Inflammation Combined with Ischemia Produces Myelin Injury and Plaque-Like Aggregates of Myelin, Amyloid-β and AβPP in Adult Rat Brain
Abstract
Background: Ischemia, white matter injury, and Alzheimer’s disease (AD) pathologies often co-exist in aging brain. How one condition predisposes to, interacts with, or perhaps causes the others remains unclear.
Objectives: To better understand the link between ischemia, white matter injury, and AD, adult rats were administered lipopolysaccharide (LPS) to serve as an inflammatory stimulus, and 24 h later subjected to 20-min focal cerebral ischemia (IS) followed by 30-min hypoxia (H).
Methods: Myelin and axonal damage, as well as amyloid-β (Aβ) and amyloid-β protein precursor (AβPP) deposition were examined by Western blot and immunocytochemistry following LPS/IS/H. Findings were compared to the 5XFAD mouse AD brain.
Results: Myelin/axonal injury was observed bilaterally in cortex following LPS/IS/H, along with an increase in IL-1, granzyme B, and LPS. AβPP deposition was present in ischemic striatum in regions of myelin loss. Aβ1-42 and AβPP were deposited in small foci in ischemic cortex that co-localized with myelin aggregates. In the 5XFAD mouse AD model, cortical amyloid plaques also co-localized with myelin aggregates.
Conclusions: LPS/IS/H produce myelin injury and plaque-like aggregates of myelin. AβPP and Aβ co-localize with these myelin aggregates.
INTRODUCTION
Stroke, Alzheimer’s disease (AD), and white matter injury (which presents on brain MRI as white matter hyperintensities, WMH) often co-exist in the aging human brain [1]. How one condition predisposes to, interacts with, or perhaps causes the others remains unclear.
Stroke is one risk factor for developing both AD and WMH [2], and infection predisposes to AD and stroke. After a stroke, dementia (including AD) develops in about 30% of patients with symptomatic infarcts AD [3–5]. Silent infarcts, which are often lacunes, also increase the risk of dementia [6]. White matter is vulnerable to ischemia [7], and WMH are frequent in stroke patients. Among stroke patients, WMH occurs more frequently in those who have lacunar infarcts than those who have cortical infarcts [8, 9]. These human data suggest that stroke, especially lacunar stroke, often precede AD and WMH. Chronic hypoxia is also a risk factor for dementia and AD [10]. Postmortem MRI and pathological studies in aging brains have suggested that white matter lesions are chronically hypoxic[11, 12]. These findings support the possibility that that ischemia/hypoxia might be causally involved in the pathogenesis of AD and WMH.
The mechanisms by which ischemia/hypoxia might contribute to AD and WMH remain unclear. Ischemia/hypoxia facilitate Aβ production by activating β-secretase that cleaves amyloid-β protein precursor (AβPP) to form Aβ and cerebral ischemia promotes amyloid plaque formation [12]. Injured vessels may impair the clearance of Aβ since Aβ is transported through perivascular pathways. White matter is very vulnerable to reduced blood flow due to its location at the distal border between the different vascular territories (watershed). In addition, Aβ is also transported through LRP1, a major receptor for endocytosis of degraded myelin. Thus, when white matter is damaged, degraded MBP may compete with Aβ to bind LRP1 and Aβ might accumulate in thebrain.
Delirium, which is often caused by infection, can lead to dementia in 50% of cognitively intact older individuals at a one year follow-up [13]. The number of infections reported over a 5-year period increased the odds of developing AD two-fold [14], and vaccination compliance reduced the risk of developing AD [15]. Though infection has not been associated with WMH, evidence of inflammation has been detected in the peripheral blood of patients with WMH compared to controls [16]. In addition, white matter injury in the neonatal brain has been associated with infection [17]. Indeed, a combination of lipopolysaccharide (LPS, a gram-negative bacteria coat protein), focal cerebral ischemia, and hypoxia is used to study white matter injury in brains of newborn rodents [18].
Given the above clinical evidence and the neonatal white matter injury model, we hypothesized that a combination of systemic inflammation/infection (LPS), focal cerebral ischemia, and systemic hypoxia would produce white matter injury in the adult rat brain and would also produce AD-like, amyloid (Aβ) neuropathology in adult rat brain.
MATERIALS AND METHODS
Animals
The University of California Animal Care Committee at Davis approved the animal protocol in accordance with NIH guidelines. Three month old Sprague-Dawley male rats (Charles River Labs, USA, n = 67) were studied. In addition, AβPP/PS1 trans-genic mice that co-express 5 familial AD mutations (5XFAD mice, n = 4) were used at 8 and 10 months of age as positive controls for Aβ and amyloid plaque staining [19].
LPS injection, focal cerebral ischemia, hypoxia
Adult rats were injected with LPS followed 1 day later by transient focal cerebral ischemia (IS) (20 min MCAO) and then 30 min later were exposed to transient hypoxia (H) (30 min 8% oxygen) (LPS/IS/H). Brains from LPS/IS/H treated animals were obtained over a 1 to 12 week period (n = 6 at 1 week, n = 4 at 2 weeks, n = 6 at 4 weeks, n = 12 at 8 weeks, and n = 17 at 12 weeks).
LPS (E. coli, 0111:B4, Sigma, St. Louis, MO) was administered intraperitoneally (3 mg/kg in 0.9% saline). One day later (24 h), rats were subjected to a temporary, 20-min middle cerebral artery occlusion (MCAO) using the suture technique [20]. Briefly, rats were anesthetized with 3% isoflurane and maintained on 1.5% isoflurane. After exposing the right common carotid artery, a 0.43 mm diameter monofilament nylon suture coated with silicon (Doccol, Redlands, CA) was threaded through the external carotid artery stump into the internal carotid artery approximately 20–23 mm beyond the carotid bifurcation until mild resistance was felt. The suture was left in place for 20 min and was then removed and anesthesia discontinued. This 20-min MCAO produces reliable infarction of the basal ganglia without any infarction of the overlying cortex [20]. Rectal temperature was maintained at 36.5°C to 37.5°C with a heating blanket throughout the procedure. Animals were allowed to recover for 30 min following MCAO and then exposed while conscious to 8% O2 in an enclosed hypoxia chamber for30 min.
To compare to the LPS/IS/H group, saline/IS/H animals and naïve animals were used as controls. In the saline/IS/H group, the same volume of 0.9% saline replaced the LPS. In the naïve group, animals did not receive any treatment, i.e., they were normalanimals.
Immunofluorescence and amyloid plaque staining
Animals were deeply anesthetized with isoflurane, and then perfused with 4% paraformaldehyde in 0.1 M phosphate buffer. Brains were sucrose protected and coronal sections cut on a cryostat (50- μm thick). After blocking nonspecific sites with a buffer containing 2% goat serum, 1% bovine serum albumin (BSA), and 0.2% Triton X-100 in PBS, sections were incubated with primary antibody. The primary antibodies were diluted at 1:500 unless otherwise stated. Antibodies included mouse monoclonals against: MBP (Millipore, 1:10 dilution); N-Sphingomyelinase2 (Abcam); Myelin-associated glycoprotein (MAG, Abbiotec); Lamp1 (Santa Cruz); Caveolin 1 (Cell Signaling); rodent Aβ (Covance); Aβ1 - 40/42 (Millipore); Aβ1 - 42 (Covance); and a rabbit polyclonal against neurofilament light chain protein (NF-L) (Millipore; 1:1000 dilutions). The primary antibody was incubated with sections at 4°C overnight. The secondary antibody was incubated with sections at room temperature for 2 h. Goat anti-mouse or goat anti-rabbit Alexa Fluor ® 488 or 594 conjugated antibodies (Invitrogen) were used for secondary antibodies depending on the species of the primary antibody. Primary antibody was omitted to assess non-specific staining. Sections were stained with FSB, a Congo red derivative [21] (Millipore, 0.5 μM in PBS, 20 min), to identify amyloid plaques in the mouse AD model.
Western blot analyses
Animals with various durations of reperfusion following LPS/IS/H or saline/IS/H were anesthetized with isoflurane and decapitated. The cortex of the ischemic hemisphere and contralateral hemisphere were dissected and frozen at –70°C. Frozen tissues were homogenized in ice-cold buffer containing a complete protease inhibitor mixture (Sigma). Homogenates were centrifuged at 14,000 g for 30 min at 4°C and the pellet was discarded. The protein in the supernatant (50 μg each) was mixed with the same volume of 2X Laemmli sample buffer, denatured at 95°C for 5 min, loaded into lanes, separated on SDS polyacrylamide gels (12% –18%) and transferred to nitrocellulose. The membranes were probed overnight at 4°C with antibodies to MBP (Millipore; 1:100 dilutions), IL-1β (Santa Cruz, 1:200 dilutions), Granzyme B (Grzm B, Millipore, 1:500 dilution), LPS (Lifespan 1:200 dilution), rodent Aβ (Covance, 1:1000 dilution), Aβ1 - 42 (Covance, 1:500 dilution), AβPP (AHP539, AbD Serotec, synthetic peptide corresponding to amino acids 672–681 of human AβPP; 1:1000 dilutions), Aβ1 - 40/42 (AB5076, Millipore, recognizes rat, human, mouse Aβ1 - 40/42, 1:1000 dilution), or β-actin (Santa Cruz, 1:10,000 dilutions). Primary antibody was detected using horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Bio-Rad). The signal was detected using the ECL chemiluminescent detection system (Thermo Fisher Scientific). The optical densities for each target protein band and β actin band were measured and the ratio used for quantification.
Statistical analysis
Differences between two groups were analyzed using a Student t-test. One-way ANOVA was performed with a Student-Neuman-Keuls post hoc test to evaluate the differences between three groups.A p value ≤0.05 was considered statistically significant.
