The GLP-1 Receptor Agonist Liraglutide Improves Memory Function and Increases Hippocampal CA1 Neuronal Numbers in a Senescence-Accelerated Mouse Model of Alzheimer’s Disease
Abstract
Recent studies indicate that glucagon-like peptide 1 (GLP-1) receptor agonists, currently used in the management of type 2 diabetes, exhibit neurotrophic and neuroprotective effects in amyloid-β (Aβ) toxicity models of Alzheimer’s disease (AD). We investigated the potential pro-cognitive and neuroprotective effects of the once-daily GLP-1 receptor agonist liraglutide in senescence-accelerated mouse prone 8 (SAMP8) mice, a model of age-related sporadic AD not dominated by amyloid plaques. Six-month-old SAMP8 mice received liraglutide (100 or 500 μg/kg/day, s.c.) or vehicle once daily for 4 months. Vehicle-dosed age-matched 50% back-crossed as well as untreated young (4-month-old) SAMP8 mice were used as control groups for normal memory function. Vehicle-dosed 10-month-old SAMP8 mice showed significant learning and memory retention deficits in an active-avoidance T-maze, as compared to both control groups. Also, 10-month-old SAMP8 mice displayed no immunohistological signatures of amyloid-β plaques or hyperphosphorylated tau, indicating the onset of cognitive deficits prior to deposition of amyloid plaques and neurofibrillary tangles in this AD model. Liraglutide significantly increased memory retention and total hippocampal CA1 pyramidal neuron numbers in SAMP8 mice, as compared to age-matched vehicle-dosed SAMP8 mice. In conclusion, liraglutide delayed or partially halted the progressive decline in memory function associated with hippocampal neuronal loss in a mouse model of pathological aging with characteristics of neurobehavioral and neuropathological impairments observed in early-stage sporadic AD.
INTRODUCTION
Alzheimer’s disease (AD) is a common neurodegenerative disease being the leading cause of dementia-associated morbidity and mortality in the elderly population, affecting more than 25 millionpeople worldwide [1]. AD is characterized by a slow but progressive cognitive decline with a defining neuropathology of abnormally high densities of amyloid-β (Aβ) senile plaques and neurofibrillary tangles, as well as brain atrophy and extensive neuronal loss [2, 3].
Type 2 diabetes (T2DM) is a risk factor for AD and the two diseases share several biochemical features of metabolic dysfunction, in particular pertaining to insufficient central and peripheral insulin function. Accordingly, molecular signatures of impaired insulin function has been consistent findings in AD which pertains to brain insulin deficiency, central insulin resistance, as well as elevated fasting plasma insulin levels and impaired peripheral insulin responses [4–8]. Furthermore, a recent proof-of-concept clinical trial in AD has demonstrated memory-improving effects of increased central insulin exposure obtained by intranasal insulin administration [9], which further emphasizes the emerging importance of counteracting deficient insulin-associated neurotransmission in the treatment of AD neuropathology and cognitive deficits.
A recent approach is to target the activity of central insulin-like hormone receptors whose signaling pathways to some degree converges with those of insulin [10], however, without posing the inherent hypoglycemia risk typically associated with insulin therapy. Specifically, preclinical studies on long-acting glucagon-like peptide 1 (GLP-1) receptor agonists have shown encouraging results in the context of disease-modifying therapeutic potential in AD. GLP-1 receptors are widely expressed in the brain [11, 12], and there is increasing evidence that blood-borne GLP-1 receptor agonists can reach the brain [13, 14], which provides a fundamental basis for direct central action of this drug class. To date, the preclinical evidence for potential therapeutic benefits of GLP-1 receptor agonists in AD is largely derived from studies on rodent models overexpressing rare, but highly penetrant, genetic mutations recognized as high-risk factors for development of early-onset familial dementia, including amyloid-β protein precursor (AβPP) and presenilin-1 (PS1). Using a transgenic AβPP/PS1 mouse model of AD to characterize the effects of long-term GLP-1 receptor agonist administration, Hölscher and colleagues have reported lowered levels of important markers associated with AD neuropathology, including amyloid plaque load, astrogliosis, microglial activation, and central insulin resistance [15–17]. Notably, these effects of GLP-1 receptor agonists are associated with improved memory function in AD models with characteristics of amyloidosis[17–19].
With regard to the reported findings of beneficial effects of GLP-1 receptor agonist treatment in transgenic AβPP/PS1 mice, it should be emphasized that early-onset familial AD accounts for less than 5% of total AD cases. AD is predominantly a sporadic (i.e., non-familial with no known genetic determinants) and late-onset disease with exponentially increasing prevalence starting at the age of 65 years [20]. Consequently, there is a need to determine effects of GLP-1 receptor agonists in other AD models which closer mimic the polygenetic and multifactorial nature of age-related sporadic AD. Importantly, it also remains to be assessed whether pro-cognitive effects of GLP-1 receptor agonist treatment also translate into effects on hippocampal neuronal numbers.
An inbred senescence-accelerated prone 8 (SAMP8) mouse model of AD-like dementia has been extensively phenotyped and shows prominent age-dependent hippocampus-driven memory deficits and neuropathology in the absence of significant amyloid plaque load, thus being considered as a model of early-stage sporadic AD [21, 22]. Hence, the non-transgenic SAMP8 mouse model may allow for the assessment of potential pro-cognitive drug effects in AD before irreversible late-stage plaque-associated toxicity occurs. To this end, we therefore employed SAMP8 mice to characterize the impact of long-term liraglutide treatment on plaque-independent memory deficits, as assessed in an active avoidance T-maze and novel object recognition task. Also, a stereological analysis was applied to address potential effects of liraglutide treatment on total hippocampal CA1 pyramidal neuron numbers.
MATERIAL AND METHODS
Animals
SAMP8 mice (6-month-old, 28.0 ± 0.6 g, n = 51) were employed. To control for age-dependent memory decline in SAMP8 mice and minimize strain differences, age-matched 50% back-crossed SAMP8 mice (6-month-old, 35.8 ± 0.6 g, n = 11) were used. 50% back-crossed SAMP8 mice is a cross between SAMP8 and CD-1 mice and display normal memory function comparable to young (4-month-old) SAMP8 mice [23]. Therefore, 4-month-old SAMP8 mice (26.6 ± 0.6 g, n = 11) were also included and served as additional controls for normal memory performance in the behavioral tests indicated below. All animals included in study were males and bred at St. Louis University Division of Geriatrics, St. Louis, MO. The mice were group-housed (n = 5 per cage) in cages equipped with solid floors and a layer of bedding and nest material. All mice received a unique identity number (ear tag) and were randomized to individual experimental groups (n = 10-11 per group) according to body weight, as GLP-1 receptor agonists, including liraglutide, show weight-reducing properties with transient food intake suppression [24, 25]. Throughout the study period, the mice had ad libitum access to chow diet (Richland #5001, LabDiet, St. Louis, MO) and tap water. The animal room environment was controlled (temperature 22 ± 2°C; relative humidity 50 ± 10% ; light/dark cycle 12 h light, 12 h dark, lights on from 6 AM to 6 PM).
Drug treatment
Mice were dosed subcutaneously (s.c.) once daily with vehicle (0.1% BSA in PBS) or liraglutide (100 or 500 μg/kg/day; equivalent to 26 or 133 nmol/kg/day) in the morning between 7 AM and 9 AM for a total duration of 4 months. The liraglutide doses used in the present study are within the dose range reported efficacious in other rodent models of neurodegenerative diseases [26–28]. A total of three control groups were employed, i.e., vehicle-dosed SAMP8 mice (6-month-old at dosing start), vehicle-dosed 50% backcrossed SAMP8 mice (6-month-old at dosing start), and non-dosed young (4-month-old) SAMP8 mice. A dose-escalation scheme was implemented to reduce expected initial effects of liraglutide treatment, as transient GLP-1 receptor induced discomfort in rodents, including taste aversion and pica behavior, is typically only observed within the first 2-3 days of treatment [29]. Therefore, the liraglutide dose was increased through daily increments, starting from 25 μg/kg/day (day 0), until reaching the dose of 100 μg/kg/day on day 2 (25–50–100 μg/kg/day) or 500 μg/kg/day on day 6 (25–50–100–150–200–300–500 μg/kg/day), respectively, with the specified target dose thereafter being maintained throughout the remainder of the 4 months of treatment.
