
Tau Proteolysis in the Pathogenesis of Tauopathies: Neurotoxic Fragments and Novel Biomarkers
Abstract
With predictions showing that 131.5 million people worldwide will be living with dementia by 2050, an understanding of the molecular mechanisms underpinning disease is crucial in the hunt for novel therapeutics and for biomarkers to detect disease early and/or monitor disease progression. The metabolism of the microtubule-associated protein tau is altered in different dementias, the so-called tauopathies. Tau detaches from microtubules, aggregates into oligomers and neurofibrillary tangles, which can be secreted from neurons, and spreads through the brain during disease progression. Post-translational modifications exacerbate the production of both oligomeric and soluble forms of tau, with proteolysis by a range of different proteases being a crucial driver. However, the impact of tau proteolysis on disease progression has been overlooked until recently. Studies have highlighted that proteolytic fragments of tau can drive neurodegeneration in a fragment-dependent manner as a result of aggregation and/or transcellular propagation. Proteolytic fragments of tau have been found in the cerebrospinal fluid and plasma of patients with different tauopathies, providing an opportunity to develop these fragments as novel disease progression biomarkers. A range of therapeutic strategies have been proposed to halt the toxicity associated with proteolysis, including reducing protease expression and/or activity, selectively inhibiting protease-substrate interactions, and blocking the action of the resulting fragments. This review highlights the importance of tau proteolysis in the pathogenesis of tauopathies, identifies putative sites during tau fragment-mediated neurodegeneration that could be targeted therapeutically, and discusses the potential use of proteolytic fragments of tau as biomarkers for different tauopathies.
INTRODUCTION
The microtubule-associated protein tau (MAPT) undergoes a range of modifications in many different types of dementia, classified as tauopathies, such as Alzheimer’s disease (AD), corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP) (reviewed by [1]). In tauopathies, tau loses its physiological role of promoting microtubule (MT) formation and cellular stability (reviewed by [2]) as a result of hyperphosphorylation. At the same time tau exerts a pathological effect in the form of both toxic tau oligomers [3] and other soluble tau species [4] which are then incorporated into neurofibrillary tangles (NFTs) [5]. Both tau oligomers and NFTs are found in postmortem brain samples from patients with tauopathies (reviewed in [6]). The MAPT gene undergoes alternative splicing to produce six different central nervous system (CNS) isoforms of tau (Fig. 1), which differ in the presence of the N-terminal inserts N1 and N2 and the second repeat domain (R2) (reviewed by [2]). N1, N2 and R2 are encoded by exons 2, 3, and 10, respectively. Mutations of the MAPT gene have been identified in specific tauopathies, such as frontotemporal dementia (FTD), and cluster in the repeat domains (reviewed by [7]). Although the structure and role of tau in disease have been extensively reviewed [2, 8–10], this review will focus on tau proteolysis, the role of tau proteolytic fragments in disease pathogenesis and their potential use as biomarkers for diagnosing different types of dementia and/or following disease progression.
Fig.1
Schematic of full-length human tau (2N4R; 441 amino acids) and the other 5 central nervous system isoforms produced due to alternative splicing of the MAPT gene. Tau is divided into an N-terminal projection domain and a C-terminal microtubule-binding domain. N1 and N2 are the N-terminal inserts, P1 and P2 are the proline-rich domains, R1, R2, R3 and R4 make up the repeat domain and R’ is the flanking domain. Within the repeat domain there are two sequences required for aggregation of the tau protein, 275VQIINK280 and 306VQIVYK311. Letters refer to the single amino acid code while the number refers to the position along the length of the tau441 isoform. The splicing name is provided on the left-hand side and these contain 0, 1 or 2 N-terminal inserts and 3 or 4 repeat domains. Figure adapted from [143–145].
![Schematic of full-length human tau (2N4R; 441 amino acids) and the other 5 central nervous system isoforms produced due to alternative splicing of the MAPT gene. Tau is divided into an N-terminal projection domain and a C-terminal microtubule-binding domain. N1 and N2 are the N-terminal inserts, P1 and P2 are the proline-rich domains, R1, R2, R3 and R4 make up the repeat domain and R’ is the flanking domain. Within the repeat domain there are two sequences required for aggregation of the tau protein, 275VQIINK280 and 306VQIVYK311. Letters refer to the single amino acid code while the number refers to the position along the length of the tau441 isoform. The splicing name is provided on the left-hand side and these contain 0, 1 or 2 N-terminal inserts and 3 or 4 repeat domains. Figure adapted from [143–145].](https://ip.ios.semcs.net:443/media/jad/2018/63-1/jad-63-1-jad170959/jad-63-jad170959-g001.jpg)
TAU PROTEOLYSIS
There are many different post-translational modifications (PTMs) that affect tau, including acetylation, deamidation, glycation, glycosylation, isomerisation, methylation, nitration, phosphorylation, proteolysis, sumoylation and ubiquitylation (reviewed by [11]). The different PTMs that map along the tau protein and their role in disease have been summarised on the online interactive tool (http://www.tauptm.org/) [12]. PTM-specific antibodies have been characterised using a range of different techniques and sources of tau such as synthetic peptides, mouse brain and induced pluripotent stem cell-derived human cortical neurons [12]. Recently mass spectrometry (MS)-based assays have been used to study the global PTM profile of tau [13]. These PTMs can alter the propensity of tau to aggregate and its ability to undertake its physiological role. Phosphorylation is the most characterised and investigated PTM of tau, due to its ability to affect tau binding to MTs and the presence of 45 phosphorylation sites along the length of the protein. The increased levels of hyperphosphorylated tau in a number of tauopathies has been thoroughly reviewed [14–16].
The proteolysis of tau has recently sparked much research interest because of its ability to exacerbate tau aggregation [17]. This is likely to occur due to an initial disruption of the primary structure of tau and/or the generation of neurotoxic tau fragments (Fig. 2). PTMs of tau can destabilise its primary structure preventing correct folding into the paper-clip like tertiary structure (reviewed by [2]) and can lead to the formation of aggregated tau species due to a disordered quaternary structure (reviewed by [18]). Proteolysis of tau could also play a beneficial role within the cell via the clearance and recycling of aberrantly modified pathological forms of tau (reviewed by [19]). Fragments of tau have been found in cerebrospinal fluid (CSF) and plasma of patients with different tauopathies, making them potentially novel biomarkers for disease progression (reviewed by [20]). Proteolytic fragments of tau can be classified by their cleavage site in full-length tau, their function, the responsible protease, their potential use as a biomarker and their presence in disease as illustrated in Table 1.
Fig.2
Schematic of tau proteolysis and its contribution to tauopathy pathogenesis. The physiological role of tau is binding to microtubules and supporting their function, during tauopathy pathogenesis, proteases cleave both 3- and 4-repeat tau into fragments which have been shown to be neurotoxic. Proteolytic fragments of tau can have an increased propensity to be phosphorylated and aggregate into neurofibrillary tangles. Proteolytic cleavage of tau can cause its detachment from the microtubule potentially leading to microtubule collapse (loss of normal function). Neurotoxic fragments, neurofibrillary tangles and other aggregated species such as oligomers and the collapse of microtubules (gain of toxic function) all contribute to the synaptic and neuronal loss underpinning tauopathy-associated neurodegeneration.
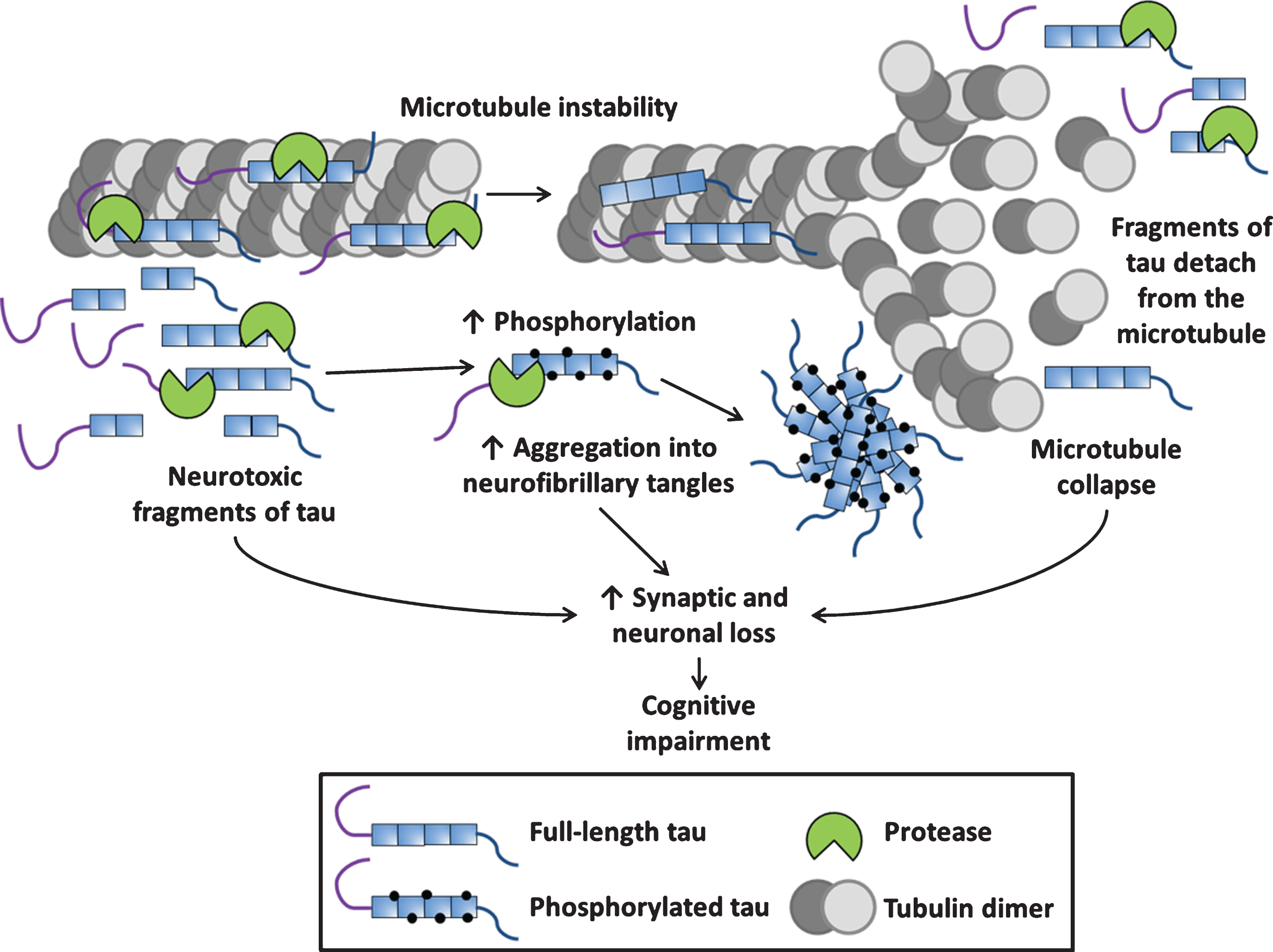
Table 1
Known fragments of tau and their cleavage site, responsible protease, function, use as a biomarker and presence in disease
| Fragment | Cleavage site | Function | Protease if known | Biomarker use | Disease presence |
| 1–13 | D13-H14 | Unknown | Caspase-6 [40]. | Unknown | Unknown |
| 14–441 | D13-H14 | Possible role in tangle maturation [134]. | Caspase-6 [40]. | Unknown | Unknown |
| 1–25 | D25-Q26 | No toxicity observed when overexpressed in neurons [135]. | Caspase-3 [135]. | Unknown | Unknown |
| 26–230 (20-22kDa fragment) | D25-Q26 and R230-T231 | Enriched in synaptic mitochondria and binds to Aβ peptides, exacerbating mitochondrial dysfunction [136]. Caused NMDAR-mediated cell death in rat CGCs [135]. | Caspase-3 [135] and Calpain-1 [33] and -2 [34]. | Increased CSF levels in AD and non-AD associated dementia compared to cognitively unimpaired neurological disease patients [128]. However, no correlation was seen between the CSF 20-22kDa fragment and cognitive decline [128]. | Present in AD11 mice [41] and in AD, DLB and PD patient brains [136]. |
| 1–314 (Δtau314) | D314-L315, unknown whether it undergoes further N-terminal cleavage [42]. | Lower propensity to aggregate compared to tau441. Exerts its effects on P301L tau, causes it to infiltrate dendritic spines, impair synaptic transmission and drive hippocampal neuronal loss, particularly in the CA1. Tau1 - 314 alone not sufficient to affect memory or synaptic function [42]. | Caspase-2, however, in vitro data suggests it preferentially cleaves at D421-S422 instead [42]. | Unknown | Elevated in brains of rTg4510 mice [42]. Also found to be increased compared to control in brains from both MCI and AD patients [42]. |
