latter specifically enrolls AD, both suggest AF-associated CVDs raise the risk of patients with AD changes progressing to dementia. Vascular factors, therefore, could be key to understanding the link between AF and AD. Further epidemiological, basic and pathological studies could work in concert to solve this complicated but important issue in a rapidly aging society.
EXPLORATION FOR TREATMENT
Warfarin, the only preventive medicine for CES due to AF, has been used for many years in cardiovascular disease treatment [55]. However, it has also recently been suggested as a potential treatment in dementia patients with AF. The reason for this is that ‘cardiovascular events’ refer to cerebral infarction and systemic embolism but few studies have examined dementia as a ‘cardiovascular event’. Nevertheless, an observational study has provided interesting findings [56]. 2,605 patients with AF receiving warfarin (mean age 73.7 years old, 46% female) were examined for new-onset dementia with time-in-therapeutic range (TTR) as an indicator of drug control for an observation period of median 4 years (maximum 9.5 years). New-onset dementia was observed in 109 patients (37 senile dementia, 1.4%; 8 vascular dementia, 0.3%; 64 AD, 2.5%), and compared to the well-controlled TTR >75% group; the hazard ratio for dementia in the TTR <25% group, 25 <TTR <50% group, and 50% <TTR <75% group were 5.34, 4.10, and 2.57, respectively. This suggests strict anticoagulation therapy can reduce the risk of dementia. Correlation analysis in dementia subtypes, however, was not conducted in this study.
Meanwhile, in a report of the Intermountain AF study, incidence of dementia subtypes in subjects who received catheter ablation for AF was examined for at least three years of observation period and compared to the dementia incidence in 16,848 age- and sex-matched patients with AF who had not received ablation and in another 16,848 age- and sex-matched controls without AF. Incidence of AD was 0.2% in patients who underwent ablation, 0.9% in those who had not received ablation, and 0.6% in those without AF, showing that AD risk was significantly reduced after ablation (p < 0.0001). Similar significant differences were observed in other dementia subtypes, namely vascular, and senile, dementia [57]. Although this was an observational study, it is interesting that catheter ablation in AF has the possibility of reducing the risk of dementia, including AD. In this study, incidence of stroke and rate of mortality were also reduced by receiving ablation and the reduction in risk of dementia should be interpreted as a result of multiple factors besides rhythm- and/or rate-control due to ablation.
The next important question relates to whether a rate-control is a promising strategy in AF in the reduction of AD incidence. Effects of ventricular response in AF and cognitive impairment were monitored for 10 years in 44 AF and 314 non-AF patients whose MMSE scores were less than 24. Those who had AF with low/high ventricular rate (average rate <50 or >90 beats per minute) showed higher rate of cognitive decline compared to those with AF plus moderate ventricular rate (50 <beats per minute <90) [58]. AF was a strong predictor of dementia (HR = 4.10) independently of other factors such as age, sex, and heart rate. Moreover, the low/high ventricular response was predictive of dementia in the presence (HR = 7.70), but not in the absence (HR = 1.85), of AF in the cognitively impaired elderly subjects. Interestingly, vascular dementia was more frequent in the presence than in the absence of AF (57.0 versus 46.0%, p < 0.01) although subjects with stroke were excluded from the analysis, suggesting preference of hypoperfusive vascular dementia over AD in AF patients with low/high ventricular rate.
A substudy of AFFIRM (Atrial Fibrillation Follow-Up Investigation of Rhythm Management) trial tested the cognitive status in 245 participants at baseline, at 2 months, then yearly. The average Mini-Mental State Examination (MMSE) scores at baseline were 28.3 in the rate-control arm and 27.3 in the rhythm-control arm, which showed no significant difference in MMSE scores for the mean follow-up time of 3.6 years [59]. This study suggests rate-control is as effective as a rhythm-control strategy in the prevention of cognitive decline, though the small sample of patients with relatively preserved cognitive function weakened the study findings. In another study enrolling 17 patients with medically refractory rapidly-conducted AF who received ablation (average age, 55.3 years) followed by pacemaker implantation increased left ventricular systolic function and brain perfusion and improved cognitive function at 3 months after operation, implying that a combination of ablation plus pacing holds promise for cognitive recovery. However, since the etiology of cognitive impairment was not stipulated in these two studies, it remains unknown whether rate-control is beneficial to AD pathology.