Body weight, food intake, and plasma sampling
Body weight and food intake was determined and recorded on a weekly basis. Food intake was determined by net food pellet consumption and expressed as cage averages. Blood samples were taken from the liraglutide-treated groups after 10 weeks of dosing, obtained in the morning prior to dosing and 4 hours post-dosing (n = 5 mice per time point), respectively. For blood collections, 50 μL of tail blood was obtained from tail vein with a 25 μL capillary glass tube in an EDTA-coated Eppendorf tube, centrifuged at 2,000 g for 15 min at 4°C, plasma was collected and samples were snap-frozen in liquid nitrogen before storage at −80°C.
Behavioral testing
Measurement of the effects of long-term liraglutide treatment on learning and memory function was performed in an active avoidance T-maze and novel object recognition (NOR) task, respectively (n = 10-11 mice per experimental group). Experiments were conducted between 7.30 AM and 11.00 AM. The T-maze consisted of a black plastic alley with a start box at one end and two goal boxes at the other. The start box was separated from the alley by a plastic guillotine door that prevented movement down the alley until raised at the onset of training. An electrifiable floor of stainless steel rods ran throughout the maze to deliver a foot-shock using a scrambled grid floor shocker (Model E13-08, Coulbourn Instruments, Whitehall, PA). Mice were not permitted to explore the maze prior to training. A block of training trials began when a mouse was placed in the start box. The guillotine door was raised and a cue buzzer sounded simultaneously (doorbell type, at 55 dB); 5 s later a mild aversive foot-shock was applied with an intensity of 0.35 mA. The arm of the maze entered on the first trial was designated “incorrect” and the mild foot-shock was continued until the mouse entered the other goal box, which in all subsequent trials was designated “correct” for the particular mouse. At the end of each trial, the mouse was returned to its home cage until the next trial. Mice were trained until they made one active avoidance. The intertrial interval was 30–35 s. The number of trials to make one active avoidance was the measure of acquisition. Retention was tested one week later by continuing training until the mice achieved the criterion of making five active avoidances in six consecutive trials. The number of trials needed to reach this criterion was the measure of retention [30, 31].
In the novel object recognition task, the mice were habituated to an empty apparatus, a 58×66×11-cm white plastic box, for 5 min a day for 3 consecutive days prior to entry of objects. On the first day of training, the mouse was placed in the testing apparatus with two identical objects (A and B), both with dimensions of 7×6.3×5.1 cm. 24 h later the animal was reintroduced to the arena but this time one of the original objects was removed and a new object (C) with dimensions of 8.2×3.8×7.4 cm added. Mice were placed in the testing apparatus for 5 min and the time each mouse spent sniffing or touching the new object was recorded. Total time spent exploring each of the two objects was recorded. The discrimination index was defined as the amount of time exploring the familiar object or the novel object over the total time spent exploring both objects multiplied by 100, and was used to measure recognition memory.
Immunohistochemical staining
Five days after the final behavioral test, animals were terminated using CO2/O2 anesthesia followed by decapitation. Brains were rapidly removed, freshly weighed to assess possible gross brain atrophy, and then immersion fixed in 4% formaldehyde. The two cerebral hemispheres were divided and the left or right hemisphere was selected randomly before dehydration and infiltration overnight in paraffin. The hemispheres were embedded in blocks of paraffin and sectioned in the horizontal plane in 40 μm thick sections for estimation of total hippocampal CA1 pyramidal neuron numbers. Using systematic uniform random sampling, every 8th section in the series was collected directly on preheated slides primed with a chromalum-gelatin solution. Sections were dried for 3 days at 37°C and stained in an optimized Giemsa stain allowing clear differentiation of cell types in thick sections.
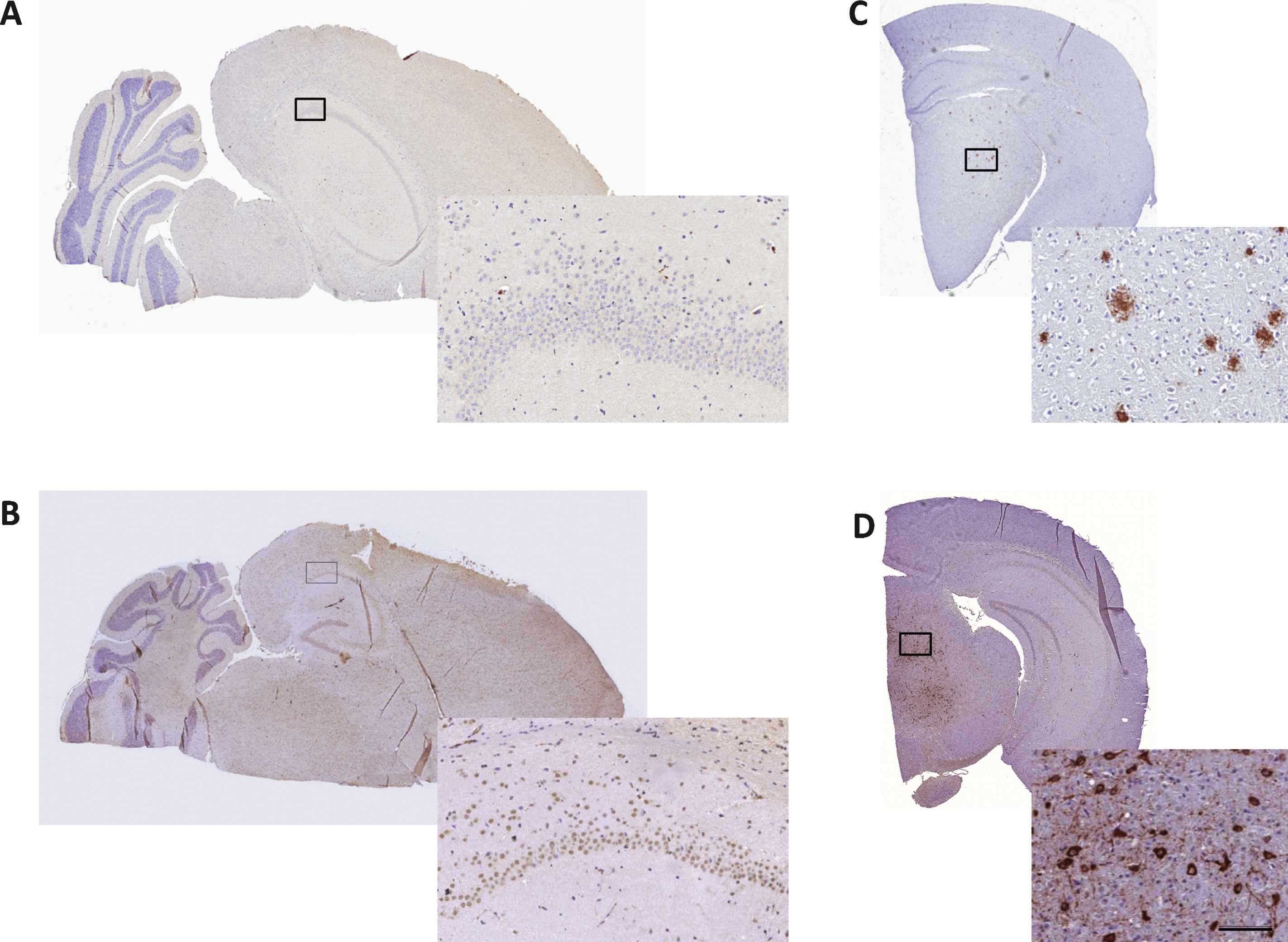
To confirm the absence of Aβ immunoreactivity in the brains of 10-month-old SAMP8 mice [32], 5- μm thick sections were additionally collected at each horizontal sampling level also sampled for quantification of neuronal numbers. Immunohistochemical analysis of Aβ and hyperphosphorylated tau was performed in a pilot study on 10-month-old control SAMP8 mice (n = 3). The sections were deparaffinized followed by antigen retrieval for 15 min in a TEG buffer (10 mM, pH 9, 90°C), endogenous peroxidase activity was blocked for 10 min in 1% H2O2+KPBS+0.3% Triton-X. Blocking of unspecific binding was obtained with 5% swine serum in KPBS+0.3% Triton-X+1% BSA for 20 min followed by incubation with a primary antibody against Aβ (1 : 250, cat. no. 10323, IBL, Minneapolis, MN) or phosphorylated tau (1 : 300, cat. no. MN1060, Thermoscientific, Rockford, IL) for 30 min. After a rinse in buffer, sections were incubated for 30 min with a horseradish peroxidase-labelled polymer conjugated with anti-mouse antibody (Envision, Dako, Glostrup, Denmark) and rinsed in buffer. Immunostaining was visualized with 3,3′-diaminobenzidine (10 min) as chromagen (Dako, Glostrup, Denmark), and sections were finally counterstained with Mayer’s hematoxylin (Dako, Glostrup, Denmark). Following mounting on cover glass with Pertex, slides were allowed to dry overnight and scanned on a digital slide scanner (Aperio ScanScope AT, Leica Biosystems, Ballerup, Denmark). Using this staining procedure, the two antibodies were also applied to brain sections from transgenic mouse models of Aβ plaque (hAβPP/PS1-delta9) and neurofibrillary tangle (hTau P301L) pathology, and found highly specific (Fig. 6).
