
The Anti-Amyloid-β and Neuroprotective Properties of a Novel Tricyclic Pyrone Molecule
Abstract
There is an urgent unmet need for new therapeutics for Alzheimer’s disease (AD), the most common cause of dementia in the elderly. Therapeutic approaches targeting amyloid-β (Aβ) and its downstream toxicities have become major strategies in AD drug development. We have taken a rational design approach and synthesized a class of tricyclic pyrone (TP) compounds that show anti-Aβ and other neuroprotective actions. The in vivo efficacy of a lead TP named CP2 to ameliorate AD-like pathologies has been shown in mouse models. Here we report the selection and initial characterization of a new lead TP70, which exhibited an anti-Aβ therapeutic index even higher than CP2. Moreover, TP70 was able to reduce oxidative stress, inhibit acyl-coenzyme A:cholesterol acyltransferase (ACAT), and upregulate the expression of ATP-binding cassette subfamily A, member 1 (ABCA1), actions considered neuroprotective in AD. TP70 further showed excellent pharmacokinetic properties, including brain penetration and oral availability. When administered to 5xFAD mice via intraperitoneal or oral route, TP70 enhanced the overall solubility and decreased the level of cerebral Aβ, including both fibrillary and soluble Aβ species. Interestingly, TP70 enhanced N-methyl-D-aspartate (NMDA) receptor-mediated excitatory post-synaptic potential (EPSP) in the hippocampal CA1 area, increased the magnitude of NMDA-dependent hippocampal long-term potentiation (LTP), a cellular model of learning and memory, and prevented the Aβ oligomer-impaired LTP. Significantly, a single dose of TP70 administered to aged 5xFAD mice was effective in mitigating the impaired LTP induction, recorded at 24 h after administration. Our results support a potential of TP70 in clinical development for AD in view of its synergistic neuroprotective actions, ability to positively modulate NMDA receptor-mediated hippocampal plasticity, and favorable pharmacokinetic properties in rodents.
INTRODUCTION
More than 5.4 million Americans and 44 million people worldwide suffer from AD, a progressive neurodegenerative disease. Currently, no FDA approved disease-modifying therapy for AD exists. Available medications include donepezil, rivastigmine, galantamine, and memantine. These drugs modulate neurotransmission and temporarily ameliorate some symptoms; however, they do not halt the progression of the disease [1–3]. Major strategies for experimental disease-modifying therapies include modifying Aβ metabolism, excitotoxicity, neuroinflammation, oxidative stress, and calcium homeostasis, for example. However, so far none of the large clinical trials have met their combined endpoints of showing cognitive changes and a meaningful difference in daily function of the subjects [4]. A major hypothesis driving therapeutic development is the amyloid cascade hypothesis, which proposes that aggregates of Aβ initiate a cascade of molecular changes leading to neurodegeneration in AD [5]. Although difficult to verify, the success of aducanumab, an Aβ-targeting immunotherapeutic, to slow clinical decline in early AD subjects in a recent small trial maintains the promises of this hypothesis [5, 6]. However, side effects such as amyloid-related imaging abnormalities and neuroinflammation remain a concern for antibody-based immunotherapy [6].
Our group has developed a series of tricyclic pyrone (TP) compounds as experimental small molecule therapeutics for AD. A lead called CP2 (see Fig. 1 for structure) has been well characterized in preclinical studies [7–12]. CP2 possesses anti-Aβ properties [8, 9, 11], able to disaggregate Aβ amyloid and protect neurons from toxicity of Aβ oligomers (AβO), widely considered the most toxic species of Aβ aggregates [13, 14]. In addition, CP2 has other direct and indirect actions potentially benefiting AD patients, such as inhibition of acyl-CoA: cholesterol acyltransferase (ACAT) [10], activation of AMP-activated protein kinase, augmentation of mitochondrial bioenergetics, inhibition of glycogen synthase kinase 3β, and restoration of axonal trafficking [11]. CP2 administered to mouse models harboring familial AD genes reduced levels of Aβ and phospho-tau and averted cognitive decline without apparent adverse effects [11].
Fig.1
Synthetic scheme leading to tricyclic pyrone molecules containing C7-[(1’-alkylamino)-2’-propyl] side chain (Method A) and C7-[(1’-heterocyclo)-2’-propyl] side chain (Method B).
![Synthetic scheme leading to tricyclic pyrone molecules containing C7-[(1’-alkylamino)-2’-propyl] side chain (Method A) and C7-[(1’-heterocyclo)-2’-propyl] side chain (Method B).](https://ip.ios.semcs.net:443/media/jad/2017/58-2/jad-58-2-jad161175/jad-58-jad161175-g001.jpg)
As the vast majority of AD, especially late-onset AD, arises from a confluence of multiple toxic insults and genetic factors, small molecules such as CP2 with anti-Aβ and various neuroprotective actions may have a better chance to achieve clinical efficacy [15–18]. Moreover, small molecules offer superior possibilities for chemical modifications and substitutions, an efficient way to rapidly gain insights into molecular interactions and drug optimization. Our goal, therefore, is to further derivatize and screen TP compounds for enhanced potency, reduced toxicity, and improved pharmacokinetic (PK) properties. The selected TP derivative(s) may serve as “development candidate” or “designate backup” compounds required for an IND (Investigational New Drug) application. From such an effort, here we report the initial characterization of a new lead TP70, which exhibits potent anti-Aβ actions as well as excellent PK properties, including brain penetration and oral availability. Moreover, TP70 is likely an anti-oxidant and enhances NMDA receptor-mediated excitatory post-synaptic potential (EPSP) in hippocampal neurons as well as increases the magnitude of NMDA-dependent hippocampal long-term potentiation (hLTP). Our results show a potential of TP70 in clinical development for AD.
MATERIALS AND METHODS
Synthesis of TP compounds
In our continuing effort on the study of tricyclic pyrone molecules as possible anti-Alzheimer lead compounds [11], we identified a potent neural protective tricyclic pyrone molecule, code name TP70, by PK and pharmacodynamic (PD) studies. TP70 was synthesized according to our reported procedure [10]. Briefly, 4-hydroxy-6-methyl-2-pyrone (1) and (S)-perillaldehyde (2) (both commercially available) were condensed to give a tricyclic pyrone, 3, that was converted to TP70 in two steps (Fig. 1). Hydroamination of 3 with borane•THF and hydroxylamine-O-sulfonic acid, followed by reductive amination with 4-hydroxybenzaldehyde and sodium cyanoborohydride in methanol yielded TP70. Similarly, three other active tricyclic pyrones, TP36, TP82, and TP101, were synthesized using different aryl aldehydes and found to have neural protective activities in the submicromolar concentrations (Fig. 1). Other TPs, such as CP2, were synthesized from a sequence of reactions: 1) hydroboration-oxidation of compound 3; 2) mesylation of the resulting alcohol with methanesulfonyl chloride and triethylamine; and 3) displacement with adenine in N,N-dimethylacetamide (DMA) at 150°C. Substitution of the mesylate group of compound 5 with 8-aminopurine and sodium hydride in N,N-dimethylformamide (DMF) afforded fluorescence active TP13. Similarly, TP4 was obtained from the displacement reaction of tricyclic pyrone 5A andadenine [19].
Cell cultures
MC65 cells were grown in the presence of 1 μg/mL tetracycline (TC), as described [7, 20]. The cell toxicity was induced by the removal of TC to induce C99 expression. Briefly, cells grown in the presence of TC were washed extensively, plated at 2.5×104 cells/well in 96-well plates, and cultured in Opti-minimal essential medium (Opti-MEM without phenol-red, Gibco/BRL, Carlsbad, CA, USA) without serum and without TC. After 72 h of incubation, the cytotoxicity was determined using a colorimetric MTT assay.
Western blot analysis
To obtain cell lysates, cells were washed with ice-cold PBS and incubated with a lysis buffer (150 mM NaCl, 10 mM NaH2PO4, 1 mM EDTA, 1% TritonX100, 0.5% SDS) containing protease inhibitor cocktail (Sigma, St. Louis, MO). Equivalent amounts of protein were analyzed by 16.5% Tris-Tricine gel electrophoresis (Bio-Rad, Hercules, CA). Proteins were transferred to polyvinylidene difluoride membranes and probed with antibodies. Visualization was performed using enhanced chemiluminescence (ECL, GE Healthcare Pharmacia). The following primary antibodies (dilutions) were used: anti-β-amyloid 1–16 antibody (6E10) (1:1000, BioLegend), anti-GAPDH (1:2000, cell signaling). Secondary antibodies were HRP-conjugated anti-rabbit or anti-mouse antibody (1:1000, GE Healthcare).
Reactive oxygen species (ROS) assay
To measure cellular ROS, cells were gently washed with PBS and incubated with CM-H2DCFDA (General Oxidative Stress Indicator) (Invitrogen) according to the manufacturer’s instructions. After 40-min incubation, photomicrographs of cell were randomly taken from each culture condition by a fluorescence microscope (Olympus). The images were analyzed by the ImageJ program. The photography and analysis of fluorescence intensity were conducted in an investigator-blinded manner.
