
Drivers: A Biologically Contextualized, Cross-Inferential View of the Epidemiology of Neurodegenerative Disorders
Abstract
Background:
Sutherland et al. (2011) suggested that, instead of risk factors for single neurodegenerative disorders (NDDs), there was a need to identify specific “drivers”, i.e., risk factors with impact on specific deposits, such as amyloid-β, tau, or α-synuclein, acting across entities.
Objectives and Methods:
Redefining drivers as “neither protein/gene- nor entity-specific features identifiable in the clinical and general epidemiology of conformational NDDs (CNDDs) as potential footprints of templating/spread/transfer mechanisms”, we conducted an analysis of the epidemiology of ten CNDDs, searching for patterns.
Results:
We identified seven potential drivers, each of which was shared by at least two CNDDs: 1) an age-at-exposure-related susceptibility to Creutzfeldt-Jakob disease (CJD) and several late-life CNDDs; 2) a relationship between age at onset, survival, and incidence; 3) shared genetic risk factors for CJD and late-life CNNDs; 4) partly shared personal (diagnostic, educational, behavioral, and social risk factors) predating clinical onset of late-life CNDDs; 5) two environmental risk factors, namely, surgery for sporadic CJD and amyotrophic lateral sclerosis, and Bordetella pertussis infection for Parkinson’s disease; 6) reticulo-endothelial system stressors or general drivers (andropause or premenopausal estrogen deficiency, APOEɛ4, and vascular risk factors) for late-life CNDDs such as dementia/Alzheimer’s disease, type-2 diabetes mellitus, and some sporadic cardiac and vascular degenerative diseases; and 7) a high, invariant incidence ratio of sporadic to genetic forms of mid- and late-life CNDDs, and type-2 diabetes mellitus.
Conclusion:
There might be a systematic epidemiologic pattern induced by specific proteins (PrP, TDP-43, SOD1, α-synuclein, amyloid-β, tau, Langerhans islet peptide, and transthyretin) or established combinations of these.
INTRODUCTION
It is increasingly accepted that failure in protein synthesis control, resulting in the aggregation and accumulation of misfolded proteins, and ultimately in impaired cell function and death in specific anatomical structures, characterizes a group of human disorders in which approximately 30 different proteins are implicated [1, 2]. Among such ailments, this paper mainly concentrates on three groups of conditions: (a) those encompassing the majority of the protein-associated sporadic, conformational neurodegenerative disorders (sCNDDs), namely Alzheimer’s disease (AD), late age-related macular degeneration (AMD), idiopathic Parkinson’s disease (PD), Lewy body disease (LBD), frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), sporadic Creutzfeldt-Jakob disease (sCJD), and sporadic rapidly-progressing neurodegenerative dementia (sRPNDd); (b) some chronic vascular disorders, i.e., cerebral amyloid-β (Aβ) angiopathy (CAA), and those characterized by transthyretin deposits, associated with sporadic senile systemic amyloidosis (SSA), heart failure, and aortic aneurism; and, (c) type 2 diabetes mellitus (T2DM) [2]. These constitute examples of the group denoted as localized or organ-limited amyloid disorders, in which different, conformationally abnormal protein extracellular deposits or intracellular deposits such as fibrils, have been identified [2] (see Table 1 for a biochemical and epidemiologic outline). In the field of neurodegenerative disorders (NDDs), this expanding knowledge has recently been articulated in a new theoretical framework that incorporates two key elements, namely, one which is biochemical, i.e., pathogenic protein deposit, and the other which is anatomical and functional, i.e., neural networks [3].
In a previous report, we discussed some implications of the theoretical proposal [3] when interpreting incidence features of the abovementioned sCNDDs [4]. The aim of this paper is to group and discuss selected findings of the descriptive and analytical, general, and clinical epidemiology of sCNDDs, relevant both from the biochemical and pathophysiological perspective and for public health. A third, unpublished manuscript and end of the whole series, incorporates experimental, laboratory data supporting specific theory and two main testable hypotheses for late-life entities and disorders with onset mid-life.
METHODS
The driver concept
The rationale of our approach relies on the driver concept (first proposed by Sutherland et al. [5]). Bearing in mind the mixed molecular phenotype of some sCNDDs represented by the AD, PD, and FTD-ALS spectrum, and the fact that some conditions share one or more risk factors, the above authors argue that, instead of risk factors for single entities, there is a need to identify specific “drivers”, i.e., risk factors or modifiers with impact on specific deposits, such as Aβ, tau, or α-synuclein, able to act across entities (i.e., history of head trauma) [5].
For the purpose of considering the presence of specific risk factors, we redefined “drivers” as “neither protein/gene- nor entity-specific epidemiologic features, i.e. time/place/person related, identifiable as potential footprints of templating/spread/transfer mechanisms, observed in the clinical and general epidemiology of several sCNDDs”. Identification of potential drivers requires a targeted, in-depth review of both NDD epidemiology and the epidemiology of some essentially non-neurological, sporadic human amyloid disorders. Here we attempt an initial approach including selected entities listed in Table 1 [6–9].
We searched for epidemiologic features shared by at least two of the abovementioned disorders, supporting the view that a causally relevant “driver” or pathophysiological mechanism might underlie them. In line with the above definition, the remainder of this paper is devoted to identifying drivers and discussing their usefulness for selecting, assessing, and combining data in research populations and registries. Methodologically, we proceeded with a biologic-context-based conceptual overview of the literature on CNDDs, disregarding aspects of T2DM and vascular disease epidemiology unrelated to NDDs. We listed a potential number of drivers linking exposures to conditions, taking a life-course approach to allow for the recently analyzed influence of age at clinical onset on incidence features of CNDDs [4]. Age at onset is a variable used to facilitate research-resource identification (see “Longitudinal cohort studies in neurodegeneration research”) [10].
Interpretation
On the basis of proposed drivers for sCNDDs, we outlined an epidemiologic complement to Warren et al.’s proposal for NDDs [3].
Ethics Statement
The study was based on reported data and neither animal nor human subject research was implicated. Accordingly, neither informed consent (Public Health General Law. BOE 2011, 240, Article 41.2 page 194613) nor ethical approval was required from the Carlos III Institute of Health Ethics Committee (Comité de é tica de la investigació n y del bienestar animal).
PROPOSED DRIVERS
See the list of seven proposed drivers in Fig. 1.
Driver 1 (D1). Age-at-exposure-related susceptibility to environmental exposure effects
Susceptibility to the effects of exposure (genetic or environmental) has been defined as the “underlying factor sufficient to make a person contract a disease following exposure” [11, 12], i.e., a precondition for causal action. While the factor determining susceptibility may be unknown, it would nonetheless appear to be partly age-related. Hence, age periods— traditionally defined and categorized as prenatal, postnatal, juvenile, and adult— constitute life-stage exposure strata that provide opportunities for experimentally or observationally detecting the presence of susceptibility by reference to a change in the stratum-specific effect measure [13, 14], i.e., relative risk.
Juvenile age at exposure to either diet containing bovine spongiform encephalopathy tissues (BSE-Diet) or treatment with human growth hormone increased the risk of variant CJD or accidentally transmitted (iatrogenic) iCJD, respectively [15–17]. High age at first-in-life whooping-cough epidemic increased risk of PD [18, 19]. Inverted V-shaped, age-at-exposure functions were suggested. Recently, a high risk of sCJD was observed for surgery undergone at ages up to 30 years, as compared to surgery undergone at ages above 31 years or non-exposure to surgery [20]. See Fig. 2 for a graphic outline [15–17, 19, 20]. This pattern, interpreted as susceptibility to exposure effects, might have diluted effect estimates in epidemiologic sCNDD research [21].
The existence of such a driver is consistent with some epidemiologic findings for other sCNDDs. For instance, smaller head size and shorter limb length associated with lower early-life socio-economic status appeared to be independent markers of risk for dementia among North Korean women [22]. The authors interpreted that these might indicate socioeconomic risk factors in childhood that affected both brain and skeletal development but were masked in men by preferential treatment of male children. A lower educational level in adulthood determined ethnic risk differences for cognitive decline or dementia [23]. Hall et al. suggested that, rather than being a risk factor of AD, low education (6 or fewer years of schooling) was a marker for associated deleterious rural socioeconomic or environmental influences in childhood [24]. Some established risk indicators, such as low educational level for AD and well water use for PD, are associated with rural residence and constitute exposures more frequent in early-life (before age 18 years) [25, 26]. These data support the contention that, as with cardiovascular disease, socioeconomic status in childhood or adolescence may constitute a risk factor for sCNDDs [27]. The presence of tauopathy, a hallmark of AD, observed in neurites as early as the second decade of life, would be consistent with an early in life induction of AD [28]. We propose that age-related susceptibility to exposure effects is a key element of an epidemiologic pattern shared by several sCNDDs.
Driver 2 (D2). Tridimensional pattern: Peak age-specific incidence (age at clinical onset), incidence magnitude, and survival (clinical disease duration)
For an outline of the tridimensional pattern, i.e., incidence, age at onset, and clinical disease duration, the reader is referred to Fig. 3 [4, 29–39], which shows data on selected sCNDDs extracted from those reported in de Pedro-Cuesta et al. [4]. The figure depicts normalized age-specific incidences, as well as selected reported age-adjusted incidences and clinical disease duration [29–31, 33, 35, 36, 38–40], suggesting that for sCJD, ALS, FTD, PD, LBD, AD, and AMD, there is a direct correlation between age at highest or peaking age-specific incidence (range 77.5 to ≥95 years), median clinical disease duration (range 0.4 to 8.9 years), and age-adjusted incidence (range ≤1.5–2589×million person-years). The driver defined by “a tridimensional correlate of incidence magnitude, age at onset, and clinical course duration” can be simplified in ordinal terms as incidence spanning 1–1000 (1 for sCJD, 10 for ALS, 100 for PD, 1000 for AD and AMD), median age at onset ranging from almost 70 years for sCJD to 80 years at first visit for AD, and disease duration ranging from <1 year for sCJD to almost 10 years for PD. The pattern can be summarized using the driver notion in four parameters, i.e., age at onset and age at death, both measured in general and clinical populations.
Driver 2 is most relevant when combined with driver 1. Firstly, this is because it may define life-course references for subclinical disease or clinical progress, i.e., how templating and self-propagation is phenotypically manifested. Secondly, the notion that early- versus late-life onset corresponds to rapidly versus slowly progressive and to short versus long clinical-course duration is not difficult to reconcile with views from neuropathology on neurodegeneration progression or cell-to-cell spread, particularly in late-life NDDs [41]. Thirdly, the fact that high sCNDD incidence requires late onset may suggest that ubiquitous or widespread, tiny exposures correspond to high attack rates and lifetime risks.
Driver 3 (D3). Shared, age-at-onset-related, genetic risk factors (APOEɛ4, BACE1, complement-system-related, and other genes)
Mutations and common polymorphisms constitute biologically similar variations in the human genome determining CNDDs. The most common pattern linking genetic variations to CNDDs is where a number of different mutations in the same or different genes determine similar familial CNDD forms. A considerable number of mutations have been described for PD [42]. However, only a few of these result in α-synuclein deposits, a feature of Lewy body lesions in PD [43]. Mutations in the Cu/Zn superoxide dismutase (SOD) gene, the FUS/TLS gene, the TDP-43 gene and, in particular, GGGGCC (G4C2) repeat expansion in the C9orf72 gene have been seen in ALS, and the latter two and the MAPT gene in FTD [44–46]. Conversely, albeit infrequently, point mutations are present in cases with different phenotypes. For instance, in human prion diseases, mutations at codon 178 of the PRNP gene have been described as generating pathologically confirmed cases of either CJD or fatal familial insomnia, depending on codon 129 status on the same allele containing the mutation [47]. In other instances, the disease phenotype appears to uncover a similar abnormal protein deposit, with the cellular subsystem being the determinant for clinical expression as cerebellar ataxia or ALS [48–51]. Mutations at different codons of the PRNP gene were identified in neuropathologically different forms of CJD or AD, with deposits of PrP or Aβ, respectively [52]. For the purposes of driver identification, some polymorphisms determining excess risk shared by sCNDDs are paramount, i.e., APOEɛ4, BACE1, and those related to the complement system [53–56].