Stereological quantification of hippocampal CA1 pyramidal neuron numbers
Neuronal loss in the hippocampal CA1 subregion has been identified as memory-associated neuropathological marker in both humans and animals, and atrophy of CA1 pyramidal neurons is a key pathological change in AD [33, 34]. We therefore estimated the total number of Giemsa-stained CA1 pyramidal neurons by the use of the unbiased optical fractionator method [35, 36]. The boundaries of the CA1 subdivision were carefully defined using a mouse brain atlas [37] and by the clear morphological indication of conspicuous smaller and more densely organized CA1 pyramidal neurons, as compared to the relatively larger and less packed neurons characterizing the CA2-CA3 subdivisions [36] (see Fig. 4). Neurons with a clear nuclear Giemsa stain were included in the counts. Glia cells were readily identified by their smaller size and were excluded from the counts. The entire CA1 pyramidal layer was delineated using the Visiopharm newCAST system (Visiopharm, Hoersholm, Denmark), and the total number of CA1 pyramidal neurons was determined using the optical disector at regular predetermined x, y positions within the subdivision. The disector height was set at 10 μm and protected by upper and lower guard zones of 10 μm, the x-y step size and the disector counting frame were adjusted allowing for an average of 150–200 neurons (ΣN) to be identified in around 100 disectors. The actual counting was performed using the optical disector (a three-dimensional probe generated with the aid of a microscope fitted with an oil immersion objective with a high numerical aperture (NA = 1.30) to observe thin focal planes in relatively thick sections). The neuron counts were performed online on a computer screen with the z-axis measured using a digital Heidenhain microcator with a precision of 0.5 μm. Neurons were counted when they came into focus within the counting frame provided they were either found entirely within the counting frame or hit at least one of the inclusion lines but none of the exclusion lines. The total number of CA1 pyramidal neurons (N) and corresponding volume reference in the unilateral sample was calculated according to previously published methods [35, 36].
Data analysis
All data were fed into Excel 2003 spread sheets and subsequently subjected to relevant statistical analyses using GraphPad Prism 5.0. Results are presented as mean ± standard error of the mean (S.E.M.), unless otherwise stated. Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Dunnet’s post-hoc analysis where applicable. Comparisons were first made between vehicle-dosed 10-month-old SAMP8 mice versus vehicle-dosed age-matched 50% back-crossed and non-dosed 4-month-old SAMP8 mice (validation of memory deficits in 10-month-old SAMP8 control mice), then between 10-month-old SAMP8 mice treated with either vehicle or liraglutide (treatment effect). A p-value less than 0.05 was considered statistically significant.
RESULTS
Body weight, food intake, and plasma levels of liraglutide
Irrespective of treatment, body weight and food intake was kept stable throughout the dosing period (data not shown). Hence, liraglutide administration did not induce body weight and food intake reduction in SAMP8 mice. This is most likely ascribed to a normal body weight regulation in SAMP8 mice, as obese mice show greater sensitivity to the such common metabolic effects of GLP-1 receptor agonists [25, 38]. Sustained systemic exposure of liraglutide was confirmed in tail vein plasma samples obtained after 10 weeks of dosing. Dose-dependent significant elevations of plasma liraglutide concentrations were observed 4 h after dosing, corresponding to three- (lowest dose) and six-fold (highest dose) elevations, as compared to the corresponding pre-dosing level on the same day (data not shown).
Liraglutide improves memory retention in an active-avoidance T-maze task
Analysis of acquisition of foot-shock avoidance in the T-maze indicated a significantly increased number of training trials to first avoidance in vehicle-dosed 10-month-old SAMP8 mice (11.9 ± 0.6 trials), as compared to both 50% back-crossed (7.7 ± 0.7 trials, p < 0.001) and 4-month-old SAMP8 mice (7.0 ± 0.3 trials, p < 0.001, Fig. 1A). Liraglutide treatment did not change the number of training trials in 10-month-old SAMP8 mice (100 μg/kg/day, 9.6 ± 0.8 trials; 500 μg/kg/day, 10.4 ± 0.8 trials; p > 0.05), as compared to vehicle-dosed 10-month-old SAMP8 mice (Fig. 1A). In the subsequent memory retention test, vehicle-dosed 10-month-old SAMP8 mice showed a significantly increased number of trials to fulfill the test criterion (14.5 ± 1.1 trials, p < 0.001), as compared to both 50% back-crossed (6.3 ± 0.3 trials, p < 0.001) and 4-month-old SAMP8 control mice (6.8 ± 0.2 trials, p < 0.001, Fig. 1B). Compared to vehicle-dosed 10-month-old SAMP8 mice, liraglutide-treated age-matched SAMP8 mice exhibited improved memory performance, as indicated by significantly fewer trials to complete the retention test (100 μg/kg/day, 9.0 ± 0.6 trials, p < 0.001; 500 μg/kg/day, 9.1 ± 0.5 trials, p < 0.001, Fig. 1B). A statistical significant difference was attained between retention trial numbers of 10-month-old SAMP8 mice treated with the highest, but not lowest, dose of liraglutide versus 50% back-crossed (p < 0.05) SAMP8 mice.
Liraglutide shows no effect in a novel object recognition task
As illustrated in Fig. 2, vehicle-dosed 10-month-old SAMP8 mice failed to discriminate between novel and familiar objects in the NOR test (discrimination index, 0.52 ± 0.02), as compared to young (4-month-old, discrimination index, 0.63 ± 0.03, p < 0.05), but not vehicle-dosed 10-month-old 50% back-crossed (discrimination index, 0.57 ± 0.04, p = 0.22), SAMP8 control mice. Liraglutide-treated 10-month-old SAMP8 mice showed a dose-dependent trend (discrimination index, 0.55 ± 0.05 (100 μg/kg/day); 0.63 ± 0.03 (500 μg/kg/day), p = 0.081) in improved discrimination index, as compared to vehicle-dosed age-matched SAMP8 mice.
SAMP8 mice exhibit reduced brain weight
The experimental groups differed on the basis of their fresh total brain weight (see Fig. 3). Accordingly, non-dosed young 4-month-old SAMP8 mice exhibited a lower brain weight (−9.9 ± 1.1% , p < 0.001) as compared to vehicle-dosed 10 month-old 50% back-crossed SAMP8 mice. Also vehicle-dosed 10 month-old SAMP8 controls showed a similar degree of reduced total brain weight (−11.9 ± 1.2% , p < 0.001 versus 50% back-crossed SAMP8 mice). The lowered brain weight in SAMP8 mice was unaffected by long-term liraglutide treatment (100 μg/kg/day, −11.1 ± 1.2% ; 500 μg/kg/day, −8.5 ± 1.2% , p < 0.001 for both groups versus 50% back-crossed SAMP8 mice).
Liraglutide-treated SAMP8 mice have preserved hippocampal CA1 pyramidal neuron numbers
Figure 4 shows a representative Giemsa-stained horizontal section of the hippocampus. Total CA1 pyramidal neuronal numbers were estimated by means of stereological counting principles. In the three control groups, mean ( ± S.E.M.) total CA1 pyramidal neuron numbers were: 192,004 ± 11,696 (50% back-crossed 10-month-old SAMP8 mice); 186,631 ± 6,834 (4-month-old SAMP8 mice), and 179,094 ± 7,116 (10-month-old vehicle-dosed SAMP8 mice) (see Fig. 5A). Although no significant quantitative difference in total CA1 pyramidal neuron numbers was attained between the three individual control groups (one-way ANOVA, overall p = 0.5570), vehicle-dosed 10-month-old SAMP8 thus showed lowest total CA1 pyramidal neuron numbers. Notably, liraglutide-treated (100 μg/kg/day) SAMP8 mice exhibited significantly higher (14.3 ± 0.3% ) total CA1 pyramidal neuron number (204,744 ± 5,442, p < 0.01), as compared to age-matched vehicle-dosed SAMP8 controls (p < 0.01), see Fig. 5A. The highest dose of liraglutide (500 μg/kg/day) had no significant effect on total CA1 pyramidal neuronal levels in SAMP8 mice (186,573 ± 4,667, p > 0.05).