Quantification of tissue TP70 levels by high performance liquid chromatography (HPLC)/mass spectrometry (MS)
For administration to animals, TP70 was completely dissolved in dimethyl sulfoxide (DMSO) and then diluted in 0.5% hydroxypropyl cellulose to the desired concentration. In the final preparations for administration, the DMSO concentration was less than 2%. For HPLC/MS analysis of TP70, a calibration graph from the ratios of HPLC peak areas of TP70 to 4-methoxyphenol (as an internal standard in the HPLC measurement) versus molar ratios of TP70 to 4-methoxyphenol was first determined. Cardiac blood collection was withdrawn from tail vein at different time points and treated with ethylenediamine tetraacetic acid. About 250 μl plasma was collected from each mouse. All the plasma or tissue samples collected were stored at –78°C until further analysis was carried. Plasma or tissue sample (cut into small pieces) was added 3 mL of deionized water and 10 ml of 9:1 mixture of ethyl acetate and 1-propanol. This solution was sonicated for 6 min. The organic layer was separated from a separatory funnel and the aqueous layer was extracted twice with 10 ml each of a 9:1 mixture of ethyl acetate and 1-propanol. The organic layers were combined, washed with 10 mL of brine, dried (anhydrous MgSO4), and concentrated to dryness. The residue was added a known amount of 4-methoxyphenol, diluted with 1 ml of 1-propanol, and filtered through a 0.2 μm filter disc (PTFE 0.2 μm, Fisherbrand). The filtrate was concentrated to dryness and dissolved in 300 μl of methanol. A 100 μl sample of this methanol solution was analyzed using HPLC and mass spectrometry (to verify the masses of TP70 and 4-methoxyphenol from isolated HPLC materials). The analysis was repeated two more times. The amount of TP70 in plasma or tissue extract was determined by calculating the peak areas ratios of TP70 to 4-methoxyphenol from the HPLC chromatogram.
5xFAD mice, TP70 treatment, and amyloid analysis
All experimental protocols were carried out with approval from the Institutional Animal Care and Use Committee of the University of California Davis. The line Tg6799 5xFAD mice co-expressing human APP695 with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) mutations, and human PS1 harboring M146L and L286V mutations was obtained from Dr. Robert Vassar at Northwestern University [21]. Three-month-old 5xFAD mice were treated with once daily intraperitoneal injection of TP70 at a dose of 25 mg/kg for six weeks. The brain tissues were collected from saline-perfused mice, snap frozen, and used for fluorescence staining (20 μm section) and protein fractionation.
For fluorescence staining, sections were fixed with 4% paraformaldehyde, and stained with 500 nM (E,E)-1-fluoro-2,5-bis(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (FSB) for amyloid [22] and propidium iodide (PI) for nucleus. The fluorescence images were analyzed by a laser scanning cytometry (LSC) as we previously described [23]. Briefly, stained sections are scanned by lasers to obtain digital images, and location-specific quantitative data regarding fluorescent intensity in defined regions were recorded. For protein fractionation, brain tissues were homogenized with ice cold TBS-containing proteinase inhibitors, followed by centrifugation (100 k×g, 30 min). The supernatant was collected as the TBS-soluble fraction. The TBS-insoluble pellet was homogenized in 1% Triton X-100 buffer containing protease inhibitors. Centrifugation yielded a supernatant collected as the Triton X-100-soluble fraction, and a Triton X-100-insoluble pellet which was further homogenized in 2% SDS buffer, followed by centrifugation. The supernatant was collected as the SDS-soluble fraction. The SDS-insoluble pellet was completely dissolved in 6 M guanidine HCl, which was saved as the guanidine-soluble fraction. Protein concentrations were measured by Pierce™ BCA Protein Assay Kit (Thermo-Fisher). Samples were analyzed by western blotting as described above.
Spontaneous activity, locomotion, and drinking behavior monitoring
The SmartCageTM system (AfaSci, Inc., Redwood City, CA) was used to assess drug effects on spontaneous activity in home cages. Automated data analysis was performed using CageScoreTM software (AfaSci, Inc.). The home cage activity was determined by infrared beam breaks (x, y, and z photo-beam break counts). Distance traveled in centimeters was obtained from the lower horizontal sensors (x and y), taking into account the path taken. Average velocity was distance traveled/second in the forward direction, averaged over the block time (variable bin). Mice (male C57/BL6, 3–4 months old, approximately 30 g) were assessed continuously for 72 h. The first 24 h was used as a habituation period. The locomotion data obtained from the second 24 h served as the baseline data for each mouse. At the completion of the second 24 h, each mouse was injected intraperitoneally with either TP70 (25 mg/kg, i.p.) or vehicle (2% DMSO in 0.5% aqueous hydroxypropyl cellulose). After receiving their respective treatments, each mouse’s locomotor activity was continually monitored over the 24 h to assess drug effects and duration of action. Drinking behavior was monitored using “LickoShock” module integrated with the SmartCageTM. Water consumption in 24 h was also manually weighed using a scale.
Rotarod test
The motor coordination and balance of the mouse was assessed by the rotarod test, which measures the time that a mouse can stay on a rotating rod with a forced motor activity controlled by the SmartCageTM system (AfaSci, Inc., Redwood City, CA). The rod has a diameter of 4.5 cm and is 10 cm long, and was operated in an adjustable constant speed (15 revolutions per min). The animals were trained three times daily for 4 consecutive days, approaching a stable level and then treated with vehicle (i.p.). In a similar circadian time of the following day, animals were injected with TP70, i.p. (see Fig. 4C). The drug effect on rotarod performance was assessed and compared to vehicle control in the same subjects using paired Student’s t-test.
Fig.2
The neuroprotective effects of TP70. A) MC65 cells were induced to express AβO by withdrawal of tetracycline (TC-) from the medium in the presence of indicated concentrations of TP70 or TP67. Cultures in the presence of TC (TC+) as well as the drug were used as controls to determine the toxicity of the drug independent of Aβ. At 72 h, viability was assessed by MTT assay. Data are expressed as mean percentage viability (n = 3) with parallel TC+ cultures without drug set at 100% viability. Error bars represent standard error. The EC50, TD50, and TI values of TP70 were calculated and listed in Fig. 1. B) MC65 cultures at 24 h after TC withdrawal were homogenized and 5 μg cellular proteins were subjected to Tris/tricine SDS-PAGE and western blot analysis with antibody 6E10 for Aβ1-16. The band pattern was comparable to our published data [8, 38]; accordingly, each band was identified. Both CP2 and TP70, but not TP67, reduced the levels of Aβ-OC, C99, and Aβ dimer without altering the level of AβPP. C) Fluorescence intensity of each culture conditions were expressed as fold change compared to the TC+ (no Aβ expression) condition. n = 7-8, *p < 0.05 compared to the TC- condition (with Aβ expression). TP70 and α-tocopherol but not CP2 or TP67 reduced ROS levels related to intracellular Aβ expression. Data are expressed as mean percentage viability (n 1/4 7) with parallel +TC cultures set at 100% viability. Error bars represent standard error.
![The neuroprotective effects of TP70. A) MC65 cells were induced to express AβO by withdrawal of tetracycline (TC-) from the medium in the presence of indicated concentrations of TP70 or TP67. Cultures in the presence of TC (TC+) as well as the drug were used as controls to determine the toxicity of the drug independent of Aβ. At 72 h, viability was assessed by MTT assay. Data are expressed as mean percentage viability (n = 3) with parallel TC+ cultures without drug set at 100% viability. Error bars represent standard error. The EC50, TD50, and TI values of TP70 were calculated and listed in Fig. 1. B) MC65 cultures at 24 h after TC withdrawal were homogenized and 5 μg cellular proteins were subjected to Tris/tricine SDS-PAGE and western blot analysis with antibody 6E10 for Aβ1-16. The band pattern was comparable to our published data [8, 38]; accordingly, each band was identified. Both CP2 and TP70, but not TP67, reduced the levels of Aβ-OC, C99, and Aβ dimer without altering the level of AβPP. C) Fluorescence intensity of each culture conditions were expressed as fold change compared to the TC+ (no Aβ expression) condition. n = 7-8, *p < 0.05 compared to the TC- condition (with Aβ expression). TP70 and α-tocopherol but not CP2 or TP67 reduced ROS levels related to intracellular Aβ expression. Data are expressed as mean percentage viability (n 1/4 7) with parallel +TC cultures set at 100% viability. Error bars represent standard error.](https://ip.ios.semcs.net:443/media/jad/2017/58-2/jad-58-2-jad161175/jad-58-jad161175-g002.jpg)
Fig.3
Some PK properties of TP70. A) Distribution of TP70 in various tissues and plasma after i.p. administration in 3-month-old male WT C57BL/6 mice (25 mg/Kg body weight) from 0–24 h. Shown are the means from three mice per time point. B) The concentrations of TP70 in plasma following i.v. and p.o. administration.