APOEɛ4
Recent reports have repeatedly indicated that APOEɛ4 is a shared genetic risk polymorphism for AD, LBD, and sCJD [53, 54, 57], for mild cognitive impairment (MCI), LBD, and vascular dementia (VaD), and for dementia in PD but not PD itself [5, 58–60]. From a genetic standpoint, AD is considered to be heterogeneous, with APOEɛ4 constituting a risk factor for the most frequent AD form able to account for 50% of the etiologic fraction [53]. Moreover, it is well established that the APOEɛ4 allele and homozygosity at polymorphic codon 129 in the PRNP gene are the major genetic risk factors for AD and human prion diseases, respectively [54]. A synergistic age-dependent interaction was seen between APOE and PRNP in both AD and sCJD, suggesting shared genetic factors paving the way for shared genetic susceptibility to environmental causes [54].
BACE1
The β-site AβPP cleaving enzyme 1 (BACE1) appears to be the rate-limiting β-secretase enzyme in the amyloidogenic processing of AβPP and Aβ formation, and thus plays a prominent role in AD pathology. Recent evidence suggests that the PrP protein interacts directly with BACE1, regulating its β-secretase activity. The BACE1 rs638405 C-allele is associated with an increased risk of developing both sCJD [61] and sporadic AD [62].
Complement-system-related genes
Multiple genetic risk factors related to the complement system have been associated with AMD (see references [58] and [63] for recent reviews). For example, the functional polymorphism rs2230199 in the complement C3 gene was linked to AMD [64]. In contrast, modest associations between such genes and AD were seen, and their direction and genetic model differed from that observed for AMD [65].
Other genetic factors
Shared genetic factors for PD and other selected NDDs are not common. Genome-wide association studies suggest that SNCA and LRRK2 polymorphisms constitute modest risk factors for PD [66–68], and that PD variants around the 17q21 are associated with gene expression suggestive of AD risk [69]. The H2 variant in the MATP (tau) gene is protective for PD [70]. C9orf72 expansion (G4C2 repeats) was strongly associated with ALS and FTD, though not with AD or PD [71]. The opposite effects of APOEɛ4 and APOEɛ2 in AD and AMD, i.e., APOEɛ2, protective for AD and causal for AMD, and APOEɛ4, causal for AD and protective for AMD, are well established [58]. Clusterin and PICALM (phosphatidylinositol binding clathrin assembly protein) behave as different risk factors for AMD and AD [58].
Genetic exposures might constitute the paradigm of the driver notion in NDDs. The idea of a univocal correspondence between genes and entities has long been abandoned. Here evidence is gathered on three genes, APOEɛ4, BACE1, and PRNP, which, separately or by interaction, cause the risk of several sCNDDs to increase, e.g., AD, CJD, LBD, and, probably mediated by serum lipids, atherosclerosis [72, 73]. Positive findings in sCJD for CALHM1-3 and BACE-1 polymorphisms underscore the interplay among AβPP, Aβ oligomers, APOE, PrP, and BACE1 in sCJD and AD, and suggest that aging, and perhaps vascular risk factors (VRF), may partly modulate disease pathologies through these key players [61, 74]. It would appear that a few genes, APOE and PRNP, constitute a driver for entities with midlife and late-life onset, with CJD, LBD, and AD constituting a genetically differentiated group.
Driver 4 (D4). Personal risk factors (diagnoses, anthropometric parameters, social factors)
Personal factors encompass a set of multiple variables, such as clinical signs and symptoms (i.e., diagnoses), behavioral patterns (i.e., health-related habits), and educational factors, which constitute the group of best-established associations with specific sCNDDs. A large number of these have been studied. Associations with multiple personal variables have been found for NDDs exhibiting peak incidences at very similar ages, e.g., PD and LBD [75]. Recently, a case-control study on LBD concluded that the risk factors were an amalgam of those described for PD and AD [59]: many of them (history of depression, educational level) were personal variables.
Diagnoses
Perhaps the first variable to be considered is a second sCNDD diagnosis. For instance, PD is associated with AD [76], and essential tremor is associated with increased risks of both PD and AD [77]. The inverse overall co-occurrence of cancer in patients with either AD or PD, shown by a recent meta-analysis of 50 studies, is consistent with a lower exposure to environmental carcinogens in early decades of life, i.e., due to rural residence [78].
Vascular risk factors
The concept of VRF of sCNDDs encompasses different forms of high blood pressure (HBP), dyslipidemia, obesity, T2DM, physical inactivity, hypothyroidism, and vascular diseases such as coronary disease, stroke, or heart failure. In all probability, the liveliest debate in the context of personal risk factors for NDDs turns on VRF. This in part may be due the association being masked by study designs’ sensitivity to bias: (a) selection secondary to impact of vascular disease on taxonomy, e.g., for vascular or mixed dementia or for vascular parkinsonism [79]; (b) use of hospital controls, as seen in sCJD studies [80], and; (c) exposure measurement after clinical onset, i.e., arterial hypertension in PD, LBD, and hereditary dysautonomy, NDDs which may result in vascular pathology or dysregulation. Finally, non-causal interpretations of positive associations can be reconciled with etiologic mechanisms acting on the vascular wall decades before neurological and vascular symptom onset for single disorders (see reference [81] for a discussion on VRF preceding sCJD onset) or shared by, say, AD and sCJD (see driver 6). Bearing such problems in mind, a brief overview now follows.
Little is known about VRF for sporadic ALS, FTD, and PD. Controversial results are reported for VRF and risk of ALS (see a review by Hardiman [82]), and for T2DM and risk of PD, with both positive [83] and negative findings [84, 85]. No cardiovascular risk factors, including APOEɛ4 [86] and hypertension [87], have been reported for PD, whereas the protective factor of smoking— a risk factor for AD, at least in APOEɛ4 carriers— has been well established (see Sutherland et al. [5] for a review). Risk of FTD from T2DM increases twofold [88]. When compared with AD and vascular parkinsonism, both FTD and PD had a lower prevalence of VRF (systolic blood pressure and APOEɛ4 allele) [79, 89].
VRF associations have traditionally been reported for dementia and AD. For instance, with regard to late-life AD, this association has been established for high midlife systolic blood pressure, elevated midlife total cholesterol, the APOEɛ4 allele [90], and T2DM [90]. Microvascular brain lesions as well as VRF underlie vascular dementia and mixed (vascular and AD) dementia [91, 92]. Obesity, arteriosclerosis, diabetes, hypertension, and hypercholesterolemia predispose to AMD [93–95]. However, a recent meta-analysis still casts doubts on links between hypertension and incident AD [96], and some reports (see Kravitz et al. [97] for a review) question its value for dementia with onset after age 90 years. A review of method- and quality-stratified studies on VRF and AD by Chui et al. concluded that VRF predicted vascular pathology linked to dementia rather than determining plaques and tangles, with such associations being attributed to selection [98]. Coronary artery disease is associated with AD neuropathology in APOEɛ4 carriers [99]. We described a significantly high frequency of coronary surgery during the ten years preceding clinical onset of sCJD [100].
In brief, various VRF, including hypertension and hypercholesterolemia in particular, have been found to be directly associated with AD and AMD, while coronary disease has been found to be associated with AD-APOEɛ4 carriers and coronary surgery with sCJD. One of these, smoking habit, has repeatedly been found to be negatively associated with PD.
Vascular disorders and proteinopathies
Discussion on VRF and NDDs largely preceded recent knowledge of proteinopathies. Similar misfolded protein deposits in nervous system and vascular structures, i.e., Aβ in AD [101, 102] and APrP in sCJD [103], challenge the concept of such entities as organ-limited amyloidoses, and suggest that some factors or a proportion of the excess vascular risk, well known for AD, might be attributable to the vasculopathy that precedes clinical expression of central nervous system lesions [104]. Therefore, associations between coronary surgery and sCJD [100], and coronary surgery and AD pathology in APOEɛ4 carriers [99] might correspond to vascular comorbidity potentially determined by similar or different causes in sCJD and AD acting at long latency periods. Furthermore, the fact that Aβ vasculopathy in neurodegenerative dementia appears to be linked to AD pathology, that a large majority of AD patients present with Aβ vasculopathy, and that AD and sCJD share genetic predictors [54, 61, 74] may support the view that there is a conformational protein-related vasculopathy in both AD and sCJD, partially accounting for the associations between vascular risk factors and each of the two disorders. To sum up, all the above indicates that there might be biologically shared mechanisms of misfolded protein accumulation in the brain and the vascular wall, affecting Aβ, PrP, and a few other proteins and, to a less clear degree, α-synuclein. In addition, the fact that T2DM is classified within the group of disorders presenting similar pathologic toxic deposits of misfolded proteins [105–107] adds evidence for existing, albeit complex, links between T2DM, angiopathy, and sCNDDs, and motivates the section below on General Drivers. It would appear that the driver concept proposed for etiologic inference in the field of shared risk-factor epidemiology [5], may be expanded to cover a proportion of vascular disease and, to a possibly different extent, selected sCNDDs, particularly sCJD, sRPNDd, LBD, AD, and AMD, entities in which a small vessel vasculopathy of the brain or retina have been described. The driver concept might provide a rationale for prevention, complementing, insofar as dementia is concerned, strategies aimed at promoting healthy lifestyles, e.g., by multi-domain interventions [108].
Driver 5 (D5). Environmental risk factors (invasive medical procedures; Bordetella pertussis infection)
Despite intensive efforts, environmental factors continue to be an elusive research field for sCNDDs. For instance, although higher exposure to or use of agrochemicals (herbicides, fertilizers, pesticides, and solvents) has been described as being linked to AD, PD, ALS, etc. [109], not a single chemical product has ever been unequivocally identified as a risk factor for AD, PD, or ALS, mainly due to poor measurements of exposure [110, 111]. Head trauma, has been singled out by Sutherland et al. as a pluripotential risk factor for several sCNDDs, and therefore proposed as a driver valid for AD, PD, and other disorders [5]. We will focus on two other drivers, namely, invasive medical procedures and, bearing recent findings in amyloid research in mind, infection.
Invasive medical procedures (surgery and blood transfusion)
Neurosurgery has been linked to CJD after a few reports on the use of instruments potentially contaminated by surgery on a CJD patient, and an experimental study based on one such report, focusing on re-used electrodes. However, this association has never been demonstrated in epidemiologic controlled studies. In contrast, a number of associations and epidemiologic features potentially underlying or masking surgery as a risk factor have been reported for sCJD and ALS. There is increasing epidemiologic evidence of significant etiologic links: in the case of sCJD, to history of general surgery [100] with long incubation periods [100, 112], and to surgery of the retina and peripheral nerves after shorter incubation intervals [112]; and in the case of ALS, to cervical compression with and without spinal surgery [113], and to occupations with high levels of physical activity [114, 115], e.g., Italian football players [116]. Whether surgical history might underlie reported associations with agricultural or non-specialized work [117], war veterans [118, 119], history of cranial or other trauma [120, 121], or history of repeated or severe trauma [122], is a matter of speculation, yet it cannot be ignored. Since none of the abovementioned approaches to studying associations between medical procedures and ALS used latency analysis, the results suggest that surgery as a risk factor for ALS may have confounded some of the reported associations. The role of surgical history in sRPNDd, PD, or AD has not been explored in depth [123].
A recent review has shown inconsistent results for blood transfusion as a potential risk factor for sCJD after a 10-year lag [80]. Blood transfusion was not linked to AD [124], and its link with PD and sRPNDd has probably not been well studied [123]. The abovementioned, recently reported significant excess risk for motor neurone disease (3 cases) in a cohort of human growth hormone recipients opens up the question of accidental transmission of ALS [7]. All the above suggests that invasive medical procedures followed by a considerable lag may constitute D5 drivers for mid-life sCNNDs.
Bordetella pertussis (BP) infection
The association between age at first major whooping cough outbreak and PD constitutes a single observation in Iceland, reinforced by negative results for birth-cohort effects in continental populations. Considered as a natural quasi-experiment, it is consistent with the high prevalence and incidence of PD among the Färoe Islanders and Greenland Inuit reported from direct surveys [125, 126]. While the excess risk of PD as a long-term biological effect of BP infection was attributed to Pertussis toxin [19], there is recent evidence to propose prion-like mechanisms. For instance: (a) PD shares protein deposits (see Sutherland et al. [5] for a review) with LBD and AD, BP; (b) amyloidosis by various amyloid fibrils is best induced by mouse AApoAII(C) amyloid [127] and A amyloidosis in mice was enhanced by cross seeding curli of Escherichia coli (a human adapted pathogen like BP) [128]; and, (c) multiple system atrophy, an α-synucleinopathy, has recently been transmitted in cell and mouse models [129].