Total hippocampal CA1 volume (mm3) was similar across all experimental groups (one-way ANOVA, overall p = 0.5853, Fig. 5B). In correspondence to total CA1 pyramidal neuron counts, SAMP8 mice treated with the low, but not high, dose of liraglutide therefore showed a significantly higher CA1 pyramidal neuron density (631,611 ± 15,719 neurons/mm3), as compared to vehicle-dosed SAMP8 mice (551,563 ± 15,432 neurons/mm3, p < 0.01, Fig. 5C).
A scrutinized analysis of brain sections from 10-month-old SAMP8 mice indicated no presence of aggregated Aβ (plaques) or hyperphosphorylated tau (neurofibrillary tangles) in any brain regions assessed, including the hippocampus and cortex (Fig. 6).
DISCUSSION
This study aimed to evaluate liraglutide-induced effects on memory function and hippocampal neuron numbers in a mouse model of age-related sporadic AD. The effects of long-term liraglutide treatment were therefore characterized in SAMP8 mice, which has been extensively phenotyped with respect to features of early-stage pathogenesis associated with age-related AD, including cognitive deficits [21, 22].
10-month-old vehicle-dosed SAMP8 mice exhibited significant and robust memory deficits in an aversive active avoidance T-maze paradigm, as compared to both 4-month-old SAMP8 mice as well as age-matched 50% back-crossed vehicle-dosed SAMP8 controls, thus being in close agreement with previous reports on the age-related decline of cognitive function in this model [31, 39]. In the NOR test, vehicle-dosed SAMP8 mice also showed impaired memory performance, however, only compared to 4-month-old SAMP8 mice. Notably, liraglutide treatment resulted in a marked improvement of memory retention in SAMP8 mice as assessed in the T-maze, although this was not paralleled by similar improvements in the preceding acquisition trial. It may thus be inferred from the T-maze data that liraglutide treatment improved spatial long-term memory rather than learning in SAMP8 mice.
The mnemonic effects of GLP-1 agonist treatment in SAMP8 mice is in agreement with previous studies in AβPP/PS1 mice and intrahippocampal Aβ toxicity rat models of AD [19, 40, 41]. In contrast to studies in AβPP/PS1 mice [16, 17, 41], liraglutide treatment did not result in significant effects on novel object recognition in SAMP8 mice, although the highest dose tended to increase the object discrimination index. While T-maze and NOR settings both involve hippocampus-dependent declarative memory tasks [42, 43], it should, however, be taken into account that the two tests engage different cognitive domains. Hence, the present T-maze task is an aversive learning-task based on working and reference memory [44], whereas NOR performance is a measure of novelty preference in a non-reinforced memory test setting [45].
Our stereological analyses indicated that SAMP8 mice receiving daily liraglutide treatment for 4 months exhibited significantly higher total neuron numbers in the hippocampal CA1 pyramidal region, as compared to age-matched vehicle-dosed SAMP8 mice. The robust liraglutide-induced improvement in spatial memory function and increased hippocampal neuron numbers in SAMP8 mice may be causally linked. In support of this view, lesion studies have indicated the critical relevance of intact hippocampal function in T-maze based spatial learning and memory performance [42, 46]. Also, the age-related decline in memory function in SAMP8 mice is correlated with the progression of pathological changes in the CA1 region of the hippocampus, which includes neuronal loss [47], gliosis [48], and increased apoptotic activity [49]. Correspondingly, several laboratories have reported GLP-1 receptor-dependent neurotrophic, proliferative, anti-inflammatory, and anti-apoptotic effects of liraglutide and other long-acting GLP-1 receptor agonists [50, 51].
GLP-1 receptors are expressed, albeit at low levels, in the rat [12], non-human primate [52], and human hippocampus [11], which could possibly indirectly argue for direct hippocampal effects of liraglutide treatment in SAMP8 mice. However, a recent study addressing central liraglutide uptake upon acute subcutaneous administration of fluorescently-labeled liraglutide showed that liraglutide was detected within the mouse and rat circumventricular organs as well as in subregions of the hypothalamus, but not in other brain regions including the hippocampus [14]. It is therefore tempting to speculate whether the mnemonic and hippocampal neuron-sparing effects of liraglutide in SAMP8 mice might be independent of hippocampal GLP-1 receptor function. Fluorescent tags are, however, not optimal for detecting low nanomolar levels of peptides, and it is therefore possible that liraglutide could be present at very low levels in regions not exhibiting a significant fluorescently-labeled liraglutide signal. Also, given that a long-term liraglutide dosing regimen was applied in the present study and SAMP8 mice show features of blood-brain barrier disruption [53, 54], it is relevant to consider the possibility that liraglutide could exhibit a wider central distribution in SAMP8 mice, perhaps dispersed via the cerebroventricular circulation as recently suggested for endogenous GLP-1 [55]. If applicable to the present findings, this may potentially also suggest more generalized central effects of liraglutide in SAMP8 mice. For example, memory deficits in SAMP8 mice have been associated with impaired cerebral glucose homeostasis and hypometabolic activity [54, 56], and as GLP-1 receptor agonists can influence central glucose uptake and metabolism [18, 57], changes in cerebrometabolic activity could potentially have contributed to the liraglutide-induced improvement of memory function in SAMP8 mice.
The present study on SAMP8 mice indicated dose-dependent plasma exposure of liraglutide, although the lowest dose of liraglutide appeared consistently more efficacious on improving memory function and increasing hippocampal CA1 neuronal levels in SAMP8 mice. Previous pharmacological studies in SAMP8 mice have shown that after the optimum memory enhancing drug dose is achieved, higher doses tend to be less effective in this model [58]. Whether this disparity in dose-responsiveness reflects, e.g., saturable kinetics of cerebral liraglutide uptake or GLP-1 receptor desensitization upon chronic administration of higher doses must await further studies.
A number of in vivo studies in AβPP/PS1 mice have associated the memory stimulatory effects of GLP-1 receptor agonists with reduced hippocampal and cortical soluble Aβ oligomer levels and amyloid plaque load [17, 40, 41], and it is suggested that GLP-1 receptor stimulation increases neuronal survival, e.g., by lowering AβPP processing [50, 59]. We did not find immunohistochemical evidence of Aβ aggregates in a pilot study on 10-month-old SAMP8 mice. This is in close accordance with the observation that amyloid plaques in SAMP8 mice are not observed before the age of 16–20 months [32], and indicates that the observed memory impairments in SAMP8 mice are not a consequence of amyloid aggregation per se. It should be noted, however, that elevated levels of AβPP and soluble amyloid oligomers, as well as presence of amyloid granules, is reported in the cortex and hippocampus of SAMP8 mice from the age of 6–8 month [32, 60]. Furthermore, antibody and antisense-mediated blockade of Aβ function improves spatial memory in 10–12-month-old SAMP mice [31, 32, 61], demonstrating the functional relevance of AβPP processing in SAMP8 mice at earlier ages being independent on following Aβ aggregation. Hence, we cannot exclude the possibility that liraglutide may have interfered with mechanisms associated with early-stage Aβ pathology in SAMP8 mice, although this needs to be addressed specifically in future studies. Furthermore, the average wet brain weight was approximately 10% higher in 10-month-old 50% back-crossed SAMP8 mice (a cross between SAMP8 and CD-1 mice) as compared to both age-matched and 4-month-old SAMP8 mice. It should be noted, however, that we did not compare to the average brain weight of age-matched CD-1 mice. It is therefore unclear whether the observed differences in brain weight are attributed to strain differences, or gross brain atrophy being manifest prior to the onset of memory deficits in SAMP8 mice.
In conclusion, we here report that liraglutide treatment delayed or, alternatively, partially halted the age-associated progressive decline in spatial memory function associated with hippocampal neuronal loss in a mouse AD model of early-stage pathological aging not dominated by amyloid plaques. The present data thus supports the notion that liraglutide-induced stimulation of GLP-1 receptor function may be a viable target in AD therapy, currently also being pursued in a proof-of-concept study in mild cognitively impaired AD patients [62].
ACKNOWLEDGMENTS
The authors would like to acknowledge Sarah Kampfeldt for skillful technical assistance. The study was funded by Novo Nordisk A/S.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/14-3090r2).