RESULTS
LPS/IS/H is associated with MBP degradation and increased inflammatory molecules
To characterize the overall changes in myelin and inflammatory molecules over time, we performed Western blot analyses for myelin basic protein (MBP), interleukin 1-β (IL-1β), and Grzm B on different rats at 4, 8, and 12 weeks following LPS/IS/H compared to control rats (Fig. 1). One animal at each time point was examined. Using an antibody that detected native MBP (MW = 19 kDa), Western blots showed two ∼19 kDa bands in control brain (Control, Fig. 1A). Following LPS/IS/H, the pattern and amount of staining changed. In the ischemic ipsilateral hemisphere, there was one native ∼19 kDa band and a second ∼14 kDa band we interpreted to be degraded MBP (dMBP) (Fig. 1A). In the contralateral non-ischemic hemisphere, there were two ∼19 kDa bands and a third ∼14 kDa band we again interpreted to be dMBP (Fig. 1A). The data show marked loss of the native ∼19 kDa MBP in the ipsilateral ischemic hemisphere (3.8 fold decrease compared to contralateral cortex, p < 0.01) and appearance of dMBP in the ipsilateral ischemic hemisphere at 4, 8, and 12 weeks following LPS/IS/H (Fig. 1A). In the non-ischemic contralateral hemisphere at 4, 8, and 12 weeks post LPS/IS/H, the ∼14 kDa dMBP was also detected at all timepoints in spite of little change in total native ∼19 kDa MBP (Fig. 1A).
Because of the increased dMBP in the ischemic and non-ischemic hemispheres at all times, we hypothesized that this was related to a persistent bilateral inflammatory state. Thus, IL-1β and Grzm B were assessed since IL1 is associated with monocytes/microglia and Grzm B with cytotoxic T cells/Natural Killer cells, and they both increase in the chronic stages of inflammation [22, 23]. Though IL-1beta and Grzm B protein were not detected in control brain (Fig. 1A), both were detected in the ipsilateral ischemic hemisphere of LPS/IS/H rats at 4 weeks, but continued to increase at 8 and 12 weeks (Fig. 1A). Moreover, both IL-1β and Grzm B were detected at 4 weeks in the contralateral, non-ischemic hemisphere and increased by 12 weeks (Fig. 1A).
The possibility that the persistent inflammatory state was due in part to continued entry of LPS into brain over time was examined next. Indeed, LPS appeared to increase in the ischemic cortex as well as in contralateral cortex from 4 to 12 weeks post LPS/IS/H (Fig. 1B). There was no LPS in control brain (Fig. 1B).
To determine if the inflammatory response was related to LPS and/or IS/H and to quantify the inflammatory response, Western blot analysis was performed on ischemic cortex at 12 weeks post LPS/IS/H versus saline/IS/H using antibodies against IL-1β and Grzm B (Fig. 2). Six animals were examined in each group. Following saline/IS/H two bands were detected with molecular weights around 150 kDa and 120 kDa using an antibody against IL-1β (Fig. 2A). Following LPS/IS/H there was expression of IL-1β though this band was around 140 kDa (up to ∼150 kDa) using the same antibody (Fig. 2A). Since the molecular weight of unprocessed IL-1β is ∼30 kDa and processed IL-1β is ∼17.5 kDa, these results suggest that saline/IS/H produces different IL-1β oligomers compared to LPS/IS/H. LPS/IS/H produced a 2.3-fold increase in the 140 kDa IL-1β oligomers compared to saline/IS/H 150 kDa IL-1β (p < 0.01) whereas the 120 kDa IL-1β oligomers were expressed only in saline/IS/H brain. IL-1β was not expressed in control brain (Fig. 1A).
Grzm B was not detected in control cortex (Fig. 1A) or in cortex at 12-weeks post saline/IS/H (Fig. 2B). In contrast, there was marked induction of Grzm B at 12 weeks following LPS/IS/H (Fig. 2B). Since the molecular weight of Grzm B is ∼26 kDa, this antibody appears to be detecting ∼144 kDa Grzm B complexes following LPS/IS/H (Fig. 2B).
MBP immunostaining associates with blood vessels at 1 week post LPS/IS/H
To determine the histological fate of MBP in cortex following LPS/IS/H, we performed MBP immunostaining at several time points. In normal cortex, MBP immunostaining showed stained axons that ran perpendicular to the cortical surface (Fig. 3A). This MBP staining co-localized with axonal NF-L staining (Fig. 3A, right panel), and was not associated with blood vessels (Fig. 3A).
By one week post LPS/IS/H, the MBP in cortex lost its normal staining pattern and instead was localized mainly to blood vessels (Fig. 3B, arrow, figure from ipsilateral hemisphere) bilaterally throughout cortex. Vascular staining of MBP occurred in 3 out of 6 animals at 1 week. There was some MBP staining of axons perpendicular to the cortical surface following LPS/IS/H (Fig. 3B), but this staining was much less than in control cortex (Fig. 3A). Double labeling showed that MBP staining at 1 week (Fig. 3C) post LPS/IS/H appeared to co-localize with Caveolin 1 positively stained endothelial cells (Fig. 3C), suggesting the MBP was either in brain endothelial cells, in adjacent pericytes and/or in spaces adjacent to these cells.
Tangled dystrophic myelinated axons at 2-12 weeks
By two weeks post LPS/IS/H, the MBP staining in or around endothelial cells had disappeared. Instead, abnormal focal tangles of MBP staining approximately 100–150 μm in diameter occurred that were detected bilaterally in cortex at 1 week (not shown), 2 weeks (Fig. 4B, white arrow), 4 weeks (Fig. 4A), 8 weeks (Fig. 4C), and 12 weeks (Fig. 4D) after LPS/IS/H. These figures were from ipsilateral ischemic cortex, but similar findings were observed in the contralateral cortex. Similar focal MBP stained tangles were detected bilaterally in hippocampus (not shown). Double-labeling for MBP (Fig. 4E) with NF-L (Fig. 4F) showed that the tangles of MBP-stained fibers were swollen myelin sheaths associated with axons (Fig. 4E-G). These structures were termed “tangled dystrophic myelinated axons” which were distributed randomly throughout both hemispheres in cortex and hippocampus following LPS/IS/H. The “tangled dystrophic myelinated axons” were present in every coronal section of neocortex and hippocampus and varied in number from 8 to 30 per coronal section. Foci of tangled dystrophic myelinated axons were noted in cortex in 5 of 6 rats (83%) at 1 week, 4 of the 4 rats (100%) at 2 weeks, 6 of the 6 rats (100%) at 4 weeks, 12 of the 16 rats (75%) at 8 weeks, and 12 of the 17 rats (70%) at 12 weeks following LPS/IS/H.
Induction of sphingomyelinase and lamp 1
Two potential mechanisms for myelin damage were examined. Levels of neuro-sphingo-myelinase 2 (NSMase2), which breaks down sphingomyelin, were low in normal cortex (Fig. 5A, C). At 4 weeks, NSMase2 was induced mainly in axons (Fig. 5A, 4 weeks) that co-localized partially with NF-L (Fig. 5B, C, 4 weeks). Note that the overall NF-L level was reduced at 4 weeks compared to normal cortex, suggesting axonal injury as well. NSMase2 was near control levels by 12 weeks (Fig. 5, 12 weeks). Lysosomal-associated membrane protein 1 (Lamp1), which is involved in lysosomal degradation of myelin, was not detected in control brains (not shown). At 4 weeks post LPS/IS/H, the Lamp1 protein was associated with the cortical foci of tangled dystrophic myelinated axons, and co-localized with MBP (Fig. 6).
Deposition of AβPP in areas of myelin loss
MBP staining of axon bundles in normal striatum (Fig. 7A) disappeared in the hemisphere ipsilateral to ischemia by 8 weeks following LPS/IS/H (Fig. 7B). Since MBP can degrade Aβ [24] and inhibit the deposition of Aβ [25] in vitro, Aβ and AβPP were examined in areas of MBP loss. Thus, using a purported rodent Aβ (R Aβ) antibody there was no R Aβ detected in control striatum (Fig. 7A) whereas R Aβ was detected in areas of MBP loss in the ischemic striatum (Fig. 7B). Western blot analysis using the same R Aβ antibody showed a 100 kDa band for R Aβ (Fig. 7C) that was significantly increased following LPS/IS/H (n = 6 animals) compared to saline/IS/H (n = 6 animals) (Fig. 7C, D, p < 0.01). Since many Aβ antibodies share the same epitopes with AβPP, an antibody specific to AβPP was used to compare with R Aβ on Western blot analysis in animals following LPS/IS/H (n = 6 animals), saline/IS/H (n = 6 animals), and in naïve control animals (n = 6 animals) (Fig. 8). The results showed that a similar ∼100 kDa protein was detected using the AβPP antibody (Fig. 8A) compared to the R Aβ antibody (Fig. 7C). These results strongly suggested that the protein detected using the R Aβ antibody is likely AβPP rather than Aβ. Notably, the expression of AβPP following LPS/IS/H was significantly increased compared to saline/IS/H (Fig. 8, p < 0.001), but the expression of AβPP following LPS/IS/H was not significantly different compared to naïve animals (Fig. 8A, B).
MBP and MAG stained myelin aggregates co-localize with Aβ1 - 42 and AβPP
Beginning at 8 weeks post LPS/IS/H there was the appearance of discrete, ovoid structures that stained for MBP in the cortex ipsilateral to ischemia that were particularly evident by 12 weeks following LPS/IS/H (Fig. 9A, left panel). These MBP stained structures varied from 5 μm to about 15 μm in diameter and were observed only in the cortex ipsilateral to the ischemia. These structures appear to be aggregates of degraded myelin. Indeed, staining for another myelin protein, myelin associated glycoprotein (MAG), demonstrated these cortical aggregates included both MBP and MAG myelin proteins (Fig. 9B, left panel). Thus, these MBP/MAG stained structures in ischemic cortex were termed “myelin aggregates”.