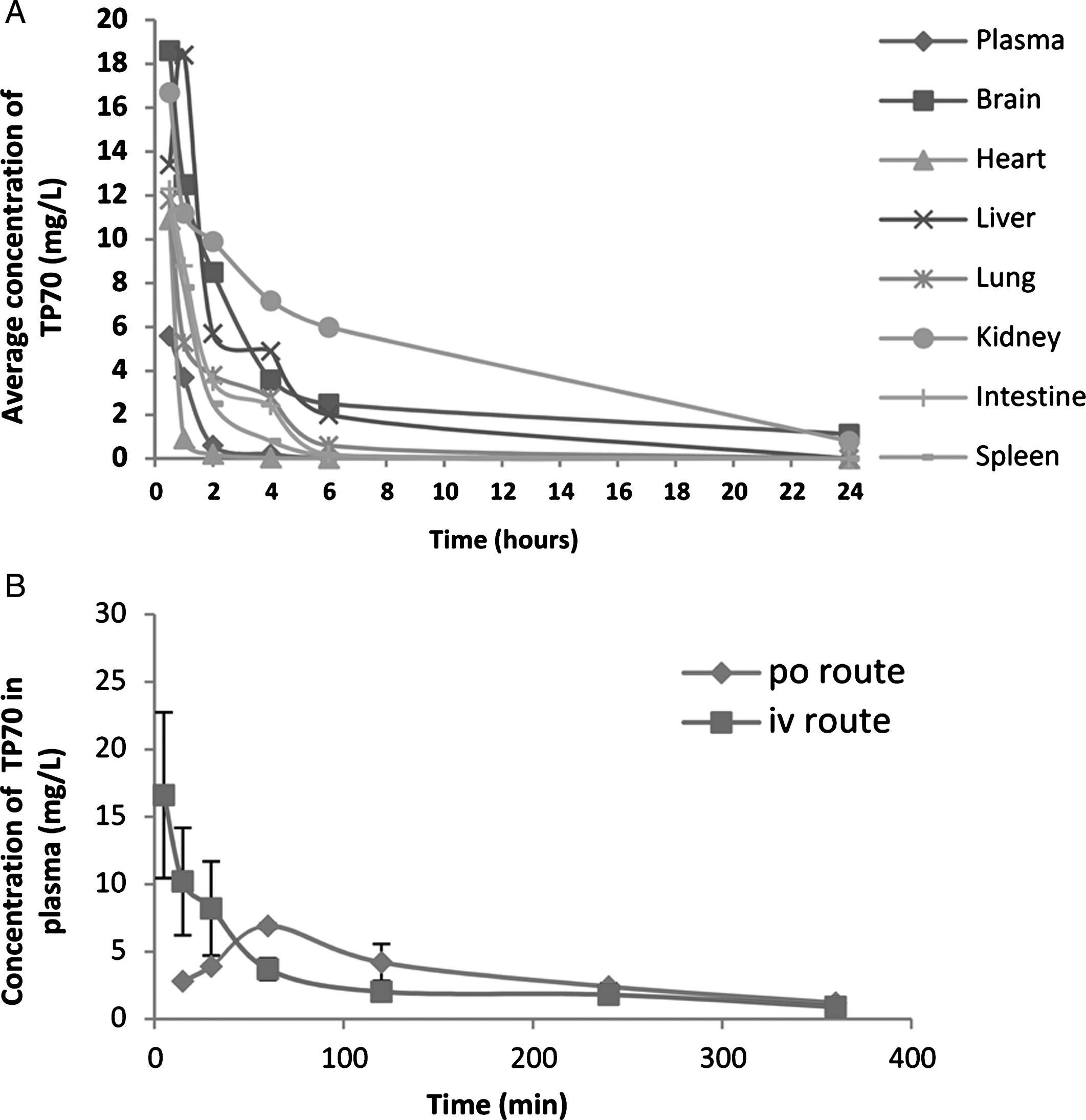
Fig.4
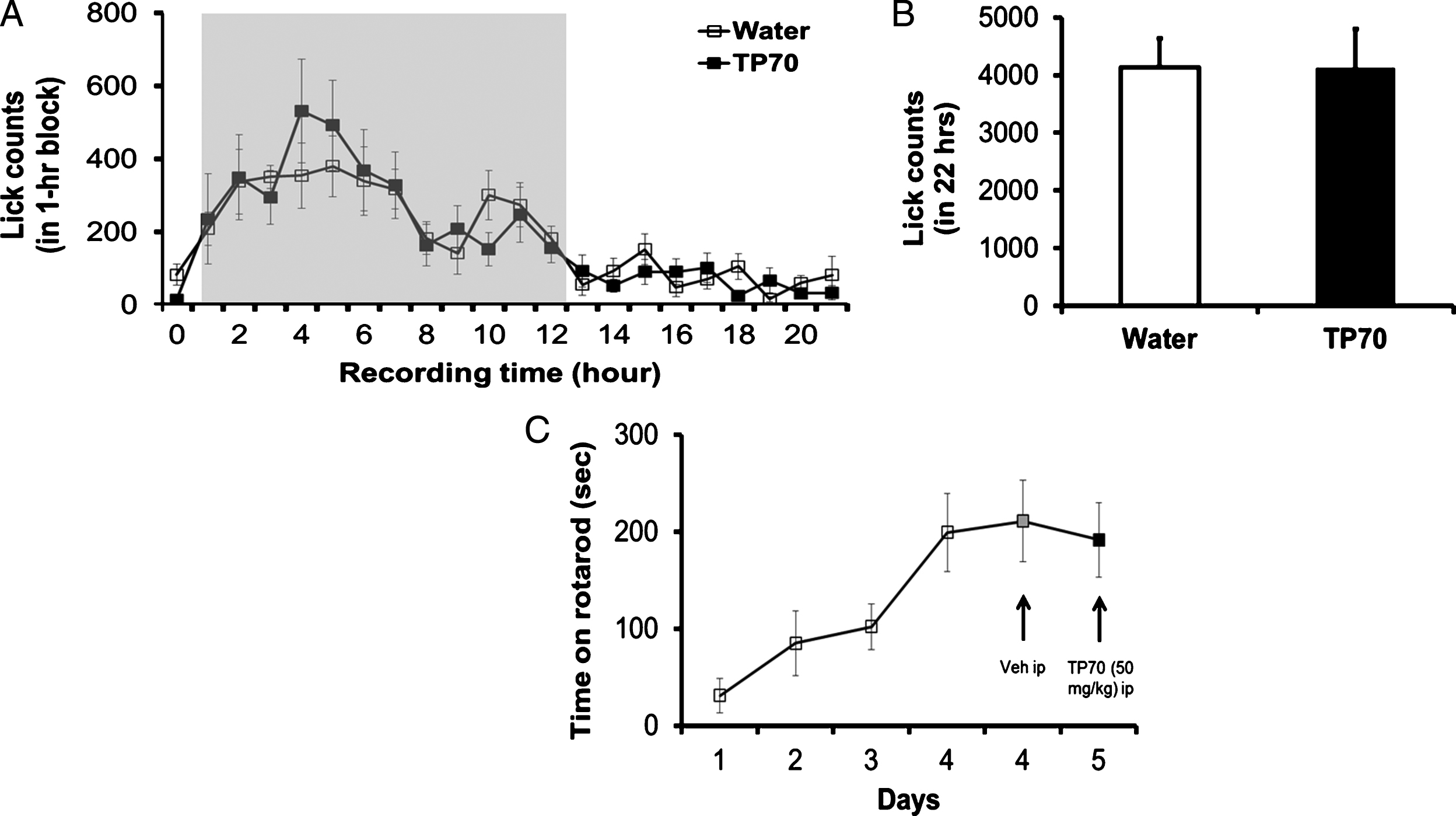
Some assessments of possible toxic effects of TP70 on animals. TP70 has no adverse effects on drinking behavior, water consumption (A and B), and motor function (C). A) Time course of lick counts. The dark phase was indicated by the gray shaded area in a 24 h light/dark cycle in home cage. B) Total lick counts in 22 h showed no difference between the two groups (n = 8 per group). C) TP70 (50 mg/kg, i.p.) caused insignificant effects on rotarod performance compared to vehicle treatment in WT mice (n = 8 per drug test).
Electrophysiology
Hippocampal slices were prepared as previously described [24]. Rats (Sprague Dawley, male, weighing 200–250 g) or mice (Harlan, C57BL/6) were deeply anesthetized with 2% isoflurane, decapitated, and the brains placed into ice-cold artificial cerebrospinal fluid (ACSF) that was continuously oxygenated with 5% CO2/95% O2. The composition of ACSF was as follows (in mM): NaCl 124.0; KCl 2.5; KH2PO4 1.2; CaCl2 2.4; MgSO4 1.3; NaHCO3 26.0; glucose 10.0 (pH 7.4). Hippocampal slices (400 μm thick) were prepared using a tissue slicer (Stoelting Co., IL) and incubated at room temperature in continuously oxygenated ACSF at least 1 h before being transferred to the recording chamber. Extracellular recordings were performed at room temperature with AxoClamp2B amplifier and pClamp 10.3 software (Molecular Device, Inc.). Submerged slices were continuously perfused with oxygenated ACSF at a flow rate of 1.5 ml/min with a peristaltic pump.
Field excitatory post-synaptic potentials (fEPSPs) were recorded using glass microelectrode filled with ACSF placed in the hippocampal CA1 field. A concentric bipolar stimulating electrode (FHC, Inc. USA) was placed in the middle of the stratum radiatum to stimulate the Schaffer collateral fibers-CA1 synapses. The stimulus was delivered through an isolator (A.M.P.I. Inc., Israel) with triggering source from computer programmed in ClampEx of pClamp 10.3. Biphasic current pulses (0.2 ms in duration) were delivered in a 10-s interval through. Stable baseline was recorded for at least 10 min before any drug application. An input-output curve (i/o curve) was established in the beginning of recording with increasing stimulus intensity using threshold as the elevating unit until the maximum response was reached. No obvious synaptic depression or facilitation was observed with this frequency stimulation. Recordings of fEPSP were obtained from the stratum radiatum of the CA1 region of the hippocampus after stimulation of the Schaffer collateral afferents. The tips of the two electrodes were separated approximately 500 μm. Slices were recorded between 1 to 6 h after dissection.
The intensity to induce about 35% of maximal response was used for basal synaptic response prior to induction of LTP. In most slices, LTP in CA1 was induced successfully using tetanic stimulation consisting of 2 trains of 100 pulses (0.2 ms pulse duration, 100 Hz) delivered with an inter-train interval of 5 s. The slope of fEPSP was measured from the initial phase of negative wave. LTP was plotted as mean percentage change in the slope of EPSP, following high frequency stimulation relative to the basal slope of fEPSP (mean±SEM). The data points were averaged from every three consecutive fEPSPs.