REFERENCES
1 | Qiu C, Kivipelto M, von Strauss E(2009) Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward interventionDialogues Clin Neurosci11: 111128 |
2 | Querfurth HW, LaFerla FM(2010) Alzheimer’s diseaseN Engl J Med362: 329344 |
3 | Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT(2011) Neuropathological alterations in Alzheimer disease.aCold Spring Harb Perspect Med1: 006189 |
4 | Rönnemaa E, Zethelius B, Sundelöf J, Sundström J, Degerman-Gunnarsson M, Berne C, Lannfelt L, Kilander L(2008) Impaired insulin secretion increases the risk of Alzheimer diseaseNeurology71: 10651071 |
5 | Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C(2010) Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signallingNeurobiol Aging31: 224243 |
6 | Schrijvers EMC, Witteman JCM, Sijbrands EJG, Hofman A, Koudstaal PJ, Breteler MMB(2010) Insulin metabolism and the risk of Alzheimer disease: The Rotterdam StudyNeurology75: 19821987 |
7 | Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM(2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes?J Alzheimers Dis7: 6380 |
8 | Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE(2012) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive declineJ Clin Invest122: 13161338 |
9 | Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B(2012) Intral insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trialJAMA69: 2938nasa |
10 | Hölscher C, Li L(2010) New roles for insulin-like hormones in neuronal signalling and protection: New hopes for novel treatments of Alzheimer’s disease?Neurobiol Aging31: 14951502 |
11 | Alvarez E, Martínez MD, Roncero I, Chowen JA, García-Cuartero B, Gispert JD, Sanz C, Váazquez P, Maldonado A, de Cáceres J, Desco M, Pozo MA, Bláazquez E(2005) The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstemJ Neurochem92: 798806 |
12 | Merchenthaler I, Lane M, Shughrue P(1999) Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous systemJ Comp Neurol403: 261280 |
13 | Orskov C, Poulsen SS, Møller M, Holst JJ(1996) Glucagon-like peptide I receptors in the subfornical organ and the area postrema are accessible to circulating glucagon-like peptide IDiabetes45: 832835 |
14 | Secher A, Jelsing J, Baquero AF, Hecksher-Sørensen J, Cowley MA, Dalbøge LS, Hansen G, Grove KL, Pyke C, Raun K, Schäffer L, Tang-Christensen M, Verma S, Witgen BM, Vrang N, Bjerre Knudsen L(2014) The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight lossJ Clin Invest124: 44734488 |
15 | Long-Smith CM, Manning S, McClean PL, Coakley MF, O’Halloran DJ, Holscher C, O’Neill C(2013) The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-β plaque and glial pathology in a mouse model of Alzheimer’s diseaseNeuromolecular Med15: 102114 |
16 | McClean PL, Hölscher C(2014) Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s diseaseNeuropharmacology86C: 241258 |
17 | McClean PL, Hölscher C(2014) Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease.Pt ANeuropharmacology76: 5767 |
18 | Bomba M, Ciavardelli D, Silvestri E, Canzoniero LMT, Lattanzio R, Chiappini P, Piantelli M, Di Ilio C, Consoli A, Sensi SL(2013) Exenatide promotes cognitive enhancement and positive brain metabolic changes in PS1-KI mice but has no effects in 3xTg-AD animals.eCell Death Dis4: 612 |
19 | Han WN, Hölscher C, Yuan L, Yang W, Wang XH, Wu MN, Qi JS(2013) Liraglutide protects against amyloid-β protein-induced impairment of spatial learning and memory in ratsNeurobiol Aging34: 576588 |
20 | Reitz C, Mayeux R(2014) Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkersBiochem Pharmacol88: 640651 |
21 | Morley JE, Farr SA, Kumar VB, Armbrecht HJ(2012) The SAMP8 mouse: a model to develop therapeutic interventions for Alzheimer’s diseaseCurr Pharm Des18: 11231130 |
22 | Pallas M, Camins A, Smith MA, Perry G, Lee HG, Casadesus G(2008) From aging to Alzheimer’s disease: Unveiling “the switch” with the senescence-accelerated mouse model (SAMP8)J Alzheimers Dis15: 615624 |
23 | Armbrecht HJ, Siddiqui AM, Green M, Farr SA, Kumar VB, Banks WA, Patrick P, Shah GN, Morley JE(2014) SAMP8 mice have altered hippocampal gene expression in long term potentiation, phosphatidylinositol signaling, and endocytosis pathwaysNeurobiol Aging35: 159168 |
24 | Sisley S, Smith K, Sandoval DA, Seeley RJ(2014) Differences in acute anorectic effects of long-acting GLP-1 receptor agonists in ratsPeptides58: 16 |
25 | Tatarkiewicz K, Sablan EJ, Polizzi CJ, Villescaz C, Parkes DG(2014) Long-term metabolic benefits of exenatide in mice are mediated solely via the known glucagon-like peptide 1 receptorAm J Physiol Regul Integr Comp Physiol306: R490R498 |
26 | Briyal S, Shah S, Gulati A(2014) Neuroprotective and anti-apoptotic effects of liraglutide in the rat brain following focal cerebral ischemiaNeuroscience281C: 269281 |
27 | DellaValle B, Hempel C, Johansen FF, Kurtzhals JA(2014) GLP-1 improves neuropathology after murine cold lesion brain traumaAnn Clin Transl Neurol1: 721732 |
28 | Xiong H, Zheng C, Wang J, Song J, Zhao G, Shen H, Deng Y(2013) The neuroprotection of liraglutide on Alzheimer-like learning and memory impairment by modulating the hyperphosphorylation of tau and neurofilament proteins and insulin signaling pathways in miceJ Alzheimers Dis37: 623635 |
29 | Kanoski SE, Rupprecht LE, Fortin SM, De Jonghe BC, Hayes MR(2012) The role of nausea in food intake and body weight suppression by peripheral GLP-1 receptor agonists, exendin-4 and liraglutideNeuropharmacology62: 19161927 |
30 | Flood JF, Farr SA, Uezu K, Morley JE(1998) Age-related changes in septal serotonergic, GABAergic and glutamatergic facilitation of retention in SAMP8 miceMech Ageing Dev105: 173188 |
31 | Morley JE, Farr SA, Flood JF(2002) Antibody to amyloid beta protein alleviates impaired acquisition, retention, and memory processing in SAMP8 miceNeurobiol Learn Mem78: 125138 |
32 | Morley JE, Kumar VB, Bernardo AE, Farr SA, Uezu K, Tumosa N, Flood JF(2000) Beta-amyloid precursor polypeptide in SAMP8 mice affects learning and memoryPeptides21: 17611767 |
33 | Llorens-Martín M, Blazquez-Llorca L, Benavides-Piccione R, Rabano A, Hernandez F, Avila J, DeFelipe J(2014) Selective alterations of neurons and circuits related to early memory loss in Alzheimer’s disease.eFront Neuroanat8: 38 |
34 | Zola-Morgan S, Squire LR(1993) Neuroanatomy of memoryAnnu Rev Neurosci16: 547563 |
35 | Slomianka L, West MJ(2005) Estimators of the precision of stereological estimates: An example based on the CA1 pyramidal cell layer of ratsNeuroscience136: 757767 |
36 | West MJ, Slomianka L, Gundersen HJ(1991) Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionatorAnat Rec231: 482497 |
37 | Paxinos G, Franklin K(2004) The Mouse Brain in Stereotaxic CoordinatesLondonAcademic Press |
