
Collapsin Response Mediator Protein-2 (CRMP2) is a Plausible Etiological Factor and Potential Therapeutic Target in Alzheimer’s Disease: Comparison and Contrast with Microtubule-Associated Protein Tau
Abstract
Alzheimer’s disease (AD) has long been viewed as a pathology that must be caused either by aberrant amyloid-β protein precursor (AβPP) processing, dysfunctional tau protein processing, or a combination of these two factors. This is a reasonable assumption because amyloid-β peptide (Aβ) accumulation and tau hyperphosphorylation are the defining histological features in AD, and because AβPP and tau mutations can cause AD in humans or AD-like features in animal models. Nonetheless, other protein players are emerging that one can argue are significant etiological players in subsets of AD and potentially novel, druggable targets. In particular, the microtubule-associated protein CRMP2 (collapsin response mediator protein-2) bears striking analogies to tau and is similarly relevant to AD. Like tau, CRMP2 dynamically regulates microtubule stability; it is acted upon by the same kinases; collects similarly in neurofibrillary tangles (NFTs); and when sequestered in NFTs, complexes with critical synapse-stabilizing factors. Additionally, CRMP2 is becoming recognized as an important adaptor protein involved in vesicle trafficking, amyloidogenesis and autophagy, in ways that tau is not. This review systematically compares the biology of CRMP2 to that of tau in the context of AD and explores the hypothesis that CRMP2 is an etiologically significant protein in AD and participates in pathways that can be rationally engaged for therapeutic benefit.
INTRODUCTION
Alzheimer’s disease (AD) is neuropathologically defined by brain regional neurodegeneration accompanied by cardinal features of amyloid-β protein (Aβ)-rich extracellular plaques and intraneuronal neurofibrillary tangles (NFTs) composed largely (but not exclusively) of aggregated, hyperphosphorylated, microtubule-associated protein (MAP) tau (MAPT) [1–7]. Decades’ worth of research attention has been devoted to both Aβ and tau, and both species have been plausibly hypothesized to either initiate AD or contribute to disease progression. Despite this intense investment, however, research into neither Aβ nor tau has yet generated any clinically proven treatments to slow human AD. The vast majority of AD pharmacology research and development efforts have focused on amyloid targeting therapies, but these strategies have wholly failed to slow human disease [8]. Failure of the “amyloid cascade hypothesis” to yield effective drugs has prompted a shift within the mainstream AD drug development community toward strategies targeting the tauopathy component instead [9, 10]. While it remains too early to pass judgment on the wisdom of this shift toward tau-targeting drugs, AD researchers risk repeating historical mistakes by concentrating therapy development so monolithically on the most obvious AD neuropathological features. Instead, research and drug development arguably should be diversified in order to consider other, less obvious protein players in AD.
With the above caveats in mind, the present review and discussion aims to explore the hypothesis that the MAP protein CRMP2 (collapsin response mediator protein 2; also known as dihydropyrimidinase-like protein-2, DRP2 or DPYSL2) is a significant factor in AD pathogenesis, which has been relatively neglected as a potential therapeutic target compared to its more famous MAP cousin, tau. The known biology of CRMP2 will be described, along with data that associates CRMP2 with AD cytopathology. It will be argued that CRMP2 post-translational modifications (PTMs) known to occur in AD likely compromise neuronal microtubule integrity, much in the same way that tau PTMs are thought to do, while simultaneously undermining the cells’ ability to conduct normal vesicular transport and competently execute autophagy programs. The discussion will critically evaluate the main argument against CRMP2 being as important a pathogenic factor as tau in neurodegenerative disease: Tau mutations are known to cause familial neurodegenerative “tauopathies”, whereas analogous mutations in CRMP2 have not been reported. Finally, the druggability of CRMP2 will be discussed with reference to emerging therapeutic strategies for either engaging CRMP2 directly or for modulating CRMP2-directed upstream signal transduction pathways through semaphorins, neuropilin receptors, and proline-directed serine/threonine kinases.
CRMP2 and tau are similarly associated with neurofibrillary tangles
NFTs are largely, but not exclusively, composed of hyperphosphorylated and aggregated tau [1–6]. Within tangles, tau aggregates form twisted ribbons called paired helical filaments (PHFs) that can be partially extracted and studied. Tau is not, however, the only component of NFTs. Laser capture microdissection–mass spectrometry revealed over 150 proteins enmeshed in NFTs, including CRMP2 [11]. Moreover, CRMP2 is a biochemically prominent component of NFTs. This is evidenced by the fact that one of the early polyclonal antibodies raised against partially purified PHFs and initially thought to recognized phospho-tau (pTau) in human AD tissue sections, was ultimately found to recognize phosphorylated CRMP2 (pCRMP2) instead [12, 13]. Many studies have since confirmed the association of pCRMP2 with AD neurons, in NFTs or NFT-like structures [14–19]. Hyperphosphorylated CRMP2 is also increased in the cortex and hippocampus of the triple transgenic mouse [presenilin-1 (PS1)M146VKI; Thy1.2-amyloid β-precursor protein (APP)swe; Thy1.2tauP301L] that develops AD-like plaques and tangles, as well as the double transgenic (PS1M146VKI; Thy1.2-APPswe) mouse, but not in AD mouse models that develop plaques only [14]. In the former mice, CRMP2 phosphorylation is an early— and hence, potentially pathogenic— event, occurring before plaque or tangle formation [14].
CRMP2 and tau are similarly regulated by phosphorylation to alter microtubule interactions and possibly affect protein scaffolding functions of the molecules
Both CRMP2 and tau are, in fact, properly designated as microtubule-associated proteins (MAPs) [20, 21]. CRMP2 associates with microtubules (MTs) through a taxol-sensitive C-terminal binding interaction, which stabilizes MTs against nocodazole-stimulated depolymerization in live cells [20]. In fact, CRMP2 was first characterized as the central protein responsible for semaphorin 3A (Sema3A)-mediated growth cone collapse in chick dorsal root ganglia (DRG) [22, 23] and further confirmed as an axon-inducing factor in mammalian neurons [24].
Like tau, CRMP2 is subject to glycogen synthase kinase-3β (GSK3β)-mediated phosphorylation on T509 and T514 residues [25–27]. Also similarly to tau phosphorylation, the GSK3β-mediated CRMP2 phosphorylation requires prior phosphorylation (priming) of CRMP2 on S522 via cyclin-dependent kinase-5 (Cdk5) [25–28]. Such phosphorylated CRMP2 loses affinity for tubulin heterodimers, thus reducing microtubule growth at the distal end of axons and encouraging axon retraction [25–28]. Conversely, neurotrophin-3 and brain-derived neurotrophic factor (BDNF) inhibit GSK3β via the phosphatidylinositol-3-kinase (PI3K)/Akt pathway, reducing phospho-CRMP2(509,514) and promoting axon growth [26, 29]. Thus, CRMP2 and tau are both phospho-regulated by a common Cdk5 and GSK3β-dependent pathway to stabilize (or destabilize) microtubules.
Both proteins have a variety of alternative phos-phorylation sites as well. For instance, a phospho-CRMP2(T555) site is targeted by Rho kinase downstream from ephrin signals [30] and also triggered by exposure to Aβ [31]. Similarly, tau has been identified as an alternative substrate for Rho kinase [32]. The functional significance of this phosphorylation is still subject to scientific investigation, but phosphorylation on T555 seems to promote CRMP2 dissociation from microtubules and growth cone collapse in a fashion analogous to T509/T514 phosphorylation [21, 30, 33].
The folded core domain of CRMP2 forms a central tetrameric structure [34], but it is highly likely that the last ∼100 C-terminal residues emerge as unfolded chains from the central core of the tetramer. Moreover, essentially all the known PTMs and protein interaction sites on CRMP2 lie within the C-terminal tail [21–27]. Remarkably, an 82 residue C-terminal region of CRMP2, unrelated to other microtubule binding motifs, is sufficient to stabilize MTs against nacodozole-mediated depolymerization in cell-based assays [20]. These facts immediately suggest a potential function for CRMP2 as a hub for kinase-regulated molecular complexes and/or MT-stabilizing complexes. Unfortunately the currently available high-resolution structures of CRMP2 or its homologs lack the unstructured C-terminal 100 amino acid residues and hence give very few hints as to the molecular interactions and PTMs that might affect them.