Given the small size and random distribution of the myelin aggregates, this raised the possibility they could be related to amyloid aggregates/amyloid-like plaques. Double staining showed that most MBP stained myelin aggregates were co-localized with R Aβ (Fig. 9A, white arrows), though some were not (Fig. 9A, yellow arrows). As noted above, this R Aβ immunostaining likely represents AβPP. To determine if Aβ co-localized with the myelin aggregates, the myelin aggregates that stained with the anti-MAG (myelin-associated glycoprotein) antibody co-localized with an antibody to Aβ1 - 42 at 12 weeks post LPS/IS/H (Fig. 9B). Western blot analysis using the Aβ1 - 42 antibody showed expression of ∼140 kDa and 150 kDa bands for Aβ1 - 42 in LPS/IS/H and saline/IS/H brains (Fig. 9C), though the expression of both Aβ1 - 42 bands (Fig. 8C) were significantly increased following LPS/IS/H compared to saline/IS/H (Fig. 9D, p < 0.001). The bands detected using the Aβ1 - 42 antibody (∼140 kDa and ∼150 kDa) were larger than that of AβPP (∼100 kDa) and R Aβ (∼100 kDa), suggesting that they were not the same protein. To further confirm this, another antibody directed against Aβ1 - 40/42 was used for Western blot analysis in animals following LPS/IS/H (n = 6 animals), saline/IS/H (n = 6 animals), and naïve (n = 6 animals) animals (Fig. 10). The results showed that a ∼150 kDa molecular weight band (perhaps two in saline/IS/H) was detected using the Aβ1 - 40/42 antibody (Fig. 10). This band had the same molecular weight (Fig. 10) as the ∼150 kDa molecular weight band detected using the Aβ1 - 42 antibody (Fig. 9C). These results suggested that the protein band detected using Aβ1 - 40/42 is likely the same as that was detected using Aβ1 - 42 but not the same as AβPP or R Aβ. The ∼150kDa bands detected by antibodies to Aβ1 - 40/42 and Aβ1 - 42 likely are oligomers of Aβ peptides or Aβ oligomers bound to another protein. The expression of Aβ1 - 40/42 oligomers following LPS/IS/H was significantly increased compared to saline/IS/H (Fig. 10; p < 0.001), though expression of Aβ1 - 40/42 following LPS/IS/H was not significantly different from naïve animals (Fig. 10A, B).
The myelin aggregates that occurred in ischemic cortex following LPS/IS/H, however, were observed in only a portion of the animals. Four out of 16 rats at 8 weeks (25%) and 8 out of 17 rats at 12 weeks (47%) had myelin aggregates in ischemic cortex following LPS/IS/H. In the rats with myelin aggregates, the numbers of myelin aggregates varied from 12 to 50 in ischemic cortex per coronal brain section. The reasons for only half of the animals showing myelin aggregates are discussed below and are under further study.
Myelin aggregates co-localize with amyloid plaques in mouse AD model
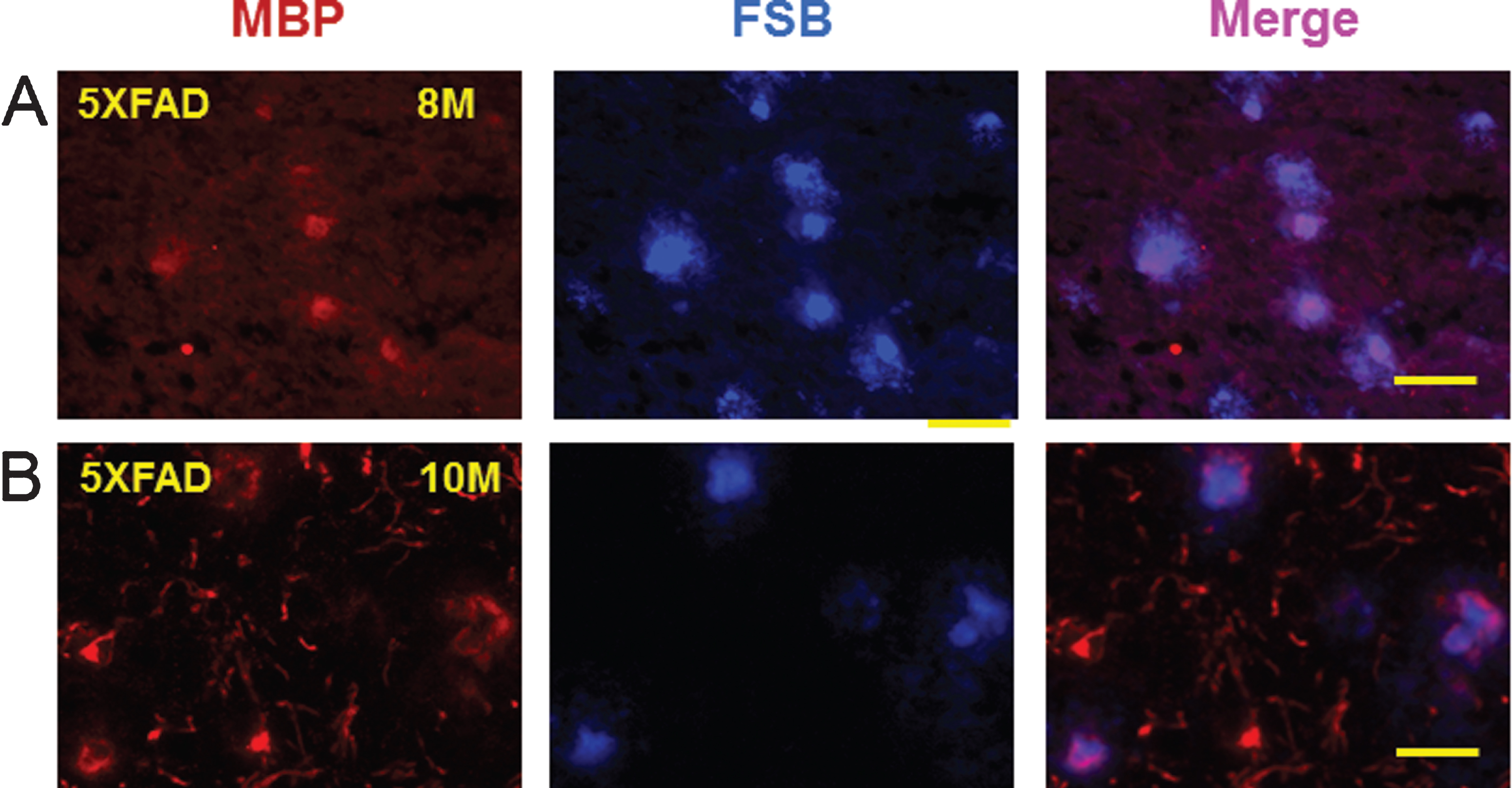
Since myelin aggregates co-localized with Aβ peptides and AβPP in the rat LPS/IS/H model, the possibility that myelin aggregates were associated with amyloid plaques in mouse AD brains was explored. Brains of AβPP/PS1 double transgenic mice that co-express five familial AD (5XFAD) mutations were used [19]. A sensitive fluorescent amyloid dye FSB ((E, E)-1-fluoro-2,5-bis (3-hydroxycarbonyl-4-hydroxy) styrylbenzene) [21], a derivative of Congo red, was used to stain amyloid plaques in cortical sections of 8-month-old and 10-month-old 5XFAD mice. These sections were then double immunostained for MBP (Fig. 11). FSB stained amyloid plaques in cortex co-localized with MBP staining in both 8-month-old (Fig. 11A, upper panels) and 10-month-old (Fig. 11B, lower panels) 5XFADmice.
DISCUSSION
The combination of inflammation and ischemic-hypoxic injury resulted in myelin and axonal injury in adult rodent forebrain. This was associated with deposition of AβPP in areas of MBP loss, and with deposition of Aβ peptides and AβPP in myelin aggregates in the ischemic cortex. Moreover, myelin aggregates were associated with amyloid plaques in the 5XFAD genetic AD mouse model. The focal deposits of Aβ and/or amyloid could produce white matter injury. Alternatively, the myelin aggregates could form the nidus for Aβ deposition in the LPS/IS/H model and formation of amyloid plaques in the mouse AD model. These data provide support for the idea that an inflammatory challenge combined with cerebral ischemia and hypoxia could contribute to the pathogenesis of white matter injury in adult human brain and possibly to Aβ/amyloid pathology in AD.
Damaged myelin may exit brain via blood vessels
The combination of LPS with focal ischemia and hypoxia led to myelin damage that occurred in the ischemic and non-ischemic, contralateral hemisphere. At early times after LPS/IS/H the MBP was localized in or near vascular endothelial cells. This could represent MBP being removed from brain perhaps by LRP1 transporters for degraded myelin located on endothelial cells. Notably, LRP1 is not only the major receptor for endocytosis of degraded myelin, but also the major receptor for Aβ peptide transport across the blood-brain barrier (BBB) from brain to blood [26]. Thus, when Aβ is produced, LRP1 could bind Aβ and MBP might compete with Aβ to bind LRP1 and thus accumulate around blood vessels. Why the vessel-related MBP staining disappears by 4 weeks is unclear since degraded MBP is detected at 4, 8, and 12 weeks following LPS/IS/H. The mechanisms for the appearance and disappearance of MBP around the blood vessels remain to be elucidated.
Prolonged, bilateral inflammation may damage myelin bilaterally
The bilateral cortical damage to myelin correlated with bilateral expression of IL-1β and Grzm B. Moreover, there was a progressive increase of these inflammatory molecules as well as progressive increase of degraded MBP over time after LPS/IS/H. Grzm B likely indicates the presence of cytotoxic T cells and/or Natural Killer cells that contain the Grzm B proteolytic protein that, in combination with perforin, produces apoptosis of targeted cells.