To record NMDA mediated fEPSP, hippocampal slices were perfused with Mg2+-free ACSF and DNQX (15 μM) to enhance NMDA receptor activation and block AMPA receptors. In initial recording of each rat hippocampal slice, the i/o curve was determined with increasing stimulus intensities. The intensity (typically 0.1 mA) evoking about 35% of maximum NMDA-mediated response was used to establish the baseline. Signals were processed through AxoClamp 2B amplifier without amplification and acquired through DigiData 1332 (Molecular Device, Inc.) with ClampEx in pClamp 10.3. Data was digitized at 10 KHz and saved in the computer. Excel (Mircosoft) and Statview were mainly used for data processing and analysis. Initial negative slope of EPSP was measured with Clampfit. Representative traces were the average of six consecutive ones, which was the data of 1 min recording. The presented data on the plots were average of every three consecutive points.
Statistics
All data are reported as mean±SEM. Paired or unpaired Student’s t-test (for two group means) or one or two-way analysis of variance (ANOVA) with post-hoc Tukey test, Scheffe’s test, or Bonferroni test, as appropriate, were conducted using the SigmaStat 3.1 (Systat Inc. Point Richmond, CA) or StatView program (version 5.0.1, SAS Institute Inc., Cary, NC). The significance level for the two-sided analyses was set at p≤0.05.
RESULTS
TP70, selected by the MC65 cell-based assay, has neuroprotective properties
The MC65 cell line was used as a screen to search for neural protective compounds against cell death induced by cellular accumulation of AβO [7, 8].This approach has been successfully used to identify compounds with anti-Aβ toxicity and neuroprotective properties via a variety of mechanisms [10, 11, 25–36]. It was engineered to conditionally produce the C99 fragment of amyloid precursor protein and subsequently oligomer-forming Aβ peptides, resulting in cell death. We employed this assay to screen over one hundred TP compounds, which were synthesized based on a structure-guided design by keeping the tricyclic pyrone skeleton intact and modifying the C7 side chain through two simple methods (Fig. 1): 1) Method A: reductive amination of tricyclic amine 4 and various aldehydes in the presence of a hydride reducing agent, such as sodium cyanoborohydride [37]; or 2) Method B: displacement reactions of mesylate 5 (or the corresponding bromide derivative) with various heterocycles such as adenine or 8-aminopurine [32]. We have identified six hits, CP2, TP4, TP36, TP70, TP82, and TP101, with ED50 (effective dose for 50% cell survival) values in the low nanomolar range. The structures and values of ED50, TD50 (median toxic dose), and TI (therapeutic index = TD50/ED50) for those hits are shown in Fig. 1. Among them, CP2 has been extensively studied [8–11]. The remaining TP analogs have ED50 ranging from 0.3 to 1.2 μM and some TPs (e.g., TP49 or TP67 (Fig. 1)), having ED50 above 5 μM, were considered inactive. It appears that the tricyclic pyrone skeleton is needed for various bioactivities, and the C7-heterocycles or aryl moieties affect the activity. For instance, other 2-nitrogen or 5-nitrogen analogs of CP2 showed much higher EC50 values in MC65 cell assays than that of CP2 [32]. Also, replacing the tricyclic pyrone to monocyclic or bicyclic pyrones resulted in a loss ofactivity.
Figure 2A shows exemplary dose-response curves of TP67 and TP70. Corresponding to its neuroprotective effect, TP70, but not inactive TP67, reduced the levels of intracellular C99 (17% reduction, p < 0.05, n = 3) and AβO, including SDS-stable Aβ dimer (72% reduction, p < 0.001, n = 3) and higher molecular weight homo- or hetero-oligomeric complexes of Aβ (Aβ-OC) (80% reduction, p < 0.001, n = 3), the identities of which were previously confirmed by various biochemical assays including mass spectrometry [8, 38] (Fig. 2B). Previously, we also found that α-tocopherol as an anti-oxidant blocked MC65 cell death [39]. Using CM-H2DCFDA to stain cellular ROS, we found that TP70, similar to α-tocopherol, significantly blocked ROS production of MC65 cells. In contrast, CP2 as well as inactive TP67 did not show such an effect (Fig. 2C).
TP70 readily enters the brain and is orally available
As more than 98% of small-molecule drugs, including many proposed or previously tested drug candidates for AD [40], do not have satisfactory brain penetrance, a major hindrance for developing effective neurotherapeutics [41], we tested the brain penetrance and other PK properties of TP70. The molecular weight of TP70 is 381. A Molinspiration program calculated the Log P (partition coefficient) and TPSA (interactive molecular polar surface area) values of 3.7 and 71 Å2, respectively. The distribution coefficient Log Doct value of TP70 from an n-octanol/PBS partitioning at pH 7.4 was 1.48. This result suggests that TP70 has brain penetration capability similar to other central nervous system drugs, such as chlorpheniramine (Log Doct = 1.25). The TPSA value of TP70 implies that it has good drug transport property for intestinal absorption and blood-brain barrier penetration. TP70 is soluble in water up to 0.72 mM.
To test the above predictions in vivo, we used HPLC/MS to quantify TP70 in plasma and organs and found that, following 25 mg/Kg intravenous administration, the TP70 concentration reached 14.7 μM in plasma and 48.8 μM in brain at 5 min, and subsequently, its levels quickly decreased. Figure 3A summarizes amounts of TP70 in plasma and key organs from 0.5–24 h. Among seven organs examined, the brain had the second highest TP70 level (Fig. 3A). Following oral and intravenous drug administration, the t1/2 values are 160 min and 30 min, respectively; the areas under the curve AUCs were 678 (mg·min/L) and 989 (mg·min/L), respectively, yielding F (oral bioavailability) = 0.68, which suggests good oral bioavailability (Fig. 3B). Taken together, the PK data indicate TP70 is orally available, has good CNS penetration, and can reach therapeutic brain drug concentrations. The good clearance and bioavailability properties suggest that TP70 can be administered orally for a long period of time, potentially suitable for treating chronic neurological disorders such as AD.
Potential drug target interaction of TP70
In an attempt to identify potential drug targets and assess selectivity of TP70, we contracted Eurofins-CEREP to conduct a target panel screen against 68 targets comprised of G protein-coupled receptors, ion channels, transporters, and enzymes using radiolabel-binding assays. Compound binding was calculated as percentage (%) inhibition of the binding of a radioactively labeled ligand specific for each target. An inhibition (or stimulation) higher than 50% is considered a significant effect of the test compound. TP70 at 10 μM did not show any significant interaction with the 68 targets, indicating its high selectivity and relative lack of off-target activities.
TP70 does not affect spontaneous activity, locomotion, drinking behavior, and motor coordination
In preparation for in vivo assessment of the effects of TP70, we first tested its effect on general neurobehavior. Spontaneous activity and drinking behavior in their home cages were monitored using our SmartCageTM system [42]. The animal’s active time, travel distance, travel speed, and rearing were monitored by infrared (IR)-beam breaks. Drinking behavior was monitored using the “LickoShock” modular device integrated with the SmartCageTM. Individual mice (4–6 months of age, n = 8) were placed into home cages with bedding, food, and water and recorded for 2–3 days under the 12:12 h light:dark cycle. Parameters include active/inactive (or ‘sleep’), locomotion (distance traveled and speed) and rearing. WT or AD transgenic mice receiving TP70 at 25 mg/kg, i.p., did not cause any obvious adverse effects on general neurobehavior compared to those receiving vehicle injection (n = 8 per group, data not shown) during the 24-h post-injection.
We also assessed whether TP70 dissolved in water would cause any changes in drinking behavior per se using the SmartCageTM described above. We compared if the lick count over time was different when the mice were given 0.12 mg/ml TP70 in drinking water. In parallel, water consumption in a 22 h period was measured to further corroborate the automated lick counts. Both measures indicate that TP70 in the drinking water did not interfere with the drinking behavior, and each mouse consumed approximately 0.6 mg TP70/5 ml/day, which, for an adult mouse weighing 25 g, is equivalent to 24 mg/kg/day (Fig. 4A, B).
We further assessed if TP70 affects motor coordination using rotarod test conducted at home cage controlled by SmartCage system [42]. Individual mice (C57BL/6J, 2 months old, n = 7), were placed onto the rod and the latency to fall was recorded. After 4 days training, the rotarod performance reached a steady state. TP70 (50 mg/kg, i.p) caused insignificant effects on rotarod performance compared to vehicle treatment (Fig. 4C).
TP70 reduces cerebral Aβ amyloid load in 5xFAD mice
Neurons in 5xFAD mice were engineered to generate a large quantity of Aβ42, which results in robust AD-like Aβ accumulation and aggregation in the brain [21]. At three months of age, the mice demonstrate numerous amyloid plaques [43]. After finding that TP70 was able to reach potentially therapeutic brain concentrations, we treated 3-month-old 5xFAD mice with TP70 at a daily i.p. dose of 25 mg/kg for a course of six weeks. We used the fluorescent amyloid dye FSB to stain amyloid plaques and objectively quantified the fluorescent intensities by automatic LSC [23]. TP70 treatment resulted in smaller and less numerous amyloid plaques (Fig. 5A). Compiled result showed a 56% reduction (n = 12-13/group, p < 0.05) of FSB fluorescence intensity in TP70-treated group compared to vehicle-treated group (Fig. 5B, C), while the two wild-type groups (TP70- and vehicle-treated) did not show any amyloid plaques (data not shown). To test oral administration, a new cohort of 5xFAD mice was chronically treated with TP70, 20–25 mg/kg daily via drinking water, for six weeks. This regimen resulted in a 31% reduction of amyloid load in the TP70-treated group (n = 8/group, p < 0.05). To determine that this chronic treatment regimen can sustain a therapeutic concentration, the brain concentrations of TP70, administered via drinking water as above for two months, were measured. The average concentrations of TP70 were 5.95±0.66 μM for wild-type (n = 4) and 4.30±0.89 μM for 5xFAD mice (n = 4), respectively. In all the above, mice chronically treated with TP70 were well groomed, displaying no observable differences compared to WT littermates. Postmortem examination of organs by an experienced pathologist (L-WJ) revealed no significant gross or histological abnormalities.