While CRMP2 and tau do not share a similar domain arrangement overall (Fig. 1A), the C-terminal tail of CRMP2 and the proline-rich domain of tau present striking sequence homology (Fig. 1B) with high concentrations of Pro and Ser/Thr in both (Fig. 1C), in line with the well documented Pro-directed phosphorylation of both proteins [21–27]. Both segments are highly basic, with pI values above 11. In addition, short sequence motifs are conserved (Fig. 1). These also concern the phosphorylation sites on CRMP2, suggesting putative common mechanisms of PTM regulation between CRMP2 and tau, also with respect to molecular interactions involving these proteins. For example, the Cdk5 site S522 in CRMP2 is functionally analogous to S235 in tau, which is preferentially targeted by Cdk5 in order to prime tau for subsequent GSK3-mediated phosphorylation at AD-associated epitopes, including T231 [5]. Similarly to CRMP2, tau phosphorylation at the Cdk5-gated, GSK3-dependent T231 alters protein:protein interactions and favors tau dissociation from microtubules [35]. As both tau and the CRMP2 tail are natively disordered, these common sequence properties, conserved motifs, and known similar PTMs strongly suggest similar pathways of action. In this sense, it may be of importance that CRMP2 is natively tetrameric, but also has a disordered tail (Fig. 1D). It is highly likely that CRMP2, with these properties, is involved in molecular networks of high neurobiological relevance, the details of which still remain to be unraveled.
CRMP2 differs from tau with respect to vesicle transport processes that are critical to maintenance of healthy neurites
CRMP2 was first discovered as a principal mediator of neurite retraction and neuron polarization (the process by which one developing neurite becomes an axon) during semaphorin signaling in chick DRGs and primary rodent hippocampal neurons [22]. With respect to this process, CRMP2 acts largely by binding and stabilizing tubulin at the plus end of microtubules, thus promoting axon extension (Fig. 2A). This is similar to the mechanism by which tau stabilizes MTs through binding between tubulin dimers to affect the MT dynamic equilibrium [36, 37]. However, CRMP2 also indirectly stabilizes actin-based microfilament networks in distal axons and at the synapse by at least two distinct anterograde transport mechanisms (Fig. 2A).
In the first major mechanism of transport-dependent neuritic stabilization, CRMP2 acts as an adaptor protein to connect the microtubule motor kinesin-1 to anterograde transport vesicles carrying the neurotrophin receptor tyrosine kinase TrkB [38, 39]. After insertion into the cell membrane, TrkB promotes axon growth by signaling for accumulation and polymerization of F-actin in distal axon shafts and growth cones [38]. GSK3β-phosphorylated CRMP2 releases kinesin-1, impeding TrkB function and reducing structural integrity of the actin-based cytoskeleton in distal axons, growth cones, and synapses [39]. CRMP2 likely also helps adapt separate sets of proteins to dynein motors for retrograde trafficking [40].
A second unique means of CRMP2-dependent neuritic remodeling operates through kinesin-1-dependent transport of the Sra1/WAVE1 complex (Fig. 2A). Analogous to the case with TrkB, CRMP2 links kinesin-1 to Sra1/WAVE1 for transport to distal axons and synapses [41]. There, WAVE1 activates the Arp2/3 complex, which in turn nucleates actin monomers, which otherwise would be kinetically impeded from polymerization into microfilaments [42]. RNA interference of CRMP2 delocalizes WAVE1 from growth cones, triggering cone collapse [41]. Similarly, knockdown of Sra1 and WAVE1 cancels CRMP2-induced axon outgrowth [41], indicating that proper connection of CRMP2 to Sra1/WAVE1 is essential to preserve integrity of distal actin networks. This is quite relevant to the case in AD, where CRMP2 appears to complex with WAVE1- and pTau-positive NFT-like structures, and these complexes may be pathologically entangled in a way that depletes the neuron of CRMP2, WAVE1, or both [16, 17]. A primary manifestation of WAVE1 sequestration in CRMP2+ tangles would be loss of synapses; this feature is more tightly correlated to AD dementia than either Aβ burden or phospho-tau [43, 44].
CRMP2 differs from tau in its involvement with endosomal-lysosomal trafficking and autophagy
Other evidence suggests additional importance of CRMP2 to endosomal-lysosomal trafficking (Fig. 2B) and, quite unexpectedly, autophagy regulation. For instance, besides directly binding kinesin motors [41], CRMP2 binds the adaptor protein Numb, thereby regulating Numb interaction with the clathrin coat protein α-adaptin [45]. In line with this, siRNA knockdown of CRMP2 or expression of truncated, dominant-negative CRMP2 inhibits endocytosis at axonal growth cones [45]. Similarly, CRMP2 regulates endosomal trafficking by linking adaptor proteins to dynein motors via an interaction with MICAL-L1 [40] (Fig. 2B).
Because most amyloidogenic conversion of amyloid-β precursor protein (AβPP) to Aβ occurs in acidic endosomal-lysosomal vesicles, whose movement is partly Numb-dependent [46], any event that slows the transit of AβPP through this system could increase its dwell-time in the same compartments as presenilin/gamma secretase and favor amyloidogenesis [47, 48] (Fig. 2B). Functional sequestration of CRMP2 in nascent NFTs, or through changes in CRMP2 interactions secondary to post-translational modifications (discussed in more detail below), would likely affect Aβ release dynamics. Indeed there is published evidence for early deficits of endosomal function in AD, though the molecular origin of the problem remains obscure [49].
We recently reported that siRNA-mediated knock-down of CRMP2 interferes with normal autophagy in SH-SY5Y neuroblastoma cells, producing a build-up of phosphatidylethanolamine-conjugated MAP 1 light chain 3 (Atg8/MAP1-LC3/LC3) form II (LC3-II), most likely caused by impeded autophagy flux in the presence of competent autophagy initiation [50]. This could be an important clue to the roles of CRMP2 in AD pathogenesis, because autophagy is increasingly being recognized as a fundamental cell homeostatic process that is profoundly impeded from completion in human AD and animal models [51–58]. What is particularly observed in AD, as well as in animal models, is that immature autophagosomal vesicles seem to accumulate, as if the autophagosome-lysosome fusion step or the autophagolysosomal clearance process were blocked [51, 52]. The levels of several autophagy-associated proteins or autophagy regulatory proteins are altered in human AD or animal models, including beclin-1, which is diminished by 35% in mild cognitive impairment and 70% in frank AD [51]. Genetic manipulation of beclin-1 up or down in AD mouse models results in changes of amyloid burden, reciprocally relating to beclin-1 protein levels [51]. Unlike CRMP2, tau has not yet been clearly implicated as a factor in normal autophagy regulation, although it is often discussed as an autophagy substrate that might accumulate pathologically under conditions of autophagy impairment [59].
Autophagy is also likely amenable to pharmacological intervention in the aging, damaged brain. Several recent studies report that genetic or pharmacological strategies aimed at stimulating autophagy, reduce behavioral deficits and histological sequelae in animal models of AD [55–58]. One of these studies notably used the CRMP2-binding small molecule lanthionine ketimine-ethyl ester (LKE), which normalized CRMP2 phosphorylation while reducing amyloid burden and phosphorylated tau in a triple-transgenic AD mouse model (3xTg-AD) [58; discussed further below]. Interestingly, prolonged LKE treatment of SH-SY5Y cells or 3xTg-AD mice increases beclin-1 protein, which negatively correlates to Aβ release by SH-SY5Y cells [50, 55–58].
CRITICAL ARGUMENTS FOR THE IMPORTANCE OF TAU OVER CRMP2 IN ALZHEIMER’S DISEASE AND THEIR REFUTATION
As described above, tau and CRMP2 share many structural and functional similarities as microtubule-stabilizing proteins. There are however two key features that tau possesses and CRMP2 apparently does not, which would seem to favor hypotheses emphasizing tau over CRMP2 as an agent of neurodegenerative stress in AD.
Tau is more prone than CRMP2 to form non-dissociable high molecular weight complexes
Given the fact that CRMP2 and tau are both prominent and analogously hyperphosphorylated MAPs that collect in AD-associated NFTs, one might then ask why CRMP2 has been so relatively understudied in AD relative to tau? One reason might be that mammalian CRMP2 was discovered in the mid-1990s [22], some 20 years after tau [3]; so CRMP2 role in MT stabilization was not known when Iqbal and Grundke-Iqbal were publishing seminal PHF work in the late 1980s [1, 2]. Another cogent argument for the relative discounting of CRMP2 versus tau importance to AD is that CRMP2 and pCRMP2 are not as prone to form detergent-insoluble filaments as pTau, so that early attempts to purify PHFs through harsh methods lost the CRMP2 component and led to early concentration on tau rather than CRMP2. Though CRMP2 itself is not prone to aggregation or filament formation, pCRMP2 does accumulate in NFTs with strong enough affinity that it can be co-immunoprecipitated with other NFT-related proteins [16, 17]. These include crucial synapse-stabilizing proteins such as Sra1/WAVE1 that collect in pCRMP2 and pTau-positive NFT-like structures, both in human AD and in pTau-expressing mouse models [16–18]. Thus, although CRMP2 might not be a core structural contributor to NFTs, its importance to downstream neuropathological events cannot be disdained. It should also be considered that some researchers now think that mature tau aggregates are not the most important players in tau-induced neural stress, however, and instead invoke pre-tangle tau oligomers or toxic proteolytic tau fragments as more critical factors [59–62]. CRMP2, like tau, is similarly subject to proteolytic generation of such potentially toxic fragments (discussed further below).