| 315–441 | D314-L315 | Unknown | Caspase-2 [42]. | Unknown | Unknown |
| 26–44 | D25-Q26 and K44-E45 | Caused NMDAR-mediated cell death in rat CGC’s [135]. | Caspase-3 [135] and calpain-1 [32]. | Unknown | Unknown |
| 1–44 | K44-E45 | Caused NMDAR-mediated cell death in rat CGC’s [135]. | Calpain-1 [32]. | Unknown | Unknown |
| 45–441 | K44-E45 | Unknown | Calpain-1 [32]. | Unknown | Unknown |
| 1–152 | A152-T153 | Unknown | ADAM10 [78]. | Unknown | Unknown |
| 153–441 (Tau-A) | A152-T153 | Unknown | ADAM10 [78]. | Found in serum from patients with AD and inversely correlates with cognitive test scores [78] and health risk factors [133]. | Found in serum from rTg4510 mice as well as patients with mild-moderate AD [78]. |
| 1–156 | G156-A157 | Caused NMDAR-mediated cell death in rat CGC’s [135]. | Unknown | Unknown | Unknown |
| 1–155 | R155-G156 | Unknown | Purified human thrombin incubated with PHF tau extracted from AD brains resulted in cleavage at multiple peptide bonds [137]. | Unknown | Unknown |
| 156–441 | R155-G156 | Unknown | Thrombin [137]. | Unknown | Unknown |
| 156–209 | R155-G156 and R209-S210 | Unknown | Thrombin [137]. | Unknown | Unknown |
| 210–441 | R209-S210 | Unknown | Thrombin [137]. | Unknown | Unknown |
| 210–230 | R209-S210 and R230-T231 | Unknown | Thrombin [137], Calpain-1 [33] and -2 [34]. | Unknown | Unknown |
| 231–441 | R230-T231 | Unknown | Thrombin [137], Calpain-1 [33] and -2 [34]. | Unknown | Unknown |
| 1–197 (Projection domain) | Y197-S198 | Unknown | Chymotrypsin [138]. | Unknown | Unknown |
| 198–441 (MT-binding domain) | Y197-S198 | Unknown | Chymotrypsin [138]. | Unknown | Unknown |
| 3–230 | A2-E3 and R230-T231 | Unknown | Calpain-2 [34] and thrombin [137], Calpain-1 [33] and -2 [34]. | Unknown | Unknown |
| 45–230 (17kDa fragment) | K44-E45 and R230-T231 | Transgenic tau45 - 230 mice expressing this fragment exhibited synapse loss and behavioural abnormalities [35]. Impairs organelle transport [36]. | Calpain-1 [32] and thrombin [137], Calpain-1 [33] and -2 [34]. | Unknown | Present in AD, CBD and PSP, but not seen in control brain samples [139]. |
| 1–242 | R242-L243 | Unknown | Calpain-1 [37]. | Unknown | Unknown |
| 243–441 (Tau-CTF24) | R242-L243 | Accelerates intracellular propagation of tau and has reduced capacity for promoting MT assembly compared to tau441 [37]. | Calpain-1 [37]. | Yes | Increased with age in Tg601 mice brains [37]. Present in brains from AD and familial FTLD containing the tau mutation (N279K) [37]. |
| 125–230 | Q124-A125 and R230-T231 | Fragment not toxic [34]. | Calpain-2 [34] and thrombin [137], Calpain-1 [33] and -2 [34]. | No | Present in some AD and control brains [34]. |
| 3–124 | A2-E3 and Q124-A125 | Unknown | Calpain-2 [34]. | Unknown | Unknown |
| 1–255 | N255-V256 | Failed to induce MT polymerization and aggregate into PHFs; however, tau1 - 255 had strong AT8 (phosphorylated tau at S202 and T205) immunoreactivity [80]. | AEP [80]. | No | No |
| 256–441 | N255-V256 | Greatly reduced ability to induce MT polymerization but tau256 - 441 had increased propensity to aggregate into PHFs compared to tau441 [80]. | AEP [80]. | No | No |
| 256–368 | N255-V256 and N368-K369 | Triggered substantial apoptosis compared to other AEP-cleaved tau fragments or tau441. Moderately increased MT polymerization, as well as increased propensity to aggregate into PHFs compared to tau441 [80]. | AEP [80]. | No | No |
| 1–368 | N368-K369 | Reduced ability to induce MT polymerization and triggered substantial apoptosis compared to other AEP-cleaved tau fragments or tau441. Increased propensity to aggregate into PHFs compared to tau441 and tau1 - 368 also had strong AT8 immunoreactivity [80]. | AEP [80]. | No | Only AEP-cleaved tau fragment present in AD but not found in control brains [80]. |
| 369–441 | N368-K369 | Unable to induce MT polymerization and aggregate into PHFs [80]. | AEP [80]. | No | No |
| 1–402 (TauΔCasp6) | D402-T403 | Unknown but acts as a marker of caspase-6-induced neurodegeneration [57]. | Caspase-6 [43]. | CSF fragment levels distinguished AD from control [53]. | Correlates with tau pathology in human AD olfactory bulb brain sections [57]. |
| 403–441 | D402-T403 | Unknown | Caspase-6 [43]. | Unknown | Unknown |
| 1–421 (Tau-C) | D421-S422 | Faster aggregation rate and forms twice the amount of filament mass compared to tau441 [44]. Cells expressing tau1 - 421 have mitochondria with fragmentation phenotypes [140], increased caspase-3 activity and cell toxicity [141]. | Caspase-1, -3, -6, -7 and -8 [44]. | Tau1 - 421 alone not sufficient as serum based biomarker for AD (p = 0.06) but significant when combined with tau153 - 441 (p = 0.006) [132]. | Found associated with NFTs in AD brains [44, 142]. Found increased in AD, FTD-tau and PSP compared to control brain samples [45]. |
| 422–441 | D421-S422 | Unknown | Caspase-1, -3, -6, -7 and -8 [44]. | Unknown | Unknown |
| 151–421 (Δtau) | K150-I151 and D421-S422 | Co-expressing tau151 - 421 and human tau441 in mice led to tau aggregation and disruption of axonal transport, mitochondria, Golgi apparatus and synaptic proteins [100]. Co-expression led to severe paralysis within 3 weeks which could be prevented by stopping tau151 - 421 expression [100]. | Caspase-1, -3, -6, -7 and -8 cleave at D421-S422 [44] but the protease that cleaves at K150-I151 is unknown. | Tau1 - 421 has been used as a biomarker [132] but fragments produced due to cleavage at K150-I151 have not. | Tau1 - 421 has been found associated with NFTs in AD brains [44, 142]. Tau1 - 421 increased in AD, FTD-tau and PSP compared to control brain samples [45]. Fragments produced due to cleavage at K150-I151 unknown. |
| 2–441 | M1-A2 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 11–441 | V10-M11 | Increased α-tubulin acetylation compared to tau441 [22]. | Unknown | Unknown | AD and control brains [22]. |
| 12–441 | M11-E12 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 103–441 | T102-A103 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 124–441 | T123-Q124 | Cells expressing tau124 - 441 had increased α-tubulin acetylation, stronger binding to MTs and less sensitive to depolymerisation compared to tau441 [22]. | Unknown | Unknown | AD and control brains [22]. |
| 127–441 | R126-M127 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 172–441 | I171-P172 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 174–441 | A173-K174 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 224–441 | P223-K224 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 238–441 | S237-S238 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 240–441 | A239-K240 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 259–441 | S258-K259 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 261–441 | I260-G261 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 280–441 | N279-K280 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 306–441 | S305-V306 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 308–441 | Q307-I308 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 309–441 | I308-V309 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 311–441 | Y310-K311 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 331–441 | H330-K331 | Unknown | Unknown | Unknown | AD and control brains [22]. |
| 1–391 | E391-I392 | Increased aggregation propensity and increased nucleation efficiency compared to tau441 [96]. | Unknown | Unknown | Present at a late stage of aggregation into NFTs [98]. |
| 392–441 | E391-I392 | Unknown | Unknown | Unknown | Unknown |
| 395–441 | Y394-K395 | Unknown | Unknown | Unknown | Unknown |
| C-terminal tau35 fragment | Cleaved somewhere between P182 and R194 [92]. | Causes a mouse model to exhibit defective motor function and spatial learning deficit. Induces alterations in kinase activity, lysosomal and synaptic function [94]. | Unknown | Unknown | Tau35 seen in the brains of patients with AGD [91], CBD, FTD [92], PSP [93], but not in AD [92]. |
Aβ, amyloid-β; AD, Alzheimer’s disease; ADAM10, a disintegrin and metalloprotease; AEP, asparagine endopeptidase; AGD, argyrophilic grain disease; CBD, corticobasal degeneration; CGC, cerebellar granule cells; DLB, dementia with Lewy bodies; FTD, frontotemporal dementia; MCI, mild cognitive impairment; MT, microtubule; NFT, neurofibrillary tangles; NMDAR, N-methyl-D-aspartate receptor; PHF, paired-helical filament; PD, Parkinson’s disease; PSP, progressive supranuclear palsy; Tg, transgenic.
For ease of comparison, we use the numbering of amino acids in the 2N4R (tau441) isoform of tau and the P1-P1’ nomenclature of Schechter and Berger to indicate the amino acids N-terminal and C-terminal to the peptide bond that is cleaved [21]. Although a number of different proteases have been identified to cleave tau (Fig. 3A), many fragments of tau have been found in the brain without an identified protease responsible for their cleavage [22] (Fig. 3B). Here, we first describe the proteases that have been identified to cleave tau, along with the properties of the resulting fragments. We then detail a few of the fragments that have been identified in the brain but for which the protease(s) responsible for generating them have yet to be identified.
Fig.3
Schematic of human tau441 showing the currently identified proteolytic cleavage sites. A) Proteolytic cleavage sites in tau indicated by the P1-P1’ residues with the protease responsible indicated. Caspase-6 [40, 43, 44, 53], caspase-3 [41, 44], calpain-1 [32, 33, 37], a disintegrin and metalloprotease 10 (ADAM10) [78], initial thrombin cleavage site [137], chymotrypsin [138], calpain-2 [34], human high temperature requirement serine protease A1 (HtrA1) [69], asparagine endopeptidase (AEP) [80], acetylation induced auto-proteolysis sites [87], caspase-2 [42], caspases-1, -7, and -8 [44]. B) Identified cleavage sites in tau where the protease responsible has yet to be identified. Data from [22] and cleavage at E391-I392 [98].