These observations suggest there is a prolonged inflammatory state in brain which could be due to damage to the BBB and allow for continued entry of LPS into brain after a single LPS/IS/H insult. Previous studies show LPS is cleared systemically by being bound to TLR4 receptors on peripheral monocytes and other leukocytes [27]. Therefore, it is possible that peripheral leukocytes containing LPS continue to enter the brain over time after LPS/IS/H and this would increase brain LPS levels. Microglial TLR4 receptors are required for leukocyte recruitment into the brain following LPS [28]. The finding of bilateral myelin injury implies that the BBB is impaired not only in the ischemic hemisphere, but also in the contralateral hemisphere. This is consistent with studies showing that systemic LPS alone can damage the BBB bilaterally at least in part via metallo-proteinases [29]. We postulate this bilateral BBB damage facilitates continued entry of LPS over time into brain and accounts for the prolonged, bilateral inflammatory state of the brain.
Pro-inflammatory cytokines can cause myelin damage. LPS induced cytotoxicity to myelin in optic nerves is prevented by blocking IL-1β [30]. Grzm B can hydrolyze MBP in myelin membranes [31]. Thus, the progressive increases of pro-inflammatory molecules in brain, that likely result from continued LPS entry in brain, may lead to myelin damage that continues for months after a single LPS/IS/H.
Grzm B is a serine protease that is found in the granules of cytotoxic T cells and natural killer cells. Although the molecular weight of Grzm B is about 27 kDa, high molecular weight of granzyme complexes have been detected for Granzyme A [32] and Granzyme K [33] in the plasma of healthy and infected patients. The sizes of the reported granzyme protein complexes include >250 kDa, ∼150 kDa, ∼120 kDa, and 90 kDa [32][33]. Our study shows a ∼150 kDa Grzm B complex in rat blood following LPS/IS/H which is consistent with these previous studies.
The proinflammatory cytokine IL-1β is synthesized as a 31 kDa non-active precursor which is processed into a 17-kDa active form by IL-1 converting enzyme [34, 35]. However, studies show that different sizes of IL-1β from 20 kDa to 28 kDa form under different conditions [36, 37]. The large 120–150 kDa complexes of IL-1β protein observed in this study have not been previously reported. Future studies will need to determine if the 120–150 kDa complexes are IL-1β aggregates or IL-1β bound to other proteins.
Mechanisms of myelin damage
Sphingomyelinases (SMases) are enzymes that hydrolyze sphingomyelin to generate ceramide. SMases are classified into acid, neutral, and alkaline subtypes [38]. Previous work shows that LPS, IL-1β, and TNFα activate neutral sphingomyelinase [39] and N-SMase2 is the predominant N-SMase in mammals that degrades sphingomyelin [38]. N-SMase2 induction following LPS/IS/H could occur via IL-1β [40]. LPS can increase N-SMase activity through IL-1 receptor activation [41] and hypoxia can also activate N-SMase [42]. N-SMase2 is upregulated in AD brain [43].
Lamp1 is part of the protein complex required for fusion of lysosomes to phagosomes [44]. Intracellular co-localization of MBP with lysosomal membrane protein LAMP1 suggests that MBP is taken up by lysosome and lysosomal degradation of myelin lipids may occur.
AβPP deposits in myelin aggregates and in areas of myelin loss
LPS/IS/H markedly induced the expression of AβPP in the ischemic hemisphere compared to saline/IS/H, suggesting a specific effect of LPS on inducing AβPP. Indeed, LPS, IL-1β, and TNFα activate N-SMase to produce ceramide [39], and C2-ceramide, an analogue of ceramide, increases AβPP mRNA and protein production [45]. In addition, a synthetic ceramide analogue increases Aβ42 production by modulating the activity of γ-secretase, an enzyme that cleaves AβPP to generate Aβ [45]. In our study, N-SMase2 is induced which suggests that the ceramide might be involved in the production of AβPP and Aβ following LPS/IS/H.
MBP itself can degrade Aβ and possibly AβPP protein [24] and inhibit Aβ deposition [25]. Thus, the presence of normal myelin may prevent or decrease AβPP deposition, and with myelin damage AβPP deposition may occur. Braak and Braak found initial amyloid deposits develop in poorly myelinated areas of neocortex in human AD brain [46]. Moreover, several studies have shown that decreases of brain blood flow and myelin damage precede the appearance of amyloid plaques and neurofibrillary tangles in animal AD models and in human AD brain [26, 47–52]. Our data supports the notion that myelin damage can precede deposition of AβPP and Aβ.
Aβ peptide deposits in myelin aggregates in ischemic cortex
We used two different antibodies to Aβ1 - 42 and Aβ1 - 40/42 to detect Aβ oligomers that did not cross react with the ∼100 kDa AβPP protein. Using the Aβ1 - 42 antibody the Aβ oligomers are shown to be deposited in myelin aggregates in ischemic cortex following LPS/IS/H. Whether these aggregates evolve into typical amyloid plaques will require further studies as these animals age. However, the FSB stained cortical amyloid plaques in the 5XFAD AD mice did co-localize with MBP stained myelin aggregates. The data could suggest that focal deposits of Aβ and/or amyloid produce focal myelin damage. Alternatively, myelin aggregates could serve as a nidus for deposition of Aβ and/or amyloid. In recent human studies, we demonstrated that dMBP was observed in dMBP vesicles in white matter of AD brains [53] and MBP/dMBP myelin aggregates were associated with amyloid plaques in the gray matter of late onset sporadic AD human brains [54].
LPS/IS/H and increases of AβPP and Aβ
LPS/IS/H markedly increased AβPP and Aβ in ischemic cortex compared to saline/IS/H. However, an unexpected finding of this study was that saline/IS/H significantly decreased AβPP and Aβ in ischemic cortex compared to naïve controls. How IS/H lead to decreases of AβPP and Aβ compared to naïve animals is unclear.
In our study, the molecular weights of Aβ and AβPP are about 150 kDa and 100 kDa, respectively. The predicted molecular weight of AβPP is ∼100 kDa. However, though the molecular weight of Aβ1 - 42 is only about 4 kDa, a ladder of Aβ oligomers from 4 kDa to 150 kDa are observed in both AD and control brains [55, 56]. Using two different antibodies, we did not detect the 4 kDa form of Aβ1 - 42. Instead, we detected a 150 kDa form of Aβ1 - 42 which suggests these are Aβ oligomers or complexes of Aβ with other proteins.
Though the levels of Aβ appeared to be similar on Western blots of naïve and LPS/IS/H brains, immunocytochemistry using the same antibodies showed staining in LPS/IS/H brains but not in the naïve brains. These two findings suggest there are similar overall levels of Aβ in naïve and LPS/IS/H brains, but that the Aβ is soluble in naïve brain and is more insoluble and thus immunostains in the LPS/IS/H brains.
Both AβPP and Aβ are synthesized and present in normal brain, so that the decreases with IS/H presumably may relate to either decreased synthesis and/or increased export of both from brain. Some previous studies of ischemia report increases of AβPP and Aβ. This might be related to combined ischemia and hypoxia in this study compared to previous ischemia studies. Alternatively, it may relate to insoluble deposited forms following ischemia compared to controls as compared to the decreased total AβPP and Aβ observed on Western blots following combined ischemia and hypoxia in this study.
How LPS increases Aβ and AβPP in brain has received considerable study. LPS alone induces Aβ production in a mouse AD model [57]. LPS can increase brain Aβ through increased influx, decreased efflux, and increased neuronal production [58]. LPS impairs Aβ efflux from brain by altered vascular sequestration, cerebrospinal fluid re-absorption, peripheral clearance, and transporter function at the BBB [59]. Ischemia and hypoxia increase brain Aβ production [60] and hypoxia can increase serum Aβ levels [61].
LRP1 (mentioned above) is the major receptor for endocytosis of degraded myelin, and also the major receptor for Aβ peptide transport across the BBB from brain to blood [26]. Thus, when myelin is damaged, LRP1 could bind MBP and Aβ might compete with MBP to bind LRP1. This could decrease Aβ clearance from the brain to blood and increase brain Aβ.
Limitations of study
All LPS/IS/H animals developed myelin injury, but only half developed myelin aggregates in the ischemic hemisphere. Reasons for this could relate to variable degrees of cortical ischemia, variable BBB damage or other factors. Developing a model in which all animals develop myelin aggregates will be a priority.
Though the myelin aggregates co-localize with Aβ oligomers following LPS/IS/H, future studies will be needed to determine if LPS/IS/H can produce typical amyloid plaques that occur with age. However, we did find that amyloid plaques in at least one mouse AD model co-localize with myelin aggregates. In addition, in a recent study we demonstrated that amyloid plaques in human AD brain are associated with degraded myelin basic protein [54]. These results suggest that there may be a connection between myelin injury, Aβ/AβPP deposition in the resulting myelin aggregates and the formation of amyloid plaques in human AD brains.
ACKNOWLEDGMENTS
These studies were supported in part by the UC Davis Alzheimer’s Disease Research Center Core (NIH/NIA P30 AG10129-21, Charles DeCarli) and the UCD Department of Neurology. We are grateful to the UC Davis Alzheimer’s Disease Research Center Core for providing a Pilot Award to Xinhua Zhan to perform these studies.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/14-3072r1).