A third major and legitimate reason that tau has been historically emphasized over CRMP2 is that mutations in tau can cause tangle-associated fronto-temporal dementia [4], whereas analogous neurodisease-causing CRMP2 mutations have not been discovered. This important issue warrants particular discussion.
Tau mutations are demonstrably neuropathic in humans whereas CRMP2 mutations are not
Mutations in tau are the causative reason for certain “tauopathies” including particular genetic subsets of frontotemporal dementia (FTD), corticobasal degeneration, and progressive supranuclear palsy [63]. Accordingly, expression of human tau containing the P301L mutation underlying FTD with Parkinson’s disease linked to chromosome 17 (FTDP-17) results in motor and behavioral deficits and anterior horn cell loss in transgenic mice, with age- and gene-dose-dependent development of NFTs [64]. Though dominant mutations in MAPT were the first causal mutations identified in familial FTD, tau is certainly not the only causal mutation in these, as other FTD subsets are associated with mutations in CHMP2B, VCP, TARDBP, and FUS genes; GRN and C9ORF72 [65]. Furthermore, certain normal haplotypes of tau may not directly cause disease, but they can influence tau expression or stability or predisposition to aggregation [62]. This can increase the risk for progressive supranuclear palsy and Parkinson’s disease, and rare variants may even influence AD risk [63]. Analogous mutations or polymorphisms in the DPYSL2 gene, encoding CRMP2, have not been genetically associated with neurodegenerative disease thus far, although CRMP2 promotor polymorphisms have been associated with schizophrenia risk in some populations [66, 67] and suggest a link to deficient autophagy signaling [65].
Human genetic examples [63] prove that tau mutations can cause neurodegeneration in certain instances, whereas the same cannot be said for CRMP2. However, it is important to recognize that no tau mutation causes AD per se; the only link is that some tau mutations cause diseases that happen to share a common cytological feature of NFTs. It is also important to consider that the lack of genetic linkage between CRMP2 and AD does not prove that CRMP2 is unimportant or even non-essential for AD. Pathogenic mutations affecting CRMP2 could cause non-CNS disease or embryonic lethality, for instance.
Indeed, the specificity of CRMP2 involvement with AD contrasts with the more prevalent association of tau mutations across the neurodegenerative spectrum in a way that may argue for the particular importance of CRMP2 in AD. Current evidence suggests that CRMP2 hyperphosphorylation is unique to AD, is not seen in human mutant tau-linked FTD, and is only observed in mouse models that have both plaques and tangles, but not in mutant tau mice that have only phosphorylated tau in the absence of Aβ deposits [14, 68]. Moreover, in animal models of AD, wherein CRMP2 becomes hyperphosphorylated, the event is very early, occurring prior to Aβ accumulation or tau hyperphosphorylation [14]. This has lead Cole, Williamson and colleagues to suggest that CRMP2 phosphorylation is a downstream consequence of altered AβPP and perhaps more of a primary player than tau in disorders involving both amyloidopathy and NFTs [68].
CRMP2 post-translational modifications may contribute to disease in the absence of obvious genetic variation
There are many epigenetic and post-transcriptional events that can render a protein product pathogenic that are not clearly reflected in genetic variations. CRMP2 is remarkable for the number of splice variants and PTMs present in the brain. Aside from varying phosphorylation site combinations, the latter include asparagine deamination products; isoaspartyl rearrangements; SUMOylation; redox disulfide switching [69, 70]; and various irreversible amino acid side chain oxidation products [71–74]. Unbiased whole brain proteomics indicate that CRMP2 is among the proteins most heavily modified by the lipid oxidation product 4-hydroxynonenal [73] and one of the brain proteins most sensitive to isoaspartate formation [71].
CRMP2 is also quite sensitive to calpain-dependent proteolysis associated with excitotoxicity or brain trauma, liberating C-terminal fragments that may limit neuritic regeneration [75]. This is strikingly analogous to circumstances, wherein tau also experiences calpain-dependent proteolysis to yield apparently neurotoxic protein fragments after exposure of neurons to Aβ peptides [60]. Each of these features begins to suggest plausible ways in which CRMP2 could become functionally compromised in AD to engender neuronal stress in a fashion that is either independent of, or downstream from, Aβ and tau-associated stress. Thus, the current lack of specific genetic association between CRMP2 and AD does not constitute reason to disregard CRMP2 as a contributing and pharmacologically accessible factor in AD.
CRMP2 AND CRMP2-DIRECTED SIGNALING PATHWAYS CAN BE PHARMACOLOGICALLY ENGAGED FOR THERAPEUTIC BENEFIT IN NEURODEGENERATION MODELS
Of paramount practical importance in AD research is the identification of protein targets that are druggable, which is to say, proteins that can bind therapeutic molecules or be affected by drugs in such a way as to block or reverse the protein’s contribution to cellular pathology in AD. CRMP2 is emerging as a druggable molecule that has been shown to bind at least three distinct pharmacological agents; whose expression level is manipulated by other drugs; and whose PTMs are subject to pharmacological control.
CRMP2 is a putative binding partner and target for the anti-epileptic drug lacosamide, which acts as a functional CRMP2 inhibitor
CRMP2 is reportedly a binding target for the anti-epileptic drug lacosamide [76]. Lacosamide reportedly binds CRMP2 with low micromolar affinity and acts as a functional CRMP2 antagonist to reduce neurite outgrowth [77–79]. While this is not a desirable phenotypic effect in neurodegenerative disease, it is a desirable outcome in epilepsy, where inappropriate neuritic sprouting is a significant pathophenomenon [76–78]. The binding is subtle, and some techniques for measuring the interaction have yielded disparate results (discussed in [79]). Lacosamide also has other effects mediated more directly through voltage-activated Na+ channels [76]. Further research is needed to better define how lacosamide interacts with CRMP2 and the extent to which the neuritic effects of lacosamide are dissociable from other pharmacological actions of the drug.
CRMP2 specifically binds particular biologically-active peptides and amino acid metabolites that act as functional CRMP2 enhancers
CRMP2 also has been evidenced to bind certain peptides and amino acid metabolites. We have shown that natural thioether ketimines exemplified by lanthionine ketimine (LK) prominently bound CRMP2 among a select set of potential binding partners in an unbiased whole-brain proteomic screen and could influence CRMP2 binding partner interactions in whole brain lysate, increasing some CRMP2 interactions (for instance increasing CRMP2 binding to neurofibromin) and decreasing others [80–81]. Subsequently, we showed that cell-penetrating ester derivatives of LK act as functional CRMP2 enhancers, to promote growth factor-dependent neurite outgrowth [80–82] and beneficial autophagy [50, 55]. An ethyl ester of LK could partially rescue the neuroanatomic phenotype of incomplete cholinergic commissure development in C. elegans expressing partial loss-of-function Unc-33, the nematode homolog of mammalian CRMP2 [83]. Work is currently ongoing in our labs to assess the sites and nature of the apparent binding between CRMP2 and LK derivatives. Meanwhile, LK ethyl ester has shown positive histological or behavioral effects when administered to a diverse set of rodent neurodisease models including the 3xTg-AD mouse model [50, 55–56]; the SOD1G93A mouse model of amyotrophic lateral sclerosis [82]; a permanent middle cerebral artery occlusion model of stroke [84]; the experimental autoimmune encephalomyelitis model of multiple sclerosis [85]; and delayed damage caused by mild traumatic brain injury [86]. Notably, LKE increases markers of autophagy in the traumatic brain injury model [86] just as it does in the 3xTg-AD model [50, 55]. With respect to peptides, Khanna and colleagues have shown that synthetic oliogopeptides based on C-terminal protein-binding motifs of CRMP2 can act as “decoy” molecules to reduce CRMP2 binding to voltage-gated calcium channels, thus reducing neuropathic pain in rodent models [87, 88].