![Schematic of human tau441 showing the currently identified proteolytic cleavage sites. A) Proteolytic cleavage sites in tau indicated by the P1-P1’ residues with the protease responsible indicated. Caspase-6 [40, 43, 44, 53], caspase-3 [41, 44], calpain-1 [32, 33, 37], a disintegrin and metalloprotease 10 (ADAM10) [78], initial thrombin cleavage site [137], chymotrypsin [138], calpain-2 [34], human high temperature requirement serine protease A1 (HtrA1) [69], asparagine endopeptidase (AEP) [80], acetylation induced auto-proteolysis sites [87], caspase-2 [42], caspases-1, -7, and -8 [44]. B) Identified cleavage sites in tau where the protease responsible has yet to be identified. Data from [22] and cleavage at E391-I392 [98].](https://ip.ios.semcs.net:443/media/jad/2018/63-1/jad-63-1-jad170959/jad-63-jad170959-g003.jpg)
TAU PROTEASES
Calpains
The calpain family is comprised of fourteen different cysteine-dependent proteases that are activated by calcium binding to a specific site that is conserved in all family members [23, 24]. Calpains are endogenously inhibited by their own substrate, calpastatin, which is able to avoid cleavage by the active site of the protease [25]. The two most common isoforms are calpain-1 and -2, which differ in their response to calcium; calpain-1 is sensitive to micromolar concentrations whereas calpain-2 is sensitive to millimolar concentrations of calcium (reviewed by [26]). Calpain-1 and -2 both cleave tau [reviewed by 19] and play opposing roles in the regulation of synaptic plasticity and inducing neurodegeneration [reviewed by 27]. Although calpains have been implicated in the cleavage of tau for several years, new insights have been uncovered recently regarding cleavage of tau by calpains and the crucial role these proteases play in disease.
Calpain-1 is a major therapeutic target for a range of neurodegenerative disorders, including Huntington’s disease and Parkinson’s disease (PD) (reviewed by [28]). Calpain-1 levels were significantly elevated in AD cortical brain tissue from Braak stages III to VI compared to control samples [29]. Due to compensatory mechanisms within the cell, increases in overall calpastatin levels were observed, the bulk of which was in the inactive form, indicative of calpastatin being proteolytically cleaved by calpain-1 resulting in its inactivation [29]. Upregulation of both calpain-1 and calpastatin preceded an increase in tau phosphorylating kinases [29]. The relationship between calpains and calpastatin is crucial, as shown by the transgenic (Tg) JNPL3 mouse model of tauopathy (P301L tau), which has reduced endogenous calpastatin levels compared to controls [30]. Increasing active calpastatin levels in the JNPL3 mice led to a 3 month lifespan extension, likely due to calpastatin inhibiting calpains which subsequently prevented the production of toxic calpain-cleaved tau fragments [30].
Much of the interest surrounding calpain cleavage of tau has focused on the 17kDa tau45 - 230 fragment, produced due to cleavage by calpain-1 at K44-E45 [32] and calpain-1 [33] or –2 [34] at R230-T231. Specific effects of the fragment were elucidated by the development of a mouse model overexpressing tau45 - 230, which had significantly increased hippocampal pyramidal cell death and synaptic loss, as early as 6 months of age [35]. Alongside the cellular phenotypes, behavioural abnormalities were noted with significantly increased freezing response in the fear conditioning test [35]. Tau45 - 230 was also shown to impair both anterograde and retrograde organelle transport, which could help to explain how it exerts its neurotoxic effects [36]. Which calpain cleaves at R230-T231 in vivo is still unclear, since both calpain-1 and –2 [34] have been shown to cleave this peptide bond in vitro. Recent data showed that tau is also cleaved by calpain-1 at R242-L243 producing a 24kDa C-terminal fragment (CTF), tau243 - 441 [37]. Tau243 - 441 increased with aging in a tauopathy mouse model (Tg601; wild-type human tau) and CTFs between 20 and 28kDa were present in AD and familial FTD brain samples [37]. Tau243 - 441 was able to efficiently propagate to other tau expressing cells, resulting in the seeding of further aggregation and phosphorylation of tau441 [37]. Tau243 - 441 is likely to be the original fragment seen by Park and Ferreira (2005) excluding cleavage at K44-E45, however, further biochemical assays are needed to clarify the exact fragment produced and the protease responsible. Interestingly, build-up of tau441 is able to activate calpain-2 leading to the degradation of the nicotinic acetylcholine receptor subunit α4 [38], which plays a crucial role in cholinergic signalling, one of the first pathways affected in AD pathogenesis. Tau441 activation of calpain-2 is indicative of a positive-feedback loop, which leads to increased cleavage of tau into neurotoxic fragments.
Caspases
Mammalian caspases play a critical role in driving apoptosis and inflammation. The caspase family is comprised of twelve different cysteine-dependent proteases that cleave substrates when aspartic acid is at the P1 position (reviewed by [39]). Cleavage of tau has been observed at the following peptide bonds D13-H14 [40], D25-Q26 [41], D314-L315 [42], D402-T403 [43], and D421-S422 [44] by different members of the caspase family [reviewed by 19]. Tau fragments produced due to cleavage at D314-L315 [42], D402-T403 [43] and D421-S422 [44] have been found in AD brains.
Caspase-2, a protease known to initiate the activation of other caspases, cleaved tau at D314-L315 resulting in the production of a soluble, toxic N-terminal fragment (NTF), termed tau1 - 314 (Δtau314) [42]. Whether this cleavage event is truly significant is still controversial as caspase-2 was shown to preferentially cleave recombinant tau at D421-S422 compared to D314-L315, however, this discrepancy may be due to the nature of the recombinant preparation [42]. In the regulatable Tg4510 (rTg4510) mouse model of tauopathy (human tau; P301L), tau1 - 314 detached from MTs and invaded healthy dendritic spines leading to disrupted synaptic signalling, but toxicity and spatial memory deficits were only observed when tau1 - 314 promoted the dendritic spine mislocalization of full-length tau [42]. These memory deficits could be ameliorated through the use of anti-caspase-2 morpholino oligonucleotides, which reduced caspase-2 levels by approximately 33% [42]. Tau1 - 314 was found to be elevated in mild cognitive impairment (MCI) and AD brains compared to healthy controls [42].
One of the effector caspases, caspase-3, that is activated by initiator caspases, such as caspase-2, cleaves tau at D25-Q26 [41] and D421-S422 [44]. Cleavage at D421-S422 results in the production of the NTF, tau1 - 421 (Tau-C), initially thought to be specific to AD brains [44], however, links to other tauopathies, such as PSP have been recently uncovered [45]. The pro-apoptotic and caspase-3 activator, appoptosin [46], is upregulated by an unknown mechanism in PSP through a single nucleotide polymorphism near the myelin-associated oligodendrocyte basic protein gene (rs1768208C/T) [47]. The T allele, not the C allele, was shown to boost appoptosin expression leading to increased caspase-3 and tau1 - 421 in the PSP cortical neuron samples [45]. The T allele frequency in the small PSP case cohort was 0.77 (n = 26) and 0.32 in the control (n = 22) [45]. To elucidate appoptosin pathological pathways, the authors overexpressed appoptosin in rat primary cortical neurons. These neurons showed increased levels of tau aggregation and increased susceptibility to cell death, which could be exacerbated by treatment with amyloid-β (Aβ) or the mitochondrial protein regulator, mitochondrial processing peptidase [45]. To test appoptosin effects in vivo, JNPL3 mice were injected with an adeno-associated virus containing appoptosin. These mice exhibited biochemical and motor defects which could be halted by the use of the selective caspase-3 inhibitor, Z-DEVD-FMK [45]. Appoptosin and tau1 - 421 levels were increased in postmortem frontal cortex samples from both AD and FTD with tauopathy, suggesting this pathway is common across a range of tauopathies [45].
Tau1 - 421 has increased aggregation propensity compared to tau441 [44], resulting in conformational changes detectable by the MC1 antibody [48], which is specific to a partially folded, pre-paired helical filament (PHF) conformation of tau. This conformational change was interrogated in the murine neuroblastoma Neuro-2A cell line and could be exacerbated by treatment with caspase-activating cell stressors, such as staurosporine, and inhibited through the use of the caspase inhibitor Z-DEVD-FMK [48]. In order to look at the effect of this fragment in vivo, a 0N4R tau expressing Tg mouse was developed, which lacked the 20 C-terminal amino acids (tau1 - 363; TauC3), equivalent to 2N4R tau1 - 421 [49]. Expression of tau1 - 363 was predominantly localised to the CA3 region of the hippocampus and layers I and II of the cerebral cortex [49], but was not observed in wild-type mice. The tau1 - 363 expressing mice did not display significant neurodegeneration or inflammation, however, they exhibited learning and memory impairments, as early as 40 days of age [49], stressing that this fragment may drive early cognitive impairment but may require other proteins, such as Aβ, to elicit further neurodegeneration. Tau1 - 421 forebrain levels in aged wild-type male mice correlated with NFT formation and swim maze test-assessed cognitive decline [50]. Young cognitively impaired mice were also found to have tau1 - 421 in their forebrain [50]. Along with its presence in mouse models and human tauopathy brain samples, tau1 - 421 has been shown to co-localise with caspase-3 within amyloid plaques, blood vessels and NFTs in samples taken from vascular dementia cases [51].
Another effector caspase, caspase-6, is sufficient to cause axonal degeneration, via the proteolytic cleavage of key cytoskeletal proteins, such as α-tubulin and tau [52]. Caspase-6 cleavage of tau at D402-T403 results in the production of the NTF, tau1 - 402, which has been tested as a CSF biomarker for AD [53]. Caspase-6 is upregulated in aging, MCI, sporadic and familial forms of AD, while closely associating with pathogenic markers such as NFTs and amyloid plaques [54, 55]. Alongside elevated levels in the CA1 of the hippocampus and entorhinal cortex, as a key driver of early memory impairments [56], caspase-6 and tau1 - 402 are present in the anterior olfactory nucleus of the olfactory bulb, correlating with tau but not Aβ pathology [57]. The anterior olfactory nucleus, which is critical for the ability of the olfactory bulb to identify odours, is one of the first areas to be affected during AD progression and suffers high levels of neuronal loss [58, 59]. Caspase-6 levels in the anterior olfactory nucleus inversely correlated with cognition, while positively correlating with disease progression [57]. The authors posited that high levels of caspase-6, originating in the anterior olfactory nucleus, could propagate degeneration to the entorhinal cortex leading to the early memory impairments observed in AD [57].
Cathepsins
There are fifteen different cathepsins belonging to the C1 family of papain-like proteases that are ubiquitously expressed within, and responsible for degradation of proteins in, lysosomes, and are further classified by their cleavage specificity, exhibiting either asparagine, cysteine or serine-dependent proteolysis [reviewed by 60]. The lysosomal pathway can become dysregulated during different types of neurodegeneration resulting in leaky lysosomal membranes [61, 62], leading to the release of lysosomal proteases (reviewed by [63]). Alongside lysosomal expression, cathepsins are also found in various other cellular compartments, such as the nucleus and cytoplasm, as well as being localised extracellularly [reviewed by 64], increasing their potential to interact with tau. The role of cathepsins in aging and neurodegeneration has recently been reviewed [64].
Cathepsins B, D, and L have been proposed to proteolytically cleave tau. Cathepsin B has been shown to associate with intracellular NFTs and its expression is elevated surrounding amyloid plaques [65], however, no direct evidence has been found for the proteolysis of tau by cathepsin B. Cathepsin D was shown to cleave recombinant tau at F8-E9, M419-V420 and L436-A437, with another potential cleavage site at either T427-L428 or L428-A429 and further additional cleavage sites in the regions D34-G161, P200-K257 and K267-D358 [66]. Interestingly, phosphorylation of tau exacerbated its proteolytic degradation [66]. Cathepsin L can cleave the mutated aggregation-prone MT-binding domain fragment of tau (tau244 - 372 without K280; tauRDΔK) in Neuro-2A murine cells [67]. Cleavage of tauRDΔK by cathepsin L is dependent on initial cleavage by an unidentified cytosolic protease at K257-S258 producing tau258 - 372, F1, which is subsequently cleaved by cathepsin L at V363-P364 producing tau258 - 363, F2, which in turn is then cleaved at I360-T361 to produce tau258 - 360, F3 [67]. Tau258 - 372 is unable to enter the lysosome and attaches to the lysosomal membrane where it is cleaved into tau258 - 363 and tau258 - 360, which can induce aggregation of intact tauRDΔK and full-length tau coinciding with lysosomal leakage in the Neuro-2A cells as shown by cytosolic release of a lysosomal hydrolase [67]. The presence of cathepsin-cleaved tau fragments in the brains of patients with AD and other tauopathies has yet to be identified, underlying the importance of further work to unravel the pathophysiological relevance of these fragments in human tauopathies.