38 | Sisley S, Gutierrez-Aguilar R, Scott M, D’Alessio DA, Sandoval DA, Seeley RJ(2014) Neuronal GLP1R mediates liraglutide’s anorectic but not glucose-lowering effectJ Clin Invest124: 24562463 |
39 | Flood JF, Morley JE(1992) Early onset of age-related impairment of aversive and appetitive learning in the SAM-P/8 mouse.B52-BJ Gerontol47: 59 |
40 | Bomfim TR1, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, Silverman MA, Kazi H, Melo HM, McClean PL, Holscher C, Arnold SE, Talbot K, Klein WL, Munoz DP, Ferreira ST, De Felice FG(2012) An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomersJ Clin Invest122: 13391353 |
41 | McClean PL, Parthsarathy V, Faivre E, Hölscher C(2011) The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s diseaseJ Neurosci31: 65876594 |
42 | Farr SA, Banks WA, La Scola ME, Flood1 JF, Morley JE(2000) Permanent and temporary inactivation of the hippocampus impairs T-maze footshock avoidance acquisition and retentionBrain Res872: 242249 |
43 | Hammond RS, Tull LE, Stackman RW(2004) On the delay-dependent involvement of the hippocampus in object recognition memoryNeurobiol Learn Mem82: 2634 |
44 | Dudchenko PA(2004) An overview of the tasks used to test working memory in rodentsNeurosci Biobehav Rev28: 699709 |
45 | Ennaceur A(2010) One-trial object recognition in rats and mice: Methodological and theoretical issuesBehav Brain Res215: 244254 |
46 | Gerlai R(1998) A new continuous alternation task in T-maze detects hippocampal dysfunction in mice. A strain comon and lesion studyBehav Brain Res95: 91101paris |
47 | Li G, Cheng H, Zhang X, Shang X, Xie H, Zhang X, Yu J, Han J(2013) Hippocampal neuron loss is correlated with cognitive deficits in SAMP8 miceNeurol Sci34: 963969 |
48 | Han S, Rudd JA, Hu ZY, Zhang L, Yew DT, Fang M(2010) Analysis of neuronal nitric oxide synthase expression and increasing astrogliosis in the brain of senescence-accelerated-prone 8 miceInt J Neurosci120: 602608 |
49 | Ma F, Wang H, Chen B, Wang F, Xu H(2011) Metallothionein 3 attenuated the apoptosis of neurons in the CA1 region of the hippocampus in the senescence-accelerated mouse/PRONE8 (SAMP8)Arq Neuropsiquiatr69: 105111 |
50 | Li Y, Tweedie D, Mattson MP, Holloway HW, Greig NH(2010) Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cellsJ Neurochem113: 16211631 |
51 | Parthsarathy V, Hölscher C(2013) Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model.ePLoS One8: 58784 |
52 | Heppner K, Kirigiti M, Secher A, Paulsen S, Buckingham R, Pyke C, Knudsen L, Vrang N, Grove K(2015) Expression and distribution of glucagon-like peptide-1 receptor mRNA, protein and binding in the male nonhuman primate (Macaca mulatta) brainEndocrinology1: 255267 |
53 | del Valle J, Duran-Vilaregut J, Manich G, Pallàs M, Camins A, Vilaplana J, Pelegrí C(2011) Cerebral amyloid angiopathy, blood-brain barrier disruption and amyloid accumulation in SAMP8 miceNeurodegener Dis8: 421429 |
54 | Zhang X, Li G, Guo L, Nie K, Jia Y, Zhao L, Yu J(2013) Age-related alteration in cerebral blood flow and energy failure is correlated with cognitive impairment in the senescence-accelerated prone mouse strain 8 (SAMP8)Neurol Sci34: 19171924 |
55 | Hsu TM, Hahn JD, Konanur VR, Lam A, Kanoski SE(2015) Hippocampal GLP-1 receptors influence food intake, meal size, and effort-based responding for food through volume transmissionNeuropsychopharmacology40: 327337 |
56 | Ohta H, Nishikawa H, Hirai K, Kato K, Miyamoto M(1996) Relationship of impaired brain glucose metabolism to learning deficit in the senescence-accelerated mouseNeurosci Lett217: 3740 |
57 | Gejl M, Rungby J, Brock B, Gjedde A(2014) At the centennial of Michaelis and Menten, competing Michaelis-Menten steps explain effect of GLP-1 on blood-brain transfer and metabolism of glucoseBasic Clin Pharmacol Toxicol115: 162171 |
58 | Flood JF, Harris FJ, Morley JE(1996) Age-related changes in hippocampal drug facilitation of memory processing in SAMP8 miceNeurobiol Aging17: 1524 |
59 | Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM, Greig NH(2003) Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and ironJ Neurosci Res72: 603612 |
60 | Del Valle J, Duran-Vilaregut J, Manich G, Casadesús G, Smith MA, Camins A, Pallàs M, Pelegrí C, Vilaplana J(2010) Early amyloid accumulation in the hippocampus of SAMP8 miceJ Alzheimers Dis19: 13031315 |
61 | Zhang Y, He JS, Wang X, Wang J, Bao FX, Pang SY, Yin F, Hu HG, Peng XL, Sun WM, Zheng YP, Hou LL, Hong T(2011) Administration of amyloid-β42 oligomer-specific monoclonal antibody improved memory performance in SAMP8 miceJ Alzheimers Dis23: 551561 |
62 | Egefjord L, Gejl M, Møller A, Brændgaard H, Gottrup H, Antropova O, Møller N, Poulsen HE, Gjedde A, Brock B, Rungby J(2012) Effects of liraglutide on neurodegeneration, blood flow and cognition in Alzheimer’s disease - protocol for a controlled, randomized double-blinded trial.ADan Med J59: 4519 |
Figures and Tables
Fig.1
Memory acquisition and retention function in SAMP8 mice assessed in an active avoidance T-maze test. Memory acquisition (A) and retention (B) performance in vehicle-dosed or liraglutide-treated 10-month-old SAMP8 mice. Vehicle-dosed 50% backcrossed SAMP8 mice and untreated 4-month-old SAMP8 control mice, respectively, served as controls for normal memory function. Long-term liraglutide treatment restored memory retention in 10-month-old SAMP8 mice, as compared to age-matched vehicle-dosed SAMP8 control mice. The number of trials to make one active avoidance was a measure of acquisition. Retention was tested one week later by continuing training until the mice achieved the criterion of making five active avoidances in six consecutive trials. The number of trials needed to reach this criterion was the measure of memory retention. **p < 0.01, ***p < 0.001 (one-way ANOVA, Dunnet’s post-hoc test).
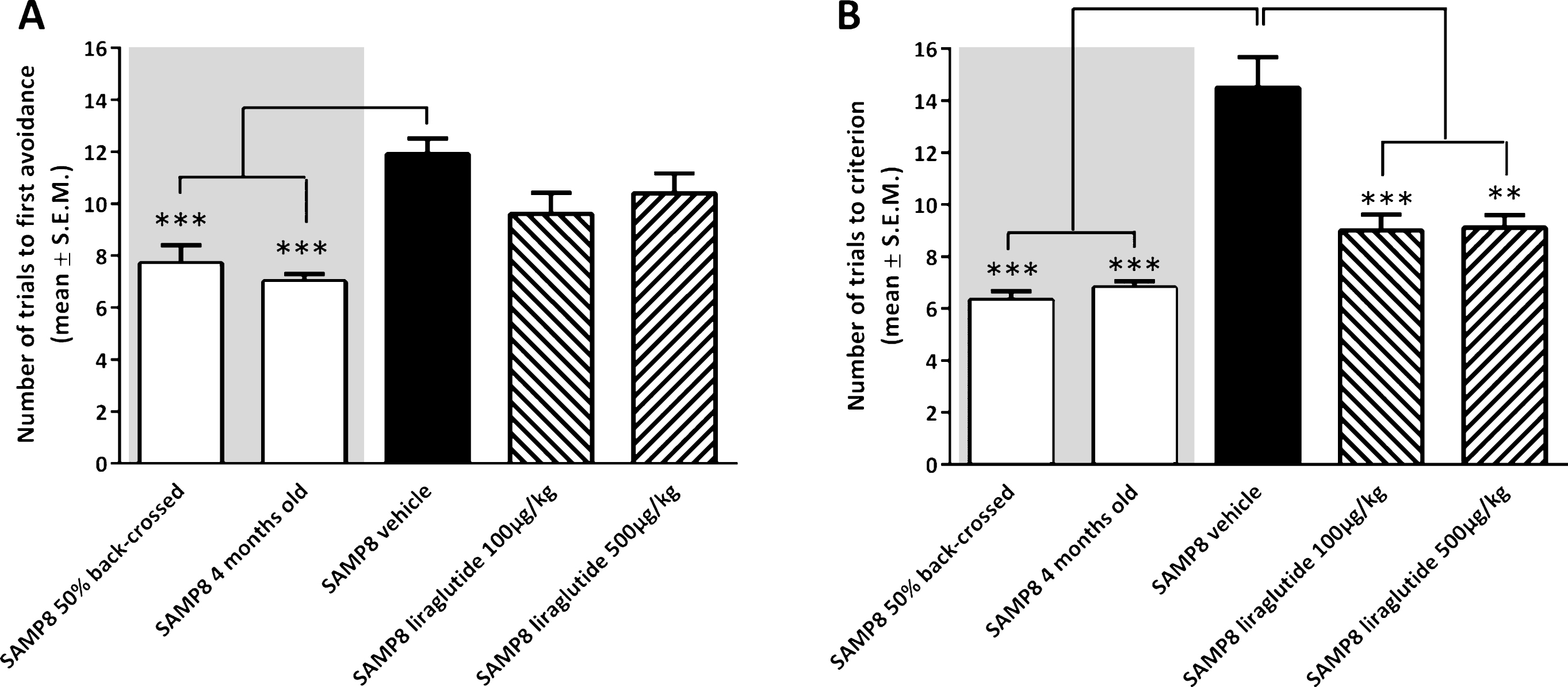
Fig.2
Recognition memory function in SAMP8 mice assessed in a novel object recognition (NOR) test. NOR performance in 10-month-old vehicle-dosed or liraglutide-treated SAMP8 mice. Vehicle-dosed 50% backcrossed SAMP8 mice and untreated 4-month-old SAMP8 control mice, respectively, served as controls for normal memory function. Vehicle-dosed 10-month-old SAMP8 mice showed poor object recognition memory performance, as compared to untreated 4-month-old SAMP8 mice. Liraglutide-treated 10-month-old SAMP8 mice did not show significant improvement of memory performance in the NOR test. The discrimination index was defined as the amount of time exploring the familiar object or the novel object over the total time spent exploring both objects multiplied by 100. *p < 0.05 (one-way ANOVA, Dunnet’s post-hoc test).