Fig.5
TP70 treatment reduces soluble and insoluble cerebral Aβ levels in 5xFAD mice. Three months old 5xFAD mice were treated with TP70 (25 mg/kg) (n = 13) or vehicle (n = 12) by daily oral gavage for 6 weeks. A, B) Four corresponding coronal sections evenly spaced through the cerebrum were taken from one hemisphere of each mouse and fluorescently stained with the amyloid dye (E,E)-1-fluoro-2,5-bis(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (FSB, blue) [22] and the nuclear dye propidium iodide (PI, red). A) Representative images. * indicated the area where the magnified image on the right panel was taken. B) Fluorescent staining was objectively quantified by LSC. LSC histograms show no difference in nuclear count (PI stain, left panel) in sections between groups and a substantial reduction of plaque count (FSB stain, right panel) in the TP70 treated group (red tracing) compared to the vehicle-treated group (blue tracing). C) LSC quantification showed a significant difference in total FSB fluorescence intensity between the two groups (n = 12-13, p < 0.05). D) A representative western blot containing the solubilized fractions of 5xFAD mouse brains was analyzed for the Aβ species by the 6E10 antibody. The area indicated by the rectangle was exposed for a longer time to reveal Aβ monomer and dimer shown below. The intensities of the bands were quantified and normalized by using the APP band as the loading control in all fractions except in the guanidine-soluble fraction, where repeated experiments and prolonged exposures did not show any trace of Aβ monomer in this fraction of TP70-treated 5xFAD mice.
![TP70 treatment reduces soluble and insoluble cerebral Aβ levels in 5xFAD mice. Three months old 5xFAD mice were treated with TP70 (25 mg/kg) (n = 13) or vehicle (n = 12) by daily oral gavage for 6 weeks. A, B) Four corresponding coronal sections evenly spaced through the cerebrum were taken from one hemisphere of each mouse and fluorescently stained with the amyloid dye (E,E)-1-fluoro-2,5-bis(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (FSB, blue) [22] and the nuclear dye propidium iodide (PI, red). A) Representative images. * indicated the area where the magnified image on the right panel was taken. B) Fluorescent staining was objectively quantified by LSC. LSC histograms show no difference in nuclear count (PI stain, left panel) in sections between groups and a substantial reduction of plaque count (FSB stain, right panel) in the TP70 treated group (red tracing) compared to the vehicle-treated group (blue tracing). C) LSC quantification showed a significant difference in total FSB fluorescence intensity between the two groups (n = 12-13, p < 0.05). D) A representative western blot containing the solubilized fractions of 5xFAD mouse brains was analyzed for the Aβ species by the 6E10 antibody. The area indicated by the rectangle was exposed for a longer time to reveal Aβ monomer and dimer shown below. The intensities of the bands were quantified and normalized by using the APP band as the loading control in all fractions except in the guanidine-soluble fraction, where repeated experiments and prolonged exposures did not show any trace of Aβ monomer in this fraction of TP70-treated 5xFAD mice.](https://ip.ios.semcs.net:443/media/jad/2017/58-2/jad-58-2-jad161175/jad-58-jad161175-g005.jpg)
To examine TP70-induced alterations of Aβ species, especially the soluble species not readily detected by FSB staining, brain homogenates from the above i.p.-injected mice were sequentially extracted into TBS-soluble, Triton X-100-soluble, SDS-soluble, and guanidine-soluble fractions. Figure 4D shows a western blot of representative samples from one vehicle-treated and one TP70-treated 5xFAD mouse. We previously identified that 5xFAD brain solubilized by RIPA buffer contained mainly Aβ dimers [43]. After sequential extraction using the above scheme, the majority of Aβ was solubilized in 2% SDS as monomer and SDS-stable dimer. As expected, the bulk of insoluble, fibrillar Aβ was completely solubilized by guanidine HCl into monomeric Aβ. Furthermore, prolonged exposure of the blot revealed Aβ monomer and dimer in the Triton X-100-soluble fraction (Fig. 5D). Comparisons between groups by quantifying band intensities showed that TP70 treatment significantly reduced the levels of Triton X-100-soluble monomer (p < 0.05) and dimer (p < 0.05), and SDS-soluble monomer (p < 0.05). Although there is a trend of reduction in SDS-soluble dimer, it did not reach statistical significance. Interestingly, TP70 treatment completely eradicated guanidine-soluble Aβ, despite the remaining presence of FSB-positive amyloid plaques in tissue sections (Fig. 4A). It is likely that TP70 enhanced the solubility of Aβ in the amyloid plaques, resulting in their shift to biochemically more soluble fractions such as from the guanidine-soluble to the SDS-soluble fraction. In summary, TP70 enhanced the overall solubility and decreased the level of cerebral Aβ in 5xFAD mice after peripheraladministration.
TP70 enhances NMDA receptor-mediated fEPSP and restores AβO-impaired hippocampal long-term potentiation
As aging- or AD-related memory deficits, especially those of episodic memory, are intimately related to NMDA receptor-mediated synaptic plasticity [44], we tested if TP70 modulates such synaptic events. Using rat hippocampal slices, we recorded NMDA receptor-mediated EPSP by perfusion of Mg2+-free ACSF, containing DNQX to block the AMPA component. After the baseline was stabilized for at least 5 min, we perfused TP70 dissolved in modified ACSF. The results showed that TP70 concentration-dependently (1–30 μM) caused a long-lasting enhancement of the NMDA receptor-mediated fEPSP (133.6±9.5% of control in 10 μM, Fig. 5A, B), but had little effects on basal fEPSP predominantly mediated by AMPA receptors (data not shown). The effect of enhancing NMDA-mediated fEPSP apparently saturated at 30 μM. Perfused standard ACSF containing 1.3 mM Mg2+ by the end of the recording almost completely eliminated the fEPSP, confirming that it was largely mediated by NMDA receptors (Fig. 5C).
hLTP is a well-established cellular model of learning and memory and has been shown to be impaired in several AD models associated with Aβ toxicity. Using mouse hippocampal slices, we tested whether TP70 could affect hLTP induction and maintenance in the Schaffer-collateral-CA1 synapses following high frequency stimulation [45]. We found that TP70 by itself significantly increased the magnitude of NMDA-dependent hLTP (246.7±20.7% versus 191.4±17.5% in control). AβO (0.1 μM, 45 min perfusion) alone significantly decreased the magnitude of hLTP (145.3±10.9%), and this effect was prevented by co-application of TP70 (1 μM, 45 min treatment prior to recordings; Fig. 5D, E).
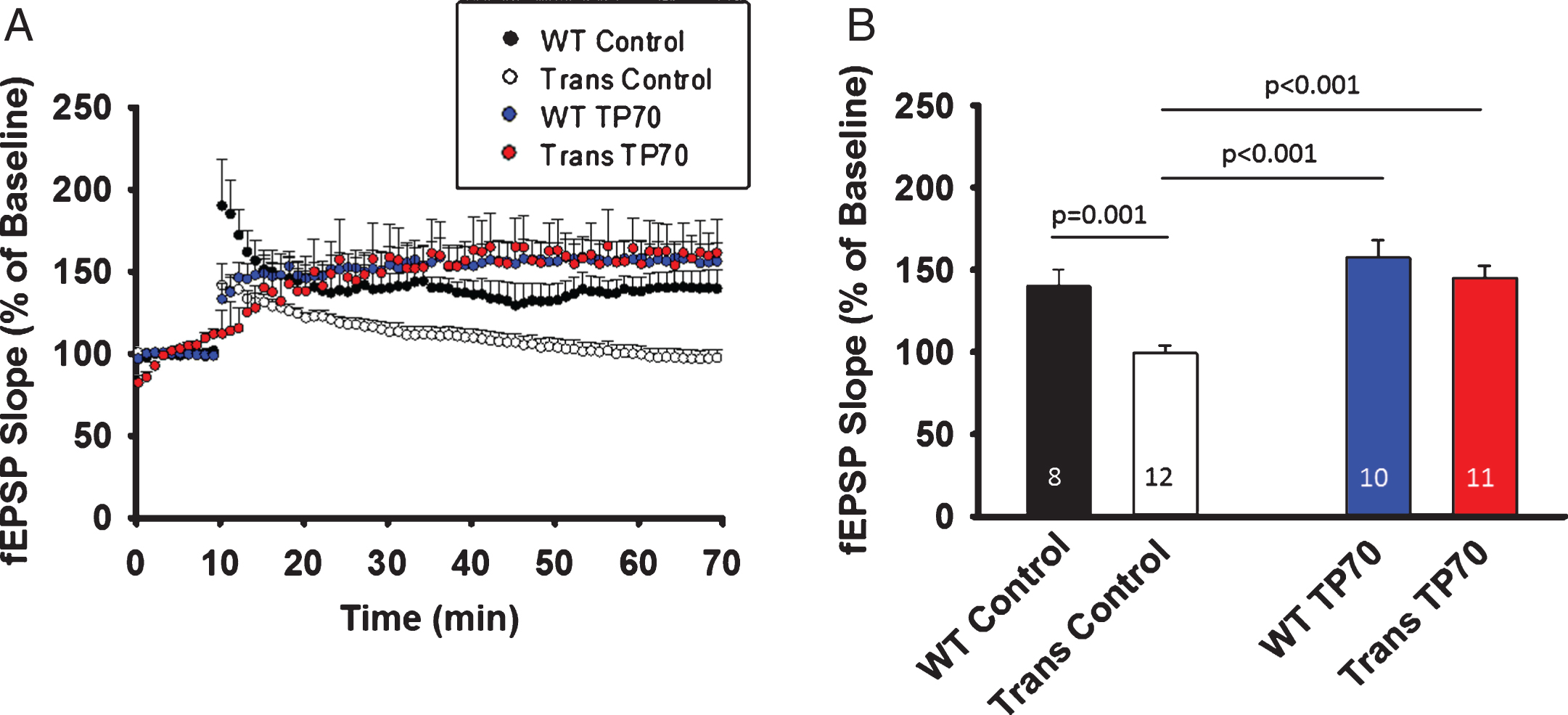
To test the in vivo effect of TP70 on hLTP in WT as well as AD transgenic animals, we i.p. injected TP70 (25 mg/kg) or equal volume of vehicle to 15-16-month-old 5xFAD mice and age-matched WT littermates, sacrificed them at 24-h post-injection, and obtained hippocampal slices for recording. The magnitude of hLTP induction was significantly reduced in 5xFAD mice compared to WT littermates, but was significantly increased after TP70 treatment (p < 0.001). There was no significant difference between TP70-treated 5xFAD and TP70-treated WT mice, or between TP70-treated 5xFAD mice and vehicle-treated WT mice. Although in the above in vitro study TP70 by itself significantly increased the magnitude of hLTP, the in vivo treatment did not enhance hLTP in WT mice (Fig. 7).