REFERENCES
1 | Schneider JA, Arvanitakis Z, Bang W, Bennett DA(2007) Mixed brain pathologies account for most dementia cases in community-dwelling older personsNeurology69: 21972204 |
2 | Mayeux R, Stern Y(2012) Epidemiology of Alzheimer diseaseCold Spring Harb Perspect Med2: pii: a006239 |
3 | Pohjasvaara T, Erkinjuntti T, Ylikoski R, Hietanen M, Vataja R, Kaste M(1998) Clinical determinants of poststroke dementiaStroke29: 7581 |
4 | Desmond DW, Moroney JT, Paik MC, Sano M, Mohr JP, Aboumatar S, Tseng CL, Chan S, Williams JB, Remien RH, Hauser WA, Stern Y(2000) Frequency and clinical determinants of dementia after ischemic strokeNeurology54: 11241131 |
5 | Henon H, Durieu I, Guerouaou D, Lebert F, Pasquier F, Leys D(2001) Poststroke dementia: Incidence and relationship to prestroke cognitive declineNeurology57: 12161222 |
6 | Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM(2003) Silent brain infarcts and the risk of dementia and cognitive declineN Engl J Med348: 12151222 |
7 | Matute C, Domercq M, Perez-Samartin A, Ransom BR(2013) Protecting white matter from stroke injuryStroke44: 12041211 |
8 | Hijdra A, Verbeeten BJr, Verhulst JA(1990) Relation of leukoaraiosis to lesion type in stroke patientsStroke21: 890894 |
9 | Leys D, Henon H, Pasquier F(1998) White matter changes and poststroke dementiaDement Geriatr Cogn Disord9: Suppl 12529 |
10 | Liu H, Le W(2014) Epigenetic modifications of chronic hypoxia-mediated neurodegeneration in Alzheimer’s diseaseTransl Neurodegener3: 7 |
11 | Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R, Kalaria RN, Forster G, Esteves F, Wharton SB, Shaw PJ, O’Brien JT, Ince PG(2006) White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injuryStroke37: 13911398 |
12 | Iadecola C(2013) The pathobiology of vascular dementiaNeuron80: 844866 |
13 | Rahkonen T, Makela H, Paanila S, Halonen P, Sivenius J, Sulkava R(2000) Delirium in elderly people without severe predisposing disorders: Etiology and 1-year prognosis after dischargeInt Psychogeriatr12: 473481 |
14 | Dunn N, Mullee M, Perry VH, Holmes C(2005) Association between dementia and infectious disease: Evidence from a case-control studyAlzheimer Dis Assoc Disord19: 9194 |
15 | Verreault R, Laurin D, Lindsay J, De Serres G(2001) Past exposure to vaccines and subsequent risk of Alzheimer’s diseaseCMAJ165: 14951498 |
16 | Xu H, Stamova B, Jickling G, Tian Y, Zhan X, Ander BP, Liu D, Turner R, Rosand J, Goldstein LB, Furie KL, Verro P, Johnston SC, Sharp FR, Decarli CS(2010) Distinctive RNA expression profiles in blood associated with white matter hyperintensities in brainStroke41: 27442749 |
17 | Volpe JJ, Kinney HC, Jensen FE, Rosenberg PA(2011) The developing oligodendrocyte: Key cellular target in brain injury in the premature infantInt J Dev Neurosci29: 423440 |
18 | Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T(2002) The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNSJ Neurosci22: 24782486 |
19 | Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R(2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formationJ Neurosci26: 1012910140 |
20 | Zhan X, Kim C, Sharp FR(2008) Very brief focal ischemia simulating transient ischemic attacks (TIAs) can injure brain and induce Hsp70 proteinBrain Res1234: 183197 |
21 | Sato K, Higuchi M, Iwata N, Saido TC, Sasamoto K(2004) Fluoro-substituted and 13C-labeled styrylbenzene derivatives for detecting brain amyloid plaquesEur J Med Chem39: 573578 |
22 | Liu L, Chan C(2014) The role of inflammasome in Alzheimer’s diseaseAgeing Res Rev15C: 615 |
23 | Hiebert PR, Granville DJ(2012) Granzyme B in injury, inflammation, and repairTrends Mol Med18: 732741 |
24 | Liao MC, Ahmed M, Smith SO, Van Nostrand WE(2009) Degradation of amyloid beta protein by purified myelin basic proteinJ Biol Chem284: 2891728925 |
25 | Hoos MD, Ahmed M, Smith SO, Van Nostrand WE(2009) Myelin basic protein binds to and inhibits the fibrillar assembly of Abeta42 in vitroBiochemistry48: 47204727 |
26 | Zlokovic BV(2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disordersNat Rev Neurosci12: 723738 |
27 | Ulevitch RJ(1993) Recognition of bacterial endotoxins by receptor-dependent mechanismsAdv Immunol53: 267289 |
28 | Zhou H, Lapointe BM, Clark SR, Zbytnuik L, Kubes P(2006) A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharideJ Immunol177: 81038110 |
29 | Mun-Bryce S, Rosenberg GA(1998) Gelatinase B modulates selective opening of the blood-brain barrier during inflammationAm J Physiol274: R1203R1211 |
30 | Sherwin C, Fern R(2005) Acute lipopolysaccharide-mediated injury in neonatal white matter glia: Role of TNF-alpha, IL-1beta, and calciumJ Immunol175: 155161 |
31 | Vanguri P, Lee E, Henkart P, Shin ML(1993) Hydrolysis of myelin basic protein in myelin membranes by granzymes of large granular lymphocytesJ Immunol150: 24312439 |
32 | Spaeny-Dekking EH, Kamp AM, Froelich CJ, Hack CE(2000) Extracellular granzyme A, complexed to proteoglycans, is protected against inactivation by protease inhibitorsBlood95: 14651472 |
33 | Rucevic M, Fast LD, Jay GD, Trespalcios FM, Sucov A, Siryaporn E, Lim YP(2007) Altered levels and molecular forms of granzyme k in plasma from septic patientsShock27: 488493 |
34 | Simi A, Lerouet D, Pinteaux E, Brough D(2007) Mechanisms of regulation for interleukin-1beta in neurodegenerative diseaseNeuropharmacology52: 15631569 |
35 | Denes A, Lopez-Castejon G, Brough D(2012) Caspase-1: Is IL-1 just the tip of the ICEberg?Cell Death Dis3: e338 |
36 | Davaro F, Forde SD, Garfield M, Jiang Z, Halmen K, Tamburro ND, Kurt-Jones E, Fitzgerald KA, Golenbock DT, Wang D(2014) 3-Hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor (statin)-induced 28-kDa interleukin-1beta interferes with mature IL-1beta signalingJ Biol Chem289: 1621416222 |
37 | Edye ME, Lopez-Castejon G, Allan SM, Brough D(2013) Acidosis drives damage-associated molecular pattern (DAMP)-induced interleukin-1 secretion via a caspase-1-independent pathwayJ Biol Chem288: 3048530494 |
38 | Shamseddine AA, Airola MV, Hannun YA(2015) Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processesAdv Biol Regul57C: 2441 |
39 | Nikolova-Karakashian M, Karakashian A, Rutkute K(2008) Role of neutral sphingomyelinases in aging and inflammationSubcell Biochem49: 469486 |
40 | Nalivaeva NN, Rybakina EG, Pivanovich I, Kozinets IA, Shanin SN, Bartfai T(2000) Activation of neutral sphingomyelinase by IL-1beta requires the type 1 interleukin 1 receptorCytokine12: 229232 |