To sum up, host-adapted human pathogens such as BP might constitute D5 drivers for PD, LBD, and AD and other late-life sCNDDs induced by infection in genetically susceptible young individuals. Invasive procedures sharing similar mechanisms might be proposed for sCJD, ALS and sRCNDd.
Driver 6 (D6). General drivers. Reticuloendotelial system (RES) stressors: 1) Endocrine factors (premenopausal estrogen deficiency; andropause); 2) APOE gene; 3) Vascular risk factors and vascular disease
We define general drivers as any driver, whether personal, environmental, or genetic, which, in accordance with (a) the theory attributing protein misfolding, aggregation, and deposit to an RES dysfunction [105, 107], and (b) the molecular nexopathy expanded paradigm, exhibits associations with variables related to onset and progression of different sCNDDs or entities of the amyloid spectrum [130]. We perceive three fields in which general drivers might be manifested: endocrine factors; the APOE gene; and risk factors or progression biomarkers potentially reflecting extraneuronal amyloid deposits in vascular wall and pancreas.
Endocrine factors
An as yet incomplete association array may be structured around the two axes, i.e., premenopausal estrogen deficiency and testosterone deficiency.
Premenopausal estrogen deficiency
From single studies and recent reviews on the interplay between natural or surgical menopause, oophorectomy, and hormonal therapies, and its potential impact on vascular risk [131] and dementia [132], it might be pointed out: (a) that bilateral oophorectomy not followed by post-menopausal estrogen therapy doubled the risk of coronary disease (two cohort studies) [133] and of cognitive decline or dementia [134]; and, (b) in the Danish national population, premenopausal bilateral oophorectomy doubled the risk of dementia, with this risk increasing with younger age at surgery [135]. In two US populations, age at surgical menopause was associated with a faster decline in global cognition, and in the case of the sample studied postmortem, was associated with AD neuritic plaque burden [136]. When controlling for estrogen therapy, analyses in most studies [133, 134, 136] indicate either an absence of associations, or associations that are in part mediated. Despite the fact that not all studies support the excess risk for cardiovascular conditions [137], premenopausal oophorectomy might illustrate the existence of drivers shared by NDDs and vascular disease.
Andropause
The combination of short androgen receptor CAG alleles with lower levels of serum testosterone increased the risk of AD in men fourfold [138]. A second line of observations linking andropause to AD and T2DM was recently reviewed [139]. Low testosterone precedes cognitive [140] and neuropathologic [141, 142] diagnoses of AD, suggesting a direct causal role in AD. Additionally, low testosterone levels predict metabolic syndrome, preceding the onset of metabolic and cardiovascular symptoms by 5–10 years [143, 144], and testosterone depletion due to prostate cancer therapy increases the incidence and prevalence of T2DM [145, 146].
The fact that estrogen and testosterone deprivation at a certain age might result in increased risk of AD, coronary disease, and T2DM would support the view that modest associations among these entities might be partly due to confounding.
APOE gene
The APOEɛ4 allele has been described as one of the genetic risk factors for different sCNDDs, namely, AD and sCJD (see Driver 4). Nevertheless, it must also be considered as a general driver, since many other associations with it as a predictor of sCNDD progression or as a risk factor for potentially conformational disorders have been described. For instance, the APOEɛ4 allele was found to be a risk factor: for VaD and mixed dementia [147]; for AMD, AD, and VaD [148]; for LBD, pure Lewy body dementia, and dementia in PD [57]; for amnestic MCI [149]; for dementia in PD; and, interacting with PRNP, for late-onset sCJD [54]. The APOEɛ4 allele predicts conversion from MCI to AD [148] as well as microbleeds in AD [150]. On the one hand, the APOEɛ4 allele has been shown to constitute a risk factor for death from coronary heart disease [151], and a risk factor for T2DM with and without coronary artery disease [152]. However, a large-sized prospective study failed to show an association between APOEɛ4 and risk of coronary heart disease, after a mean follow-up of 11 years [153]. It is possible that the APOEɛ4 allele effect in coronary heart disease or T2DM may require a longer latency, as seen for late-life NDDs. The APOEɛ4 allele, whether alone or interacting with other cholesterol genes, affects memory [154]. On the other hand, a role for the APOEɛ4 allele in T2DM and AD is recognized, since the presence of APOEɛ4 allele in T2DM patients raises the risk of AD [155]; and in T2DM patients with AD, APOEɛ4 is associated with increased neurofibrillary tangles, amyloid plaques, and CAA [156]. In brief, the APOEɛ4 allele, in addition to constituting a driver shared by AD and CJD, may play a wider role in determining risks of T2DM and vascular disease.
Vascular risk factors and vascular disease
An arteriosclerotic disease indicator for AD [157], midlife untreated high diastolic blood pressure and high systolic blood pressure for AD and VaD, respectively, in the Honolulu study [158], or the set of high body mass index, blood pressure, serum cholesterol, or impaired glucose regulation in midlife for AD [90], constitute examples of multiple VRF associations. On the other hand, VRF have been associated with conversion from MCI to dementia [148] and have predicted natural history patterns from normal cognition to dementia [159]. Other examples include: (a) white matter hyperintensities and conversion from MCI to dementia; (b) T2DM, carotid stenosis, HBP, white matter lesions, a VRF score, and vascular disease scores and conversion from MCI to dementia [160, 161]; and, (c) untreated arterial hypertension, cholesterol, and diabetes and conversion from MCI to AD [162]. When examined from a different, possibly statistically unopposed view, obesity, metabolic syndrome, and T2DM not only contribute to impaired cognitive function, but also increase the risk of AD [139].
Drawing on Finnish studies on the prevalence at death of biochemically characterized vascular lesions, two highly frequent, unassociated angiopathies, SSA and CAA (linked to wild-type transthyretin deposit and heart failure/myocardial infarction, and associated with Aβ, dementia, or AD, respectively), illustrate different mechanisms underlying atherosclerosis among the very old [163–165].
To sum up, VRF constitute a complex array of exposures or vascular effects of exposures, and some VRF constitute a complex group of elements acting midlife as risk factors for— and, at a later stage, as confounding and competing risk associations with— a number of dementing NDDs, and with AD and mixed dementia in particular. The failure of controlled experiments to prevent AD through targeting major cardiovascular risk factors (e.g., using antihypertensive drugs, lowering cholesterol, and anti-inflammatory therapies reviewed by Qiu [166]), despite imaging and other evidence suggesting a role for VRF in dementia, would support the view that translation of NDD proteomics to epidemiologic research is required. We outline an incomplete epidemiologic/etiologic scenario where different amyloid deposits (wild-type transthyretin, lactadherin, and Aβ), sometimes shared by sCNDDs, vascular disorders, and a number of endocrine entities or functions, might determine late-life human pathology. The complex D6 might thus represent the puzzle resulting from the multiple effects of RES stressors.
Driver 7 (D7). Invariant ratio of sCNDD incidence/genetic CNDD incidence, across entities
Some authors have given special attention, early as well as recently, to the invariant ratio of sporadic to genetic sCNDD forms [167]. This ratio would correspond to the inverse value of the proportion of genetic forms in CJD, MND, PD, and AD in community-based case series including sporadic and familial/genetic forms, usually reported to range from approximately 10% to 20%, amounting to a four- to ninefold higher number of sporadic than familial cases [168]. Incidence, as a proxy mortality-based figure of purely familial/genetic forms, has been reported for PrP-related NDDs [169]. The expected annual incidence range of genetic CNDD forms corresponding to the sCNDD forms in Fig. 2 would be approximately 0.1–100 per million.
Maturity-onset diabetes of the young (MODY) and autosomal dominant, early-onset T2DM unlinked to known MODY genes (MODY gene-negative families) constitute more than 5% of all cases of T2DM [170]. The most common form of CAA is due to Aβ deposition, which occurs sporadically in the elderly or in association with AD [171]. Aβ-CAA may also be prominent in variants of familial AD, with mutations of the AβPP, presenilin-1 (PSEN1) or presenilin-2 (PSEN2) genes. Other familial CAA forms present protein deposits different from Aβ. Hereditary forms of CAA constitute a non-negligible proportion of both AD and sCAA. Familial forms of retinal atrophy have been traditionally described as retinal dystrophies. Mutations have been described in the EFMP1, SEMA4A, and other genes [172], with the deposit being semaphorin. More than one hundred mutations have been described in the transthyretin gene [173]. In essence, driver 7 appears to be shared by sCNDDs, T2DM, and a substantial proportion of atherosclerosis lesions represented by angiopathies resulting from protein elimination failure.
Despite the fact that the invariant ratio of genetic/sporadic cases across entities has not been formally studied and that observations rely on case series or unrefined figures, this type of ecological observation (absent or rare in some entities, such as Huntington’s disease) warrants detailed attention: phenotypical similarities between genetic and sporadic forms are evident. An etiologic link between the genetic and sporadic forms would require the presence of vectors of transferred human pathologic genetic material from human ancestors resulting in prion-like processes driven by human adapted organisms. A tantalizing field of knowledge anticipating the role of driver 7 in sCNDDs and other conformational disorders is horizontal gene transfer from human to prokaryotic and other human pathogens, which constituted a major force in prokaryotic evolution [174] perceived at an early stage [175]. The final step would require the presence of infectious natural protein aggregates resulting from human transferred active sequences, acting as inoculum able to cross the species barrier back to the original human source.
SUGGESTIONS FROM NDD EPIDEMIOLOGY
Neuroepidemiologic knowledge in the field of NDDs is ultimately a form of biological interpretation of the findings yielded by social science (i.e., population) research, currently in its initial stages. Relations may emerge that will erase the limits of the initial outline. Here a number of sCNDDs have been selected, though the number of entities to be included for inference remains open. For instance, one may consider the appropriateness of AMD, generally neglected when discussing NDDs. Similarly, a number of relationships or elements have to be proposed in order to define the boundaries of such knowledge. Arguably, analysis of epidemiologic events will never be definitive or absolutely valid. The proposal set forth here should be useful for designing non-experimental research aimed at testing the epidemiologic dimensions of an expanded NDD paradigm [3], thereby providing reoriented cumulative knowledge. Traditional work on the epidemiology of NDD clinical entities may benefit from the proposed biochemical perspective, by providing a more stable structure, i.e., in Foucault’s terminology, “from the tangled mass of discontinuities to the great, uninterrupted unities” [176]. The above observations may suggest that the traditionally accepted epidemiologic traits of single NDDs need to be revised toward the epidemiology of a continuum, and redefined by including links with T2DM and atherosclerosis. On the basis of our recently systematic incidence review [4] and this analysis, we propose that:
1) Incidence of single sCNDDs in the general population increases with age at onset.
2) Age-specific incidence of single sCNDDs increases with age at onset following an inverted V-shaped function, only in part perceived for late-life sCNDDs.
3) Normalized age-specific incidences of acquired CNDDs and sCNDDs follow an age-at-onset (peak incidence) and protein-related pattern. Differences for the same protein, i.e., for CJD, would reflect different causal mechanisms or protein strain.
4) Environmental risk factors may preferably act when exposed at infantile and juvenile ages or in early adulthood through being mediated by genes and shared susceptibility to effects.
5) Not being entity-specific, genetic risk factors for sCNDDs do not systematically refer to genes affected in the corresponding genetic form.
6) Risk factors and preclinical or clinical biomarkers of disease progression may correspond -albeit not systematically- to the same parameter, i.e., genes, and determine incidence as well as progression of subclinical and clinical course.
7) Age determines risk as a result of age at exposure, age-related susceptibility, and latency.
8) Sex is a risk modifier for specific sCNDDs. Some endocrine drivers may be sex- and age-at-onset specific, i.e., for late-life NDDs. The male versus female sex incidence ratio for different late-life sCNDDs may vary reflecting occupation, endocrine, and other factors. Endocrine disorders might also constitute genuine endocrine proteinopathies, such as T2DM sharing biological RES pathophysiological mechanisms.