Human high-temperature requirement serine protease A1 (HtrA1)
Human high-temperature requirement serine protease A1 (HtrA1) is one of the four HtrA family members, which are all ATP-independent serine proteases, that play key roles in protein quality control [reviewed by 68]. HtrA1 cleaves tau within the region essential for aggregation, between residues A239 and V399, with a preference shown for leucine, valine and isoleucine at the P1 position, producing 45 different fragments ranging between 8 and 22 amino acids in length [69]. High levels of HtrA1 correlated with decreased tau, NFTs and amyloid plaques in contralateral frontal cortex AD samples; no such correlation was observed in control patient brains [69]. HtrA1 was later shown to act in a two-step mechanism; initially untangling tau fibrils to expose sites for subsequent cleavage, suggesting that HtrA1 plays a crucial role in initiating tau degradation and preventing aggregate formation [70].
Recently, HtrA1 has been shown to cleave different members of the lipid transporting apolipoprotein E (ApoE) family [71], one of the major genetic risk factors for sporadic AD, with the ApoE4 variant conferring increased risk for AD compared to ApoE3 [reviewed by 72]. This cleavage event occurs in an allele specific manner, with ApoE4 being cleaved more efficiently than ApoE3 [71]. This was then shown to have implications for tau degradation, as HtrA1 cleavage of tau was impaired to a much greater extent by ApoE4 compared to ApoE3, likely due to an allele-dependent competition with tau for degradation by HtrA1 [71].
Puromycin-sensitive aminopeptidase (PSA)
Puromycin-sensitive aminopeptidase (PSA) localises predominantly within the cytoplasm of neuronal cells and was not detected within glial cells [73]. Genomic screens in P301L mice and in Drosophila determined that PSA was able to protect against tau aggregation-induced neurotoxicity [74]. Surprisingly for a protease with aminopeptidase action, recombinant human PSA purified from E. coli cleaved recombinant tau within 4 h, reducing it to approximately 65% of its original size, and within 16 h tau was completely digested [74]. This cleavage was inhibited with the PSA inhibitor, puromycin and a general aminopeptidase inhibitor, bestatin [74]. Further work by Sengupta et al., 2006 [75] showed that PSA cleaved recombinant human tau441 at D13-H14 [75] which is also a known caspase-6 cleavage site [40]. PSA was more efficient in digesting soluble human tau from control brains compared to both soluble and phosphorylated tau from AD brains [75].
However, Chow et al., 2010 [76] have shown that PSA does not directly cleave tau and proposed that it was cleaved as a result of contaminants in the PSA preparations. In contradiction to previous work [74, 75], MS showed that purified PSA from Sf9 cells cleaved tau internally within the MT-binding domain which is atypical for an aminopeptidase and that proteolysis was not blocked by puromycin or bestatin [76]. To test whether this was specific only to this preparation, three more PSA preparations from different sources were generated, however, none of these could cleave tau [76]. No reduction in full-length tau, or the production of any PSA-cleaved tau fragments, was observed in HEK-293 cells transfected with both PSA and tau441 [76]. The authors suggest that an endogenous protease from the Sf9 cells contaminated the preparation and was responsible for the proteolytic cleavage of tau [76]. However, it is important to note that increased expression of PSA is seen in FTD patient brains compared to controls [74], suggesting a potential link to disease pathology, which needs further exploration.
Thrombin
Thrombin is a serine protease, which cleaves tau at multiple peptide bonds (reviewed by [19]). In AD brains, thrombin was upregulated and colocalized with amyloid plaques, microglia and NFTs [77]. However, thrombin-cleaved tau fragments have yet to be found in the brains of patients with AD and other tauopathies, thus questioning the pathophysiological relevance of thrombin cleavage of tau.
A disintegrin and metalloprotease 10 (ADAM10)
The zinc metalloprotease, a disintegrin and metalloprotease 10 (ADAM10), has been reported to cleave tau441 at A152-T153, resulting in the production of the CTF, tau153 - 441 (Tau-A) [78]. Understanding this cleavage event is crucial as a small subset of drugs currently in development for treating AD involve upregulation of ADAM10, the main protease that cleaves amyloid-β protein precursor (AβPP) into non-amyloidogenic species (reviewed by [79]). However, it is currently unclear how the extracellular, membrane-bound ADAM10 physically interacts with intracellular tau, as well as whether tau153 - 441 has any downstream effects. Further experiments to elucidate the mechanism of ADAM10 cleavage of tau as well as studies involving tau153 - 441 transfected cell lines and animal models are required to determine whether this fragment is relevant and has any pathophysiological effects. Probing with the tau153 - 441 neo-epitope specific antibody [78] or with MS analysis in different brain regions are required to determine whether this cleavage event is relevant in AD pathogenesis and whether it correlates with Braak staging.
Asparagine endopeptidase (AEP)
Asparagine endopeptidase (AEP) is a lysosomal cysteine-dependent protease that is only activated under acidic conditions. Acidification of the brain occurs during AD, resulting in increased AEP levels [80]. In addition, AEP translocates from the neuronal lysosomes into the cytoplasm [81], promoted by phosphorylation of AEP by serine-arginine protein kinase 2 resulting in enhanced proteolytic activity [82], where it can interact with, and cleave, alpha-synuclein [83], AβPP [84], and tau [80]. AEP cleavage of tau occurs at both N255-V256 and N368-K369, resulting in the production of five tau fragments: tau1 - 255, tau1 - 368, tau256 - 368, tau256 - 441 and tau369 - 441, although only tau1 - 368 has been found in AD brains [80]. Aβ oligomers activated AEP in a concentration-dependent manner which paralleled increased production of tau1 - 368, leading to neuronal cell loss and reduced MT stability [80]. However, the subcellular location of this cleavage event is yet to be fully elucidated.
Transfection of AEP-cleaved tau fragments into rat primary neurons caused substantial apoptosis, however, this effect was fragment-dependent as tau1 - 368 and tau256 - 368 were the only significant drivers of increased apoptosis; an effect which could be diminished through site-directed mutagenesis of the critical asparagine residues at the P1 position (N255A/N368A) [80]. Tau1 - 368 and tau256 - 368 were shown to aggregate with a higher propensity into PHFs [80]. Tau1 - 255 and tau1 - 368 showed increased levels of phosphorylation at S202 and T205 compared to tau441 [80]. Tau1 - 368 was observed in parallel with synaptic damage and memory deficits in the Tau P301S mouse, which could be blocked by deletion of the AEP gene, lgmn, or mutation (N255A/N368A) of the AEP cleavage sites in tau441 [80].
The potential benefit of inhibiting AEP in a range of neurodegenerative diseases has been thoroughly reviewed [85]. Recently, a natural novel small-molecule AEP inhibitor termed compound 11 has been discovered and characterised [86]. Compound 11 targets both the active site and allosteric modulatory sites on AEP, preventing efficient AEP-substrate interactions [86]. Compound 11 inhibited AEP in a concentration- and time-dependent manner, resulting in a concurrent reduction of tau and APP cleavage both in vitro and in vivo [86]. To further characterise effects in vivo, both the Tau P301S mouse and 5XFAD mouse (APP; K670N/M671L, I716V, V717I and presenilin 1; M146L, L286V), were injected with 10 mg/kg-1 of compound 11 daily from two to five months of age [86]. Compound 11 treatment led to significant reductions in AEP activity, AEP-cleaved tau fragments, synapse loss and electrophysiological impairments, resulting in improved performance in the Morris water maze [86]. This drug meets many of the criteria for further investigation in human clinical trials as it can cross the blood-brain barrier (BBB), exhibits a high degree of selectivity and potency for AEP, was detectable in both serum and brain tissue after chronic dosing, and had no observed systemic toxicity [86].
Auto-proteolysis of tau
Recently, it has been demonstrated that tau self-degrades by acetyl coenzyme A-induced auto-acetylation at cysteine residues without the need for a separate protease [87]. This would occur physiologically when tau dissociates from MTs [87, 88]. Acetylation of tau occurs across the length of the protein and plays an important role in regulating both normal, and disease-associated, properties of tau, due to its ability to impair MT interactions and promote aggregation [89, 90]. Auto-acetylation of tau occurs in an isoform-dependent manner, predominantly within the MT-binding domains at C291 and C322, leading to auto-proteolysis at both K281-L282 and K340-S341 (Fig. 3A). Auto-proteolysis results in the production of two CTFs, tau282 - 441 and tau341 - 441 [87]. Auto-proteolysis could be prevented via modifying C291 and C322 acetylation levels with both pharmacological and genetic techniques [87]. It will be interesting to further investigate, with cleavage site-specific antibodies or MS, whether tau282 - 441 and tau341 - 441 are present in brain, CSF or plasma during tauopathy pathogenesis, and if these CTFs have any downstream effects. Auto-proteolysis highlights how one PTM (acetylation) can alter the propensity of tau to undergo further PTM (auto-proteolysis), highlighting that other proteolytic cleavage events in tau may be modulated by other PTMs such as phosphorylation, sumoylation and ubiquitylation.
Disease relevant fragments without identified proteases
Recently, a major characterisation study of tau CTFs was undertaken in twelve brain samples, taken from both control and late Braak stage AD cortices [22]. This work identified twenty-one novel proteolytic fragments of tau (Fig 3B) [22] which do not, as yet, have a protease identified for their generation. The cleavage sites were uncovered through the use of liquid chromatography coupled to tandem MS analysis, but further work is needed to develop cleavage site-specific antibodies coupled to a MS screen to dissociate which fragments are present in particular tauopathies, and relevant to disease progression. In order to investigate whether any of the identified fragments had biological effects, two CTFs, tau11 - 441 and tau124 - 441, were transfected into the mouse neuroblastoma N1E-115 cell line, resulting in altered tau fragment phosphorylation and α-tubulin acetylation levels, with tau124 - 441 exerting a more prominent phenotype [22]. Tau124 - 441 bound more strongly to MTs compared to tau441 and tau11 - 441, providing more protection against treatment with the MT destabilising drug, nocodazole [22]. Another CTF, termed tau35, due to its predicted size from sodium dodecyl sulfate-polyacrylamide gel electrophoresis, has been found in the brains of patients with argyrophilic grain disease [91], CBD, FTD [92] and PSP [93], but not in AD [92]. The CTF tau35, expressed at low levels in Tau35 mice, induces progressive cognitive and motor deficits, as well as reducing lifespan, which could be reversed by treatment with the histone deacetylase inhibitor, sodium 4-phenylbutyrate [94] that is currently in trials to treat a range of neurodegenerative disorders [95]. The protease responsible for cleavage, as well as the exact cleavage site are unknown, however, tau35 is known to contain all repeat domains of tau and is extensively phosphorylated at a range of C-terminal residues [92].
ROLE OF TAU PROTEOLYTIC FRAGMENTS IN DISEASE
There are upwards of 50 different proteolytic fragments of tau that have been proposed to play a role in the pathogenesis of tauopathies (Table 1). Fragments of tau have been shown to transcellularly propagate and aggregate in a manner dependent on their amino acid composition [37, 80, 96]. Fragments of tau make up the protease-resistant PHF and straight filament (SF) core of NFTs [97]. Proteolytic fragments of the core typically contain the third and fourth repeat domains, but sometimes also have the first and second repeat domains, depending on the 3R:4R ratio of tau in the core [97]. These fragments of the core are approximately 12kDa in size [98]. The fragment composition of the protease-resistant cores varies between the different tauopathies [99]. The SF and PHF core extracted from an AD brain has recently been resolved to atomic level detail through cryo-electron microscopy, indicating a β-sheet helical core between residues 306–378 that defines the seed for aggregation [97].