41 | Wong ML, Xie B, Beatini N, Phu P, Marathe S, Johns A, Gold PW, Hirsch E, Williams KJ, Licinio J, Tabas I(2000) Acute systemic inflammation up-regulates secretory sphingomyelinase in vivo: A possible link between inflammatory cytokines and atherogenesisProc Natl Acad Sci U S A97: 86818686 |
42 | Cogolludo A, Moreno L, Frazziano G, Moral-Sanz J, Menendez C, Castaneda J, Gonzalez C, Villamor E, Perez-Vizcaino F(2009) Activation of neutral sphingomyelinase is involved in acute hypoxic pulmonary vasoconstrictionCardiovasc Res82: 296302 |
43 | Filippov V, Song MA, Zhang K, Vinters HV, Tung S, Kirsch WM, Yang J, Duerksen-Hughes PJ(2012) Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseasesJ Alzheimers Dis29: 537547 |
44 | Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S(2007) LAMP proteins are required for fusion of lysosomes with phagosomesEMBO J26: 313324 |
45 | Rossner S, Ueberham U, Schliebs R, Perez-Polo JR, Bigl V(1998) p75 and TrkA receptor signaling independently regulate amyloid precursor protein mRNA expression, isoform composition, and protein secretion in PC12 cellsJ Neurochem71: 757766 |
46 | Braak H, Braak E(1997) Frequency of stages of Alzheimer-related lesions in different age categoriesNeurobiol Aging18: 351357 |
47 | Bartzokis G, Cummings JL, Sultzer D, Henderson VW, Nuechterlein KH, Mintz J(2003) White matter structural integrity in healthy aging adults and patients with Alzheimer disease: A magnetic resonance imaging studyArch Neurol60: 393398 |
48 | Bartzokis G, Sultzer D, Lu PH, Nuechterlein KH, Mintz J, Cummings JL(2004) Heterogeneous age-related breakdown of white matter structural integrity: Implications for cortical “disconnection” in aging and Alzheimer’s diseaseNeurobiol Aging25: 843851 |
49 | Bartzokis G(2011) Alzheimer’s disease as homeostatic responses to age-related myelin breakdownNeurobiol Aging32: 13411371 |
50 | Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME, Bowers WJ(2009) Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathologyGlia57: 5465 |
51 | Braak H, Del Tredici K, Schultz C, Braak E(2000) Vulnerability of select neuronal types to Alzheimer’s diseaseAnn N Y Acad Sci924: 5361 |
52 | Englund E, Brun A, Alling C(1988) White matter changes in dementia of Alzheimer’s typeBiochemical and neuropathological correlates. Brain111: Pt 614251439 |
53 | Zhan X, Jickling GC, Ander BP, Liu D, Stamova B, Cox C, Jin LW, DeCarli C, Sharp FR(2014) Myelin injury and degraded myelin vesicles in Alzheimer’s diseaseCurr Alzheimer Res11: 232238 |
54 | Zhan X, Jickling GC, Ander BP, Stamova B, Liu DZ, Kao PF, Zelin MA, Jin L-W, DeCarli C, Sharp FR(2015) Myelin basic protein associates with AβPP, Aβ 1-42, and amyloid plaques in cortex of Alzheimer brainJ Alzheimers Dis44: 12131229 |
55 | Upadhaya AR, Lungrin I, Yamaguchi H, Fandrich M, Thal DR(2012) High-molecular weight Abeta oligomers and protofibrils are the predominant Abeta species in the native soluble protein fraction of the AD brainJ Cell Mol Med16: 287295 |
56 | Sokolow S, Henkins KM, Bilousova T, Miller CA, Vinters HV, Poon W, Cole GM, Gylys KH(2012) AD synapses contain abundant Abeta monomer and multiple soluble oligomers, including a 56-kDa assemblyNeurobiol Aging33: 15451555 |
57 | Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE(2003) Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic miceNeurobiol Dis14: 133145 |
58 | Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, Shah GN, Price TO, Fleegal-Demotta MA, Butterfield DA, Banks WA(2009) Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: A mechanism for inflammation in therogression of Alzheimer’s diseaseBrain Behav Immun23: 507517 |
59 | Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA (2012) Lipopolysaccharide impairs amyloid beta efflux from brain: Altered vascular sequestration, cerebrosinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier J Neuroinflammation 9: 150 |
60 | Li J, Sheng W, Feng C, Zuo Z(2012) Pyrrolidine dithiocarbamate attenuates brain Abeta increase and improves long-term neurological outcome in rats after transient focal brain ischemiaNeurobiol Dis45: 564572 |
61 | Zetterberg H, Mortberg E, Song L, Chang L, Provuncher GK, Patel PP, Ferrell E, Fournier DR, Kan CW, Campbell TG, Meyer R, Rivnak AJ, Pink BA, Minnehan KA, Piech T, Rissin DM, Duffy DC, Rubertsson S, Wilson DH, Blennow K(2011) Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid beta levels in humansPLoS One6: e28263 |
Figures and Tables
Fig.1
Western blot analysis of brain samples of animals subjected to lipopolysaccharide /ischemia /hypoxia (LPS/IS/H). Animals were subjected to LPS/IS/H and sacrificed 4 weeks, 8 weeks, and 12 weeks later. Naïve animals were used as control. One animal in each time point was examined. The cortex, basal ganglia, and hippocampus ipsilateral to the focal ischemia and contralateral to the ischemia were taken for Western blotting. A) Staining of brain samples was performed using antibodies to myelin basic protein (MBP), interleukin-1β (IL-1β), and Granzyme B (Grzm B). Note decreased MBP in the ipsilateral hemisphere compared to control and contralateral hemisphere; appearance of degraded MBP (dMBP) in the ipsilateral and contralateral hemisphere; and the gradual induction of both IL-1β and Grzm B in the ipsilateral and contralateral hemisphere from 4 to 12 weeks after LPS/IS/H. β-actin was used as the loading control. B) LPS (MW ∼30 kDa) was detected on Western blot analysis in cortex ipsilateral and contralateral to the focal cerebral ischemia at 4 weeks, 8 weeks, and 12 weeks after the LPS/IS/H. Note increased LPS at 12 weeks compared to that at 4 or 8 weeks. LPS was absent in control brain. β-actin was used as loading control, showing somewhat less loading in the 8 week samples.
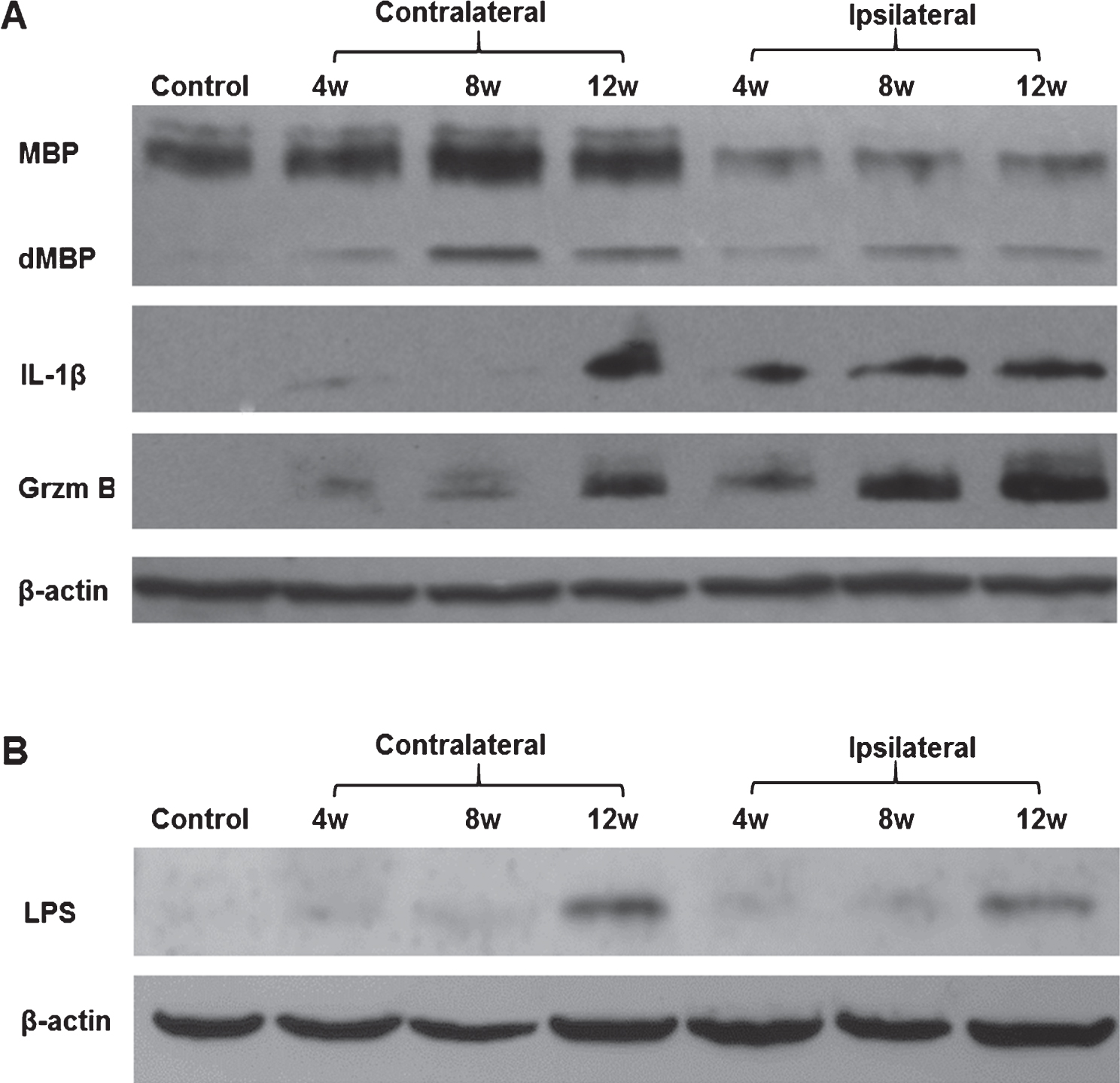
Fig.2
Western blot analysis for IL-1β and granzyme B in the cortex ipsilateral to ischemia at 12 weeks following LPS/IS/H and saline/IS/H. A) Interleukin-1β (IL-1β) was induced after saline/IS/H and LPS/IS/H. Note that two bands of proteins (around 150 kDa and 120 kDa) were detected following saline/IS/H and only one protein band (around 140 kDa) was detected following LPS/IS/H. B) Granzyme B was not detectable following saline/IS/H but was markedly induced after LPS/IS/H. β-actin was used as the loading control.