CRMP2 expression can be affected by small molecules and neurotrophic factors acting at both transcriptional and post-transcriptional levels
CRMP2 expression can also be affected pharmacologically. Glia-derived neurotrophic factor (GDNF) can increase CRMP2 expression in neurons, as can the atypical tricyclic antidepressant, tianeptine [89, 90]. Increased CRMP2 further translates to microtubule stabilization and enhanced neurite outgrowth [89, 90]. From the few studies that have been published about CRMP2 expression modifiers, it is not clear at what level(s) protein expression of CRMP2 is most subject to control (transcription, translation, or turnover). The tianeptine studies did not measure mRNA, but GDNF-stimulated CRMP2 expression was tracked to promotor-level control via signaling through RET tyrosine kinase and ERK (extracellular signal-regulated kinase) [89]. The SP1, E2F, and GATA1/2 binding sites in the CRMP2 promotor appeared to play some roles in regulation of CRMP-2 expression upon GDNF stimulation [89].
Other, very recent research indicates that CRMP2 translation can be affected by the autophagy control center, mTORC1 (mammalian target of rapamycin complex-I) [91] in a manner that is sensitive to systemic drug exposure. In particular high-dose alcohol administration to rats reportedly induced mTORC1 activation in the nucleus accumbens (NAc) with concomitant activation of the mTORC1 targets, eukaryotic translation initiation factors 4E binding protein (4E-BP) and ribosomal protein S6 kinase (S6K). Activation of this translation control system stimulated increased CRMP2 translation, which could be blocked by the mTOR inhibitor rapamycin [91]. CRMP2 protein could be increased by more than 50% by alcohol administration and additionally, GSK3β-mediated CRMP2 phosphorylation decreased in the same paradigm, leading to increased microtubule content [91]. Moreover, systemic administration of the functional CRMP2 inhibitor lacosamide reversed the behavioral phenomenon of excessive alcohol consumption in alcohol-trained rats indicating that (a) CRMP2 is involved in behavior or motivation at the level of NAc plasticity and (b) both CRMP2 expression and phosphorylation state are amenable to pharmacological regulation at a number of levels. Taken together with our own finding that CRMP2 affects mTORC1 activity by means of localization [50], this work further underscores what appears to be an intimate reciprocal interaction of CRMP2 with key autophagy control junctions and particularly mTORC1.
CRMP2 phosphorylation state is amenable to kinase inhibitors and natural products
CRMP2 phosphorylation status can be pharmacologically manipulated by inhibiting kinases or blocking cell surface receptors upstream in the CRMP2 signaling pathway. As discussed above, CRMP2 binding to tubulin and other partners is influenced by phosphorylation on T509 and T514. Pharmacological inhibitors of GSK3β can block this phosphorylation [92]. Pharmacological inhibitors of Cdk5 that may also target GSK3β are currently under development with AD in mind [93]. These compounds have the potential advantage that they might reduce both tau and CRMP2 hyperphosphorylation.
Moving further upstream, CRMP2 is canonically activated by the axon repulsion factor semaphorin 3A (Sema3A) binding to the neuropilin-1 receptor (NRP1) and plexin-A co-receptors [20, 25]. We have shown that NRP1-blocking antibodies effectively antagonize Sema3A signaling to CRMP2, stabilizing motor neuron axons to slow neuromuscular junction denervation in the SOD1G93A mouse model of amyotrophic lateral sclerosis [94]. The antibody used in our previous study is currently undergoing clinical trials as an angiogenesis inhibitor for solid tumors [95], thus arguing for the feasibility of clinical translation for such biopharmaceuticals.
Certain natural products, chief amongst which is the microbial product xanthofulvin, appear to block Sema3A-mediated CRMP2 phosphorylation at steps between the NRP1 receptor and downstream kinases [96, 97]. These compounds have shown promise as mediators of spinal cord regeneration following injury [97]. Further research and screening of natural product libraries might well uncover other lead compounds that antagonize axon repulsion factor pathways impinging upon CRMP2. The mechanism-of-action for xanthofulvin needs to be refined in order to better understand how this fungal product alters the mammalian Sema3A-stimulated pathway and how more medically practical small molecule drugs might be created that would do the same.
It is not clear why CRMP2 is hyperphosphorylated in AD, or whether Sema3A signaling might be implicated. It is worth noting, however, that one study reports increased Sema3A within vulnerable hippocampal neurons of AD subjects [98]. More recently, SNP associations have been noted between plexin A4 isoforms and AD, and these were linked to aberrant tau phosphorylation; but CRMP2 was not investigated [99]. Taken together, these findings suggest that further studies on semaphorin signaling to CRMP2 in AD are justified with a view toward identifying and testing pharmacological inhibitors of this recently implicated pathway.
SUMMARY AND CONCLUSIONS
Both CRMP2 and tau are high-abundance microtubule-associated proteins that substantially overlap in terms of functional regulation and association with AD pathology (Table 1). In addition, CRMP2 serves important functions in synaptic plasticity, vesicular trafficking, and autophagy regulation that appear quite distinct from the known functions of tau. This distinction is significant because synaptic compromise, defective vesicular trafficking, and dysfunctional autophagy are becoming recognized as important aspects of AD-associated neurodegeneration [51–55].
CRMP2 has a complex and subtle biology that offers many points of known or potential druggability (Fig. 3). More research is needed to determine whether CRMP2 manipulations, especially those which functionally alter protein level or affect specific aspects of CRMP2 post-translational modification (phosphorylation, redox regulation, or proteolysis), affect the phenotypes of rodent AD models. Once accomplished, such work could lead to rational development of CRMP2-targeting small molecules. Meanwhile, derivatives of natural products, such as LK or xanthofulvin analogs, might be explored in detail as modulators for AD-related neural pathways.
Amyloid peptides and tau species are both likely to be involved in AD-associated neurotoxicology. However, decades of effort have thus far totally failed to exploit Aβ or tau in order to create clinically proven treatments for AD. Given these incontrovertible facts, it seems reasonable that more effort should be directed towards identifying novel targets for AD pharmacotherapy that do not rely upon any assumptions about the role of Aβ or tau in AD pathogenesis. Consideration of CRMP2 offers one such specific and credible target.
ACKNOWLEDGMENTS
KH is funded for CRMP2 studies through the National Institutes of Health (NS082283 and NS093594). KH is the inventor on U.S. patent 7,683,055 (other patents pending) covering composition and use of LK derivatives including LKE and holds equity in a company engaged in commercial development of the technology.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0076r1).