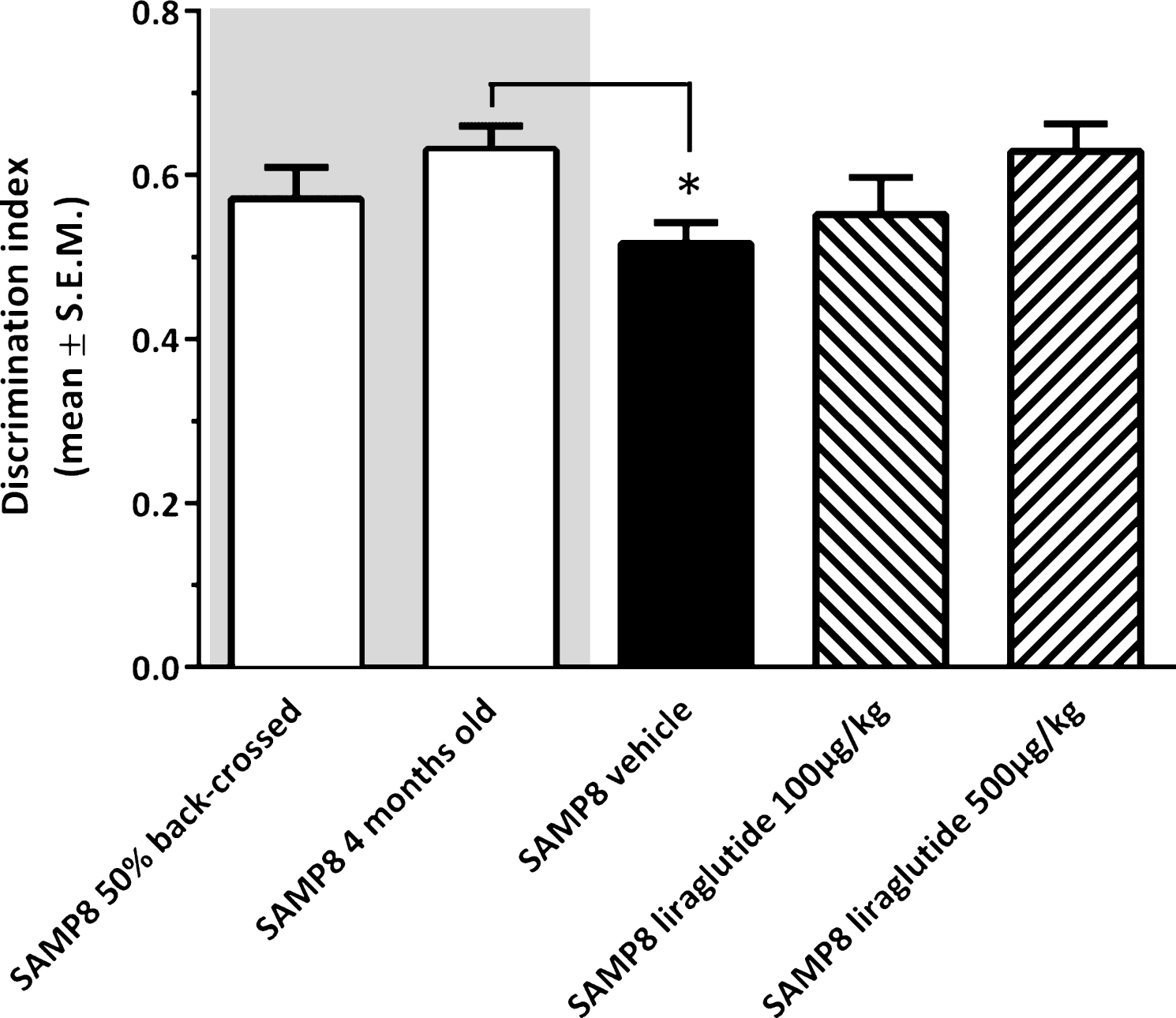
Fig.3
Brain weight of SAMP8 mice. Total brain wet weight was significantly higher in vehicle-dosed 10 month-old 50% back-crossed SAMP8 control mice, as compared to all other experimental groups. ***p < 0.001 (one-way ANOVA, Newman-Keuls post-hoc test).
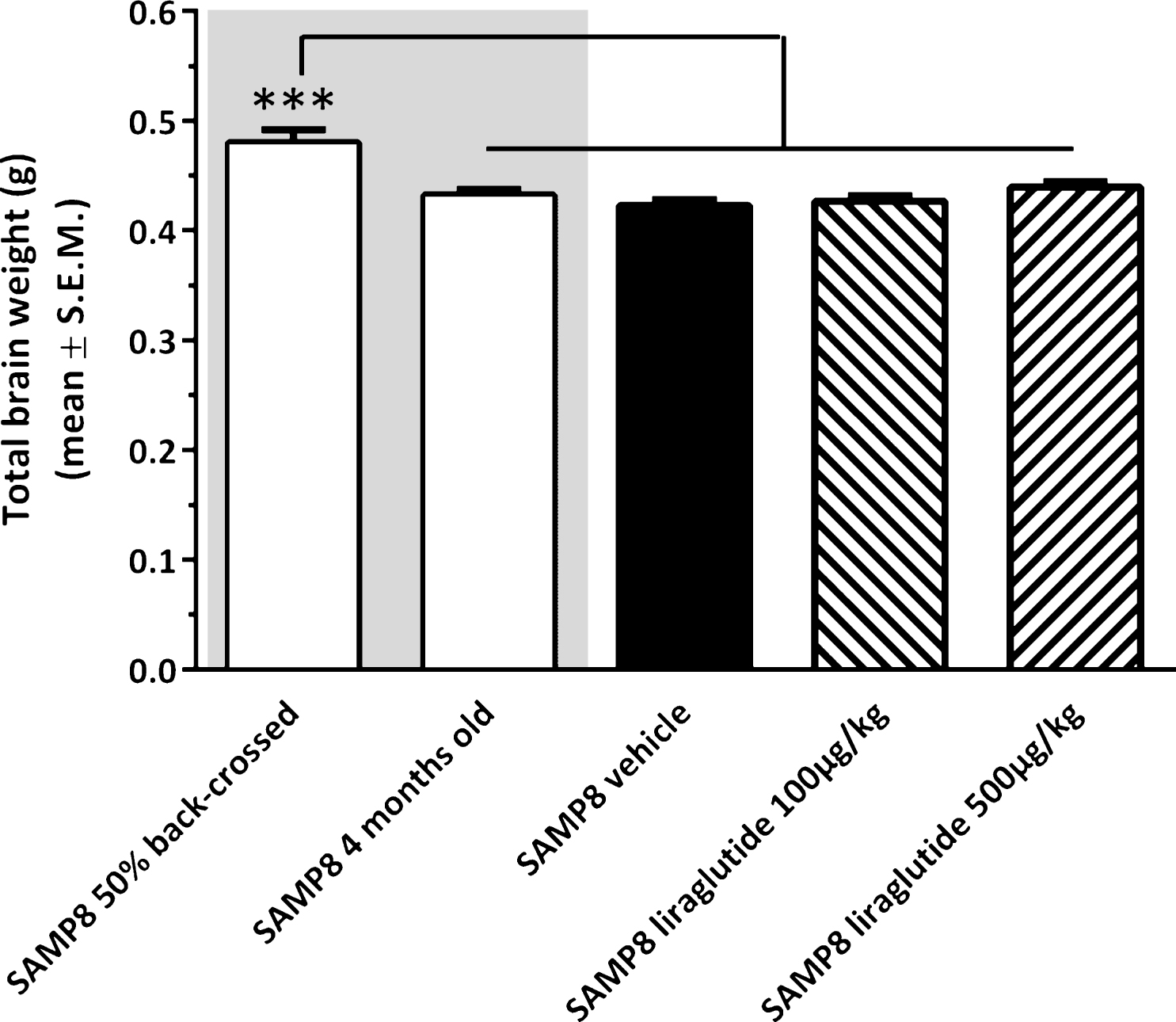
Fig.4
Histological assessment of hippocampal CA1 pyramidal neuron numbers. Brains were sampled in the horizontal plane using systematic uniform random sampling to ensure representation of the entire hippocampus. The major subfields of the mouse hippocampus are indicated in panel A (2x magnification). Panel B shows the border between CA1 and CA2. The CA1 region (Giemsa stained) was delineated on average in 8 sections per animal using a 10x objective based on cell morphology (B). CA1 pyramidal neurons are smaller and more densely organized than CA2 neurons (C; white arrows = CA1 pyramidal neuron, black arrows = CA2 pyramidal neuron; 40x magnification).