9) Etiologic or pathophysiological mechanisms may be shared by different proteinopathies, including sCNDDs. Consequently, prevention strategies might be shared, not only for NDDs and mixed NDD forms, but also for T2DM and a proportion of atherosclerotic lesions.
10) Control of vascular risk factors may or may not determine sCNDD prevention. The causal weight of selected vascular risk factors in sCNDDs would require reassessment.
11) The present view of sCNDDs reinforces the notion of a set of neurodegenerative processes in which continuity as a form of overlap, and competition— with earlier and more lethal NDDs, T2DM, or vascular disease, removing persons at high risk of other less lethal NDDs— act on the age axis. Potential chronic RES stressors, i.e., obesity, may have acted simultaneously on different target cells.
12) Should early-age susceptibility drivers be present in sCNDDs with long latency periods, this may imply that there could be a short lifetime for primary, and a protracted lifetime for secondary sCNDD prevention.
As a summary outline, we propose the epidemiologic model shown in Fig. 4. Accordingly, we would give particular consideration to research lines, such as: (a) well-assessed incidence studies on sRPNDd, FTD, and LBD, supported where possible by postmortem biochemical analysis; (b) incidence and analytical studies of combined forms, such as MND + FTD, in the same population; (c) taxonomy-oriented studies facilitating a converging, interrelated view of sCNDDs, likely within the field of organ-limited amyloidoses, e.g., sRPNDd, including sCJD and vascular dementia; (d) studies on environmental factors, where epistasis with selected genes and epigenetic aspects can be explored; (e) targeted cohort availability and resource access [10], despite the fact that diagnostic criteria, and exposure variables may not have been well defined at baseline; and, (f) studies on specific causal hypotheses, supported where possible by experimental data, requiring tailored combinations of cohorts and registries for testing.
CONCLUSION
There might be a systematic epidemiologic pattern induced by specific proteins (PrP, TDP-43, SOD1, α-synuclein, Aβ, tau, Langerhans islet peptide, and transthyretin) or established combinations of these, as a result of molecular templating. New biologic paradigms proposed for CNDDs may encompass other organ-limited disorders associated with amyloid deposits.
ACKNOWLEDGMENTS
The authors would like to thank Javier Damian of the National Center for Epidemiology, Carlos III Institute of Health (ISCIII), for his comments and meticulous review of the text, and Michael Benedict for his help with the English-language version of the manuscript. Funding was received from the Consortium for Biomedical Research in Neurodegenerative Diseases (Centro de Investigació n Biomé dica en Red sobre Enfermedades Neurodegenerativas/CIBERNED) as part of the 2014-2015 annual budget of the CIBERNED 509-group. Mr. Alcalde received support from the ISCIII Field Epidemiology Program during 2014 and 2015. The study was partially supported by a grant from the EU Joint Program – Neurodegenerative Disease Research (JPND - DEMTEST (Spanish Health Research Fund, FIS PI11/03021 and PI12/00045)). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No author other than EAC received specific funding for this work.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-0884r2).
REFERENCES
[1] | Frost B , Diamond MI ((2009) ) The expanding realm of prion phenomena in neurodegenerative disease. Prion 3: , 74–77. |
[2] | Sipe JD , Benson MD , Buxbaum JN , Ikeda S , Merlini G , Saraiva MJ , Westermark P ((2014) ) Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 21: , 221–224. |
[3] | Warren JD , Rohrer JD , Schott JM , Fox NC , Hardy J , Rossor MN ((2013) ) Molecular nexopathies: A new paradigm of neurodegenerative disease. Trends Neurosci 36: , 561–569. |
[4] | de Pedro-Cuesta J , Rabano A , Martinez-Martin P , Ruiz-Tovar M , Alcalde-Cabero E , Almazan-Isla J , Avellanal F , Calero M ((2015) ) Comparative incidence of conformational, neurodegenerative disorders.e. PLoS One 10: , e0137342. |
[5] | Sutherland GT , Siebert GA , Kril JJ , Mellick GD ((2011) ) Knowing me, knowing you: Can a knowledge of risk factors for Alzheimer’s disease prove useful in understanding the pathogenesis of Parkinson’s disease?. J Alzheimers Dis 25: , 395–415. |
[6] | Gunnarsson LG , Lygner PE , Veiga-Cabo J , de Pedro-Cuesta J ((1996) ) An epidemic-like cluster of motor neuron disease in a Swedish county during the period 1973-1984. Neuroepidemiology 15: , 142–152. |
[7] | Irwin DJ , Abrams JY , Schonberger LB , Leschek EW , Mills JL , Lee VM , Trojanowski JQ ((2013) ) Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol 70: , 462–468. |
[8] | Marmorstein LY , Munier FL , Arsenijevic Y , Schorderet DF , McLaughlin PJ , Chung D , Traboulsi E , Marmorstein AD ((2002) ) Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc Natl Acad Sci U S A 99: , 13067–13072. |
[9] | Peng S , Glennert J , Westermark P ((2005) ) Medin-amyloid: A recently characterized age-associated arterial amyloid form affects mainly arteries in the upper part of the body. Amyloid 12: , 96–102. |
[10] | Buckle R , Moody C , Aarsland D , Brayne C , Breteler M , Cappa S , Dubois B , Lawlor B , de Pedro-Cuesta J ((2013) ) Longitudinal cohort studies in neurodegeneration research, http://www.neurodegenerationresearch.eu/uploads/media/JPNDAGLCS_Final_Report_Oct_2013-version_07_01_14.pdf, Accessed 24-, 8–2015. |
[11] | Greenland S ((1987) ) Book Review: Theoretical epidemiology. Olli S, Miettinen, Wiley, New York, 1985. No. of pages: 359, Price: $32.95. Stat Med 6: , 209–211. |
[12] | Khoury MJ , Flanders WD , Greenland S , Adams MJ ((1989) ) On the measurement of susceptibility in epidemiologic studies. Am J Epidemiol 129: , 183–190. |
[13] | Armitage P , Doll R ((1954) ) The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer 8: , 1–12. |
[14] | Barton HA , Cogliano VJ , Flowers L , Valcovic L , Setzer RW , Woodruff TJ ((2005) ) Assessing susceptibility from early-life exposure to carcinogens. Environ Health Perspect 113: , 1125–1133. |
[15] | Ghani AC , Ferguson NM , Donnelly CA , Anderson RM ((2003) ) Factors determining the pattern of the variant Creutzfeldt-Jakob disease (vCJD) epidemic in the UK. Proc Biol Sci 270: , 689–698. |
[16] | Swerdlow AJ , Higgins CD , Adlard P , Jones ME , Preece MA ((2003) ) Creutzfeldt-Jakob disease in United Kingdom patients treated with human pituitary growth hormone. Neurology 61: , 783–791. |
[17] | Boelle PY , Cesbron JY , Vallerón AJ ((2004) ) Epidemiological evidence of higher susceptibility to vCJD in the young. BMC Infect Dis 4: , 26. |
[18] | de Pedro-Cuesta J , Wermuth L , Abraira V , Stawiarz L ((1995) ) Levodopa use in Denmark: High levels in Greenland and the Faroe Islands. Europarkinson Preparatory Activity Research Group. Acta Neurol Scand 91: , 89–97. |
[19] | de Pedro-Cuesta J , Gudmundsson G , Abraira V , Gudmundsson G , Love A , Tulinius H , Veiga J , Almazán J , Petersen IJ ((1996) ) Whooping cough and Parkinson’s disease. The Europarkinson Preparatory Activity Research Group. Int J Epidemiol 25: , 1301–1311. |
[20] | de Pedro-Cuesta J , Mahíllo-Fernández I , Calero M , Rábano A , Cruz M , Siden A , Martínez-Martín P , Laursen H , Ruiz-Tovar M , Molbak K ((2014) ) Towards an age-dependent transmission model of acquired and sporadic Creutzfeldt-Jakob disease.e. PLoS One 9: , e109412. |
[21] | Rothman KJ , Greenland S , Lash TL ((2008) ) Applications of stratified analysis methods. In pp. Modern Epidemiology. Third Edition 283–302. |
[22] | Kim JM , Stewart R , Shin IS , Kim SW , Yang SJ , Yoon JS ((2008) ) Associations between head circumference, leg length and dementia in a Korean population. Int J Geriatr Psychiatry 23: , 41–48. |
[23] | Sachs-Ericsson N , Blazer DG ((2005) ) Racial differences in cognitive decline in a sample of community-dwelling older adults: The mediating role of education and literacy. Am J Geriatr Psychiatry 13: , 968–975. |
[24] | Hall KS , Gao S , Unverzagt FW , Hendrie HC ((2000) ) Low education and childhood rural residence: Risk for Alzheimer’s disease in African Americans. Neurology 54: , 95–99. |
[25] | Moceri VM , Kukull WA , Emanuel I , van Belle G , Larson EB ((2000) ) Early-life risk factors and the development of Alzheimer’s disease. Neurology 54: , 415–420. |
[26] | Priyadarshi A , Khuder SA , Schaub EA , Priyadarshi SS ((2001) ) Environmental risk factors and Parkinson’s disease: A metaanalysis. Environ Res 86: , 122–127. |
[27] | Cohen S , Janicki-Deverts D , Chen E , Matthews KA ((2010) ) Childhood socioeconomic status and adult health. Ann N Y Acad Sci 1186: , 37–55. |
[28] | Braak H , Thal DR , Ghebremedhin E , del Tredici K ((2011) ) Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 70: , 960–969. |
[29] | Granieri E , Carreras M , Casetta I , Govoni V , Tola MR , Paolino E , Monetti VC , De Bastiani P ((1991) ) Parkinson’s disease in Ferrara, Italy, 1967 through 1987. Arch Neurol 48: , 854–857. |
[30] | Gao S , Hendrie HC , Hall KS , Hui S ((1998) ) The relationships between age, sex, and the incidence of dementia and Alzheimer disease: A meta-analysis. Arch Gen Psychiatry 55: , 809–815. |
[31] | Baldereschi M , Di Carlo A , Rocca WA , Vanni P , Maggi S , Perissinotto E , Grigoletto F , Amaducci L , Inzitari D ((2000) ) Parkinson’s disease and parkinsonism in a longitudinal study: Two-fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology 55: , 1358–1363. |
[32] | Will RG , Zeidler M , Stewart GE , Macleod MA , Ironside JW , Cousens SN , Mackenzie J , Estibeiro K , Green AJ , Knight RS ((2000) ) Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol 47: , 575–582. |
[33] | Benito-León J , Bermejo-Pareja F , Morales-González JM , Porta-Etessam J , Trincado R , Vega S , Louis ED ((2004) ) Incidence of Parkinson disease and parkinsonism in three elderly populations of central Spain. Neurology 62: , 734–741. |
[34] | de Lau LM , Giesbergen PC , de Rijk MC , Hofman A , Koudstaal PJ , Breteler MM ((2004) ) Incidence of parkinsonism and Parkinson disease in a general population: The Rotterdam Study. Neurology 63: , 1240–1244. |
[35] | Pocchiari M , Puopolo M , Croes EA , Budka H , Gelpi E , Collins S , Lewis V , Sutcliffe T , Guilivi A , Delasnerie-Laupretre N , Brandel JP , Alperovitch A , Zerr I , Poser S , Kretzschmar HA , Ladogana A , Rietvald I , Mitrova E , Martínez-Martín P , de Pedro-Cuesta J , Glatzel M , Aguzzi A , Cooper S , Mackenzie J , van Duijn CM , Will RG ((2004) ) Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain 127: , 2348–2359. |
[36] | Fang F , Valdimarsdottir U , Bellocco R , Ronnevi LO , Sparen P , Fall K , Ye W ((2009) ) Amyotrophic lateral sclerosis in Sweden 1991-2005. Arch Neurol 66: , 515–519. |
[37] | Chen YY , Lai CH ((2010) ) Nationwide population-based epidemiologic study of Huntington’s Disease in Taiwan. Neuroepidemiology 35: , 250–254. |