REFERENCES
[1] | Grundke-Iqbal I , Iqbal K , Quinlan M , Tung YC , Zaidi MS , Wisniewski HM ((1986) ) Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261: , 6084–6089. |
[2] | Grundke-Iqbal I , Iqbal K , Tung YC , Quinlan M , Wisniewski HM , Binder LI ((1986) ) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83: , 4913–4917. |
[3] | Weingarten MD , Lockwood AH , Hwo SY , Kirschner MW ((1975) ) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72: , 1858–1862. |
[4] | Spillantini MG , Goedert M ((2013) ) Tau pathology and neurodegeneration. Lancet Neurol 12: , 609–622. |
[5] | Braak H , Braak E ((1996) ) Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol Scand Suppl 165: , 3–12. |
[6] | Iqbal K , Grundke-Iqbal I ((2006) ) Discoveries of tau, abnormally hyperphosphorylated tau and others of neurofibrillary degeneration: A personal historical perspective. J Alzheimers Dis 9: (4 Suppl), 219–242. |
[7] | Duyckaerts C , Delatour B , Potier M-C ((2009) ) Classification and basic pathology of Alzheimer’s disease. Acta Neuropathol 118: , 5–36. |
[8] | Toyn J ((2015) ) What lessons can be learned from failed Alzheimer’s disease trials? Expert Rev Clin Pharmacol 8: , 267–269. |
[9] | Giacobini E , Gold G ((2013) ) Alzheimer disease therapy: Moving from amyloid-beta to tau. Nat Rev Neurol 9: , 677–686. |
[10] | Igbal K , Liu F , Gong CX ((2014) ) Alzheimer’s disease therapeutics: Focus on the disease and not just plaques and tangles. Biochem Pharmacol 88: , 631–691. |
[11] | Wang Q , Woltier RL , Cimino PJ , Pan C , Montine KS , Zhang J , Montine TF ((2005) ) Proteomic analysis of neurofibrillary tangles in Alzheimer disease identifies GAPDH as a detergent-insoluble paired helical filament tau binding protein. FASEB J 19: , 869–871. |
[12] | Yoshida H , Watanabe A , Ihara Y ((1998) ) Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer’s disease. J Biol Chem 273: , 9761–9768. |
[13] | Gu Y , Hamajima N , Ihara Y ((2000) ) Tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, Ser-522. Biochemistry 39: , 4267–4275. |
[14] | Cole AR , Noble W , van Aalten L , Plattner F , Meimaridou R , Hogan D , Taylor M , LaFrancois J , Gunn-Moore F , Verkhratski A , Oddo S , LaFerla F , Giese KP , Dineley KT , Duff K , Richardson JC , Yan SD , Hanger DP , Allan SM , Sutherland C ((2007) ) Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J Neurochem 103: , 1132–1144. |
[15] | Soutar MP , Thornhill P , Cole AR , Sutherland C ((2009) ) Increased CRMP2 phosphorylation is observed in Alzheimer’s disease: Does this tell us anything about disease development? Curr Alzheimer Res 6: , 269–278. |
[16] | Takata K , Kitamura Y , Nakata Y , Matsuoka Y , Taomimoto H , Taniguchi T , Shimohama S ((2009) ) Involvement of WAVE1 accumulation in Aβ/APP pathology-dependent tangle modification in Alzheimer’s disease. Am J Pathol 175: , 17–24. |
[17] | Watamura N , Toba J , Yoshii A , Nikkuni M , Ohshima T ((2016) ) Colocalization of phosphorylated forms of WAVE1, CRMP2, and tau in Alzheimer’s disease model mice: Involvement of Cdk5 phosphorylation and the effect of ATRA treatment. J Neurosci Res 94: , 15–26. |
[18] | Hensley K , Venkova K , Christov A , Gunning W , Park J ((2011) ) Collapsin response mediator protein-2: An emerging pathologic feature and therapeutic target for neurodisease indications. Mol Neurobiol 43: , 180–191. |
[19] | Triplett JC , Swomley A , Cai J , Klein JB , Butterfield DA ((2016) ) Quantitative phosphoproteomic analyses of the inferior parietal lobule from three different pathological stages of Alzheimer’s disease. J Alzheimers Dis 49: , 45–62. |
[20] | Lin PC , Chan PM , Hall C , Manser E ((2011) ) Collapsin response mediator proteins (CRMPs) are a new class of microtubule-associated protein (MAP) that selectively interacts with assembled microtubules via a taxol-sensitive binding interaction. J Biol Chem 286: , 41466–41478. |
[21] | Khanna R , Wilson SM , Brittain JM , Weimer J , Sultana R , Butterfield A , Hensley K ((2012) ) Opening Pandora’s jar: A primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future Neurol 7: , 749–771. |
[22] | Goshima Y , Nakamura F , Strittmatter P , Strittmatter SM ((1995) ) Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 376: , 509–514. |
[23] | Deo RC , Schmidt EF , Elhabazi A , Togashi H , Burley SK , Strittmatter SM ((2004) ) Structural bases for CRMP function in plexin-dependent semaphorin 3A signaling. EMBO J 23: , 9–22. |
[24] | Inagaki N , Chihara K , Arimura N , Menager C , Kawano Y , Matsuo N , Nishimura T , Amano M , Kaibuchi K ((2001) ) CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci 4: , 781–782. |
[25] | Cole AR , Knebel A , Morrice NA , Robertson LA , Irving AJ , Connolly CN , Sutherland C ((2004) ) GSK-3 phosphorylation of the Alzheimer epitope within collapsing response mediator proteins regulates axon elongation in primary neurons. J Biol Chem 279: , 50176–40180. |
[26] | Yoshimura T , Kawano Y , Arimura N , Kawabata S , Kikuchi A , Kaibuchi K ((2005) ) GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120: , 137–149. |
[27] | Uchida Y , Ohshima T , Sasaki Y , Suzuki H , Yanai S , Yamashita N , Nakamura F , Takei K , Ihara Y , Mikoshiba K , Kolattukudy P , Honnorat J , Goshima Y ((2005) ) Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: Implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 10: , 165–179. |
[28] | Li T , Hawkes C , Qureshi HY , Kar S , Paudel HK ((2006) ) Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3-beta. Biochemistry 45: , 3134–3145. |
[29] | Quach TT , Duchemin A-M , Rogemond V , Aguera M , Honnorat J , Belin M-F , Kolattukudy PE ((2004) ) Involvement of collapsing response mediator proteins in the neurite extension induced by neurotrophins in dorsal root ganglion neurons. Mol Cell Neurosci 25: , 433–443. |
[30] | Arimura N , Inagaki N , Chihara K , Menager C , Nakamura N , Iwamatsu A ((2000) ) Phosphorylation of CRMP-2 by Rho kinase: Evidence for two separate signaling pathways for growth cone collapse. J Biol Chem 275: , 23973–23980. |
[31] | Petratos S , Li QX , George AJ , Hou X , Kerr ML , Unabia SE , Hatzinisiriou I , Maksel D , Aguilar MI , Small DH ((2008) ) The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 131: , 90–108. |
[32] | Amano M , Kaneko T , Maeda A , Nakayama M , Ito M , Yamauchi T , Goto H , Fukata Y , Oshiro N , Shinohara A , Iwamatsu A , Kaibuchi K ((2003) ) Identification of tau and MAP2 as novel substrates of Rho-kinase and myosin phosphatase. Neurochem 87: , 780–790. |
[33] | Arimura N , Menager C , Kawano Y , Yoshimura T , Kawabata S , Hattori A , Fukata Y , Amano M , Goshima Y , Inagaki M , Morone N , Usukura J , Kaibuchi K ((2005) ) Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol Cell Biol 25: , 9973–9984. |
[34] | Stenmark P , Ogg D , Flodin S , Flores A , Kotenyova T , Nyman T , Nordlund P , Kursula P ((2007) ) The structure of human collapsin response mediator protein 2, a regulator of axonal growth. J Neurochem 101: , 906–917. |
[35] | Sengupta A , Novak M , Grundke-Iqbal I , Igbal K ((2006) ) Regulation of phosphorylation of tau by cyclin-dependent kinase 5 and glycogen synthase kinase-3 at substrate level. FEBS Lett 580: , 5925–5933. |
[36] | Panda D , Goode BL , Feinstein SC , Wilson L ((1995) ) Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry 34: , 11117–11127. |
[37] | Kadavath H , Hofele RV , Biernat J , Kumar S , Tepper K , Urlaub H , Mandelkow E , Zweckstetter M ((2015) ) Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A 112: , 7501–7506. |
[38] | Lykissas MG , Batistatou AK , Konstantinos A , Charalabopoulos KA , Beris AE ((2007) ) The role of neurotrophins in axon guidance, growth and regeneration. Curr Neurovas Res 4: , 143–151. |