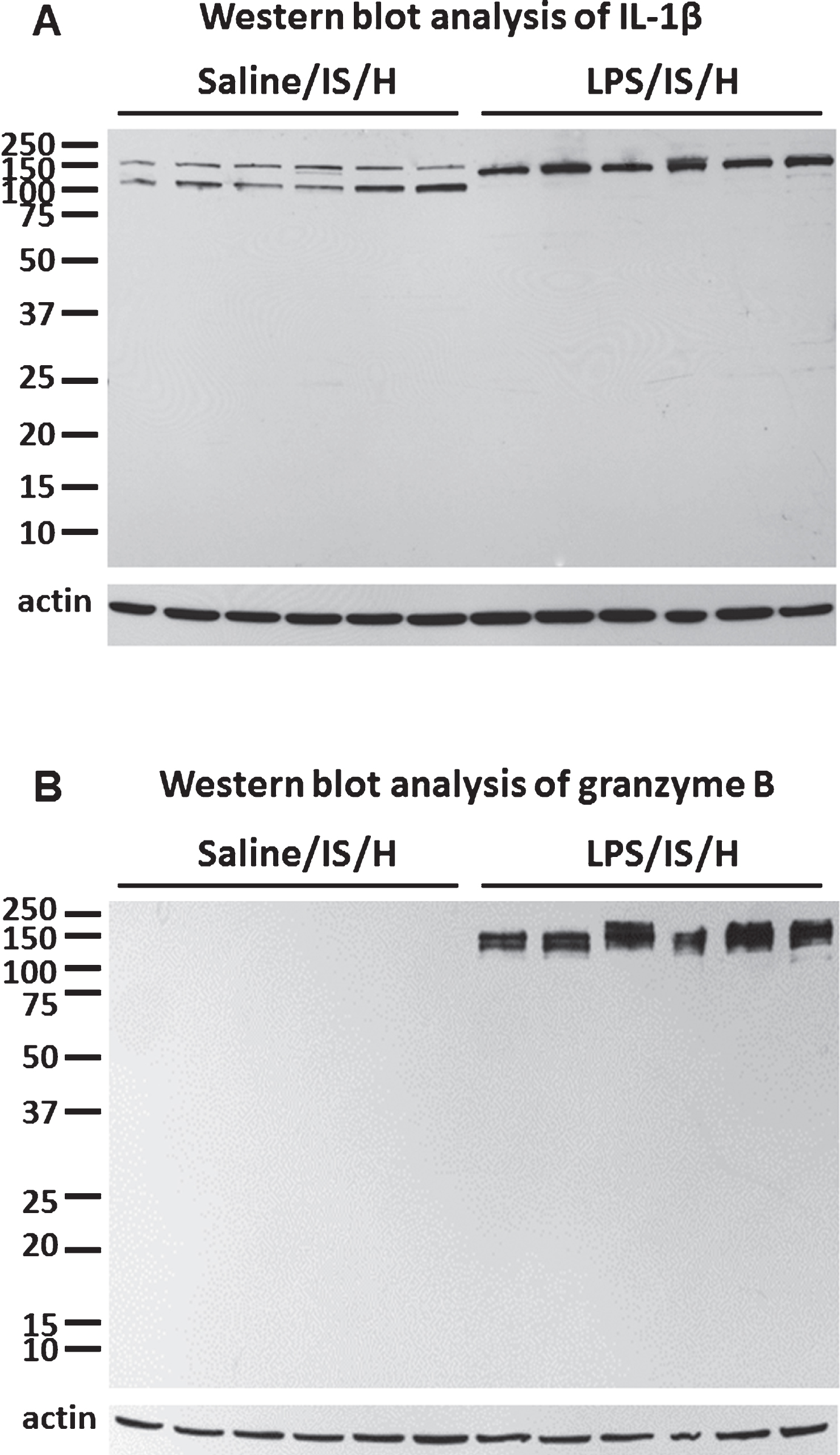
Fig.3
MBP staining in control cortex and one week following LPS/IS/H. A) Myelin basic protein (MBP) immunostaining of cortex from a control animal, and double immunostaining for axonal neurofilament light chain protein (NF-L) show that they co-localize for the most part (Merge). B) MBP immunostaining of cortex ipsilateral to the focal ischemia at one week post LPS/IS/H. Note staining in a pattern resembling blood vessels (white arrow) and lighter staining of axons throughout (figure from ipsilateral hemisphere but similar findings bilaterally). C) MBP and caveolin-1 (CVL1) double staining show that MBP is co-localized with endothelial cells (Merge) in cortex at 1 week post LPS/IS/H. Bar = 50 μm.
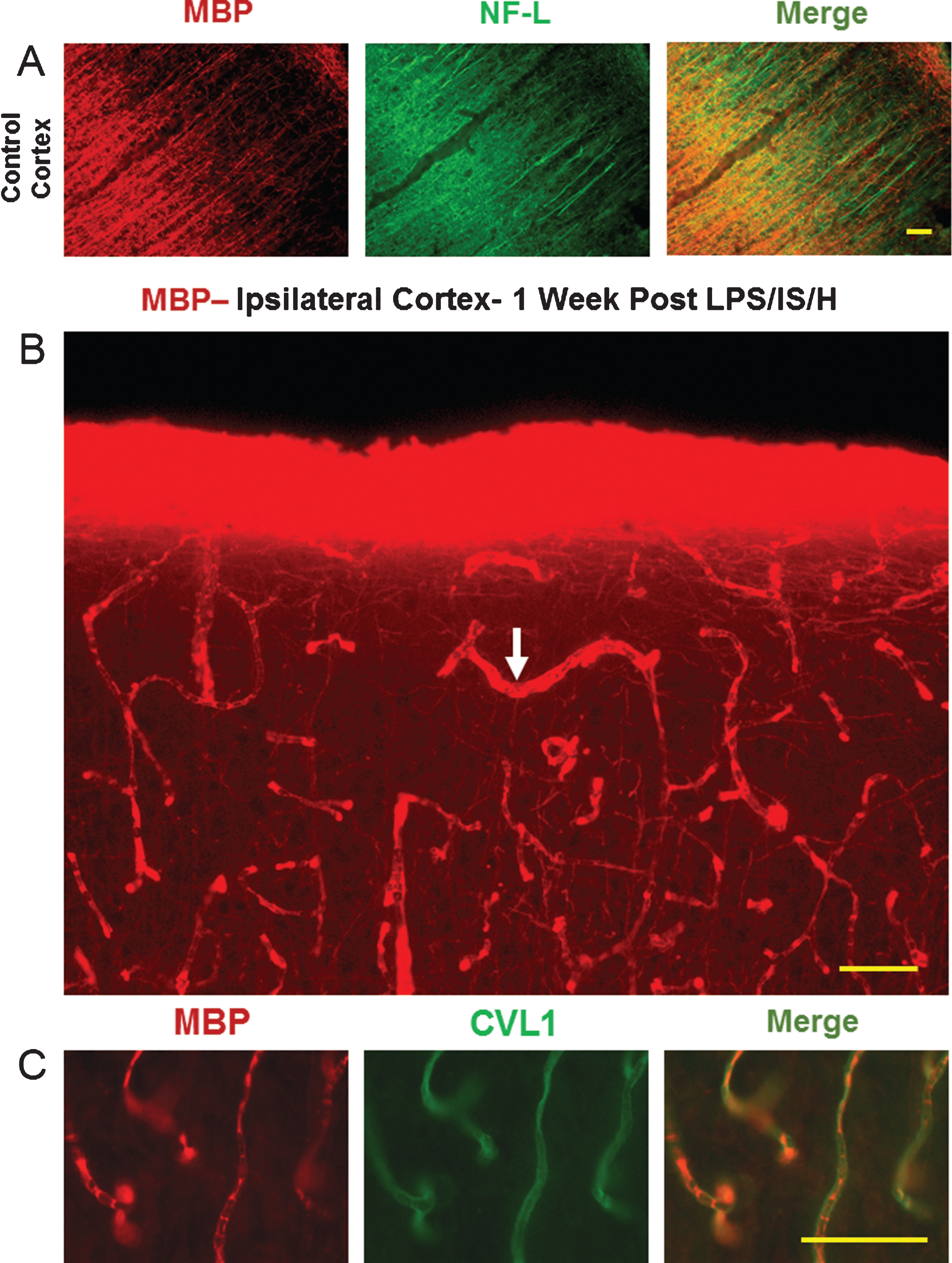
Fig.4
Tangles of dystrophic myelinated axons in cortex after LPS/IS/H. Myelin basic protein (MBP) immunostaining of cortex ipsilateral to the focal ischemia at 2 weeks (B), 4 weeks (A), 8 weeks (C), and 12 weeks (D) following LPS/IS/H. Note a tangle of enlarged MBP immunostained fibers that are focal and disorganized (A-D, white arrow). MBP staining of axons perpendicular and parallel to the cortical surface is observed which appears to be normal (A, figure from ipsilateral hemisphere but similar findings bilaterally). MBP immunostaining of the fibers in the tangle (E) co-localizes with axonal neurofilament light chain protein (NF-L) (F) as shown by the orange fibers in the merged image (G). These data suggest the tangles are composed of enlarged fibers that appear to be enlarged myelinated axons that are focal and disorganized. Bar = 50 μm.
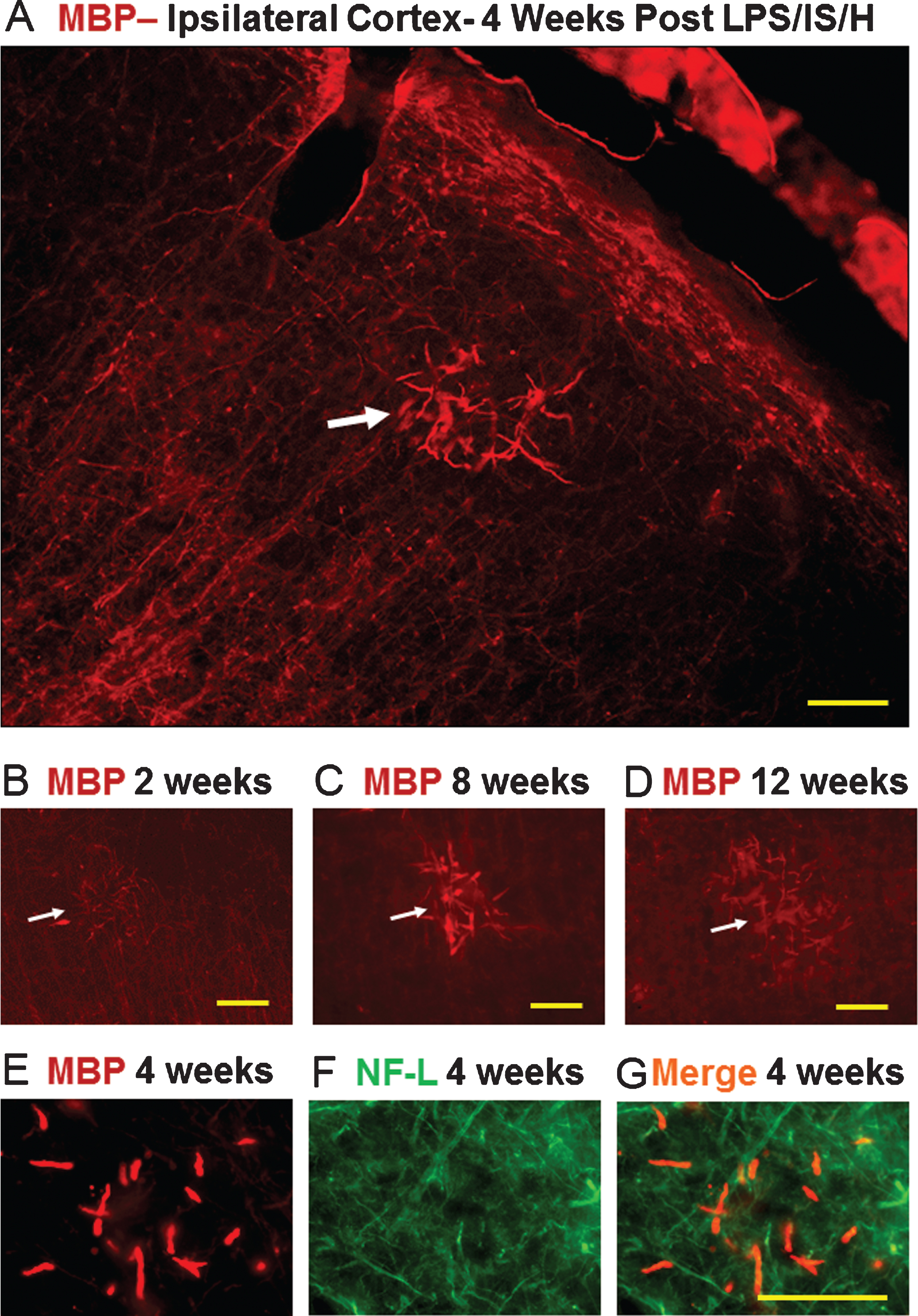
Fig.5
Localization of N-SMase2 in cortex of control rat and in rats 4 weeks and 12 weeks following LPS/IS/H. In control, N-SMase2 was localized to occasional cell bodies and to occasional axons, with N-SMase2 and neurofilament light chain protein (NF-L) co-localizing in these axons (N-SMase2/NF-L). At 4 weeks post LPS/IS/H, N-SMase2 was localized to large numbers of abnormal linear structures and some of these structures co-localized with NF-L (N-SMase2/NF-L). By 12 weeks post LPS/IS/H, N-SMase2 was again observed in a few scattered axons in cortex that co-localized with NF-L and appeared similar to controls. Bars = 50 μm.