Now that non-vitamin K antagonist oral anticoagulants (NOACs) are available for prevention of CES caused by AF [55], is there any evidence that NOACs are superior to warfarin in terms of prevention of dementia? In an observational study, 5,254 patients aged 18 years or more who started anticoagulation therapy (NOACs or warfarin) were examined for composite outcome of dementia/stroke/TIA, and superiority of NOACs to warfarin was shown for the composite outcome [60]. This study also suggests that dementia-free survival rate was significantly greater in NOACs compared to warfarin (p = 0.02) although dementia subtypes were not described. A recent retrospective registry study of 444,106 patients with a hospital diagnosis of AF showed that patients on anticoagulant treatment at baseline had a significantly lower risk of incident dementia than patients without anticoagulant treatment (HR 0.71; 95% CI, 0.68–0.74) [61]. Importantly, direct comparison between NOACs and warfarin showed no difference in terms of the lowered risk of dementia (HR 0.97; 95% CI, 0.67–1.40). Further evidence is anticipated to compare incidence of AD or all-cause dementia in patients receiving either NOACs or warfarin.
CONCLUSION
Although warfarin and NOACs are effective in preventing CES in patients with AF, future research is needed to ascertain whether anticoagulation therapy is also effective for dementia. In particular, insufficient evidence is currently available on whether warfarin and NOACs are effective in the prevention of onset and progression of AD. This is also the case in efficacy of rhythm-, or rate-control, therapy for the prevention of AD in AF patients, requiring multiple observational studies with long-term follow-up and large-scale randomized controlled trials with comprehensive neurocognitive assessment and brain imaging. However, as many believe we are entering a ‘super-aging society’, there may be little time to wait for more robust evidence. Anticoagulation therapy and rhythm- or rate-control conducted for prevention of AF-related symptoms, such as CES, is expected to lead to reduction of vascular dementia and even AD. For the time being, we must wait for the establishment of such evidence while treating AF patients for prevention of cardiovascular events associated with AF.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Ahmad Khundakar (Newcastle University, UK) for his editorial assistance. Supported by JSPS KAKENHI 15H04271 (MI), and by AMED 15ek0109130s0101 (MI).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0970r1).
REFERENCES
[1] | Marini C , De Santis F , Sacco S , Russo T , Olivieri L , Totaro R , Carolei A ((2005) ) Contribution of atrial fibrillation to incidence and outcome of ischemic stroke: Results from a population-based study. Stroke 36: , 1115–1159. |
[2] | Friberg L , Rosenqvist M , Lindgren A , Terént A , Norrving B , Asplund K ((2014) ) High prevalence of atrial fibrillation among patients with ischemic stroke. Stroke 45: , 2599–2605. |
[3] | Ihara M , Kalaria RN ((2014) ) Understanding and preventing the development of post-stroke dementia. Expert Rev Neurother 14: , 1067–1077. |
[4] | Gross AF , Stern TA ((2013) ) The cognitive impact of atrial fibrillation. Prim Care Companion CNS Disord 15: , pii: PCC.12f01471. |
[5] | Rusanen M , Kivipelto M , Levälahti E , Laatikainen T , Tuomilehto J , Soininen H , Ngandu T ((2014) ) Heart diseases and long-term risk of dementia and Alzheimer’s disease: A population-based CAIDE study. J Alzheimers Dis 42: , 183–191. |
[6] | Ott A , Breteler MM , de Bruyne MC , van Harskamp F , Grobbee DE , Hofman A ((1997) ) Atrial fibrillation and dementia in a population-based study. The Rotterdam Study. Stroke 28: , 316–321. |
[7] | Dublin S , Anderson ML , Haneuse SJ , Heckbert SR , Crane PK , Breitner JCS , McCormick W , Bowen JD , Teri L , McCurry SM , Larson EB ((2011) ) Atrial fibrillation and risk of dementia: A prospective cohort study. J Am Geriatr Soc 59: , 1369–1375. |