[38] | Steenland K , MacNeil J , Seals R , Levey A ((2010) ) Factors affecting survival of patients with neurodegenerative disease. Neuroepidemiology 35: , 28–35. |
[39] | Owen CG , Jarrar Z , Wormald R , Cook DG , Fletcher AE , Rudnicka AR ((2012) ) The estimated prevalence and incidence of late stage age related macular degeneration in the UK. Br J Ophthalmol 96: , 752–756. |
[40] | Pérez F , Helmer C , Dartigues JF , Auriacombe S , Tison F ((2010) ) A 15-year population-based cohort study of the incidence of Parkinson’s disease and dementia with Lewy bodies in an elderly French cohort. J Neurol Neurosurg Psychiatry 81: , 742–746. |
[41] | Polymenidou M , Cleveland DW ((2012) ) Prion-like spread of protein aggregates in neurodegeneration. J Exp Med 209: , 889–893. |
[42] | Trinh J , Farrer M ((2013) ) Advances in the genetics of Parkinson disease. Nat Rev Neurol 9: , 445–454. |
[43] | Hardy J , Revesz T ((2012) ) The spread of neurodegenerative disease. N Engl J Med 366: , 2126–2128. |
[44] | Gitcho MA , Baloh RH , Chakraverty S , Mayo K , Norton JB , Levitch D , Hatanpaa KJ , White CL III , Bigio EH , Caselli R , Baker M , Al-Lozi MT , Morris JC , Pestronk A , Rademakers R , Goate AM , Cairns NJ ((2008) ) TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 63: , 535–538. |
[45] | Kwiatkowski TJ Jr , Bosco DA , LeClerc AL , Tamrazian E , Vanderburg CR , Russ C , Davis A , Gilchrist J , Kasarskis EJ , Munsat T , Valdmanis P , Rouleau GA , Hosler BA , Cortelli P , de Jong PJ , Yoshinaga Y , Haines JL , Pericak-Vance MA , Yan J , Ticozzi N , Siddique T , Kenna-Yasek D , Sapp PC , Horvitz HR , Landers JE , Brown RH Jr ((2009) ) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205–1208. |
[46] | Yener GG , Rosen HJ , Papatriantafyllou J ((2010) ) Frontotemporal degeneration. Continuum (Minneap Minn) 16: , 191–211. |
[47] | Zarranz JJ , Digon A , Atares B , Rodríguez-Martínez AB , Arce A , Carrera N , Fernández-Manchola I , Fernández-Martínez M , Fernández-Maiztegui C , Forcadas I , Galdós L , Gómez-Esteban JC , Ibáñez A , Lezcano E , López de Munain A , Martí-Masso JF , Mendibe MM , Urtasun M , Uterga JM , Saracibar N , Velasco F , de Pancorbo MM ((2005) ) Phenotypic variability in familial prion diseases due to the D178N mutation. J Neurol Neurosurg Psychiatry 76: , 1491–1496. |
[48] | Ghoshal N , Cali I , Perrin RJ , Josephson SA , Sun N , Gambetti P , Morris JC ((2009) ) Codistribution of amyloid beta plaques and spongiform degeneration in familial Creutzfeldt-Jakob disease with the E200K-129M haplotype. Arch Neurol 66: , 1240–1246. |
[49] | Yoshida H , Terada S , Ishizu H , Ikeda K , Hayabara T , Ikeda K , Deguchi K , Touge T , Kitamoto T , Kuroda S ((2010) ) An autopsy case of Creutzfeldt-Jakob disease with a V180I mutation of the PrP gene and Alzheimer-type pathology. Neuropathology 30: , 159–164. |
[50] | Jayadev S , Nochlin D , Poorkaj P , Steinbart EJ , Mastrianni JA , Montine TJ , Ghetti B , Schellenberg GD , Bird TD , Leverenz JB ((2011) ) Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype. Ann Neurol 69: , 712–720. |
[51] | Heinemann U , Krasnianski A , Meissner B , Grasbon-Frodl EM , Kretzschmar HA , Zerr I ((2008) ) Novel PRNP mutation in a patient with a slow progressive dementia syndrome. Med Sci Monit 14: , CS41–CS43. |
[52] | Muñoz-Nieto M , Ramonet N , López-Gastón JI , Cuadrado-Corrales N , Calero O , Díaz-Hurtado M , Ipiens JR , Ramón y Cajal S , de Pedro-Cuesta J , Calero M ((2013) ) A novel mutation I215V in the PRNP gene associated with Creutzfeldt-Jakob and Alzheimer’s diseases in three patients with divergent clinical phenotypes. J Neurol 260: , 77–84. |
[53] | Ashford JW ((2004) ) APOE genotype effects on Alzheimer’s disease onset and epidemiology. J Mol Neurosci 23: , 157–165. |
[54] | Calero O , Bullido MJ , Clarimon J , Frank-García A , Martínez-Martín P , Lleo A , Rey MJ , Rábano A , Blesa R , Gómez-Isla T , Valdivieso F , de Pedro-Cuesta J , Ferrer I , Calero M ((2011) ) Genetic cross-interaction between APOE and PRNP in sporadic Alzheimer’s and Creutzfeldt-Jakob diseases. PLoS One 6: , e22090. |
[55] | Allen M , Kachadoorian M , Quicksall Z , Zou F , Chai HS , Younkin C , Crook JE , Pankratz VS , Carrasquillo MM , Krishnan S , Nguyen T , Ma L , Malphrus K , Lincoln S , Bisceglio G , Kolbert CP , Jen J , Mukherjee S , Kauwe JK , Crane PK , Haines JL , Mayeux R , Pericak-Vance MA , Farrer LA , Schellenberg GD , Parisi JE , Petersen RC , Graff-Radford NR , Dickson DW , Younkin SG , Ertekin-Taner N ((2014) ) Association of MAPT haplotypes with Alzheimer’s disease risk and MAPT brain gene expression levels. Alzheimers Res Ther 6: , 39. |
[56] | Desikan RS , Schork AJ , Wang Y , Witoelar A , Sharma M , McEvoy LK , Holland D , Brewer JB , Chen CH , Thompson WK , Harold D , Williams J , Owen MJ , O’Donovan MC , Pericak-Vance MA , Mayeux R , Haines JL , Farrer LA , Schellenberg GD , Heutink P , Singleton AB , Brice A , Wood NW , Hardy J , Martínez M , Choi SH , DeStefano A , Ikram MA , Bis JC , Smith A , Fitzpatrick AL , Launer L , van Duijn C , Seshadri S , Ulstein ID , Aarsland D , Fladby T , Djurovic S , Hyman BT , Snaedal J , Stefansson H , Stefansson K , Gasser T , Andreassen OA , Dale AM ((2015) ) Genetic overlap between Alzheimer’s disease and Parkinson’s disease at the MAPT locus. Mol Psychiatry 20: , 1588–1595. |
[57] | Tsuang D , Leverenz JB , López OL , Hamilton RL , Bennett DA , Schneider JA , Buchman AS , Larson EB , Crane PK , Kaye JA , Kramer P , Woltjer R , Trojanowski JQ , Weintraub D , Chen-Plotkin AS , Irwin DJ , Rick J , Schellenberg GD , Watson GS , Kukull W , Nelson PT , Jicha GA , Neltner JH , Galasko D , Masliah E , Quinn JF , Chung KA , Yearout D , Mata IF , Wan JY , Edwards KL , Montine TJ , Zabetian CP ((2013) ) APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 70: , 223–228. |
[58] | Kaarniranta K , Salminen A , Haapasalo A , Soininen H , Hiltunen M ((2011) ) Age-related macular degeneration (AMD): Alzheimer’s disease in the eye?. J Alzheimers Dis 24: , 615–631. |
[59] | Boot BP , Orr CF , Ahlskog JE , Ferman TJ , Roberts R , Pankratz VS , Dickson DW , Parisi J , Aakre JA , Geda YE , Knopman DS , Petersen RC , Boeve BF ((2013) ) Risk factors for dementia with Lewy bodies: A case-control study. Neurology 81: , 833–840. |
[60] | Borenstein AR , Wu Y , Bowen JD , McCormick WC , Uomoto J , McCurry SM , Schellenberg GD , Larson EB ((2014) ) Incidence rates of dementia, Alzheimer disease, and vascular dementia in the Japanese American population in Seattle, WA: The Kame Project. Alzheimer Dis Assoc Disord 28: , 23–29. |
[61] | Calero O , Bullido MJ , Clarimon J , Frank-García A , Martínez-Martín P , Lleo A , Rey MJ , Sastre I , Rábano A , de Pedro-Cuesta J , Ferrer I , Calero M ((2012) ) A common BACE1 polymorphism is a risk factor for sporadic Creutzfeldt-Jakob disease. PLoS One 7: , e43926. |
[62] | Llorca J , Rodríguez-Rodríguez E , Dierssen-Sotos T , Delgado-Rodríguez M , Berciano J , Combarros O ((2008) ) Meta-analysis of genetic variability in the beta-amyloid production, aggregation and degradation metabolic pathways and the risk of Alzheimer’s disease. Acta Neurol Scand 117: , 1–14. |
[63] | Weber BH , Charbel IP , Pauly D , Herrmann P , Grassmann F , Holz FG ((2014) ) The role of the complement system in age-related macular degeneration. Dtsch Arztebl Int 111: , 133–138. |
[64] | Yates JR , Sepp T , Matharu BK , Khan JC , Thurlby DA , Shahid H , Clayton DG , Hayward C , Morgan J , Wright AF , Armbrecht AM , Dhillon B , Deary IJ , Redmond E , Bird AC , Moore AT ((2007) ) Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med 357: , 553–561. |
[65] | Proitsi P , Lupton MK , Dudbridge F , Tsolaki M , Hamilton G , Daniilidou M , Pritchard M , Lord K , Martín BM , Johnson J , Craig D , Todd S , McGuinness B , Hollingworth P , Harold D , Kloszewska I , Soininen H , Mecocci P , Velas B , Gill M , Lawlor B , Rubinsztein DC , Brayne C , Passmore PA , Williams J , Lovestone S , Powell JF ((2012) ) Alzheimer’s disease and age-related macular degeneration have different genetic models for complement gene variation. Neurobiol Aging 33: , 1843 e9-17. |
[66] | Satake W , Nakabayashi Y , Mizuta I , Hirota Y , Ito C , Kubo M , Kawaguchi T , Tsunoda T , Watanabe M , Takeda A , Tomiyama H , Nakashima K , Hasegawa K , Obata F , Yoshikawa T , Kawakami H , Sakoda S , Yamamoto M , Hattori N , Murata M , Nakamura Y , Toda T ((2009) ) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41: , 1303–1307. |
[67] | Simón-Sánchez J , Schulte C , Bras JM , Sharma M , Gibbs JR , Berg D , Paisán-Ruiz C , Lichtner P , Scholz SW , Hernández DG , Kruger R , Federoff M , Klein C , Goate A , Perlmutter J , Bonin M , Nalls MA , Illig T , Gieger C , Houlden H , Steffens M , Okun MS , Racette BA , Cookson MR , Foote KD , Fernández HH , Traynor BJ , Schreiber S , Arepalli S , Zonozi R , Gwinn K , van der Brug M , López G , Chanock SJ , Schatzkin A , Park Y , Hollenbeck A , Gao J , Huang X , Wood NW , Lorenz D , Deuschl G , Chen H , Riess O , Hardy JA , Singleton AB , Gasser T ((2009) ) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41: , 1308–1312. |
[68] | Edwards TL , Scott WK , Almonte C , Burt A , Powell EH , Beecham GW , Wang L , Zuchner S , Konidari I , Wang G , Singer C , Nahab F , Scott B , Stajich JM , Pericak-Vance M , Haines J , Vance JM , Martín ER ((2010) ) Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 74: , 97–109. |
[69] | Liu G , Bao X , Jiang Y , Liao M , Jiang Q , Feng R , Zhang L , Ma G , Chen Z , Wang G , Wang R , Zhao B , Li K ((2015) ) Identifying the association between Alzheimer’s disease and Parkinson’s disease using genome-wide association studies and protein-protein interaction network. Mol Neurobiol 52: , 1629–1636. |
[70] | Bertram L , McQueen MB , Mullin K , Blacker D , Tanzi RE ((2007) ) Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat Genet 39: , 17–23. |
[71] | Xi Z , Zinman L , Grinberg Y , Moreno D , Sato C , Bilbao JM , Ghani M , Hernández I , Ruiz A , Boada M , Morón FJ , Lang AE , Marras C , Bruni A , Colao R , Maletta RG , Puccio G , Rainero I , Pinessi L , Galimberti D , Morrison KE , Moorby C , Stockton JD , Masellis M , Black SE , Hazrati LN , Liang Y , van Haersma de WJ , Fornazzari L , Villagra R , Rojas-García R , Clarimon J , Mayeux R , Robertson J , St George-Hyslop P , Rogaeva E ((2012) ) Investigation of c9orf72 in 4 neurodegenerative disorders. Arch Neurol 69: , 1583–1590. |
[72] | Wilson PW , Myers RH , Larson MG , Ordovás JM , Wolf PA , Schaefer EJ ((1994) ) Apolipoprotein E alleles, dyslipidemia, and coronary heart disease. The Framingham Offspring Study. JAMA 272: , 1666–1671. |
[73] | Zende PD , Bankar MP , Kamble PS , Momin AA ((2013) ) Apolipoprotein e gene polymorphism and its effect on plasma lipids in arteriosclerosis. J Clin Diagn Res 7: , 2149–2152. |