Fig.6
Electrophysiological effects of TP70 on NMDA-mediated fEPSPs. A) Representative traces and time course showing that perfusion of TP70 (10 μM) enhanced NMDA-mediated fEPSPs. B) Bar plot showing that the effects of TP70 on NMDA-mediated EPSPs was in a concentration-dependent manner. n = 3–9 for each group; *p < 0.05 compared to the vehicle-treated group. C) The NMDA-mediated fEPSPs were blocked by perfusion of ACSF containing 1.3 mM Mg2+. D) TP70 prevented the inhibitory effect of AbO on amplitude of LTP induced with high frequency stimulation. The representative traces show the fEPSP of baseline and at 45 min after high frequency stimulation. Slices were perfused with AbO (100 nM) for 45 min before recording, and perfused continuously throughout the recording, in the presence of TP70 (1 μM) or vehicle control. AbO alone significantly inhibited the level of LTP and this effect was prevented by co-application of TP70. TP70 alone significantly enhanced the magnitude of LTP. E) Summary bar plot showing the average amplitudes of LTP in the last 5 min. n = 6–12 for each group; *p < 0.05, **p < 0.0005, and ns: no significant difference.
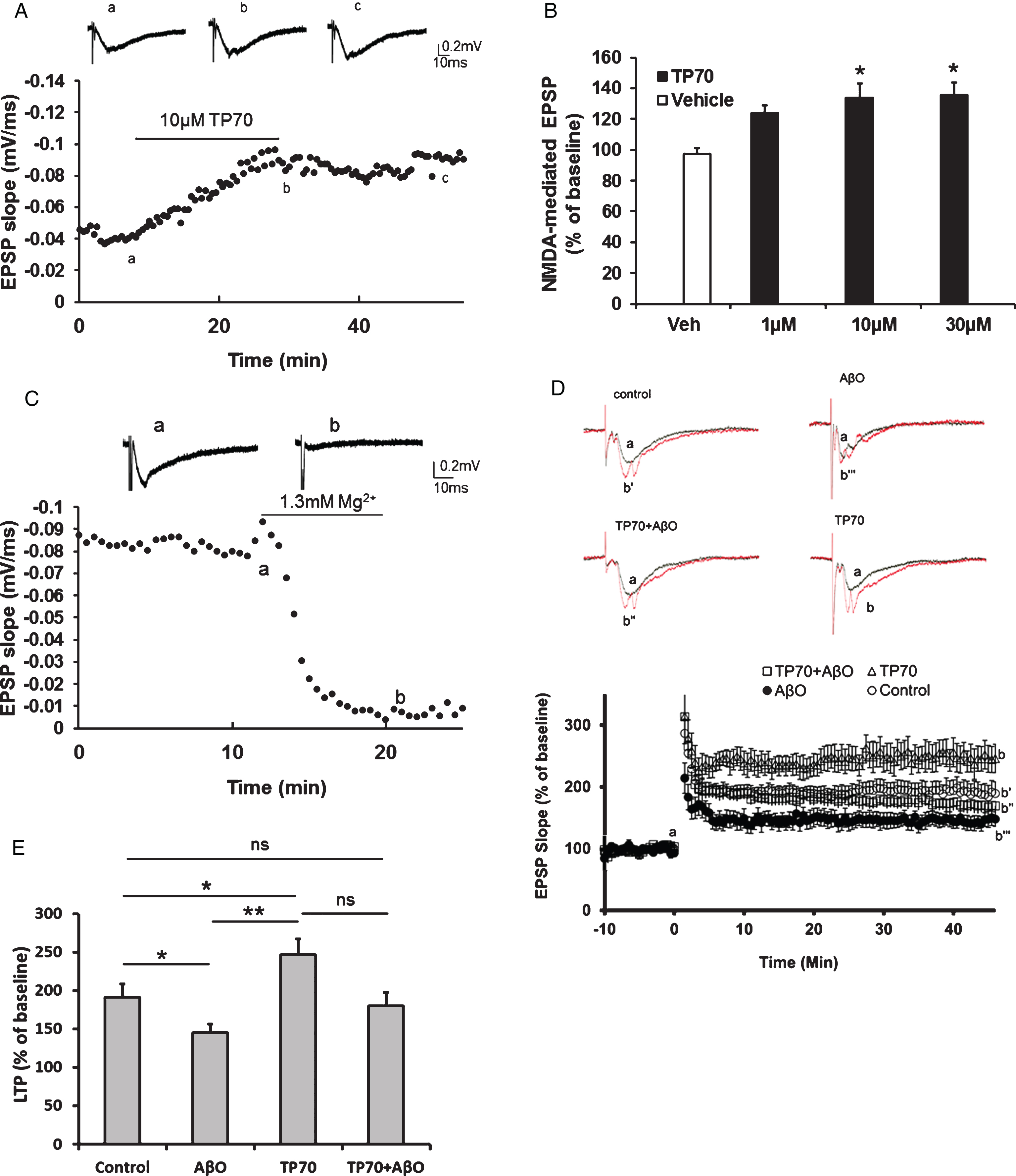
Fig.7
TP70 treatment mitigates hippocampal LTP deficits in 5xFAD mice. 5xFAD (Trans) mice and WT littermates at 15-16 months of age were intraperitoneally injected either 25 mg/kg TP70 or equal volume of vehicle. The mice were sacrificed 24 h later and hippocampal slices obtained for recording. A) Traces and time course of LTP induction showing that TP70 treatment of 5xFAD mice enhanced the amplitude of LTP to the levels not different from those of WT mice. B) Summary bar plot showing the average fEPSP slope at 45 min after high frequency stimulation. Data were compiled from recordings using slices obtained from 3 mice per group. The number within each bar indicates the number of slices used for recording.
DISCUSSION
Using the MC65 cell-based assay, we selected and conducted initial characterization of a new lead tricyclic pyrone compound, TP70. The translational advantage of this cell-based assay as a screening tool has been previously discussed [8, 12, 33]. We found that in vivo, TP70, like CP2 [9, 11], was well-tolerated and showed high oral bioavailability, excellent blood-brain barrier permeability, and low toxicity following chronic oral administration. Chronic peripheral administrations of TP70 caused an up to 56% reduction of cerebral amyloid load in 5xFAD mice. The result from differential extraction of Aβ suggests that TP70 is able to enhance the solubility of Aβ aggregates and/or prevent the formation of insoluble aggregates in vivo, an action similar to CP2 [9]. Significantly, TP70 treatment, both in vitro and in vivo, resulted in reductions of the levels of soluble Aβ oligomers, including dimers, which are considered highly toxic and potent activators of microglia [14, 46]. These results suggest a clinical potential of TP70 as an orally available neuroprotective agent for AD or mild cognitiveimpairment.
Various observations suggest that TP70 at therapeutic dosages appears not toxic. One may consider that a brain concentration of 48.8 μM, which was reached immediately after intravenous administration, is likely toxic since the TD50 value of TP70 in cell culture is 49.5 μM (Fig. 1). However, a drug’s efficiency and toxicity is usually affected by the degree to which it binds to proteins within blood plasma or tissue, and many clinical drugs have a protein binding of 60–90%. Therefore, we do not consider the TD50 measured in cell culture in a low serum condition an accurate assessment of toxic concentrations in vivo. Indeed, several sets of mice receiving chronic TP70 treatment did not show any signs of toxicity revealed by behavioral and pathological observations. Moreover, TP70 treatment did not affect spontaneous activity, locomotion, drinking behavior, and motor coordination. In an in vitro screen against 68 targets comprised of G protein-coupled receptors, ion channels, transporters, and enzymes, TP70 did not show significant off-target effects via interactions with these common targets.
While further studies are needed to understand its mechanisms of action, TP70, compared to the previously characterized CP2, shows some superior or unique features. TP70 is more potent in the MC65 cell assay, has a much higher TI level (Fig. 1). TP70, but not CP2, suppressed the ROS production of MC65 cells. While the overall PK profile is comparable, TP70 reached higher brain concentrations at 18.6 mg/L (in 30 min; i.p.) than the 2.9 mg/L (in 30 min; i.p.) of CP2. Hence, it is possible to use lesser amounts of TP70 than CP2 in vivo to reach similar cerebral concentrations.