[8] | Saito S , Ihara M ((2014) ) New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front Aging Neurosci 6: , 290. |
[9] | Bunch TJ , Weiss JP , Crandall BG , May HT , Bair TL , Osborn JS , Anderson JL , Muhlestein JB , Horne BD , Lappe DL , Day JD ((2010) ) Atrial fibrillation is independently associated with senile, vascular, and Alzheimer’s dementia. Hear Rhythm 7: , 433–437. |
[10] | Di Nisio M , Prisciandaro M , Rutjes AW , Russi I , Maiorini L , Porreca E ((2015) ) Dementia in patients with atrial fibrillation and the value of the Hachinski ischemic score. Geriatr Gerontol Int 15: , 770–777. |
[11] | Marengoni A , Qiu C , Winblad B , Fratiglioni L ((2011) ) Atrial fibrillation, stroke and dementia in the very old: A population-based study. Neurobiol Aging 32: , 1336–1337. |
[12] | Kalantarian S , Stern TA , Mansour M , Ruskin JN ((2013) ) Cognitive impairment associated with atrial fibrillation: A meta-analysis. Ann Intern Med 158: , 338–346. |
[13] | Schneider JA , Arvanitakis Z , Bang W , Bennett DA ((2007) ) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69: , 2197–2204. |
[14] | Rastas S , Verkkoniemi A , Polvikoski T , Juva K , Niinistö L , Mattila K , Länsimies E , Pirttilä T , Sulkava R ((2007) ) Atrial fibrillation, stroke, and cognition: A longitudinal population-based study of people aged 85 and older. Stroke 38: , 1454–1460. |
[15] | Pendlebury ST , Rothwell PM ((2009) ) Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and meta-analysis. Lancet Neurol 8: , 1006–1018. |
[16] | Poggesi A , Inzitari D , Pantoni L ((2015) ) Atrial fibrillation and cognition. Stroke 46: , 3316–3321. |
[17] | Marzona I , O’Donnell M , Teo K , Gao P , Anderson C , Bosch J , Yusuf S ((2012) ) Increased risk of cognitive and functional decline in patients with atrial fibrillation: Results of the ONTARGET and TRANSCEND studies. CMAJ 184: , 329–E336. |
[18] | Kalaria RN , Akinyemi R , Ihara M ((2016) ) Stroke Injury, cognitive impairment and vascular dementia. Biochim Biophys Acta 1862: , 915–925. |
[19] | Shibata M , Ohtani R , Ihara M , Tomimoto H ((2004) ) White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 35: , 2598–2603. |
[20] | Ihara M , Tomimoto H ((2011) ) Lessons from a mouse model characterizing features of vascular cognitive impairment with white matter changes. J Aging Res 2011: , 978761. |
[21] | Yao H , Sadoshima S , Kuwabara Y , Ichiya Y , Fujishima M ((1990) ) Cerebral blood flow and oxygen metabolism in patients with vascular dementia of the Binswanger type. Stroke 21: , 1694–1699. |
[22] | Ihara M, Tomimoto H, Ishizu K, Mukai T, Yoshida H, Sawamoto N, Inoue M, Doi T, Hashikawa K, Konishi J, Shibasaki H, Fukuyama H ((2004) ) Decrease in cortical benzodiazepine receptors in symptomatic patients with leukoaraiosis: A positron emission tomography study. Stroke 35: , 942–947. |
[23] | Pullicino P , Raynor S ((2013) ) Is low cardiac ejection fraction a risk factor for stroke? Malta Med J 25: , 10–17. |
[24] | Pai RG , Varadarajan P ((2007) ) Prognostic significance of atrial fibrillation is a function of left ventricular ejection fraction. Clin Cardiol 30: , 349–354. |
[25] | Dublin S , Anderson ML , Heckbert SR , Hubbard RA , Sonnen JA , Crane PK , Montine TJ , Larson EB ((2014) ) Neuropathologic changes associated with atrial fibrillation in a population-based autopsy cohort. J Gerontol A Biol Sci Med Sci 69: , 609–615. |
[26] | Akinyemi RO , Mukaetova-Ladinska EB , Attems J , Ihara M , Kalaria RN ((2013) ) Vascular risk factors and neurodegeneration in ageing related dementias: Alzheimer’s disease and vascular dementia. Curr Alzheimer Res 10: , 642–653. |
[27] | Lavy S , Stern S , Melamed E , Cooper G , Keren A , Levy P ((1980) ) Effect of chronic atrial fibrillation on regional cerebral blood flow. Stroke 11: , 35–38. |