Tau151 - 421 (Δtau) consists of the first proline region through to a caspase cleavage site at D421-S422 [100]. Tau151 - 421 was expressed in C57BL/6J mouse oocytes under control of an inducible transcriptional silencer element (Tau62) [100]. These mice were then crossed with P301S, ALZ17 (2N4R) or ALZ31 (0N3R) Tg mice [100]. Only P301S crossed with Tau62 mice exhibited paralysis within 3 weeks of age, but all double Tg mice showed deposits of aggregated tau as well as a range of cellular defects, such as the formation of non-filamentous tau aggregates, and disruption of axonal transport, mitochondria, Golgi apparatus and synaptic proteins [100]. These defects could be prevented by turning off tau151 - 421 expression in the presence of 4R tau but not in the presence of 3R tau [100]. These behavioural and biochemical effects were not observed in the mice that only expressed the full-length human tau isoforms, indicative of the disruptive nature of tau151 - 421 in the presence of the full-length protein [100]. However, tau151 - 421 has not been detected in the brains of tauopathy patients. The exact disease relevance of this work is still to be determined but it stresses how tau fragments and full-length tau can work synergistically to induce severe tauopathy-related phenotypes.
Another fragment, tau45 - 230, has recently been implicated in amyotrophic lateral sclerosis, a neurological disease not currently recognised as a tauopathy. However, tau45 - 230 aggregates were shown to accumulate in both upper and lower motor neurons [101]. Tau26 - 230 is produced due to cleavage at D25-Q26 by caspase-3 [41] and by calpain-1 [33] or –2 [34] at R230-T231. When applied extracellularly to rat hippocampal neuronal cultures, the smallest sequence of the tau26 - 230 fragment known to drive neurotoxicity, tau26 - 44, could impair presynaptic signalling, reduce presynaptic protein levels, and breakdown MTs and neurites, in its native monomeric form without the need for aggregation and neurodegeneration [102]. Importantly, the effects could not be replicated with the reverse sequence, tau44 - 26 [102]. Tau26 - 44 represents a therapeutic target, as passive immunisation with N-terminally targeting antibodies reduced tau hyperphosphorylation and improved cognition in 3xTgAD mice (APP; KM670/671NL, MAPT; P301L and presenilin 1; M146V) [103]. Antibodies against tau, including ones targeting the N-terminal region, are currently being tested in human clinical trials for tauopathies [104].
SECRETION OF TAU FRAGMENTS
Full-length and various proteolytic fragments of tau have been shown to be secreted from cells in a variety of different cell and animal models (reviewed by [105]), potentially underlying the distinct spread of neurofibrillary tangle pathology seen in tauopathies (reviewed by [106]). Fragments of tau are secreted in a proline-rich region-dependent manner compared to full-length tau in COS-7 kidney fibroblast cells; fragments lacking this region were not secreted or were secreted via a different mechanism to full-length tau [107]. The sequence-dependent secretion effects were further tested in COS-7 cells, with tau1 - 249 shown to be secreted whilst the MT-binding domain containing tau250 - 370 was not [107]. The authors also found that the N-terminal domain of tau was not responsible for secretion, as the 0N3R isoform was secreted as efficiently as the 2N4R isoform [107]. Further fragment-dependent effects have been observed in HeLa cells showing that, tau1 - 421 undergoes enhanced aggregation and secretion compared to tau441, an effect which could be exacerbated with phosphorylation [108]. The secretion of both tau1 - 421 and tau441 was not due to encapsulation within extracellular vesicles such as exosomes or microvesicles, as removal of this fraction did not alter the amount of secreted tau [108]. This backs up work showing that between 2-3% of extracellular tau is exosomal [109, 110], 7% is ectosomal and 90% comprises free tau [110]. The secretion of different species of tau by mouse neuroblastoma N2a cells, induced pluripotent stem cell-derived human cortical neurons and primary rat hippocampal neurons was investigated [111]. The authors showed that in all cell types tested, the majority of actively and passively secreted tau by living and dead cells, respectively, was in the form of NTFs, while CTFs were only passively secreted within these cultures [111]. In order to mimic more disease relevant scenarios, primary rat hippocampal neurons were treated with a partially aggregated preparation of Aβ42 which caused neuronal compromise and cell death resulting in a time-dependent increase of all species of extracellular tau [111]. The mechanism of secretion of NTFs into the extracellular space was not dependent on exosomes as <0.2% was encapsulated in exosomes [111], in agreement with [108]. To investigate whether tau secretion occurs between the synapses in the brain, intact resealed nerve terminals, synaptosomes, were prepared from postmortem human brains. The synaptosomes from the AD brains had increased levels of a 20-22 kDa fragment of tau and dimerised tau as compared to control samples, and the majority of the tau detected lacked the C-terminus [112]. Depolarization induced by potassium chloride significantly increased the release of tau from synaptosomes in a concentration-dependent manner. Interestingly, this effect was only observed in synaptosomes from AD brains, suggesting increased sensitivity to the depolarization-induced tau release in the diseased brain [112]. The ability of tau fragments to spread to neighbouring cells stresses the importance of such fragments in disease pathogenesis. Further work is needed using both microfluidic devices and biosensor cell lines to fully elucidate the mechanisms of tau secretion, with emphasis on understanding the role of extracellular vesicles in this process.
PROTEOLYTIC FRAGMENTS OF TAU AS BIOMARKERS FOR DEMENTIA
The secretion of tau causes the release of cytosolic tau into the interstitial fluid [113], where it can further spread into CSF, or plasma, in an activity-dependent manner [114, 115]. Increased CSF concentrations of total tau (all tau isoforms independent of phosphorylation; including both full-length and fragmented species), as assessed by an ELISA [116], reflect AD neurodegeneration [reviewed by 117] while the presence or increase in phospho-tau (T181 and T231) reflects NFT build-up [118]. Changes in the levels of tau occur up to 15 years before symptom onset (reviewed by [117]). Thus total and phospho-tau (predominantly T181) have been analysed as potential biomarkers for MCI and AD in both CSF and plasma or serum samples [reviewed by 119]. Current recommendations suggest using both CSF total tau and phospho-tau, in combination with low CSF Aβ42 levels, to aid in the prediction of disease progression in AD [120].
Tau in the CSF is dependent on the clinically manifested tauopathy and stage of disease progression [121, 122]. Using western blots, the presence of full-length tau in the CSF has been reported [123], while Johnson et al. [121] showed only fragmented species which predominantly contained the N-terminus and Meredith et al. [122] showed with tau epitope-specific antibodies (recognising between amino acids 9–18 and 159–163) the existence of 20–40 kDa fragments of tau but no full-length species. The variability in results highlights the difficultly in using Western blotting to analyse CSF tau species which is likely due to the differential antibody reactivity, even though this technique has the advantage of allowing you to determine the size of the tau species present. It has also become standard to analyse tau in the CSF using both ELISAs and MS, with numerous commercial ELISA-based assays established [reviewed by 124]. There are limitations to ELISAs as they are dependent on the antibody pairing used to develop them and they are unable to discriminate between full-length tau and tau fragments containing the antibody epitopes. Interestingly, using MS, a distinct lack of C-terminal peptides is observed, indicative of the existence of fragmented species [125]. These studies highlight the variability in the sensitivity of the different techniques employed.
The potential of using tau fragments in CSF as a biomarker for tauopathy progression has been investigated (Table 1), with decreases observed in CTF, NTF and total tau levels in PSP, while increases were seen in AD [126]. Why both total and cleaved tau levels decrease in PSP while tau-mediated neurodegeneration is observed is unclear. One possibility is that the predominance of the 4R tau isoform potentially has impaired secretion into CSF due to factors such as PTMs [127].
Individual fragments of tau in CSF have been posited as AD biomarkers. The caspase-6 cleaved tau fragment, tau1 - 402, was able to distinguish between severe AD and control, but not between the milder disease manifestations [53]. Tau1 - 402 did not increase in multiple sclerosis and PD [53]. Another fragment, tau26 - 230, increased in the CSF of both AD and non-AD associated dementia compared to non-cognitively impaired neurological disease patients but was unable to correlate with cognitive decline [128]. However, tau26 - 230 did not correlate with full-length tau, indicating that this fragment is not produced due to degradation of full-length protein in the CSF but is actively secreted into the CSF upon neuronal injury [128]. Tau26 - 44 [102], is also seen at the lower end of detection in a range of CSF samples containing both normal and elevated total tau levels [125]. However, the most quantifiable tau peptides in CSF contain the mid-region (residues 156–224) of the protein, likely due to resistance to degradation by proteases, while the N- and C-terminal peptides usually fall below the limit of detection [125], likely due to the multiple proteolytic cleavage events that occur in these regions reducing the N- and C-terminal peptide levels.
The work on CSF biomarkers has been pivotal in improving disease diagnosis; however, there are still costs and stigma associated with this procedure. This has led to many researchers now focusing on plasma, which is much less invasive and easier to harvest. Plasma proteins such as tau [129] have been shown to change during dementia [reviewed by 130], however, more research is needed to understand and characterise the plasma-tau profile. Increased plasma-tau levels partially reflect AD pathogenesis by correlating with higher CSF total tau and decreased CSF Aβ42. However, these correlations were weak and varied between cohorts [129]. Recently, both tau441 and fragments of tau have been shown to cross the BBB in a manner dependent on their amino acid composition [131]. Tau fragments (Table 1) are more likely to be present in plasma compared to full-length tau441 due to their smaller size allowing them to more easily cross the BBB (reviewed by [20]).
Tau-A [78] and Tau-C [44] fragments produced by ADAM10 and caspase-3 cleavage, respectively, have shown potential to act as plasma biomarkers for AD through positive correlations with cognitive deficits [132]. These fragments have also been linked to modifiable health risk factors, with positive correlations shown with body mass index, cholesterol, platelets and white blood cell count [133].
CONCLUDING REMARKS
From the above it is apparent that the identification of additional proteases that cleave tau and the presence of proteolytic fragments of tau in a range of different tauopathies adds greater complexity to the contribution of, and role of, tau in a range of neurodegenerative diseases. Research into the role proteolysis of tau plays in dementia pathogenesis is imperative to determine not only potential therapeutic targets and novel biomarkers but also to unravel the cellular and molecular mechanisms of tau-mediated neurodegeneration. More work is needed to determine the exact aggregation, seeding and spreading mechanisms of both full-length and fragments of tau.
However, recent studies on tau proteolysis also raise many specific questions still to be answered. For example, how do other PTMs such as phosphorylation and acetylation impact on different proteolytic sites in tau? What are the cumulative effects of a range of different PTMs on tau proteolysis? What are the key sequence features of certain fragments that cause them to exert neurotoxicity or have a higher propensity to undergo aggregation? Will fragments of tau act in a prion-like manner, spreading to and seeding further PTM or aggregation of tau in neighbouring cells? Can fragments of tau activate other proteases and kinases, exacerbating the PTM status of endogenous tau? Are plasma-tau or plasma-tau fragments sensitive and specific enough to serve as a biomarker to distinguish between different tauopathy sub-types? Could steps in tau fragment-mediated neurodegeneration be potential therapeutic targets for tauopathies? Answering these questions will help to unravel the role of tau proteolysis in disease pathogenesis, enabling the development of novel biomarkers and the identification of potential proteolytic-cleavage sites, as well as their associated proteolytic tau fragments, which could be targeted therapeutically.
ACKNOWLEDGMENTS
We gratefully acknowledge the financial support of the University of Manchester and the Dr. Donald Dean Fund in Dementia Research.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0959r1).