[74] | Calero O , Bullido MJ , Clarimon J , Hortiguela R , Frank-García A , Martínez-Martín P , Lleo A , Rey MJ , Sastre I , Rábano A , de Pedro-Cuesta J , Ferrer I , Calero M ((2012) ) Genetic variability of the gene cluster CALHM 1-3 in sporadic Creutzfeldt-Jakob disease. Prion 6: , 407–412. |
[75] | Frigerio R , Fujishiro H , Maraganore DM , Klos KJ , DelleDonne A , Heckman MG , Crook JE , Josephs KA , Parisi JE , Boeve BF , Dickson DW , Ahlskog JE ((2009) ) Comparison of risk factor profiles in incidental Lewy body disease and Parkinson disease. Arch Neurol 66: , 1114–1119. |
[76] | Bermejo-Pareja F ((2011) ) An old problem not yet resolved: The association of several neurodegenerative disorders. Neuroepidemiology 37: , 11–12. |
[77] | LaRoia H , Louis ED ((2011) ) Association between essential tremor and other neurodegenerative diseases: What is the epidemiological evidence?. Neuroepidemiology 37: , 1–10. |
[78] | Catalá-López F , Suárez-Pinilla M , Suárez-Pinilla P , Valderas JM , Gómez-Beneyto M , Martínez S , Balanza-Martínez V , Climent J , Valencia A , McGrath J , Crespo-Facorro B , Sánchez-Moreno J , Vieta E , Tabares-Seisdedos R ((2014) ) Inverse and direct cancer comorbidity in people with central nervous system disorders: A meta-analysis of cancer incidence in 577,013 participants of 50 observational studies. Psychother Psychosom 83: , 89–105. |
[79] | Benítez-Rivero S , Marín-Oyaga VA , García-Solís D , Huertas-Fernández I , García-Gómez FJ , Jesús S , Cáceres MT , Carrillo F , Ortiz AM , Carballo M , Mir P ((2013) ) Clinical features and 123I-FP-CIT SPECT imaging in vascular parkinsonism and Parkinson’s disease. J Neurol Neurosurg Psychiatry 84: , 122–129. |
[80] | de Pedro-Cuesta J , Ruiz-Tovar M , Ward H , Calero M , Smith A , Verduras CA , Pocchiari M , Turner ML , Forland F , Palm D , Will RG ((2012) ) Sensitivity to biases of case-control studies on medical procedures, particularly surgery and blood transfusion, and risk of Creutzfeldt-Jakob disease. Neuroepidemiology 39: , 1–18. |
[81] | Cruz M , Mahíllo-Fernández I , Rábano A , Siden A , Calero M , Laursen H , Molbak K , Almazán J , de Pedro-Cuesta J ((2013) ) Late-in-life surgery associated with Creutzfeldt-Jakob disease: A methodological outline for evidence-based guidance. Emerg Themes Epidemiol 10: , 5. |
[82] | Hardiman O ((2011) ) Amyotrophic lateral sclerosis and vascular risk: A metabolic conundrum. J Neurol Neurosurg Psychiatr 82: , 591. |
[83] | Cereda E , Barichella M , Pedrolli C , Klersy C , Cassani E , Caccialanza R , Pezzoli G ((2011) ) Diabetes and risk of Parkinson’s disease: A systematic review and meta-analysis. Diabetes Care 34: , 2614–2623. |
[84] | Driver JA , Smith A , Buring JE , Gaziano JM , Kurth T , Logroscino G ((2008) ) Prospective cohort study of type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 31: , 2003–2005. |
[85] | Palacios N , Gao X , McCullough ML , Jacobs EJ , Patel AV , Mayo T , Schwarzschild MA , Ascherio A ((2011) ) Obesity, diabetes, and risk of Parkinson’s disease. Mov Disord 26: , 2253–2259. |
[86] | Scigliano G , Musicco M , Soliveri P , Piccolo I , Ronchetti G , Girotti F ((2006) ) Reduced risk factors for vascular disorders in Parkinson disease patients: A case-control study. Stroke 37: , 1184–1188. |
[87] | Qiu C , Hu G , Kivipelto M , Laatikainen T , Antikainen R , Fratiglioni L , Jousilahti P , Tuomilehto J ((2011) ) Association of blood pressure and hypertension with the risk of Parkinson disease: The National FINRISK Study. Hypertension 57: , 1094–1100. |
[88] | Golimstok A , Campora N , Rojas JI , Fernández MC , Elizondo C , Soriano E , Cristiano E ((2014) ) Cardiovascular risk factors and frontotemporal dementia: A case-control study. Transl Neurodegener 3: , 13. |
[89] | Atkins ER , Bulsara MK , Panegyres PK ((2012) ) Cerebrovascular risk factors in early-onset dementia. J Neurol Neurosurg Psychiatry 83: , 666–667. |
[90] | Kivipelto M , Helkala EL , Laakso MP , Hanninen T , Hallikainen M , Alhainen K , Iivonen S , Mannermaa A , Tuomilehto J , Nissinen A , Soininen H ((2002) ) Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med 137: , 149–155. |
[91] | Schneider JA , Bennett DA ((2010) ) Where vascular meets neurodegenerative disease. Stroke 41: , S144–S146. |
[92] | Kling MA , Trojanowski JQ , Wolk DA , Lee VM , Arnold SE ((2013) ) Vascular disease and dementias: Paradigm shifts to drive research in new directions. Alzheimers Dement 9: , 76–92. |
[93] | Klein R , Peto T , Bird A , Vannewkirk MR ((2004) ) The epidemiology of age-related macular degeneration. Am J Ophthalmol 137: , 486–495. |
[94] | Buch H , Vinding T , la Cour M , Jensen GB , Prause JU , Nielsen NV ((2005) ) Risk factors for age-related maculopathy in a 14-year follow-up study: The Copenhagen City Eye Study. Acta Ophthalmol Scand 83: , 409–418. |
[95] | Tan JS , Mitchell P , Smith W , Wang JJ ((2007) ) Cardiovascular risk factors and the long-term incidence of age-related macular degeneration: The Blue Mountains Eye Study. Ophthalmology 114: , 1143–1150. |
[96] | Power MC , Weuve J , Gagne JJ , McQueen MB , Viswanathan A , Blacker D ((2011) ) The association between blood pressure and incident Alzheimer disease: A systematic review and meta-analysis. Epidemiology 22: , 646–659. |
[97] | Kravitz E , Schmeidler J , Beeri MS ((2012) ) Cognitive decline and dementia in the oldest-old. Rambam Maimonides Med J 3: , e0026. |
[98] | Chui HC , Zheng L , Reed BR , Vinters HV , Mack WJ ((2012) ) Vascular risk factors and Alzheimer’s disease: Are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review. Alzheimers Res Ther 4: , 1. |
[99] | Beeri MS , Rapp M , Silverman JM , Schmeidler J , Grossman HT , Fallon JT , Purohit DP , Perl DP , Siddiqui A , Lesser G , Rosendorff C , Haroutunian V ((2006) ) Coronary artery disease is associated with Alzheimer disease neuropathology in APOE4 carriers. Neurology 66: , 1399–1404. |
[100] | Mahíllo-Fernández I , de Pedro-Cuesta J , Bleda MJ , Cruz M , Molbak K , Laursen H , Falkenhorst G , Martínez-Martín P , Siden A ((2008) ) Surgery and risk of sporadic Creutzfeldt-Jakob disease in Denmark and Sweden: Registry-based case-control studies. Neuroepidemiology 31: , 229–240. |
[101] | Roher AE , Maarouf CL , Daugs ID , Kokjohn TA , Hunter JM , Sabbagh MN , Beach TG ((2011) ) Neuropathology and amyloid-beta spectrum in a bapineuzumab immunotherapy recipient. J Alzheimers Dis 24: , 315–325. |
[102] | Yarchoan M , Xie SX , Kling MA , Toledo JB , Wolk DA , Lee EB , van Deerlin V , Lee VM , Trojanowski JQ , Arnold SE ((2012) ) Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain 135: , 3749–3756. |
[103] | Koperek O , Kovacs GG , Ritchie D , Ironside JW , Budka H , Wick G ((2002) ) Disease-associated prion protein in vessel walls. Am J Pathol 161: , 1979–1984. |
[104] | Luchsinger JA , Mayeux R ((2004) ) Cardiovascular risk factors and Alzheimer’s disease. Curr Atheroscler Rep 6: , 261–266. |
[105] | Kaufman RJ ((2002) ) Orchestrating the unfolded protein response in health and disease. J Clin Invest 110: , 1389–1398. |
[106] | Soto C ((2003) ) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4: , 49–60. |
[107] | Chiti F , Dobson CM ((2006) ) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75: , 333–366. |
[108] | Solomon A , Mangialasche F , Richard E , Andrieu S , Bennett DA , Breteler M , Fratiglioni L , Hooshmand B , Khachaturian AS , Schneider LS , Skoog I , Kivipelto M ((2014) ) Advances in the prevention of Alzheimer’s disease and dementia. J Intern Med 275: , 229–250. |
[109] | Blanc-Lapierre A , Bouvier G , Garrigou A , Canal-Raffin M , Raherison C , Brochard P , Baldi I ((2012) ) Effets chroniques des pesticides sur le système nerveux central: état des connaissances épidémiologiques. Rev Epidemiol Sante Publique 60: , 389–400. |
[110] | Pezzoli G , Cereda E ((2013) ) Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 80: , 2035–2041. |
[111] | Tanner CM , Goldman SM , Ross GW , Grate SJ ((2014) ) The disease intersection of susceptibility and exposure: Chemical exposures and neurodegenerative disease risk. Alzheimers Dement 10: , S213–S225. |
[112] | de Pedro-Cuesta J , Mahíllo-Fernández I , Rábano A , Calero M , Cruz M , Siden A , Laursen H , Falkenhorst G , Molbak K ((2011) ) Nosocomial transmission of sporadic Creutzfeldt-Jakob disease: Results from a risk-based assessment of surgical interventions. J Neurol Neurosurg Psychiatry 82: , 204–212. |
[113] | Yamada M , Furukawa Y , Hirohata M ((2003) ) Amyotrophic lateral sclerosis: Frequent complications by cervical spondylosis. J Orthop Sci 8: , 878–881. |
[114] | Longstreth WT , McGuire V , Koepsell TD , Wang Y , van Belle G ((1998) ) Risk of amyotrophic lateral sclerosis and history of physical activity: A population-based case-control study. Arch Neurol 55: , 201–206. |
[115] | Beghi E , Logroscino G , Chio A , Hardiman O , Millul A , Mitchell D , Swingler R , Traynor BJ ((2010) ) Amyotrophic lateral sclerosis, physical exercise, trauma and sports: Results of a population-based pilot case-control study. Amyotroph Lateral Scler 11: , 289–292. |
[116] | Chio A , Benzi G , Dossena M , Mutani R , Mora G ((2005) ) Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain 128: , 472–476. |
[117] | Armon C , Kurland LT , Daube JR , O’Brien PC ((1991) ) Epidemiologic correlates of sporadic amyotrophic lateral sclerosis. Neurology 41: , 1077–1084. |
[118] | Horner RD , Kamins KG , Feussner JR , Grambow SC , Hoff-Lindquist J , Harati Y , Mitsumoto H , Pascuzzi R , Spencer PS , Tim R , Howard D , Smith TC , Ryan MA , Coffman CJ , Kasarskis EJ ((2003) ) Occurrence of amyotrophic lateral sclerosis among Gulf War veterans. Neurology 61: , 742–749. |
[119] | Weisskopf MG , O’Reilly EJ , McCullough ML , Calle EE , Thun MJ , Cudkowicz M , Ascherio A ((2005) ) Prospective study of military service and mortality from ALS. Neurology 64: , 32–37. |
[120] | Chen H , Richard M , Sandler DP , Umbach DM , Kamel F ((2007) ) Head injury and amyotrophic lateral sclerosis. Am J Epidemiol 166: , 810–816. |
[121] | Turner MR , Abisgold J , Yeates DG , Talbot K , Goldacre MJ ((2010) ) Head and other physical trauma requiring hospitalisation is not a significant risk factor in the development of ALS. J Neurol Sci 288: , 45–48. |
[122] | Pupillo E , Messina P , Logroscino G , Zoccolella S , Chio A , Calvo A , Corbo M , Lunetta C , Micheli A , Millul A , Vitelli E , Beghi E ((2012) ) Trauma and amyotrophic lateral sclerosis: A case-control study from a population-based registry. Eur J Neurol 19: , 1509–1517. |
[123] | Wirdefeldt K , Adami HO , Cole P , Trichopoulos D , Mandel J ((2011) ) Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur J Epidemiol 26: (Suppl 1), S1–58. |
[124] | O’Meara ES , Kukull WA , Schellenberg GD , Bowen JD , McCormick WC , Teri L , Pfanschmidt M , Thompson JD , Larson EB ((1997) ) Alzheimer’s disease and history of blood transfusion by apolipoprotein-E genotype. Neuroepidemiology 16: , 86–93. |
[125] | Wermuth L , Pakkenberg H , Jeune B ((2002) ) High age-adjusted prevalence of Parkinson’s disease among Inuits in Greenland. Neurology 58: , 1422–1425. |