[39] | Arimura N , Kimura T , Nakamuta S , Taya S , Funahashi Y , Hattori A , Shimada A , Menager C , Kawabata S , Jujii K , Iwamatsu A , Segal RA , Fukuda M , Kaibuchi K ((2009) ) Anterograde transport of TrkB in axons is mediated by direct interaction with Slp1 and Rab27. Dev Cell 15: , 675–686. |
[40] | Rahajeng J , Giridharan SS , Naslavsky N , Caplan S ((2010) ) . Collapsin response mediator protein-2 (CRMP2) regulates trafficking by linking endocytic regulatory proteins to dynein motors. J Biol Chem 285: , 31918–31922. |
[41] | Kawano Y , Yoshimura T , Tsuboi D , Kawabata S , Kaneko-Kawano T , Shirataki H , Takenawa T , Kaibuchi K ((2005) ) CRMP-2 is involved in kinesin-1-dependent transport of the Swa-1/WAVE1 complex and axon formation. Mol Cell Biol 25: , 9920–9935. |
[42] | Pollitt A , Insall RH ((2009) ) WASP and SCAR/WAVE1 proteins: The drivers of actin assembly. J Cell Sci 122: , 2575–2578. |
[43] | Arendt T ((2009) ) Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol 118: , 167–179. |
[44] | Scheff SW , Price DA , Schmitt FA , DeKosky ST , Mufson EJ ((2007) ) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurology 68: , 1501–1508. |
[45] | Nishimura T , Fukata Y , Kato K , Yamaguchi T , Matsuura Y , Kamiguchi H , Kaibuchi K ((2003) ) CRMP-2 regulates polarized Numb-mediated endocytosis for axon growth. Nat Cell Biol 5: , 819–826. |
[46] | Kyriazis GA , Wei Z , Vandermey M , Jo DG , Xin O , Mattson MP , Chan SL ((2008) ) Numb endocytic adaptor proteins regulate the transport and processing of the amyloid precursor protein in an isoform-dependent manner: Implications for Alzheimer disease pathogenesis. J Biol Chem 283: , 25492–25502. |
[47] | Peraus GC , Masters CL , Beyreuther K ((1997) ) Late compartments of amyloid precursor protein transport in SY5Y cells are involved in beta amyloid secretion. J Neurosci 17: , 7714–7724. |
[48] | Thinakaran G , Koo EH ((2008) ) Amyloid precursor protein trafficking, processing and function. J Biol Chem 283: , 29615–29619. |
[49] | Ginsberg SD , Alldred MJ , Counts SE , Cataldo AM , Neve RL , Jiang Y , Wuu J , Chao MV , Mufson EJ , Nixon RA , Che S ((2010) ) Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry 68: , 885–893. |
[50] | Harris-White ME , Ferbas KG , Johnson MF , Eslami P , Poteshkina A , Venkova K , Christov A , Hensley K ((2015) ) A cell-penetrating ester of the neural metabolite lanthionine ketimine stimulates autophagy through the mTORC1 pathway: Evidence for a mechanism of action with pharmacological implications for neurodegenerative pathologies. Neurobiol Dis 84: , 60–68. |
[51] | Pickford F , Masliah E , Britschgi M , Lucin K , Marasimhan R , Jaeger PA , Small S , Spencer B , Rockenstein E , Levine B , Wyss-Coray T ((2008) ) The autophagy-related protein beclin shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118: , 2190–2199. |
[52] | Nixon RA , Yang DS ((2011) ) Autophagy failure in Alzheimer’s disease – Locating the primary defect. Neurobiol Dis 43: , 38–45. |
[53] | Wolfe DM , Lee JH , Kumar A , Lee S , Orenstein SJ , Nixon RA ((2013) ) Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci 37: , 1949–1961. |
[54] | Tramutola A , Triplett JC , Di Domenico F , Niedowicz DM , Murphy MP , Coccia R , Perluigi M , Butterfield DA ((2015) ) Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with preclinical AD, amnestic mild cognitive impairment and late-stage AD (2015). J Neurochem 133: , 739–749. |
[55] | Hensley K , Harris-White ME ((2015) ) Redox regulation of autophagy in healthy brain and neurodegeneration. Neurobiol Dis 84: , 50–59. |
[56] | Hensley K , Venkova K , Christov A , Johnson M , Eslami P , Gabbitta SP , Harris-White M ((2013) ) A derivative of the brain metabolite lanthionine ketimine improves cognition and diminishes pathology in the 3xTg-AD mouse model of Alzheimer’s disease. J Neuropathol Exp Neurol 72: , 955–969. |
[57] | Jiang T , Yu JT , Zhu XC , Tan MS , Wang HF , Cao L , Zhang QQ , Shi JQ , Gao L , Qin H , Zhang YD , Tan L ((2014) ) Temsirolimus promotes autophagic clearance of amyloid-beta and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol Res 81: , 54–63. |
[58] | Caccamo A , De Pinto V , Messina A , Branca C , Oddo S ((2014) ) Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological defecits by restoring hippocampal gene expression signature. J Neurosci 34: , 7988–7998. |
[59] | Cardenas-Aguayo MC , Gomez-Virgilio L , DeRosa S , Meraz-Rios MA ((2014) ) The role of tau oligomers in the onset of Alzheimer’s disease neuropathology. ACS Chem Neurosci 5: , 1178–1191. |
[60] | Park SY , Ferreira A ((2005) ) The generation of a 17 kDa neurotoxic fragment: An alternative mechanism by which tau mediates beta amyloid-induced neurodegeneration. J Neurosci 25: , 5365–5375. |
[61] | Guzman-Martinez L , Farias GA , Maccioni RB ((2013) ) Tau oligomers as potential targets for Alzheimer’s diagnosis and novel drugs. Front Neurol 4: , 167. |
[62] | Mufson EJ , Ward S , Binder S ((2014) ) Prefibrillar tau oligomers in mild cognitive impairment and Alzheimer’s disease. Neurodegener Dis 13: , 151–153. |
[63] | Coppola G , Chinnathambi S , Li JJ , Dombroski BA , Baker MC , Soto-Ortolaza AI , Lee SE , Klein E , Huang A , Sears R , Lane JR , Karydas AM , Kennet RO , Biernat J , Want L-S , Cotman CW , DeCarli CS , Levey AI , Ringman JM , Mendez MF , Chui HC , Le Ber I , Brice A , Lupton MK , Preza E , Lovestone S , Powell J , Graff-Radford N , Petersen RC , Boeve BF , Lippa CF , Bigio EH , MacKenzi I , Finger E , Kertesz A , Caselli RJ , Gearing M , Juncos JL , Ghetti B , Spina S , Bordelon YM , Tourtellotte WW , Frosch MP , Vonsattel JPG , Zarow C , Beach TG , Albin RL , Lieberman AP , Lee VM , Trojanowski JQ , Van Deerlin VM , Bird TD , Galasko DR , Masliah E , White CL , Troncoso JC , Hannequin D , Boxer AL , Geschwind MD , Kumar S , Mandelkow E-M , Wszolek ZK , Uitti RJ , Dickson Dw , Haines JL , Mayeux R , Pericak-Vance MA , Farrer LA , Alzheimer’s Disease Genetics Consortium, Ross OA , Rademakers R , Schellenberg GD , Miller BL , Mandelkow E , Geschwind DH ((2012) ) Evidence for a role of the rare p.A152T variant in MAPT in increasing risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Gen 21: , 3500–3512. |
[64] | Lewis J , McGowan E , Rockwood J , Melrose H , Nacharaju P , Van Slegtenhorst M , Gwinn-Hardy K , Murphy PM , Baker M , Yu X , Duff K , Hardy J , Corral A , Lin WL , Yen SH , Dickson DW , Davies P , Hutton M ((2000) ) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: , 402–405. |
[65] | Rohrer J , Warren J ((2011) ) Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol 24: , 542–540. |
[66] | Liu Y , Pham X , Zhang L , Chen PL , Burzynski G , McGaughey DM , He S , McGrath JA , Wolyniec P , Fallin MD , Pierce MS , McCallion AS , Pulver AE , Avramopoulos D , Valle D ((2014) ) Functional variants in DPYSL2 sequence increase risk of schizophrenia and suggest a link to mTOR signaling. G3 (Bethesda) 5: , 61–72. |
[67] | Nakata K , Ujike H , Sakai A , Takaki M , Imamura T , Tanaka Y , Kuroda S ((2003) ) The human dihydropyrimidinase-like protein 2 gene on chromosome 8p21 is associated with paranoid-type schizophrenia. Biol Psychiatry 53: , 571–576. |
[68] | Williamson R , van Aalten L , Mann DM , Platt B , Plattner F , Bedford L , Mayer J , Howlett D , Usardi D , Sutherland C , Cole AR ((2011) ) CRMP2 hyperphosphorylation is characteristic of Alzheimer’s disease and not a feature common to other neurodegenerative diseases. J Alzheimers Dis 27: , 615–625. |
[69] | Morinaka A , Yamada M , Itofusa R , Funato Y , Yoshimura Y , Nakamura F , Yoshimura T , Kaibuchi K , Goshima Y , Hoshino M , Kamiguchi H , Miki H ((2011) ) Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci Signal 4: , ra26. |
[70] | Gellert M , Venz S , Mitlohner J , Cott C , Hanschmann EM , Lillig CH ((2013) ) Identification of a dithiol-disulfide switch in collapsing response mediator protein 2 (CRMP2) that is toggled in a model of neuronal differentiation. J Biol Chem 288: , 35117–35125. |