REFERENCES
[1] | Lebouvier T , Pasquier F , Buee L ((2017) ) Update on tauopathies. Curr Opin Neurol 30: , 589–598. |
[2] | Wang Y , Mandelkow E ((2015) ) Tau in physiology and pathology. Nat Rev Neurosci 17: , 22–35. |
[3] | Lasagna-Reeves CA , Castillo-Carranza DL , Sengupta U , Clos AL , Jackson GR , Kayed R ((2011) ) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener 6: , 1–14. |
[4] | Fox LM , William CM , Adamowicz DH , Pitstick R , Carlson GA , Spires-Jones TL , Hyman BT ((2011) ) Soluble tau species, not neurofibrillary aggregates, disrupt neural system integration in a tau transgenic model. J Neuropathol Exp Neurol 70: , 588–595. |
[5] | Brion JP , Couck AM , Passareiro E , Flament-Durand J ((1985) ) Neurofibrillary tangles of Alzheimer’s disease: An immunohistochemical study. J Submicrosc Cytol 17: , 89–96. |
[6] | Takashima A ((2013) ) Tauopathies and tau oligomers. J Alzheimers Dis 37: , 565–568. |
[7] | Ghetti B , Oblak AL , Boeve BF , Johnson KA , Dickerson BC , Goedert M ((2015) ) Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol 41: , 24–46. |
[8] | Iqbal K , Liu F , Gong CX ((2015) ) Tau and neurodegenerative disease: The story so far. Nat Rev Neurol 12: , 15–27. |
[9] | Guo T , Noble W , Hanger DP ((2017) ) Roles of tau protein in health and disease. Acta Neuropathol 133: , 655–704. |
[10] | Sotiropoulos I , Galas MC , Silva JM , Skoulakis E , Wegmann S , Maina MB , Blum D , Sayas CL , Mandelkow EM , Mandelkow E , Spillantini MG , Sousa N , Avila J , Medina M , Mudher A , Buee L ((2017) ) Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol Commun 5: , 1–11. |
[11] | Martin L , Latypova X , Terro F ((2011) ) Post-translational modifications of tau protein: Imlications for Alzheimer’s disease. Neurochem Int 58: , 458–471. |
[12] | Ercan E , Eid S , Weber C , Kowalski A , Bichmann M , Behrendt A , Matthes F , Krauss S , Reinhardt P , Fulle S , Ehrnhoefer DE ((2017) ) A validated antibody panel for the characterization of tau post-translational modifications. Mol Neurodegener 12: , 1–19. |
[13] | Mair W , Muntel J , Tepper K , Tang S , Biernat J , Seeley WW , Kosik KS , Mandelkow E , Steen H , Steen JA ((2016) ) FLEXITau: Quantifying post-translational modifications of tau protein in vitro and in human disease. Anal Chem 88: , 3704–3714. |
[14] | Hanger DP , Anderton BH , Noble W ((2009) ) Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol Med 15: , 112–119. |
[15] | Wang JZ , Xia YY , Grundke-Iqbal I , Iqbal K ((2013) ) Abnormal hyperphosphorylation of tau: Sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis 33: (Suppl 1), S123–S139. |
[16] | Simic G , Babic Leko M , Wray S , Harrington C , Delalle I , Jovanov-Milosevic N , Bazadona D , Buee L , de Silva R , Di Giovanni G , Wischik C , Hof PR ((2016) ) Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6: , 1–28. |
[17] | Wang YP , Biernat J , Pickhardt M , Mandelkow E , Mandelkow EM ((2007) ) Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc Natl Acad Sci U S A 104: , 10252–10257. |
[18] | Avila J , Jimenez JS , Sayas CL , Bolos M , Zabala JC , Rivas G , Hernandez F ((2016) ) Tau structures. Front Aging Neurosci 8: , 1–10. |
[19] | Chesser AS , Pritchard SM , Johnson GV ((2013) ) Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front Neurol 4: , 1–12. |
[20] | Inekci D , Jonesco DS , Kennard S , Karsdal MA , Henriksen K ((2015) ) The potential of pathological protein fragmentation in blood-based biomarker development for dementia - with emphasis on Alzheimer’s disease. Front Neurol 6: , 1–14. |
[21] | Schechter I , Berger A ((1967) ) On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 27: , 157–162. |
[22] | Derisbourg M , Leghay C , Chiappetta G , Fernandez-Gomez FJ , Laurent C , Demeyer D , Carrier S , Buee-Scherrer V , Blum D , Vinh J , Sergeant N , Verdier Y , Buee L , Hamdane M ((2015) ) Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci Rep 5: , 1–10. |
[23] | Dear TN , Boehm T ((2001) ) Identification and characterization of two novel calpain large subunit genes. Gene 274: , 245–252. |
[24] | Moldoveanu T , Hosfield CM , Lim D , Elce JS , Jia Z , Davies PL ((2002) ) A Ca(2+) switch aligns the active site of calpain. Cell 108: , 649–660. |
[25] | Hanna RA , Campbell RL , Davies PL ((2008) ) Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature 456: , 409–412. |
[26] | Goll DE , Thompson VF , Li H , Wei W , Cong J ((2003) ) The calpain system. Physiol Rev 83: , 731–801. |
[27] | Baudry M , Bi X ((2016) ) Calpain-1 and calpain-2: The yin and yang of synaptic plasticity and neurodegeneration. Trends Neurosci 39: , 235–245. |
[28] | Siklos M , BenAissa M , Thatcher GR ((2015) ) Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm Sin B 5: , 506–519. |
[29] | Kurbatskaya K , Phillips EC , Croft CL , Dentoni G , Hughes MM , Wade MA , Al-Sarraj S , Troakes C , O’Neill MJ , Perez-Nievas BG , Hanger DP , Noble W ((2016) ) Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer’s disease brain. Acta Neuropathol Commun 4: , 1–15. |
[30] | Rao MV , McBrayer MK , Campbell J , Kumar A , Hashim A , Sershen H , Stavrides PH , Ohno M , Hutton M , Nixon RA ((2014) ) Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. J Neurosci 34: , 9222–9234. |
[31] | Lewis J , McGowan E , Rockwood J , Melrose H , Nacharaju P , Van Slegtenhorst M , Gwinn-Hardy K , Paul Murphy M , Baker M , Yu X , Duff K , Hardy J , Corral A , Lin WL , Yen SH , Dickson DW , Davies P , Hutton M ((2000) ) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: , 402–405. |
[32] | Yang LS , Ksiezak-Reding H ((1995) ) Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. Eur J Biochem 233: , 9–17. |
[33] | Park SY , Ferreira A ((2005) ) The generation of a 17 kDa neurotoxic fragment: An alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci 25: , 5365–5375. |
[34] | Garg S , Timm T , Mandelkow EM , Mandelkow E , Wang Y ((2010) ) Cleavage of Tau by calpain in Alzheimer’s disease: The quest for the toxic 17 kD fragment. Neurobiol Aging 32: , 1–14. |
[35] | Lang AE , Riherd Methner DN , Ferreira A ((2014) ) Neuronal degeneration, synaptic defects, and behavioral abnormalities in tau(4)(5)(-)(2)(3)(0) transgenic mice. Neuroscience 275: , 322–339. |
[36] | Afreen S , Riherd Methner DN , Ferreira A ((2017) ) Tau45-230 association with the cytoskeleton and membrane-bound organelles: Functional implications in neurodegeneration. Neuroscience 362: , 104–117. |
[37] | Matsumoto SE , Motoi Y , Ishiguro K , Tabira T , Kametani F , Hasegawa M , Hattori N ((2015) ) The twenty-four KDa C-terminal tau fragment increases with aging in tauopathy mice: Implications of prion-like properties. Hum Mol Genet 24: , 6403–6416. |
[38] | Yin Y , Wang Y , Gao D , Ye J , Wang X , Fang L , Wu D , Pi G , Lu C , Zhou XW , Yang Y , Wang JZ ((2016) ) Accumulation of human full-length tau induces degradation of nicotinic acetylcholine receptor alpha4 via activating calpain-2. Sci Rep 6: , 27283. |
[39] | McIlwain DR , Berger T , Mak TW ((2013) ) Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol 5: , a008656. |
[40] | Horowitz PM , Patterson KR , Guillozet-Bongaarts AL , Reynolds MR , Carroll CA , Weintraub ST , Bennett DA , Cryns VL , Berry RW , Binder LI ((2004) ) Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J Neurosci 24: , 7895–7902. |
[41] | Corsetti V , Amadoro G , Gentile A , Capsoni S , Ciotti MT , Cencioni MT , Atlante A , Canu N , Rohn TT , Cattaneo A , Calissano P ((2008) ) Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer’s disease models. Mol Cell Neurosci 38: , 381–392. |
[42] | Zhao X , Kotilinek LA , Smith B , Hlynialuk C , Zahs K , Ramsden M , Cleary J , Ashe KH ((2016) ) Caspase-2 cleavage of tau reversibly impairs memory. Nat Med 22: , 1268–1276. |
[43] | Guo H , Albrecht S , Bourdeau M , Petzke T , Bergeron C , LeBlanc AC ((2004) ) Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am J Pathol 165: , 523–531. |
[44] | Gamblin TC , Chen F , Zambrano A , Abraha A , Lagalwar S , Guillozet AL , Lu M , Fu Y , Garcia-Sierra F , LaPointe N , Miller R , Berry RW , Binder LI , Cryns VL ((2003) ) Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A 100: , 10032–10037. |
[45] | Zhao Y , Tseng IC , Heyser CJ , Rockenstein E , Mante M , Adame A , Zheng Q , Huang T , Wang X , Arslan PE , Chakrabarty P , Wu C , Bu G , Mobley WC , Zhang YW , St George-Hyslop P , Masliah E , Fraser P , Xu H ((2015) ) Appoptosin-mediated caspase cleavage of tau contributes to progressive supranuclear palsy pathogenesis. Neuron 87: , 963–975. |
[46] | Zhang H , Zhang YW , Chen Y , Huang X , Zhou F , Wang W , Xian B , Zhang X , Masliah E , Chen Q , Han JD , Bu G , Reed JC , Liao FF , Chen YG , Xu H ((2012) ) Appoptosin is a novel pro-apoptotic protein and mediates cell death in neurodegeneration. J Neurosci 32: , 15565–15576. |
[47] | Hoglinger GU , Melhem NM , Dickson DW , Sleiman PM , Wang LS , Klei L , Rademakers R , de Silva R , Litvan I , Riley DE , van Swieten JC , Heutink P , Wszolek ZK , Uitti RJ , Vandrovcova J , Hurtig HI , Gross RG , Maetzler W , Goldwurm S , Tolosa E , Borroni B , Pastor P , Group PSPGS , Cantwell LB , Han MR , Dillman A , van der Brug MP , Gibbs JR , Cookson MR , Hernandez DG , Singleton AB , Farrer MJ , Yu CE , Golbe LI , Revesz T , Hardy J , Lees AJ , Devlin B , Hakonarson H , Muller U , Schellenberg GD ((2011) ) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43: , 699–705. |
[48] | Mead E , Kestoras D , Gibson Y , Hamilton L , Goodson R , Jones S , Eversden S , Davies P , O’Neill M , Hutton M , Szekeres P , Wolak J ((2016) ) Halting of caspase activity protects tau from MC1-conformational change and aggregation. J Alzheimers Dis 54: , 1521–1538. |
[49] | Kim Y , Choi H , Lee W , Park H , Kam TI , Hong SH , Nah J , Jung S , Shin B , Lee H , Choi TY , Choo H , Kim KK , Choi SY , Kayed R , Jung YK ((2015) ) Caspase-cleaved tau exhibits rapid memory impairment associated with tau oligomers in a transgenic mouse model. Neurobiol Dis 87: , 19–28. |
[50] | Means JC , Gerdes BC , Kaja S , Sumien N , Payne AJ , Stark DA , Borden PK , Price JL , Koulen P ((2016) ) Caspase-3-dependent proteolytic cleavage of tau causes neurofibrillary tangles and results in cognitive impairment during normal aging. Neurochem Res 41: , 2278–2288. |
[51] | Day RJ , Mason MJ , Thomas C , Poon WW , Rohn TT ((2015) ) Caspase-cleaved tau co-localizes with early tangle markers in the human vascular dementia brain. PLoS One 10: , e0132637. |