[126] | Wermuth L , Bech S , Petersen MS , Joensen P , Weihe P , Grandjean P ((2008) ) Prevalence and incidence of Parkinson’s disease in The Faroe Islands. Acta Neurol Scand 118: , 126–131. |
[127] | Fu X , Korenaga T , Fu L , Xing Y , Guo Z , Matsushita T , Hosokawa M , Naiki H , Baba S , Kawata Y , Ikeda S , Ishihara T , Mori M , Higuchi K ((2004) ) Induction of AApoAII amyloidosis by various heterogeneous amyloid fibrils. FEBS Lett 563: , 179–184. |
[128] | Lundmark K , Westermark GT , Olsen A , Westermark P ((2005) ) Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc Natl Acad Sci U S A 102: , 6098–6102. |
[129] | Prusiner SB , Woerman AL , Mordes DA , Watts JC , Rampersaud R , Berry DB , Patel S , Oehler A , Lowe JK , Kravitz SN , Geschwind DH , Glidden DV , Halliday GM , Middleton LT , Gentleman SM , Grinberg LT , Giles K ((2015) ) Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 112: , E5308–E5317. |
[130] | Brown MK , Naidoo N ((2012) ) The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol 3: , 263. |
[131] | Barrett-Connor E ((2013) ) Menopause, atherosclerosis, and coronary artery disease. Curr Opin Pharmacol 13: , 186–191. |
[132] | Rocca WA , Grossardt BR , Shuster LT ((2014) ) Oophorectomy, estrogen, and dementia: A 2014 update. Mol Cell Endocrinol 389: , 7–12. |
[133] | Colditz GA , Willett WC , Stampfer MJ , Rosner B , Speizer FE , Hennekens CH ((1987) ) Menopause and the risk of coronary heart disease in women. N Engl J Med 316: , 1105–1110. |
[134] | Rocca WA , Bower JH , Maraganore DM , Ahlskog JE , Grossardt BR , de Andrade M , Melton LJ III , ((2007) ) Increased risk of cognitive impairment or dementia in women who underwent oophorectomy before menopause. Neurology 69: , 1074–1083. |
[135] | Phung TK , Waltoft BL , Laursen TM , Settnes A , Kessing LV , Mortensen PB , Waldemar G ((2010) ) Hysterectomy, oophorectomy and risk of dementia: A nationwide historical cohort study. Dement Geriatr Cogn Disord 30: , 43–50. |
[136] | Bove R , Secor E , Chibnik LB , Barnes LL , Schneider JA , Bennett DA , De Jager PL ((2014) ) Age at surgical menopause influences cognitive decline and Alzheimer pathology in older women. Neurology 82: , 222–229. |
[137] | Jacoby VL , Grady D , Wactawski-Wende J , Manson JE , Allison MA , Kuppermann M , Sarto GE , Robbins J , Phillips L , Martin LW , O’Sullivan MJ , Jackson R , Rodabough RJ , Stefanick ML ((2011) ) Oophorectomy vs ovarian conservation with hysterectomy: Cardiovascular disease, hip fracture, and cancer in the Women’s Health Initiative Observational Study. Arch Intern Med 171: , 760–768. |
[138] | Lehmann DJ , Hogervorst E , Warden DR , Smith AD , Butler HT , Ragoussis J ((2004) ) The androgen receptor CAG repeat and serum testosterone in the risk of Alzheimer’s disease in men. J Neurol Neurosurg Psychiatry 75: , 163–164. |
[139] | Jayaraman A , Pike CJ ((2014) ) Alzheimer’s disease and type 2 diabetes: Multiple mechanisms contribute to interactions. Curr Diab Rep 14: , 476. |
[140] | Moffat SD , Zonderman AB , Metter EJ , Kawas C , Blackman MR , Harman SM , Resnick SM ((2004) ) Free testosterone and risk for Alzheimer disease in older men. Neurology 62: , 188–193. |
[141] | Rosario ER , Chang L , Stanczyk FZ , Pike CJ ((2004) ) Age-related testosterone depletion and the development of Alzheimer disease. JAMA 292: , 1431–1432. |
[142] | Rosario ER , Chang L , Head EH , Stanczyk FZ , Pike CJ ((2011) ) Brain levels of sex steroid hormones in men and women during normal aging and in Alzheimer’s disease. Neurobiol Aging 32: , 604–613. |
[143] | Hougaku H , Fleg JL , Najjar SS , Lakatta EG , Harman SM , Blackman MR , Metter EJ ((2006) ) Relationship between androgenic hormones and arterial stiffness, based on longitudinal hormone measurements. Am J Physiol Endocrinol Metab 290: , E234–E242. |
[144] | Goncharov NP , Katsya GV , Chagina NA , Gooren LJ ((2008) ) Three definitions of metabolic syndrome applied to a sample of young obese men and their relation with plasma testosterone. Aging Male 11: , 118–122. |
[145] | Kapoor D , Goodwin E , Channer KS , Jones TH ((2006) ) Testosterone replacement therapy improves insulin resistance, glycaemic control, visceral adiposity and hypercholesterolaemia in hypogonadal men with type 2 diabetes. Eur J Endocrinol 154: , 899–906. |
[146] | Lage MJ , Barber BL , Markus RA ((2007) ) Association between androgen-deprivation therapy and incidence of diabetes among males with prostate cancer. Urology 70: , 1104–1108. |
[147] | Skoog I ((1998) ) Status of risk factors for vascular dementia. Neuroepidemiology 17: , 2–9. |
[148] | Prins ND , van der Flier WM , Brashear HR , Knol DL , van de Pol LA , Barkhof F , Scheltens P ((2013) ) Predictors of progression from mild cognitive impairment to dementia in the placebo-arm of a clinical trial population. J Alzheimers Dis 36: , 79–85. |
[149] | Sasaki M , Kodama C , Hidaka S , Yamashita F , Kinoshita T , Nemoto K , Ikejima C , Asada T ((2009) ) Prevalence of four subtypes of mild cognitive impairment and APOE in a Japanese community. Int J Geriatr Psychiatry 24: , 1119–1126. |
[150] | Benedictus MR , Goos JD , Binnewijzend MA , Muller M , Barkhof F , Scheltens P , Prins ND , van der Flier WM ((2013) ) Specific risk factors for microbleeds and white matter hyperintensities in Alzheimer’s disease. Neurobiol Aging 34: , 2488–2494. |
[151] | Stengard JH , Zerba KE , Pekkanen J , Ehnholm C , Nissinen A , Sing CF ((1995) ) Apolipoprotein E polymorphism predicts death from coronary heart disease in a longitudinal study of elderly Finnish men. Circulation 91: , 265–269. |
[152] | Chaudhary R , Likidlilid A , Peerapatdit T , Tresukosol D , Srisuma S , Ratanamaneechat S , Sriratanasathavorn C ((2012) ) Apolipoprotein E gene polymorphism: Effects on plasma lipids and risk of type 2 diabetes and coronary artery disease. Cardiovasc Diabetol 11: , 36. |
[153] | Ward H , Mitrou PN , Bowman R , Luben R , Wareham NJ , Khaw KT , Bingham S ((2009) ) APOE genotype, lipids, and coronary heart disease risk: A prospective population study. Arch Intern Med 169: , 1424–1429. |
[154] | Engelman CD , Koscik RL , Jonaitis EM , Okonkwo OC , Hermann BP , La Rue A , Sager MA ((2013) ) Interaction between two cholesterol metabolism genes influences memory: Findings from the Wisconsin Registry for Alzheimer’s Prevention. J Alzheimers Dis 36: , 749–757. |
[155] | Irie F , Fitzpatrick AL , López OL , Kuller LH , Peila R , Newman AB , Launer LJ ((2008) ) Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: The Cardiovascular Health Study Cognition Study. Arch Neurol 65: , 89–93. |
[156] | Peila R , Rodríguez BL , Launer LJ ((2002) ) Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 51: , 1256–1262. |
[157] | Hofman A , Ott A , Breteler MM , Bots ML , Slooter AJ , van Harskamp F , van Duijn CN , van Broeckhoven C , Grobbee DE ((1997) ) Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet 349: , 151–154. |
[158] | Launer LJ , Ross GW , Petrovitch H , Masaki K , Foley D , White LR , Havlik RJ ((2000) ) Midlife blood pressure and dementia: The Honolulu-Asia aging study. Neurobiol Aging 21: , 49–55. |
[159] | Lecci F , Junker B , Kuller LH , López OL , Becker JT ((2015) ) Empirically derived trajectories to dementia over 15 years of follow-up identified by using mixed membership models. Am J Epidemiol 182: , 366–374. |
[160] | Li L , Wang Y , Yan J , Chen Y , Zhou R , Yi X , Shi Q , Zhou H ((2012) ) Clinical predictors of cognitive decline in patients with mild cognitive impairment: The Chongqing aging study. J Neurol 259: , 1303–1311. |
[161] | Clerici F , Caracciolo B , Cova I , Fusari IS , Maggiore L , Galimberti D , Scarpini E , Mariani C , Fratiglioni L ((2012) ) Does vascular burden contribute to the progression of mild cognitive impairment to dementia?. Dement Geriatr Cogn Disord 34: , 235–243. |
[162] | Li J , Wang YJ , Zhang M , Xu ZQ , Gao CY , Fang CQ , Yan JC , Zhou HD ((2011) ) Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology 76: , 1485–1491. |
[163] | Tanskanen M , Kiuru-Enari S , Tienari P , Polvikoski T , Verkkoniemi A , Rastas S , Sulkava R , Paetau A ((2006) ) Senile systemic amyloidosis, cerebral amyloid angiopathy, and dementia in a very old Finnish population. Amyloid 13: , 164–169. |
[164] | Tanskanen M , Peuralinna T , Polvikoski T , Notkola IL , Sulkava R , Hardy J , Singleton A , Kiuru-Enari S , Paetau A , Tienari PJ , Myllykangas L ((2008) ) Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: A population-based autopsy study. Ann Med 40: , 232–239. |
[165] | Tanskanen M , Makela M , Myllykangas L , Notkola IL , Polvikoski T , Sulkava R , Kalimo H , Paetau A ((2012) ) Prevalence and severity of cerebral amyloid angiopathy: A population-based study on very elderly Finns (Vantaa 85+). Neuropathol Appl Neurobiol 38: , 329–336. |
[166] | Qiu C ((2012) ) Preventing Alzheimer’s disease by targeting vascular risk factors: Hope and gap. J Alzheimers Dis 32: , 721–731. |
[167] | Soto C , Estrada LD ((2008) ) Protein misfolding and neurodegeneration. Arch Neurol 65: , 184–189. |
[168] | Prusiner SB ((2013) ) Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 47: , 601–623. |
[169] | Ladogana A , Puopolo M , Croes EA , Budka H , Jarius C , Collins S , Klug GM , Sutcliffe T , Giulivi A , Alperovitch A , Delasnerie-Laupretre N , Brandel JP , Poser S , Kretzschmar H , Rietveld I , Mitrova E , de Pedro-Cuesta J , Martínez-Martín P , Glatzel M , Aguzzi A , Knight R , Ward H , Pocchiari M , van Duijn CM , Will RG , Zerr I ((2005) ) Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology 64: , 1586–1591. |
[170] | Doria A , Yang Y , Malecki M , Scotti S , Dreyfus J , O’Keeffe C , Orban T , Warram JH , Krolewski AS ((1999) ) Phenotypic characteristics of early-onset autosomal-dominant type 2 diabetes unlinked to known maturity-onset diabetes of the young (MODY) genes. Diabetes Care 22: , 253–261. |
[171] | Revesz T , Holton JL , Lashley T , Plant G , Frangione B , Rostagno A , Ghiso J ((2009) ) Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 118: , 115–130. |
[172] | Abid A , Ismail M , Mehdi SQ , Khaliq S ((2006) ) Identification of novel mutations in the SEMA4A gene associated with retinal degenerative diseases. J Med Genet 43: , 378–381. |
[173] | Ton VK , Mukherjee M , Judge DP ((2014) ) Transthyretin cardiac amyloidosis: Pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol 8: , 39–44. |
[174] | Eisen JA ((2000) ) Assessing evolutionary relationships among microbes from whole-genome analysis. Curr Opin Microbiol 3: , 475–480. |
[175] | Relman DA , Falkow S ((2001) ) The meaning and impact of the human genome sequence for microbiology. Trends Microbiol 9: , 206–208. |
[176] | Foucault M ((1972) ) Part I. Introduction. In The archaeology of knowledge and discourse on language Books Pantheon, Eds. Tavistock Publication Limited, New York p. 6. |
Figures and Tables
Fig.1
Drivers proposed in this study.