[28] | Anselmino M , Scarsoglio S , Saglietto A , Gaita F , Ridolfi L ((2016) ) Transient cerebral hypoperfusion and hypertensive events during atrial fibrillation: A plausible mechanism for cognitive impairment. Sci Rep 6: , 28635. |
[29] | Jefferson AL , Beiser AS , Himali JJ , Seshadri S , O’Donnell CJ , Manning WJ , Wolf PA , Au R , Benjamin EJ ((2015) ) Low cardiac index is associated with incident dementia and Alzheimer disease: The Framingham Heart Study. Circulation 131: , 1333–1339. |
[30] | Troncone L , Luciani M , Coggins M , Wilker EH , Ho C-Y , Codispoti KE , Frosch MP , Kayed R , del Monte F ((2016) ) Aβ amyloid pathology affects the hearts of patients with Alzheimer’s disease. J Am Coll Cardiol 68: , 2395–2407. |
[31] | Rosenberg MA , Manning WJ ((2012) ) Diastolic dysfunction and risk of atrial fibrillation. Circulation 126: , 2353–2362. |
[32] | Pluta R , Furmaga-Jabłońska W , Maciejewski R , Ułamek-Kozioł M , Jabłoński M ((2013) ) Brain ischemia activates β- and γ-secretase cleavage of amyloid precursor protein: Significance in soradic Alzheimer’s disease. Mol Neurobiol 47: , 425–434. |
[33] | Velliquette RA , O’Connor T , Vassar R ((2005) ) Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: Possible early events in Alzheimer’s disease pathogenesis. J Neurosci 25: , 10874–10883. |
[34] | Sun X , He G , Qing H , Zhou W , Dobie F , Cai F , Staufenbiel M , Huang LE , Song W ((2006) ) Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A 103: , 18727–18732. |
[35] | Fukumoto H , Rosene DL , Moss MB , Raju S , Hyman BT , Irizarry MC ((2004) ) Beta-secretase activity increases with aging in human, monkey, and mouse brain. Am J Pathol 164: , 719–725. |
[36] | Niwa K , Kazama K , Younkin L , Younkin SG , Carlson GA , Iadecola C ((2002) ) Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol 283: , 315–H323. |
[37] | Andin U , Gustafson L , Passant U , Brun A ((2005) ) A clinico-pathological study of heart and brain lesions in vascular dementia. Dement Geriatr Cogn Disord 19: , 222–228. |
[38] | Hawkes CA , Härtig W , Kacza J , Schliebs R , Weller RO , Nicoll JA , Carare RO ((2011) ) Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol 121: , 431–443. |
[39] | Okamoto Y , Yamamoto T , Kalaria RN , Senzaki H , Maki T , Hase Y , Kitamura A , Washida K , Yamada M , Ito H , Tomimoto H , Takahashi R , Ihara M ((2012) ) Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol 123: , 381–394. |
[40] | Rennels ML , Gregory TF , Blaumanis OR , Fujimoto K , Grady PA ((1985) ) Evidence for a “paravascular” fluid circulation in the mammalian central nervous system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res 326: , 47–63. |
[41] | Arbel-Ornath M , Hudry E , Eikermann-Haerter K , Hou S , Gregory JL , Zhao L , Betensky RA , Frosch MP , Greenberg SM , Bacskai BJ ((2013) ) Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol 126: , 353–364. |
[42] | Iliff JJ , Wang M , Zeppenfeld DM , Venkataraman A , Plog BA , Liao Y , Deane R , Nedergaard M ((2013) ) Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci 33: , 18190–18199. |
[43] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[44] | Yao Z , Zhang J , Xie X ((2012) ) Enriched environment prevents cognitive impairment and tau hyperphosphorylation after chronic cerebral hypoperfusion. Curr Neurovasc Res 9: , 176–184. |
[45] | Gudala K , Bansal D , Schifano F , Bhansali A ((2013) ) Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J Diabetes Investig 4: , 640–650. |
[46] | Abner EL , Nelson PT , Kryscio RJ , Schmitt FA , Fardo DW , Woltjer RL , Cairns NJ , Yu L , Dodge HH , Xiong C , Masaki K , Tyas SL , Bennett DA , Schneider JA , Arvanitakis Z ((2016) ) Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement 12: , 882–889. |
[47] | Pruzin JJ , Schneider JA , Capuano AW , Leurgans SE , Barnes LL , Ahima RS , Arnold SE , Bennett DA , Arvanitakis Z ((2016) ) Diabetes, hemoglobin A1C, and regional Alzheimer disease and infarct pathology. Alzheimer Dis Assoc Disord 31: , 1. |