[52] | Sokolowski JD , Gamage KK , Heffron DS , Leblanc AC , Deppmann CD , Mandell JW ((2014) ) Caspase-mediated cleavage of actin and tubulin is a common feature and sensitive marker of axonal degeneration in neural development and injury. Acta Neuropathol Commun 2: , 1–14. |
[53] | Ramcharitar J , Albrecht S , Afonso VM , Kaushal V , Bennett DA , Leblanc AC ((2013) ) Cerebrospinal fluid tau cleaved by caspase-6 reflects brain levels and cognition in aging and Alzheimer disease. J Neuropathol Exp Neurol 72: , 824–832. |
[54] | Albrecht S , Bogdanovic N , Ghetti B , Winblad B , LeBlanc AC ((2009) ) Caspase-6 activation in familial alzheimer disease brains carrying amyloid precursor protein or presenilin i or presenilin II mutations. J Neuropathol Exp Neurol 68: , 1282–1293. |
[55] | Albrecht S , Bourdeau M , Bennett D , Mufson EJ , Bhattacharjee M , LeBlanc AC ((2007) ) Activation of caspase-6 in aging and mild cognitive impairment. Am J Pathol 170: , 1200–1209. |
[56] | LeBlanc AC , Ramcharitar J , Afonso V , Hamel E , Bennett DA , Pakavathkumar P , Albrecht S ((2014) ) Caspase-6 activity in the CA1 region of the hippocampus induces age-dependent memory impairment. Cell Death Differ 21: , 696–706. |
[57] | Foveau B , Albrecht S , Bennett DA , Correa JA , LeBlanc AC ((2016) ) Increased Caspase-6 activity in the human anterior olfactory nuclei of the olfactory bulb is associated with cognitive impairment. Acta Neuropathol Commun 4: , 1–13. |
[58] | Esiri MM , Wilcock GK ((1984) ) The olfactory bulbs in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 47: , 56–60. |
[59] | Zou YM , Lu D , Liu LP , Zhang HH , Zhou YY ((2016) ) Olfactory dysfunction in Alzheimer’s disease. Neuropsychiatr Dis Treat 12: , 869–875. |
[60] | Turk V , Stoka V , Vasiljeva O , Renko M , Sun T , Turk B , Turk D ((2012) ) Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim Biophys Acta 1824: , 68–88. |
[61] | Cataldo AM , Barnett JL , Berman SA , Li J , Quarless S , Bursztajn S , Lippa C , Nixon RA ((1995) ) Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: Evidence for early up-regulation of the endosomal-lysosomal system. Neuron 14: , 671–680. |
[62] | Nixon RA , Cataldo AM ((2006) ) Lysosomal system pathways: Genes to neurodegeneration in Alzheimer’s disease. J Alzheimers Dis 9: , 277–289. |
[63] | Yamashima T ((2013) ) Reconsider Alzheimer’s disease by the ‘calpain-cathepsin hypothesis’–a perspective review. Prog Neurobiol 105: , 1–23. |
[64] | Stoka V , Turk V , Turk B ((2016) ) Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev 32: , 22–37. |
[65] | Ii K , Ito H , Kominami E , Hirano A ((1993) ) Abnormal distribution of cathepsin proteinases and endogenous inhibitors (cystatins) in the hippocampus of patients with Alzheimer’s disease, parkinsonism-dementia complex on Guam, and senile dementia and in the aged. Virchows Arch A Pathol Anat Histopathol 423: , 185–194. |
[66] | Kenessey A , Nacharaju P , Ko LW , Yen SH ((1997) ) Degradation of tau by lysosomal enzyme cathepsin D: Implication for Alzheimer neurofibrillary degeneration. J Neurochem 69: , 2026–2038. |
[67] | Wang Y , Martinez-Vicente M , Kruger U , Kaushik S , Wong E , Mandelkow EM , Cuervo AM , Mandelkow E ((2009) ) Tau fragmentation, aggregation and clearance: The dual role of lysosomal processing. Hum Mol Genet 18: , 4153–4170. |
[68] | Clausen T , Kaiser M , Huber R , Ehrmann M ((2011) ) HTRA proteases: Regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol 12: , 152–162. |
[69] | Tennstaedt A , Popsel S , Truebestein L , Hauske P , Brockmann A , Schmidt N , Irle I , Sacca B , Niemeyer CM , Brandt R , Ksiezak-Reding H , Tirniceriu AL , Egensperger R , Baldi A , Dehmelt L , Kaiser M , Huber R , Clausen T , Ehrmann M ((2012) ) Human high temperature requirement serine protease A1 (HTRA1) degrades tau protein aggregates. J Biol Chem 287: , 20931–20941. |
[70] | Poepsel S , Sprengel A , Sacca B , Kaschani F , Kaiser M , Gatsogiannis C , Raunser S , Clausen T , Ehrmann M ((2015) ) Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat Chem Biol 11: , 862–869. |
[71] | Chu Q , Diedrich JK , Vaughan JM , Donaldson CJ , Nunn MF , Lee KF , Saghatelian A ((2016) ) HtrA1 Proteolysis of ApoE In Vitro Is Allele Selective. J Am Chem Soc 138: , 9473–9478. |
[72] | Yu JT , Tan L , Hardy J ((2014) ) Apolipoprotein E in Alzheimer’s disease: An update. Annu Rev Neurosci 37: , 79–100. |
[73] | Tobler AR , Constam DB , Schmitt-Graff A , Malipiero U , Schlapbach R , Fontana A ((1997) ) Cloning of the human puromycin-sensitive aminopeptidase and evidence for expression in neurons. J Neurochem 68: , 889–897. |
[74] | Karsten SL , Sang TK , Gehman LT , Chatterjee S , Liu J , Lawless GM , Sengupta S , Berry RW , Pomakian J , Oh HS , Schulz C , Hui KS , Wiedau-Pazos M , Vinters HV , Binder LI , Geschwind DH , Jackson GR ((2006) ) A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron 51: , 549–560. |
[75] | Sengupta S , Horowitz PM , Karsten SL , Jackson GR , Geschwind DH , Fu Y , Berry RW , Binder LI ((2006) ) Degradation of tau protein by puromycin-sensitive aminopeptidase in vitro. Biochemistry 45: , 15111–15119. |
[76] | Chow KM , Guan H , Hersh LB ((2010) ) Aminopeptidases do not directly degrade tau protein. Mol Neurodegener 5: , 1–10. |
[77] | Arai T , Miklossy J , Klegeris A , Guo JP , McGeer PL ((2006) ) Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J Neuropathol Exp Neurol 65: , 19–25. |
[78] | Henriksen K , Wang Y , Sorensen MG , Barascuk N , Suhy J , Pedersen JT , Duffin KL , Dean RA , Pajak M , Christiansen C , Zheng Q , Karsdal MA ((2013) ) An enzyme-generated fragment of tau measured in serum shows an inverse correlation to cognitive function. PLoS One 8: , e64990. |
[79] | Andrew RJ , Kellett KA , Thinakaran G , Hooper NM ((2016) ) A Greek Tragedy: The Growing Complexity of Alzheimer Amyloid Precursor Protein Proteolysis. J Biol Chem 291: , 19235–19244. |
[80] | Zhang Z , Song M , Liu X , Kang SS , Kwon IS , Duong DM , Seyfried NT , Hu WT , Liu Z , Wang JZ , Cheng L , Sun YE , Yu SP , Levey AI , Ye K ((2014) ) Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med 20: , 1254–1262. |
[81] | Basurto-Islas G , Grundke-Iqbal I , Tung YC , Liu F , Iqbal K ((2013) ) Activation of asparaginyl endopeptidase leads to Tau hyperphosphorylation in Alzheimer disease. J Biol Chem 288: , 17495–17507. |
[82] | Wang ZH , Liu P , Liu X , Manfredsson FP , Sandoval IM , Yu SP , Wang JZ , Ye K ((2017) ) Delta-secretase phosphorylation by SRPK2 enhances its enzymatic activity, provoking pathogenesis in Alzheimer’s disease. Mol Cell 67: , 812–825.e5. |
[83] | Zhang Z , Kang SS , Liu X , Ahn EH , Zhang Z , He L , Iuvone PM , Duong DM , Seyfried NT , Benskey MJ , Manfredsson FP , Jin L , Sun YE , Wang JZ , Ye K ((2017) ) Asparagine endopeptidase cleaves alpha-synuclein and mediates pathologic activities in Parkinson’s disease. Nat Struct Mol Biol 24: , 632–642. |
[84] | Zhang Z , Song M , Liu X , Su Kang S , Duong DM , Seyfried NT , Cao X , Cheng L , Sun YE , Ping Yu S , Jia J , Levey AI , Ye K ((2015) ) Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat Commun 6: , 8762. |
[85] | Zhang Z , Xie M , Ye K ((2016) ) Asparagine endopeptidase is an innovative therapeutic target for neurodegenerative diseases. Expert Opin Ther Targets 20: , 1237–1245. |
[86] | Zhang Z , Obianyo O , Dall E , Du Y , Fu H , Liu X , Kang SS , Song M , Yu SP , Cabrele C , Schubert M , Li X , Wang JZ , Brandstetter H , Ye K ((2017) ) Inhibition of delta-secretase improves cognitive functions in mouse models of Alzheimer’s disease. Nat Commun 8: , 14740. |
[87] | Cohen TJ , Constance BH , Hwang AW , James M , Yuan CX ((2016) ) Intrinsic tau acetylation is coupled to auto-proteolytic tau fragmentation. PLoS One 11: , e0158470. |
[88] | Cohen TJ , Friedmann D , Hwang AW , Marmorstein R , Lee VM ((2013) ) The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat Struct Mol Biol 20: , 756–762. |
[89] | Cook C , Stankowski JN , Carlomagno Y , Stetler C , Petrucelli L ((2014) ) Acetylation: A new key to unlock tau’s role in neurodegeneration. Alzheimers Res Ther 6: , 1–8. |
[90] | Min SW , Chen X , Tracy TE , Li Y , Zhou Y , Wang C , Shirakawa K , Minami SS , Defensor E , Mok SA , Sohn PD , Schilling B , Cong X , Ellerby L , Gibson BW , Johnson J , Krogan N , Shamloo M , Gestwicki J , Masliah E , Verdin E , Gan L ((2015) ) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21: , 1154–1162. |
[91] | Ikeda C , Yokota O , Nagao S , Ishizu H , Oshima E , Hasegawa M , Okahisa Y , Terada S , Yamada N ((2016) ) The relationship between development of neuronal and astrocytic tau pathologies in subcortical nuclei and progression of argyrophilic grain disease. Brain Pathol 26: , 488–505. |
[92] | Wray S , Saxton M , Anderton BH , Hanger DP ((2008) ) Direct analysis of tau from PSP brain identifies new phosphorylation sites and a major fragment of N-terminally cleaved tau containing four microtubule-binding repeats. J Neurochem 105: , 2343–2352. |
[93] | Arai T , Ikeda K , Akiyama H , Nonaka T , Hasegawa M , Ishiguro K , Iritani S , Tsuchiya K , Iseki E , Yagishita S , Oda T , Mochizuki A ((2004) ) Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol 55: , 72–79. |
[94] | Bondulich MK , Guo T , Meehan C , Manion J , Rodriguez Martin T , Mitchell JC , Hortobagyi T , Yankova N , Stygelbout V , Brion JP , Noble W , Hanger DP ((2016) ) Tauopathy induced by low level expression of a human brain-derived tau fragment in mice is rescued by phenylbutyrate. Brain 139: , 2290–2306. |
[95] | Iannitti T , Palmieri B ((2011) ) Clinical and experimental applications of sodium phenylbutyrate. Drugs R D 11: , 227–249. |
[96] | Yin H , Kuret J ((2006) ) C-terminal truncation modulates both nucleation and extension phases of tau fibrillization. FEBS Lett 580: , 211–215. |
[97] | Fitzpatrick AWP , Falcon B , He S , Murzin AG , Murshudov G , Garringer HJ , Crowther RA , Ghetti B , Goedert M , Scheres SHW ((2017) ) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547: , 185–190. |
[98] | Novak M , Kabat J , Wischik CM ((1993) ) Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J 12: , 365–370. |
[99] | Taniguchi-Watanabe S , Arai T , Kametani F , Nonaka T , Masuda-Suzukake M , Tarutani A , Murayama S , Saito Y , Arima K , Yoshida M , Akiyama H , Robinson A , Mann DM , Iwatsubo T , Hasegawa M ((2016) ) Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol 131: , 267–280. |