Fig.2
Reported age-at-exposure-related patterns. (top) (left) Reported Parkinson’s disease (PD) incidence and prevalence of levodopa users (left -Iceland- triennial moving averages) [19], and main whooping cough notifications in Iceland; and (right) risk of PD for lowest age at first whooping cough epidemic [19]. (bottom) (a) age-susceptibility function for variant Creutzfeldt-Jakob disease (vCJD) in the UK [15] and (b) risks after adjustment for dietary exposure to bovine material in the UK [17]; (a and b) risk of sporadic Creutzfeldt-Jakob disease (sCJD) from age at first hospital discharge associated with a registered main surgical procedure at a lag of ≥20 years, using an anatomical and etiologic classification [20]; and, (c and d) age at first treatment with pituitary growth hormone with the Hartree-modified Wilhelmi method and accidentally transmitted Creutzfeldt-Jakob disease (iCJD) [16].
![Reported age-at-exposure-related patterns. (top) (left) Reported Parkinson’s disease (PD) incidence and prevalence of levodopa users (left -Iceland- triennial moving averages) [19], and main whooping cough notifications in Iceland; and (right) risk of PD for lowest age at first whooping cough epidemic [19]. (bottom) (a) age-susceptibility function for variant Creutzfeldt-Jakob disease (vCJD) in the UK [15] and (b) risks after adjustment for dietary exposure to bovine material in the UK [17]; (a and b) risk of sporadic Creutzfeldt-Jakob disease (sCJD) from age at first hospital discharge associated with a registered main surgical procedure at a lag of ≥20 years, using an anatomical and etiologic classification [20]; and, (c and d) age at first treatment with pituitary growth hormone with the Hartree-modified Wilhelmi method and accidentally transmitted Creutzfeldt-Jakob disease (iCJD) [16].](https://ip.ios.semcs.net:443/media/jad/2016/51-4/jad-51-4-jad150884/jad-51-jad150884-g002.jpg)
Fig.3
Normalized age-specific incidence, incidence per million, and survival for selected neurodegenerative disorders. Modified from de Pedro-Cuesta et al. [4]. Normalized age-specific incidence, age-adjusted incidence, and median clinical disease duration of different sporadic protein-associated neurodegenerative disorders (sCNDDs), obtained either from reported data (amyotrophic lateral sclerosis (ALS), personally modified by Fang F, sporadic Creutzfeldt-Jakob disease (sCJD)) or from registries [rapid progressive neurodegenerative dementia (sRPNDd) notified as suspected sCJD in Spain for 1995–2011, obtained from the Spanish CJD surveillance registry]. References for Fig. 3 [29–39]. (a) 85–89 years is equivalent to 85 years and older for sCJD, ALS, Lewy body disease (LBD), Parkinson’s disease (PD), and sRPNDd; (b) 90–94 years is equivalent to 90 years and older for age-related macular degeneration (AMD) and frontotemporal dementia (FTD).
![Normalized age-specific incidence, incidence per million, and survival for selected neurodegenerative disorders. Modified from de Pedro-Cuesta et al. [4]. Normalized age-specific incidence, age-adjusted incidence, and median clinical disease duration of different sporadic protein-associated neurodegenerative disorders (sCNDDs), obtained either from reported data (amyotrophic lateral sclerosis (ALS), personally modified by Fang F, sporadic Creutzfeldt-Jakob disease (sCJD)) or from registries [rapid progressive neurodegenerative dementia (sRPNDd) notified as suspected sCJD in Spain for 1995–2011, obtained from the Spanish CJD surveillance registry]. References for Fig. 3 [29–39]. (a) 85–89 years is equivalent to 85 years and older for sCJD, ALS, Lewy body disease (LBD), Parkinson’s disease (PD), and sRPNDd; (b) 90–94 years is equivalent to 90 years and older for age-related macular degeneration (AMD) and frontotemporal dementia (FTD).](https://ip.ios.semcs.net:443/media/jad/2016/51-4/jad-51-4-jad150884/jad-51-jad150884-g003.jpg)
Fig.4
Outline of an epidemiologic model for sCNDDs on the basis of drivers. Here we assume that there is an age-at-onset-related continuum for various late-age neurodegenerative disorders, and that the unfolded protein response explains, at least in part, the reported associations for diverse conformational neurodegenerative, vascular degenerative, and metabolic (T2DM) disorders. Arrows represent different potential types of associations, i.e., inverse (dashed line), bidirectional, etc.
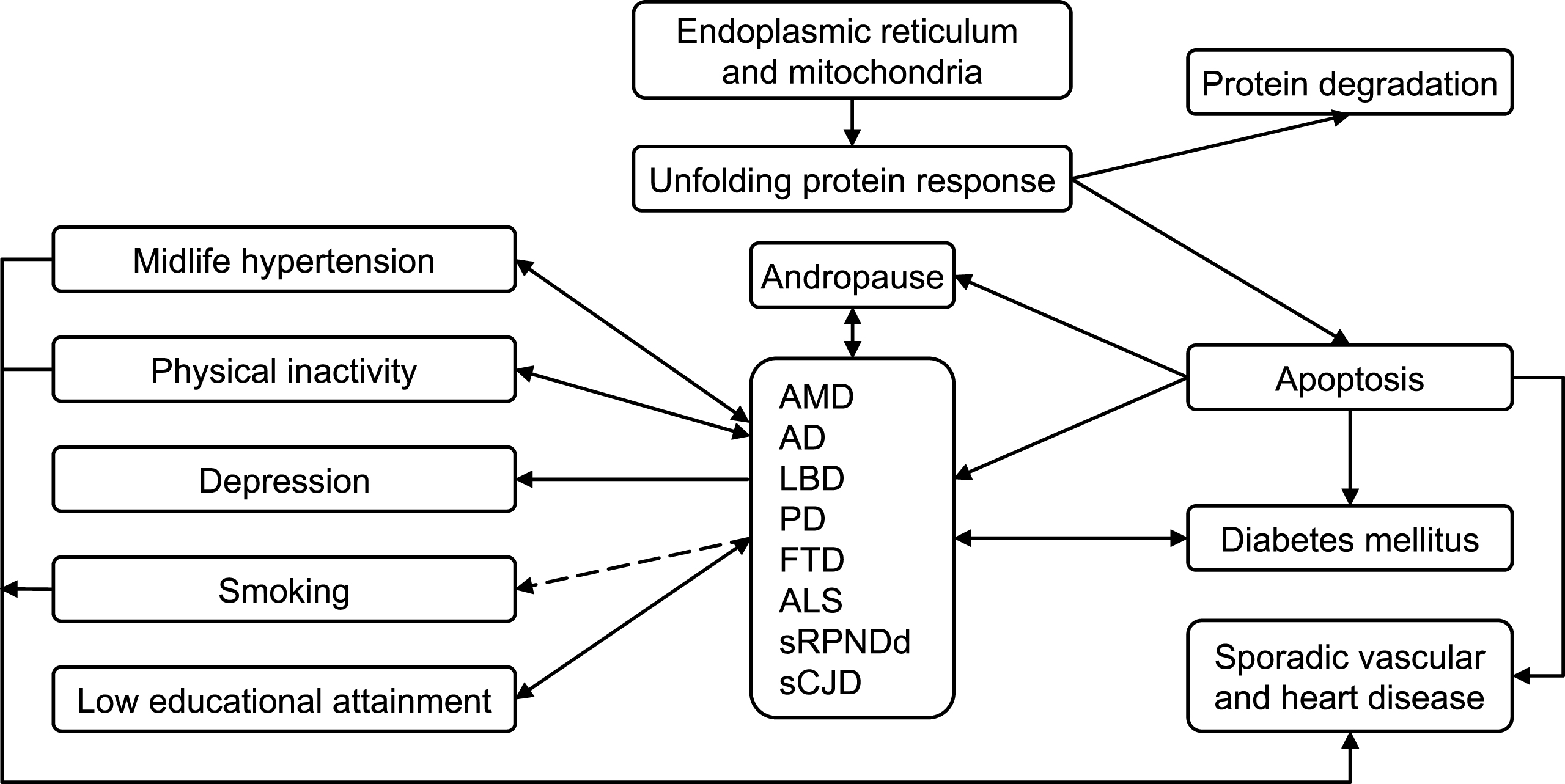
Table 1
Main biochemical, epidemiologic and inter-individual transmissibility features of sCNDDs and other sporadic human amyloid disorders
| Entity or neuropathologically-related entities | Main protein deposit | Reported outbreaks | Annual incidence per million person-years or lesion prevalence at death* | M/F | Evidence of individual transmission | ||
| Sporadic | Genetic | ||||||
| Creutzfeldt-Jakob Disease (CJD) | APrP | vCJD | UK, Ireland, France, Spain, others | 1 | 0.1 | 1.1/1 | Yes |
| Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia | Ubiquitin, MAPT, SOD, TDP-43/FUS | ALS | Skaraborg county, Sweden [6]. US human growth hormone treated cohort [7]. | 10 | 1 | 1.5–2 / 1 | No |
| Parkinson’s disease and Lewy body disease | α synuclein | – | – | 100 | 10 | 1.5–2.5 / 1 | Yes** |
| Alzheimer’s disease | Amyloid-β, Tau | – | – | 1000 | 100 | 0.92 / 1 | |
| Late age-related macular degeneration | EFEMP1 wild-type [8] | – | 1000 | Unknown | 0.95 / 1 | No | |
| T2 diabetes mellitus | Langerhans Islet peptid | – | – | – | – | – | No |
| Sporadic cerebral amyloid angiopathy | Amyloid-β wild-type | – | – | – | – | – | |
| Senile systemic amyloidosis aortic aneurism | Transthyretin wild-type | – | – | – | 25% ≥85y* | – | No |
| Medin arteriopathy [9] | Lactadherin | – | – | – | – | – | – |
*Postmortem studies only. **From patients suffering from Parkinson’s disease to fetal grafted cells.