[71] | Zhu JX , Doyle HA , Mamula HJ , Aswad DW ((2006) ) Protein repair in the brain, proteomic analysis of endogenous substrates for protein L-isoaspartyl methyltransferase in mouse brain. J Biol Chem 281: , 33802–33813. |
[72] | Majava V , Loytynoja N , Chen W-Q , Lubec G , Kursula P ((2008) ) Crystal and solution structure, stability, and post-translational modifications of collapsin response mediator protein-2. FEBS J 275: , 4583–4596. |
[73] | Reed TT , Pierce WM , Markesbery WR , Butterfield DA ((2009) ) Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res 1274: , 66–76. |
[74] | Dustrude ET , Wilson SM , Ju W , Xiao Y , Khanna R ((2013) ) CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J Biol Chem 288: , 24215–24331. |
[75] | Zhang Z , Ottens AK , Sadasivan S , Kobeissy FH , Fang T , Hayes RL , Wang KK ((2007) ) Calpain-mediated collapsing response mediator protein-1, -2, and -4 proteolysis after neurotoxic and traumatic brain injury. J Neurotrauma 24: , 460–472. |
[76] | Kelemen A , Halasz P ((2010) ) Lacosamide for the prevention of partial onset seizures in epileptic adults. Neuropsychiatr Dis Treat 6: , 465–471. |
[77] | Wilson SM , Xiong W , Wang Y , Ping X , Head JD , Brittain JM , Gagare PD , Ramachandran PV , Jin X , Khanna R ((2012) ) Prevention of posttraumatic axon sprouting by blocking collapsin response mediator protein-2-mediated neurite outgrowth and tubulin polymerization. Neuroscience 17: , 451–466. |
[78] | Wilson SM , Moutal A , Melemedjian OK , Wang Y , Ju W , Francois-Moutal L , Khanna M , Khanna R ((2014) ) The functionalized amino acid (S)-lacosamide subverts CRMP2-mediated tubulin polymerization to prevent constitutive and activity-dependent increase in neurite outgrowth. Front Cell Neurosci 8: , 196. |
[79] | Wilson SM , Khanna R ((2015) ) Specific binding of lacosamide to collapsing response mediator protein 2 (CRMP2) and direct impairment of its canonical function: Implications for the therapeutic potential of lacosamide. Mol Neurobiol 51: , 599–609. |
[80] | Hensley K, Christov A, Kamat S, Zhang XC, Jackson KW, Snow S, Post J ((2010) ) Proteomic identification of binding partners for the brain metabolite lanthionine ketimine (LK) and documentation of LK effects on microglia and motoneuron cell cultures. J Neurosci 30: , 2979–2988. |
[81] | Hensley K , Denton TT ((2015) ) Alternative functions of the brain transsulfuration pathway represent an underappreciated aspect of brain redox biochemistry with significant potential for therapeutic manipulation. Free Radic Biol Med 78: , 123–134. |
[82] | Hensley K ((2010) ) Emerging biological importance of central nervous system lanthionines. Molecules 15: , 5581–5594. |
[83] | Hubbard C , Benda E , Hardin T , Baxter T , St. John E , O’Brien S , Hensley K , Holgado A ((2013) ) Lanthionine ketimine ethyl ester partially rescues neuro-developmental defects in unc-33 (DPYSL2/CRMP2) mutants. J Neurosci Res 91: , 1183–1190. |
[84] | Nada SE , Raghava A , Tulsulkar J , Hensley K , Shah ZA ((2012) ) A derivative of the CRMP2 binding compound lanthionine ketimine provides neuroprotection in a mouse model of cerebral ischemia. Neurochem Int 61: , 1357–1363. |
[85] | Dupree JL , Polak PE , Hensley K , Pelligrino D , Feinstein DL ((2015) ) Lanthionine ketimine ester provides benefit in a mouse model of multiple sclerosis. J Neurochem 134: , 302–314. |
[86] | Hensley K , Poteshkina A , Johnson MF , Eslami P , Hristov A , Venkova K , Harris-White ME ((2015) ) Autophagy modulation by lanthionine ketimine improves longterm outcome following central fluid percussion injury in the mouse.. J Neurotrauma, doi: 10.1089/neu.2015.4196 |
[87] | Brittain JM , Piekarz AD , Wang Y , Kondo T , Cummins TR , Khanna R ((2009) ) An atypical role for collapsing response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem 284: , 31375–31390. |
[88] | Feldman P , Khanna R ((2013) )Neurosci Lett 557: (Pt A), 27–36 Challenging the catechism of therapeutics for chronic neuropathic pa, Targeting CaV2.2 interaction with CRM2 peptides.. |
[89] | Kodama Y , Murakumo Y , Ichihara M , Kawai K , Shimono Y , Takahashi M ((2004) ) Induction of CRMP-2 by GDNF and analysis of the CRMP-2 promoter region. Biochem Biophys Res Commun 320: , 108–115. |
[90] | Chu CC , Wang JJ , Chen KT , Shieh JP , Wang LK , Shui HA , Ho ST ((2010) ) Neurotrophic effects of tianeptine on hippocampal neurons: A proteomic approach. J Proteome Res 9: , 936–944. |
[91] | Liu F , Legastelois R , Morisot N , Ben Hamida S , Ron D ((2016) ) mTORC1-dependent translation of collapsing response mediator protein-2 drives neuroadaptations underlying excessive alcohol-drinking behaviors.. Mol Psychiatry, doi: 10.1038/mp.2016.12 |
[92] | Caberlotto L , Carboni L , Zanderigo F , Andreetta F , Andreoli M , Gentile G , Razzoli M ((2013) ) Differential effects of glycogen synthase kinase 3 (GSK3) inhibition or selective inhibitors in the central nervous system. Naunyn Schmiedebergs Arch Pharmacol 386: , 893–903. |
[93] | Zhang X , Hernandez I , Rei D , Mair W , Laha JK , Cornwell ME , Cuny GD , Tsai LH , Steen JA , Kosik KS ((2013) ) Diaminothiazoles modify tau phosphorylation and improve the tauopathy in mouse models. J Biol Chem 288: , 22042–22056. |
[94] | Venkova K , Christov A , Kamaludin Z , Kobalka P , Siddique S , Hensley K ((2014) ) Semaphorin-3A (Sema3A) signaling through neuropilin-1 (NRP1) is an early trigger for distal axonopathy in the SOD1G93A mouse model of familial amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 73: , 702–713. |
[95] | Weekes CD , Beeram M , Tolcher AW , Papadopoulos KP , Gore L , Hegde P , Xin Y , Yu R , Shih LM , Xiang H , Brachmann RK , Patnaik K ((2014) ) A phase I study of the human monoclonal anti-NRP1 antibody MNRP1685A in patients with advanced solid tumors. Invest New Drugs 32: , 653–660. |
[96] | Kikuchi K , Kishino A , Konishi O , Kumagai K , Hosatani N , Saji I , Nakayama C , Kimura T ((2003) ) In vitro and in vivo characterization of a novel semaphoring 3A inhibitor, SM-216289 or xanthofulvin. J Biol Chem 278: , 42985–42991. |
[97] | Kaneko S , Iwanami A , Nakamura M , Kishino A , Kikuchi K , Shibata S , Okano HJ , Ikegami T , Moriya A , Konishi O , Nakayama C , Kumagai K , Kimura T , Sato Y , Goshima Y , Taniguchi M , Ito M , He Z , Toyama Y , Okano H ((2006) ) A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat Med 12: , 1380–1389. |
[98] | Good PF , Alapat D , Hsu A , Chu C , Perl D , Wen X , Burstein DE , Kohtz DS ((2004) ) A role for semaphoring 3A signaling in the degeneration of hippocampal neurons during Alzheimer’s disease. J Neurochem 91: , 716–736. |
[99] | Jun G , Asai H , Zeldich E , Drapeau E , Chen C , Chung J , Park JH , Kim S , Haroutunian V , Foroud T , Kuwano R , Haines JL , Pericak-Vance MA , Schellenberg GD , Lunetta KL , Kim JW , Buxbaum JD , Mayeux R , Ikezu T , Abraham CR , Farrer LA ((2014) ) PLXNA4 is associated with Alzheimer disease and modulates tau phosphorylation. Ann Neurol 76: , 379–392. |
Figures and Tables
Fig.1
Structural features and homology between CRMP2 and tau. A) Domain structure of tau and CRMP2. The Pro-rich domain of tau is homologous to the C-terminal tail of CRMP2 (blue). B) Sequence alignment between the homologous regions. Known phosphorylation sites on CRMP2 (blue arrowheads) and tau (orange arrowheads) are marked below the sequences. C) Amino acid composition of the tau Pro-rich region and the CRMP2 tail. D) Structural model for full-length CRMP2, in which four disordered C-terminal tails are protruding out of the central tetramer. Note the actual structure of full-length CRMP2 including the C-terminal tail has not been formally determined; panel D represents a proposed model based on the known C-terminal truncated X-ray crystal structure [34].