[48] | Nelson PT , Abner EL , Schmitt FA , Kryscio RJ , Jicha GA , Smith CD , Davis DG , Poduska JW , Patel E , Mendiondo MS , Markesbery WR ((2010) ) Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol 20: , 66–79. |
[49] | Sposato LA , Ruíz Vargas E , Riccio PM , Toledo JB , Trojanowski JQ , Kukull WA , Cipriano LE , Nucera A , Whitehead SN , Hachinski V ((2017) ) Milder Alzheimer’s disease pathology in heart failure and atrial fibrillation. Alzheimers Dement 13: , 770–777. |
[50] | Bangen KJ , Nation DA , Delano-Wood L , Weissberger GH , Hansen LA , Galasko DR , Salmon DP , Bondi MW ((2015) ) Aggregate effects of vascular risk factors on cerebrovascular changes in autopsy-confirmed Alzheimer’s disease. Alzheimers Dement 11: , 394–403. |
[51] | Sanna T , Diener H-C , Passman RS , Di Lazzaro V , Bernstein RA , Morillo CA , Rymer MM , Thijs V , Rogers T , Beckers F , Lindborg K , Brachmann J ((2014) ) Cryptogenic stroke and underlying atrial fibrillation. N Engl J Med 370: , 2478–2486. |
[52] | Neuropathology, Group, Medical Research Council Cognitive Function, Aging (MRC, CFAS) ((2001) ) Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Lancet 357: , 169–175. |
[53] | van Veluw SJ , Shih AY , Smith EE , Chen C , Schneider JA , Wardlaw JM , Greenberg SM , Biessels GJ ((2017) ) Detection, risk factors, and functional consequences of cerebral microinfarcts. Lancet Neurol 16: , 730–740. |
[54] | Okamoto Y , Ihara M , Fujita Y , Ito H , Takahashi R , Tomimoto H ((2009) ) Cortical microinfarcts in Alzheimer’s disease and subcortical vascular dementia. Neuroreport 20: , 990–996. |
[55] | Salazar CA , del Aguila D , Cordova EG ((2014) ) Direct thrombin inhibitors versus vitamin K antagonists for preventing cerebral or systemic embolism in people with non-valvular atrial fibrillation. In Cochrane Database of Systematic Reviews, Salazar CA, ed. John Wiley & Sons, Ltd, Chichester, UK, pp. CD009893. |
[56] | Jacobs V , Woller SC , Stevens S , May HT , Bair TL , Anderson JL , Crandall BG , Day JD , Johanning K , Long Y , Mallender C , Olson JL , Osborn JS , Weiss JP , Bunch TJ ((2014) ) Time outside of therapeutic range in atrial fibrillation patients is associated with long-term risk of dementia. Hear Rhythm 11: , 2206–2213. |
[57] | Bunch TJ , Crandall BG , Weiss JP , May HT , Bair TL , Osborn JS , Anderson JL , Muhlestein JB , Horne BD , Lappe DL , Day JD ((2011) ) Patients treated with catheter ablation for atrial fibrillation have long-term rates of death, stroke, and dementia similar to patients without atrial fibrillation. J Cardiovasc Electrophysiol 22: , 839–845. |
[58] | Cacciatore F , Testa G , Langellotto A , Galizia G , Della-Morte D , Gargiulo G , Bevilacqua A , Del Genio MT , Canonico V , Rengo F , Abete P ((2012) ) Role of ventricular rate response on dementia in cognitively impaired elderly subjects with atrial fibrillation: A 10-year study. Dement Geriatr Cogn Disord 34: , 143–148. |
[59] | Chung MK , Shemanski L , Sherman DG , Greene HL , Hogan DB , Kellen JC , Kim SG , Martin LW , Rosenberg Y , Wyse DG , Investigators AFFIRM ((2005) ) Functional status in rate- versus rhythm-control strategies for atrial fibrillation: Results of the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) Functional Status Substudy. J Am Coll Cardiol 46: , 1891–1899. |
[60] | Jacobs V , May HT , Bair TL , Crandall BG , Cutler MJ , Day JD , Mallender C , Osborn JS , Stevens SM , Weiss JP , Woller SC , Bunch TJ ((2016) ) Long-term population-based cerebral ischemic event and cognitive outcomes of direct oral anticoagulants compared with warfarin among long-term anticoagulated patients for atrial fibrillation. Am J Cardiol 118: , 210–214. |
[61] | Friberg L , Rosenqvist M ((2017) ) Less dementia with oral anticoagulation in atrial fibrillation. Eur Heart J, doi: 10.1093/eurheartj/ehx579 |