[100] | Ozcelik S , Sprenger F , Skachokova Z , Fraser G , Abramowski D , Clavaguera F , Probst A , Frank S , Muller M , Staufenbiel M , Goedert M , Tolnay M , Winkler DT ((2016) ) Co-expression of truncated and full-length tau induces severe neurotoxicity. Mol Psychiatry 21: , 1790–1798. |
[101] | Vintilescu CR , Afreen S , Rubino AE , Ferreira A ((2016) ) The neurotoxic TAU45-230 fragment accumulates in upper and lower motor neurons in amyotrophic lateral sclerosis subjects. Mol Med 22: , 477–486. |
[102] | Florenzano F , Veronica C , Ciasca G , Ciotti MT , Pittaluga A , Olivero G , Feligioni M , Iannuzzi F , Latina V , Sciacca MFM , Sinopoli A , Milardi D , Pappalardo G , De Spirito M , Papi M , Atlante A , Bobba A , Borreca A , Calissano P , Amadoro G ((2017) ) Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 8: , 64745–64778. |
[103] | Dai CL , Chen X , Kazim SF , Liu F , Gong CX , Grundke-Iqbal I , Iqbal K ((2015) ) Passive immunization targeting the N-terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J Neural Transm (Vienna) 122: , 607–617. |
[104] | Pedersen JT , Sigurdsson EM ((2015) ) Tau immunotherapy for Alzheimer’s disease. Trends Mol Med 21: , 394–402. |
[105] | Mudher A , Colin M , Dujardin S , Medina M , Dewachter I , Naini SMA , Mandelkow EM , Mandelkow E , Buee L , Goedert M , Brion JP ((2017) ) What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun 5: , 1–20. |
[106] | Clavaguera F , Hench J , Goedert M , Tolnay M ((2015) ) Invited review: Prion-like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol 41: , 47–58. |
[107] | Perez M , Cuadros R , Hernandez F , Avila J ((2016) ) Secretion of full-length tau or tau fragments in a cell culture model. Neurosci Lett 634: , 63–69. |
[108] | Plouffe V , Mohamed NV , Rivest-McGraw J , Bertrand J , Lauzon M , Leclerc N ((2012) ) Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One 7: , e36873. |
[109] | Wang Y , Balaji V , Kaniyappan S , Kruger L , Irsen S , Tepper K , Chandupatla R , Maetzler W , Schneider A , Mandelkow E , Mandelkow EM ((2017) ) The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 12: , 1–25. |
[110] | Dujardin S , Begard S , Caillierez R , Lachaud C , Delattre L , Carrier S , Loyens A , Galas MC , Bousset L , Melki R , Auregan G , Hantraye P , Brouillet E , Buee L , Colin M ((2014) ) Ectosomes: A new mechanism for non-exosomal secretion of tau protein. PLoS One 9: , e100760. |
[111] | Kanmert D , Cantlon A , Muratore CR , Jin M , O’Malley TT , Lee G , Young-Pearse TL , Selkoe DJ , Walsh DM ((2015) ) C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J Neurosci 35: , 10851–10865. |
[112] | Sokolow S , Henkins KM , Bilousova T , Gonzalez B , Vinters HV , Miller CA , Cornwell L , Poon WW , Gylys KH ((2015) ) Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J Neurochem 133: , 368–379. |
[113] | Yamada K , Cirrito JR , Stewart FR , Jiang H , Finn MB , Holmes BB , Binder LI , Mandelkow EM , Diamond MI , Lee VM , Holtzman DM ((2011) ) In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci 31: , 13110–13117. |
[114] | Yamada K , Holth JK , Liao F , Stewart FR , Mahan TE , Jiang H , Cirrito JR , Patel TK , Hochgrafe K , Mandelkow EM , Holtzman DM ((2014) ) Neuronal activity regulates extracellular tau in vivo. J Exp Med 211: , 387–393. |
[115] | Pooler AM , Noble W , Hanger DP ((2014) ) A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology 76 pt A: , 1–8. |
[116] | Blennow K , Wallin A , Agren H , Spenger C , Siegfried J , Vanmechelen E ((1995) ) Tau protein in cerebrospinal fluid: A biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol 26: , 231–245. |
[117] | Masters CL , Bateman R , Blennow K , Rowe CC , Sperling RA , Cummings JL ((2015) ) Alzheimer’s disease. Nat Rev Dis Primers 1: , 1–18. |
[118] | Buerger K , Ewers M , Pirttila T , Zinkowski R , Alafuzoff I , Teipel SJ , DeBernardis J , Kerkman D , McCulloch C , Soininen H , Hampel H ((2006) ) CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain 129: , 3035–3041. |
[119] | Olsson B , Lautner R , Andreasson U , Ohrfelt A , Portelius E , Bjerke M , Holtta M , Rosen C , Olsson C , Strobel G , Wu E , Dakin K , Petzold M , Blennow K , Zetterberg H ((2016) ) CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol 15: , 673–684. |
[120] | Dubois B , Feldman HH , Jacova C , Hampel H , Molinuevo JL , Blennow K , DeKosky ST , Gauthier S , Selkoe D , Bateman R , Cappa S , Crutch S , Engelborghs S , Frisoni GB , Fox NC , Galasko D , Habert MO , Jicha GA , Nordberg A , Pasquier F , Rabinovici G , Robert P , Rowe C , Salloway S , Sarazin M , Epelbaum S , de Souza LC , Vellas B , Visser PJ , Schneider L , Stern Y , Scheltens P , Cummings JL ((2014) ) Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol 13: , 614–629. |
[121] | Johnson GV , Seubert P , Cox TM , Motter R , Brown JP , Galasko D ((1997) ) The tau protein in human cerebrospinal fluid in Alzheimer’s disease consists of proteolytically derived fragments. J Neurochem 68: , 430–433. |
[122] | Meredith JE Jr , Sankaranarayanan S , Guss V , Lanzetti AJ , Berisha F , Neely RJ , Slemmon JR , Portelius E , Zetterberg H , Blennow K , Soares H , Ahlijanian M , Albright CF ((2013) ) Characterization of novel CSF Tau and ptau biomarkers for Alzheimer’s disease. PLoS One 8: , e76523. |
[123] | Hanisch K , Soininen H , Alafuzoff I , Hoffmann R ((2010) ) Analysis of human tau in cerebrospinal fluid. J Proteome Res 9: , 1476–1482. |
[124] | Blennow K , Dubois B , Fagan AM , Lewczuk P , de Leon MJ , Hampel H ((2015) ) Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement 11: , 58–69. |
[125] | Barthelemy NR , Fenaille F , Hirtz C , Sergeant N , Schraen-Maschke S , Vialaret J , Buee L , Gabelle A , Junot C , Lehmann S , Becher F ((2016) ) Tau protein quantification in human cerebrospinal fluid by targeted mass spectrometry at high sequence coverage provides insights into its primary structure heterogeneity. J Proteome Res 15: , 667–676. |
[126] | Wagshal D , Sankaranarayanan S , Guss V , Hall T , Berisha F , Lobach I , Karydas A , Voltarelli L , Scherling C , Heuer H , Tartaglia MC , Miller Z , Coppola G , Ahlijanian M , Soares H , Kramer JH , Rabinovici GD , Rosen HJ , Miller BL , Meredith J , Boxer AL ((2015) ) Divergent CSF tau alterations in two common tauopathies: Alzheimer’s disease and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 86: , 244–250. |
[127] | Karch CM , Jeng AT , Goate AM ((2012) ) Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies. J Biol Chem 287: , 42751–42762. |
[128] | Amadoro G , Corsetti V , Sancesario GM , Lubrano A , Melchiorri G , Bernardini S , Calissano P , Sancesario G ((2014) ) Cerebrospinal fluid levels of a 20-22 kDa NH2 fragment of human tau provide a novel neuronal injury biomarker in Alzheimer’s disease and other dementias. J Alzheimers Dis 42: , 211–226. |
[129] | Mattsson N , Zetterberg H , Janelidze S , Insel PS , Andreasson U , Stomrud E , Palmqvist S , Baker D , Tan Hehir CA , Jeromin A , Hanlon D , Song L , Shaw LM , Trojanowski JQ , Weiner MW , Hansson O , Blennow K , Investigators ADNI ((2016) ) Plasma tau in Alzheimer disease. Neurology 87: , 1827–1835. |
[130] | Keshavan A , Heslegrave A , Zetterberg H , Schott JM ((2017) ) Blood biomarkers for Alzheimer’s disease: Much promise, cautious progress. Mol Diagn Ther 21: , 13–22. |
[131] | Banks WA , Kovac A , Majerova P , Bullock KM , Shi M , Zhang J ((2016) ) Tau proteins cross the blood-brain barrier. J Alzheimers Dis 55: , 411–419. |
[132] | Henriksen K , Byrjalsen I , Christiansen C , Karsdal MA ((2015) ) Relationship between serum levels of tau fragments and clinical progression of Alzheimer’s disease. J Alzheimers Dis 43: , 1331–1341. |
[133] | Neergaard JS , Dragsbaek K , Christiansen C , Nielsen HB , Workman CT , Brix S , Henriksen K , Karsdal MA ((2017) ) Modifiable risk factors promoting neurodegeneration is associated with two novel brain degradation markers measured in serum. Neurochem Int 108: , 303–308. |
[134] | Horowitz PM , LaPointe N , Guillozet-Bongaarts AL , Berry RW , Binder LI ((2006) ) N-terminal fragments of tau inhibit full-length tau polymerization in vitro. Biochemistry 45: , 12859–12866. |
[135] | Amadoro G , Ciotti MT , Costanzi M , Cestari V , Calissano P , Canu N ((2006) ) NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci U S A 103: , 2892–2897. |
[136] | Amadoro G , Corsetti V , Atlante A , Florenzano F , Capsoni S , Bussani R , Mercanti D , Calissano P ((2012) ) Interaction between NH(2)-tau fragment and Abeta in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol Aging 33: , 833 e831–825. |
[137] | Arai T , Guo JP , McGeer PL ((2005) ) Proteolysis of non-phosphorylated and phosphorylated tau by thrombin. J Biol Chem 280: , 5145–5153. |
[138] | Steiner B , Mandelkow EM , Biernat J , Gustke N , Meyer HE , Schmidt B , Mieskes G , Soling HD , Drechsel D , Kirschner MW , et al. ((1990) ) Phosphorylation of microtubule-associated protein tau: Identification of the site for Ca2(+)-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J 9: , 3539–3544. |
[139] | Ferreira A , Bigio EH ((2011) ) Calpain-mediated tau cleavage: A mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med 17: , 676–685. |
[140] | Quintanilla RA , Matthews-Roberson TA , Dolan PJ , Johnson GV ((2009) ) Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: Implications for the pathogenesis of Alzheimer disease. J Biol Chem 284: , 18754–18766. |
[141] | Matthews-Roberson TA , Quintanilla RA , Ding H , Johnson GV ((2008) ) Immortalized cortical neurons expressing caspase-cleaved tau are sensitized to endoplasmic reticulum stress induced cell death. Brain Res 1234: , 206–212. |
[142] | Rissman RA , Poon WW , Blurton-Jones M , Oddo S , Torp R , Vitek MP , LaFerla FM , Rohn TT , Cotman CW ((2004) ) Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 114: , 121–130. |
[143] | Mandelkow EM , Mandelkow E ((2012) ) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2: , a006247. |
[144] | Mukrasch MD , Bibow S , Korukottu J , Jeganathan S , Biernat J , Griesinger C , Mandelkow E , Zweckstetter M ((2009) ) Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol 7: , e34. |
[145] | von Bergen M , Friedhoff P , Biernat J , Heberle J , Mandelkow EM , Mandelkow E ((2000) ) Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci U S A 97: , 5129–5134. |