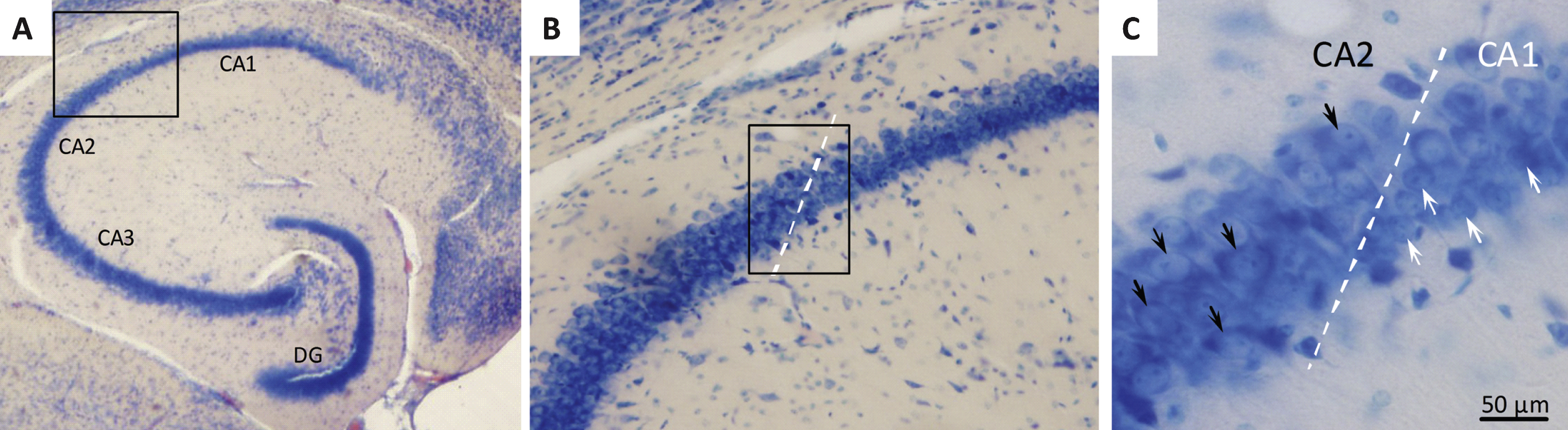
Fig.5
Stereological quantification of total hippocampal CA1 pyramidal neuron numbers in SAMP8 mice. Total hippocampal CA1 pyramidal neuron numbers were assessed in vehicle-dosed or liraglutide-treated 10-month-old SAMP8 mice, vehicle-dosed 10-month-old 50% backcrossed SAMP8 control mice, and untreated 4-month-old (young) SAMP8 control mice, respectively. Liraglutide-treated 10-month-old SAMP8 mice exhibited higher total pyramidal neuron numbers and neuronal density in the hippocampal CA1 region, as compared to age-matched vehicle-dosed SAMP8 mice. (A) total CA1 pyramidal neuron number; (B) total CA1 volume (mm3); (C) CA1 pyramidal neuron density (neurons per mm3). **p < 0.01 (one-way ANOVA, Dunnet’s post-hoc test).
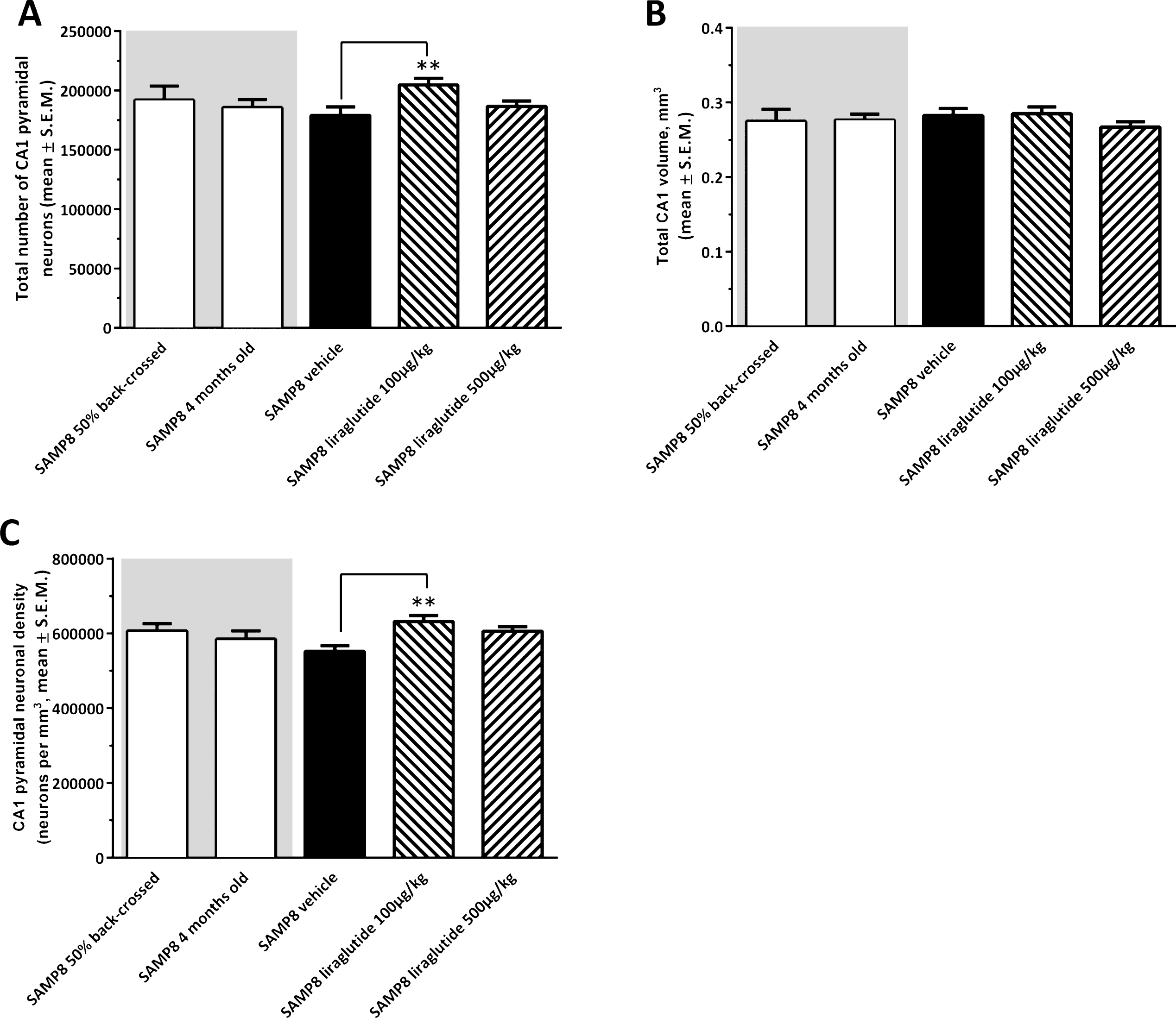
Fig.6
Immunohistochemical analysis of Aβ and neurofibrillary tangles in SAMP8 mouse brains. 10-month-old SAMP8 mice showed no presence of amyloid plaques or neurofibrillary tangles. Representative photomicrographs of Aβ (A) and phospho-tau (B) immunohistochemical staining of 5- μm thick brain sections from 10-month-old SAMP8 mice. Immunohistochemical control samples for amyloid plaques (C, AβPP/PS1 mouse) and neurofibrillary tangles (D, hTau P301L mouse) are inserted. Enlarged photos represent corresponding framed area on each specimen (scale bar = 100 μm).