In a prior study, we found that TP70, like CP2, inhibited the activity of ACAT and upregulated the expression of ATP-binding cassette subfamily A, member 1 (ABCA1), with TP70 being a more potent inhibitor of ACAT than CP2 (IC50 value of ACAT inhibition: TP70, 0.3±0.008 and CP2, 1.2±0.2) [10]. ACAT has been proposed as a therapeutic target for AD, as ACAT inhibitors were shown to induce cholesterol efflux, reduce amyloid pathology, and promote multiple beneficial effects on AD models. ACAT mainly resides in the endoplasmic reticulum and converts cholesterol to cholesteryl esters. It therefore regulates local cholesterol content in the endoplasmic reticulum membranes and affects autophagosome formation, mechanisms that potentially affect neuronal activities and alter the metabolism of several key AD pathological molecules, such as AβPP and tau [47, 48].Loss of ABCA1, a major cholesterol transporter, has also been shown to pose risk for AD development [49]. The combined effect of ACAT inhibition and ABCA1 induction is likely one of the mechanisms via which TP70 reduces cerebral amyloidpathology [50].
NMDA receptor-mediated neurotransmission plays a critical role in the pathophysiology of AD [51, 52]. We have now shown that TP70 positively modulates NMDA-mediated fEPSP, enhanced hippocampal LTP, and prevents the AbO-impaired LTP in hippocampal CA1 area. This action of TP70 appears opposite to the synaptic modulation of memantine, the clinical effects of which are thought to be associated with activity-dependent inhibition of NMDA receptors. However, the actual actions of memantine and the pathological roles of NMDA receptors in AD remain controversial [53–55]. It was shown that memantine decreased the frequency and amplitude of spontaneous inhibitory postsynaptic currents in pyramidal neurons and decreased the amplitude of NMDA receptor-mediated synaptic responses in interneurons, thus enhancing the overall excitatory tone [56]. Moreover, several lines of evidence from genetic, animal model, and human studies support that NMDA receptor hypofunction is involved in the progression of aging brain from mild cognitive impairment to AD [57]. Therefore, NMDA-enhancing agents such as TP70 may be useful in improving cognitive function. Moreover, the novel effects of TP70 may provide an opportunity to study the chemical biology behind the NMDA receptor-mediated hippocampal plasticity using TP derivatives.
Ideally, NMDA receptor-mediated neurotransmission needs to be modulated in a way to achieve cognitive enhancement while avoiding NMDA-related excitotoxicity [57], for which TP70 could be a useful compound. The potential of TP70 in enhancing memory is evidenced by its ability to enhance hippocampal LTP if applied alone and avert the LTP deficit induced by the co-applied AβO. Significantly, a single dose of TP70 administered to aged 5xFAD mice was effective in mitigating the impaired LTP induction, recorded at 24 h after administration, suggesting the promise of TP70 in restoring Aβ-related, hippocampus-dependent memory deficits, even with a short-term treatment. Various mechanisms have been proposed for alterations of hLTP mediated by Aβ, in particular by AβO. AβO has a high affinity to synapses, where it can disrupt a variety of signal transduction, decrease β2-adrenergic receptors, interact with NMDA receptors, activate microglia, or even directly act as a pathogenic ligand through several putative receptors [58–60]. It is imperative in future studies to identify TP70 targets in order to understand the mechanisms via which TP70 protects the synapse from AβO toxicity. TP70 also has synergistic capacities in anti-oxidation, promoting cholesterol efflux, enhancing amyloid solubility, and reducing its accumulation. With its multiple complementary cellular actions and favorable PK profile, TP70 is a preclinical candidate for AD therapy.
ACKNOWLEDGMENTS
This work was supported by the UC Davis Alzheimer’s Disease Center (P30 AG010129), and NIH grants R01 AG025500, R43 AG043203, and R44 AG043203. I.M., X.S.X., D.H.H., and L-W.J. are co-inventors of TPs under U.S. patent application serial no. 14/503262.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1175r2).
REFERENCES
[1] | Selkoe DJ ((2001) ) Alzheimer’s disease: Genes, proteins, and therapy. Physiol Rev 81: , 741–766. |
[2] | Raina P , Santaguida P , Ismaila A , Patterson C , Cowan D , Levine M , Booker L , Oremus M ((2008) ) Effectiveness of cholinesterase inhibitors and memantine for treating dementia: Evidence review for a clinical practice guideline. Ann Intern Med 148: , 379–397. |
[3] | Aaseth J , Alexander J , Bjørklund G , Hestad K , Dusek P , Roos PM , Alehagen U ((2016) ) Treatment strategies in Alzheimer’s disease: A review with focus on selenium supplementation. Biometals 29: , 827–839. |
[4] | Bennett DA , Yu L , De Jager PL ((2014) ) Building a pipeline to discover and validate novel therapeutic targets and lead compounds for Alzheimer’s disease. Biochem Pharmacol 88: , 617–630. |
[5] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[6] | Sevigny J , Chiao P , Bussière T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , O’Gorman J , Qian F , Arastu M , Li M , Chollate S , Brennan MS , Quintero-Monzon O , Scannevin RH , Arnold HM , Engber T , Rhodes K , Ferrero J , Hang Y , Mikulskis A , Grimm J , Hock C , Nitsch RM , Sandrock A ((2016) ) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537: , 50–56. |
[7] | Jin LW , Hua DH , Shie FS , Maezawa I , Sopher B , Martin GM ((2002) ) Novel tricyclic pyrone compounds prevent intracellular APP C99-induced cell death. J Mol Neurosci 19: , 57–61. |
[8] | Maezawa I , Hong HS , Wu HC , Battina SK , Rana S , Iwamoto T , Radke GA , Pettersson E , Martin GM , Hua DH , Jin LW ((2006) ) A novel tricyclic pyrone compound ameliorates cell death associated with intracellular amyloid-beta oligomeric complexes. J Neurochem 98: , 57–67. |
[9] | Hong HS , Rana S , Barrigan L , Shi A , Zhang Y , Zhou F , Jin LW , Hua DH ((2009) ) Inhibition of Alzheimer’s amyloid toxicity with a tricyclic pyrone molecule in vitro and in vivo. J Neurochem 108: , 1097–1108. |
[10] | Pokhrel L , Maezawa I , Nguyen TD , Chang KO , Jin LW , Hua DH ((2012) ) Inhibition of Acyl-CoA: Cholesterol acyltransferase (ACAT), overexpression of cholesterol transporter gene, and protection of amyloid β (Aβ) oligomers-induced neuronal cell death by tricyclic pyrone molecules. J Med Chem 55: , 8969–8973. |
[11] | Zhang L , Zhang S , Maezawa I , Trushin S , Minhas P , Pinto M , Jin LW , Prasain K , Nguyen TD , Yamazaki Y , Kanekiyo T , Bu G , Gateno B , Chang KO , Nath KA , Nemutlu E , Dzeja P , Pang YP , Hua DH , Trushina E ((2015) ) Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s Disease. EBioMedicine 2: , 294–305. |
[12] | Hong HS , Maezawa I , Yao N , Xu B , Diaz-Avalos R , Rana S , Hua DH , Cheng RH , Lam KS , Jin LW ((2007) ) Combining the rapid MTT formazan exocytosis assay and the MC65 protection assay led to the discovery of carbazole analogs as small molecule inhibitors of Abeta oligomer-induced cytotoxicity. Brain Res 1130: , 223–234. |
[13] | Lesné S , Koh MT , Kotilinek L , Kayed R , Glabe CG , Yang A , Gallagher M , Ashe KH ((2006) ) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440: , 352–357. |
[14] | Shankar GM , Li S , Mehta TH , Garcia-Munoz A , Shepardson NE , Smith I , Brett FM , Farrell MA , Rowan MJ , Lemere CA , Regan CM , Walsh DM , Sabatini BL , Selkoe DJ ((2008) ) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14: , 837–842. |
[15] | Schubert D , Maher P ((2012) ) An alternative approach to drug discovery for Alzheimer’s disease dementia. Future Med Chem 4: , 1681–1688. |
[16] | Reitz C , Brayne C , Mayeux R ((2011) ) Epidemiology of Alzheimer disease. Nat Rev Neurol 7: , 137–152. |
[17] | Mondragon-Rodriguez S , Basurto-Islas G , Lee HG , Perry G , Zhu X , Castellani RJ , Smith MA ((2010) ) Causes versus effects: The increasing complexities of Alzheimer’s disease pathogenesis. Expert Rev Neurother 10: , 683–691. |
[18] | Maezawa I , Jenkins DP , Jin BE , Wulff H ((2012) ) Microglial KCa3.1 channels as a potential therapeutic target for Alzheimer’s disease. Int J Alzheimers Dis 2012: , 868972. |
[19] | Trushina E , Rana S , McMurray CT , Hua DH ((2009) ) Tricyclic pyrone compounds prevent aggregation and reverse cellular phenotypes caused by expression of mutant huntingtin protein in striatal neurons. BMC Neurosci 10: , 73. |