![Structural features and homology between CRMP2 and tau. A) Domain structure of tau and CRMP2. The Pro-rich domain of tau is homologous to the C-terminal tail of CRMP2 (blue). B) Sequence alignment between the homologous regions. Known phosphorylation sites on CRMP2 (blue arrowheads) and tau (orange arrowheads) are marked below the sequences. C) Amino acid composition of the tau Pro-rich region and the CRMP2 tail. D) Structural model for full-length CRMP2, in which four disordered C-terminal tails are protruding out of the central tetramer. Note the actual structure of full-length CRMP2 including the C-terminal tail has not been formally determined; panel D represents a proposed model based on the known C-terminal truncated X-ray crystal structure [34].](https://ip.ios.semcs.net:443/media/jad/2016/53-1/jad-53-1-jad160076/jad-53-jad160076-g001.jpg)
Fig.2
CRMP2 is a MAP that also interacts with cargo adaptor proteins involved with receptor endocytosis and intracellular vesicle trafficking. Panel A is a conceptual diagram illustrating known interactions of CRMP2 with microtubules, and also with kinesin and dynein-associated adaptor proteins. In this model, CRMP2 dynamically equilibrates among its binding partners in a fashion that is likely dependent upon CRMP2 isoform and post-translational modifications. Panel B illustrates AβPP trafficking in a manner that highlights points at which CRMP2 and its binding partners, Numb and MICAL-L1, are involved with intracellular vesicle movements. It is speculated that CRMP2 becomes functionally depleted in AD, by a combination of hyperphosphorylation; sequestration into nascent neurofibrillary tangles; and by oxidative post-translational modifications. This would impede multiple CRMP2 dependent processes including AβPP trafficking through early and recycling endosomal compartments. Additionally, the author and his colleagues find that shRNA suppression of CRMP2 impedes autophagy flux [50], possibly suggesting a role for CRMP2 in the efficient traffic of LC3-II containing autophagic vesicles. Thus functional depletion of CRMP2 could contribute to multiple aspects of AD neuropathology.
![CRMP2 is a MAP that also interacts with cargo adaptor proteins involved with receptor endocytosis and intracellular vesicle trafficking. Panel A is a conceptual diagram illustrating known interactions of CRMP2 with microtubules, and also with kinesin and dynein-associated adaptor proteins. In this model, CRMP2 dynamically equilibrates among its binding partners in a fashion that is likely dependent upon CRMP2 isoform and post-translational modifications. Panel B illustrates AβPP trafficking in a manner that highlights points at which CRMP2 and its binding partners, Numb and MICAL-L1, are involved with intracellular vesicle movements. It is speculated that CRMP2 becomes functionally depleted in AD, by a combination of hyperphosphorylation; sequestration into nascent neurofibrillary tangles; and by oxidative post-translational modifications. This would impede multiple CRMP2 dependent processes including AβPP trafficking through early and recycling endosomal compartments. Additionally, the author and his colleagues find that shRNA suppression of CRMP2 impedes autophagy flux [50], possibly suggesting a role for CRMP2 in the efficient traffic of LC3-II containing autophagic vesicles. Thus functional depletion of CRMP2 could contribute to multiple aspects of AD neuropathology.](https://ip.ios.semcs.net:443/media/jad/2016/53-1/jad-53-1-jad160076/jad-53-jad160076-g002.jpg)
Fig.3
CRMP2 and CRMP2-directed pathways are potential targets for pharmacotherapy. As described in the text, examples exist wherein CRMP2 signaling through axon repulsion-factor receptors can be blocked using anti-NRP1 antibodies and the fungal metabolite xanthofulvin. Other small molecules such as tianeptine significantly increase CRMP2 protein expression, apparently by sensitizing growth factor-dependent gene expression. Small molecules including lanthionine ketimine derivatives and lacosamide bind CRMP2 to functionally enhance or inhibit CRMP2 neurotrophic actions, respectively. Calpain-specific inhibitors could decrease p25 and hence, Cdk5/p25 activity upon CRMP2. One or more of these pathway components might offer value for study in development of AD-directed therapeutics whose action need not rely upon any explicit assumptions about Aβ or tau. Examples of potential therapy target sites are indicated in red font.
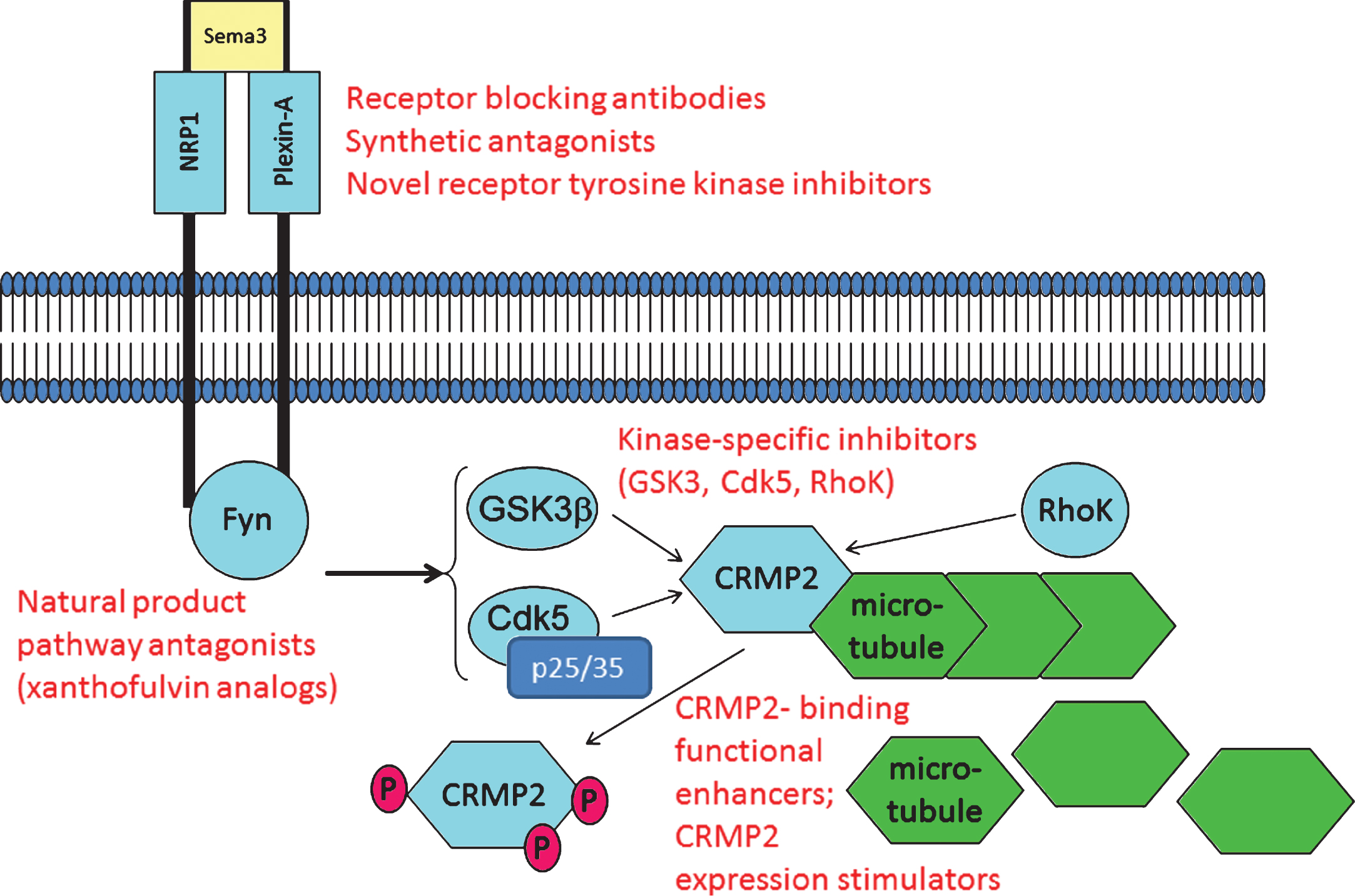
Table 1
Comparison of CRMP2 and Tau
| Property or feature | Tau | CRMP2 |
| Microtubule-associated protein (MAP) | ✓ | ✓ |
| Primed by Cdk5 then phosphorylated by GSK3β | ✓ | ✓ |
| Alternatively phosphorylated by Rho K and other kinases to alter protein:protein interactions | ✓ | ✓ |
| Forms dissociation-resistant high molecular weight structures (PHF tau, tangles) | ✓ | × |
| Collects with p-Tau in neurofibrillary tangles in AD | ✓ | ✓ |
| Depletion destabilizes microtubules and neuritis | ✓ | ✓ |
| Post-translational modifications implicated in mechanisms of neurotoxicity | ✓ | ✓ |
| Calpain-cleaved fragments implicated in neurotoxicity | ✓ | ✓ |
| Genetically associated with neurodegeneration | ✓ | × |
| Functions actively in normal vesicular transport processes | × | ✓ |
| Implicated in normal function of autophagy | × | ✓ |
| Evidence for druggability | ✓ | ✓ |
Salient features that are plausibly relevant to AD pathogenesis, and that offer potential avenues for therapy development, are compared.