[20] | Maezawa I , Jin LW , Woltjer RL , Maeda N , Martin GM , Montine TJ , Montine KS ((2004) ) Apolipoprotein E isoforms and apolipoprotein AI protect from amyloid precursor protein carboxy terminal fragment-associated cytotoxicity. J Neurochem 91: , 1312–1321. |
[21] | Oakley H , Cole SL , Logan S , Maus E , Shao P , Craft J , Guillozet-Bongaarts A , Ohno M , Disterhoft J , Van Eldik L , Berry R , Vassar R ((2006) ) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J Neurosci 26: , 10129–10140. |
[22] | Sato K , Higuchi M , Iwata N , Saido TC , Sasamoto K ((2004) ) Fluoro-substituted and 13C-labeled styrylbenzene derivatives for detecting brain amyloid plaques. Eur J Med Chem 39: , 573–578. |
[23] | Maezawa I , Swanberg S , Harvey D , LaSalle JM , Jin LW ((2009) ) Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci 29: , 5051–5061. |
[24] | Xie X , Lancaster B , Peakman T , Garthwaite J ((1995) ) Interaction of the antiepileptic drug lamotrigine with recombinant rat brain type IIA Na+ channels and with native Na+ channels in rat hippocampal neurones. Pflugers Arch 430: , 437–446. |
[25] | Gray NE , Zweig JA , Kawamoto C , Quinn JF , Copenhaver PF ((2016) ) STX, a novel membrane estrogen receptor ligand, protects against amyloid-β toxicity. J Alzheimers Dis 51: , 391–403. |
[26] | Saathoff JM , Liu K , Chojnacki JE , He L , Chen Q , Lesnefsky EJ , Zhang S ((2016) ) Mechanistic insight of bivalent compound 21MO as potential neuroprotectant for Alzheimer’s disease. Molecules 21: , 412. |
[27] | Chojnacki JE , Liu K , Saathoff JM , Zhang S ((2015) ) Bivalent ligands incorporating curcumin and diosgenin as multifunctional compounds against Alzheimer’s disease. Bioorg Med Chem 23: , 7324–7331. |
[28] | Liu K , Chojnacki JE , Wade EE , Saathoff JM , Lesnefsky EJ , Chen Q , Zhang S ((2015) ) Bivalent compound 17MN exerts neuroprotection through interaction at multiple sites in a cellular model of Alzheimer’s disease. J Alzheimers Dis 47: , 1021–1033. |
[29] | Revilla S , Ursulet S , Álvarez-López MJ , Castro-Freire M , Perpiñá U , García-Mesa Y , Bortolozzi A , Giménez-Llort L , Kaliman P , Cristòfol R , Sarkis C , Sanfeliu C ((2014) ) Lenti-GDNF gene therapy protects againstAlzheimer’s disease-like neuropathology in 3xTg-AD mice and MC65cells. CNS Neurosci Ther 20: , 961–972. |
[30] | Gray NE , Sampath H , Zweig JA , Quinn JF , Soumyanath A ((2015) ) Centella asiatica attenuates amyloid-β-induced oxidative stress and mitochondrial dysfunction. J Alzheimers Dis 45: , 933–946. |
[31] | Gray NE , Morré J , Kelley J , Maier CS , Stevens JF , Quinn JF , Soumyanath A ((2014) ) Caffeoylquinic acids in Centella asiaticaprotect against amyloid-β toxicity. J Alzheimers Dis 40: , 359–373. |
[32] | Rana S , Hong HS , Barrigan L , Jin LW , Hua DH ((2009) ) Syntheses of tricyclic pyrones and pyridinones and protection of Abeta-peptide induced MC65 neuronal cell death. Bioorg Med Chem Lett 19: , 670–674. |
[33] | Copenhaver PF , Anekonda TS , Musashe D , Robinson KM , Ramaker JM , Swanson TL , Wadsworth TL , Kretzschmar D , Woltjer RL , Quinn JF ((2011) ) A translational continuum of model systems for evaluating treatment strategies in Alzheimer’s disease: Isradipine as a candidate drug. Dis Model Mech 4: , 634–648. |
[34] | Anekonda TS , Wadsworth TL , Sabin R , Frahler K , Harris C , Petriko B , Ralle M , Woltjer R , Quinn JF ((2011) ) Phytic acid as a potential treatment for Alzheimer’s pathology: Evidence from animal and in vitro models. J Alzheimers Dis 23: , 21–35. |
[35] | Anekonda TS , Quinn JF , Harris C , Frahler K , Wadsworth TL , Woltjer RL ((2011) ) L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol Dis 41: , 62–70. |
[36] | Lin CL , Chen TF , Chiu MJ , Way TD , Lin JK ((2009) ) Epigallocatechin gallate (EGCG) suppresses beta-amyloid-induced neurotoxicity through inhibiting c-Abl/FE65 nuclear translocation and GSK3 beta activation. Neurobiol Aging 30: , 81–92. |
[37] | Pokhrel L , Kim Y , Nguyen TD , Prior AM , Lu J , Chang KO , Hua DH ((2012) ) Synthesis and anti-norovirus activity of pyranobenzopyrone compounds. Bioorg Med Chem Lett 22: , 3480–3484. |
[38] | Woltjer RL , Nghiem W , Maezawa I , Milatovic D , Vaisar T , Montine KS , Montine TJ ((2005) ) Role of glutathione in intracellular amyloid-alpha precursor protein/carboxy-terminal fragment aggregation and associated cytotoxicity. J Neurochem 93: , 1047–1056. |
[39] | Woltjer RL , Maezawa I , Ou JJ , Montine KS , Montine TJ ((2003) ) Advanced glycation endproduct precursor alters intracellular amyloid-beta/A beta PP carboxy-terminal fragment aggregation and cytotoxicity. J Alzheimers Dis 5: , 467–476. |
[40] | Golde TE , Schneider LS , Koo EH ((2011) ) Anti-aβ therapeutics in Alzheimer’s disease: The need for a paradigm shift. Neuron 69: , 203–213. |
[41] | Pardridge WM ((2005) ) The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2: , 3–14. |
[42] | Khroyan TV , Zhang J , Yang L , Zou B , Xie J , Pascual C , Malik A , Xie J , Zaveri NT , Vazquez J , Polgar W , Toll L , Fang J , Xie X ((2012) ) Rodent motor and neuropsychological behaviour measured in home cages using the integrated modular platform SmartCage™. Clin Exp Pharmacol Physiol 39: , 614–622. |
[43] | Hong HS , Maezawa I , Budamagunta M , Rana S , Shi A , Vassar R , Liu R , Lam KS , Cheng RH , Hua DH , Voss JC , Jin LW ((2010) ) Candidate anti-A beta fluorene compounds selected from analogs of amyloid imaging agents. Neurobiol Aging 31: , 1690–1699. |
[44] | Foster TC ((2012) ) Dissecting the age-related decline on spatial learning and memory tasks in rodent models: N-methyl-D-aspartate receptors and voltage-dependent Ca2+ channels in senescent synaptic plasticity. Prog Neurobiol 96: , 283–303. |
[45] | Chapman CA , Perez Y , Lacaille JC ((1998) ) Effects of GABA(A) inhibition on the expression of long-term potentiation in CA1 pyramidal cells are dependent on tetanization parameters. Hippocampus 8: , 289–298. |
[46] | Maezawa I , Zimin PI , Wulff H , Jin LW ((2011) ) Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem 286: , 3693–3706. |
[47] | Shibuya Y , Chang CC , Chang TY ((2015) ) ACAT1/SOAT1 as a therapeutic target for Alzheimer’s disease. Future Med Chem 7: , 2451–2467. |
[48] | Hutter-Paier B , Huttunen HJ , Puglielli L , Eckman CB , Kim DY , Hofmeister A , Moir RD , Domnitz SB , Frosch MP , Windisch M , Kovacs DM ((2004) ) The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron 44: , 227–238. |
[49] | Nordestgaard LT , Tybjærg-Hansen A , Nordestgaard BG , Frikke-Schmidt R ((2015) ) Loss-of-function mutation in ABCA1 andrisk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement 11: , 1430–1438. |
[50] | Puglielli L , Tanzi RE , Kovacs DM ((2003) ) Alzheimer’sdisease: The cholesterol connection. Nat Neurosci 6: , 345–351. |
[51] | Lipton SA , Rosenberg PA ((1994) ) Excitatory amino-acids as a final common pathway for neurologic disorders. N Eng J Med 330: , 613–622. |
[52] | Farber NB , Newcomer JW , Olney JW ((1998) ) The glutamate synapse in neuropsychiatric disorders. Focus on schizophrenia and Alzheimer’s disease. Prog Brain Res 116: , 421–437. |
[53] | Hu NW , Ondrejcak T , Rowan MJ ((2012) ) Glutamate receptors in preclinical research on Alzheimer’s disease: Update on recent advances. Pharmacol Biochem Behav 100: , 855–862. |
[54] | Johnson JW , Kotermanski SE ((2006) ) Mechanism of action of memantine. Currt Opin Pharmacol 6: , 61–67. |
[55] | Foster TC , Kyritsopoulos C , Kumar A ((2017) ) Central role for NMDA receptors in redox mediated impairment of synaptic function during aging and Alzheimer’s disease. Behav Brain Res 322: , 223–232. |
[56] | Povysheva NV , Johnson JW ((2016) ) Effects of memantine on the excitation-inhibition balance in prefrontal cortex. Neurobiol Dis 96: , 75–83. |
[57] | Huang YJ , Lin CH , Lane HY , Tsai GE ((2012) ) NMDA neurotransmission dysfunction in behavioral and psychological symptoms of Alzheimer’s disease. Curr Neuropharmacol 10: , 272–285. |
[58] | Pozueta J , Lefort R , Shelanski ML ((2013) ) Synaptic changes in Alzheimer’s disease and its models. Neuroscience 251: , 51–65. |
[59] | Jarosz-Griffiths HH , Noble E , Rushworth JV , Hooper NM ((2016) ) Amyloid-beta receptors: The good, the bad, and the prion protein. J Biol Chem 291: , 3174–3183. |
[60] | Yang T , Li S , Xu H , Walsh DM , Selkoe DJ ((2017) ) Large soluble oligomers of amyloid β-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci 37: , 152–163. |