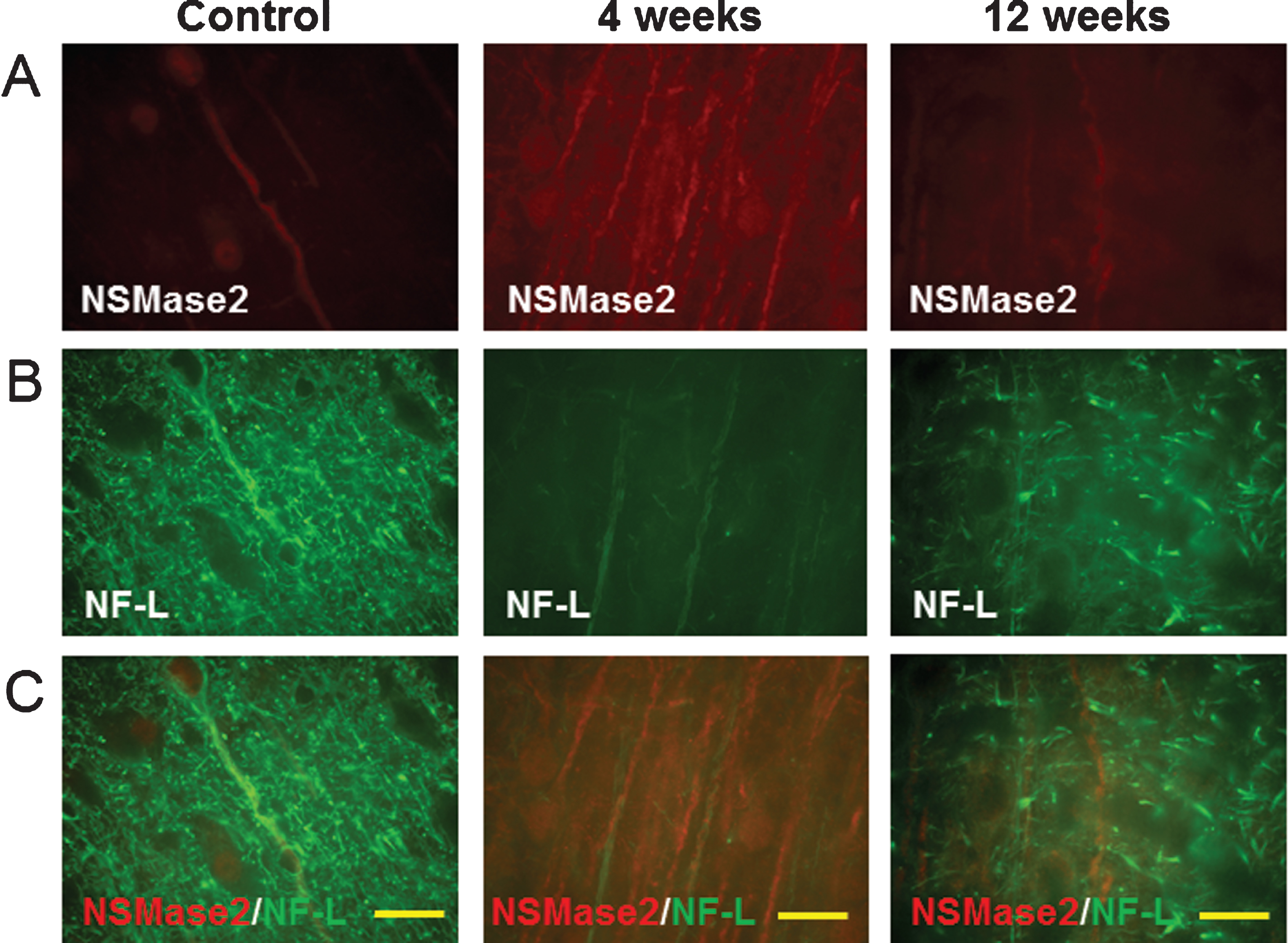
Fig.6
Double staining of myelin basic protein (MBP) with lysosomal-associated membrane protein 1 (Lamp1) in the cortex following LPS/IS/H. At 4 weeks post LPS/IS/H, focal areas of Lamp1 immunostaining and MBP immunostaining were co-localized (Merge). Bar = 50 μm.
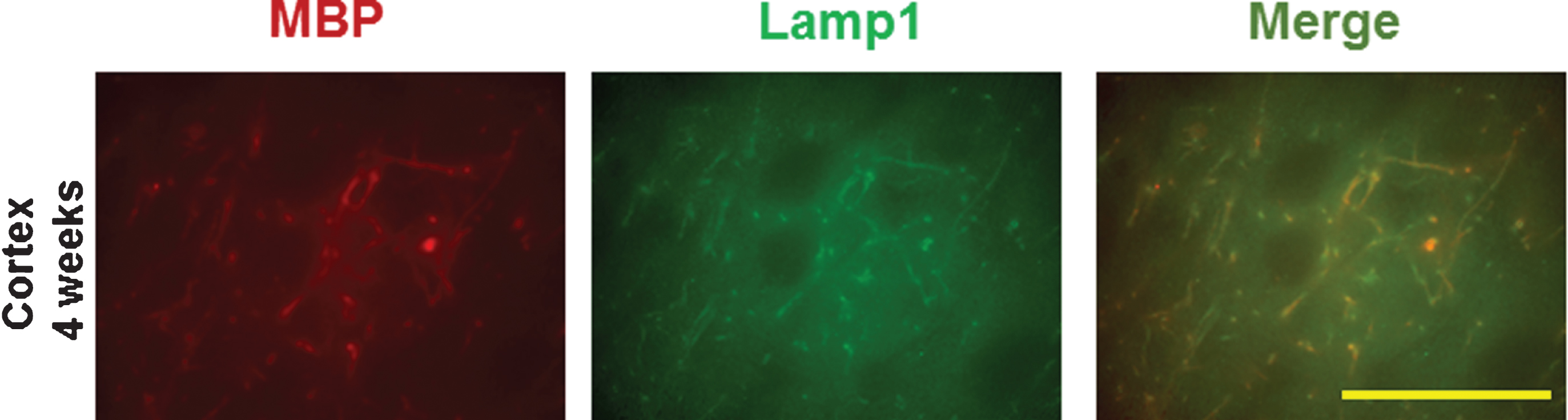
Fig.7
Myelin basic protein (MBP)/myelin loss and rodent Aβ deposition (AβPP) in the hemisphere ipsilateral to ischemia. A) Naïve control striatum. MBP immunostaining revealed large axon bundles in control striatum. There was no rodent Aβ (R Aβ) staining in normal brain including striatum. B) Striatum at 8 weeks post LPS/IS/H showed loss of normal MBP staining, and with deposition of R Aβ (which is likely AβPP protein) in these areas of MBP loss. Bars = 50 μm. C) Western blot analysis for R Aβ at 12 weeks following saline/IS/H and LPS/IS/H. A 100 kDa protein band was detected on Western blot for R Aβ either following saline/IS/H or following LPS/IS/H. Given this 100 kDa molecular weight, this protein is probably AβPP. β-actin was used as the loading control. D) The expression of R Aβ/AβPP following LPS/IS/H was significantly increased compared to that following saline/IS/H. **p < 0.01.
Fig.8
Western blot analysis for amyloid-β protein precursor (AβPP) in the hemisphere ipsilateral to ischemia at 12 weeks following LPS/IS/H and saline/IS/H compared to naïve control. A) A 100 kDa molecular weight protein was detected on Western blot analysis for AβPP either following saline/IS/H or following LPS/IS/H and in the naïve control. B) The expression of AβPP decreased in saline/IS/H animals compared to naïve, and LPS/IS/H increased AβPP compared to saline/IS/H. The expression of AβPP following LPS/IS/H was not significantly different compared to naïve controls. β-actin was used as the loading control. ***p < 0.001.
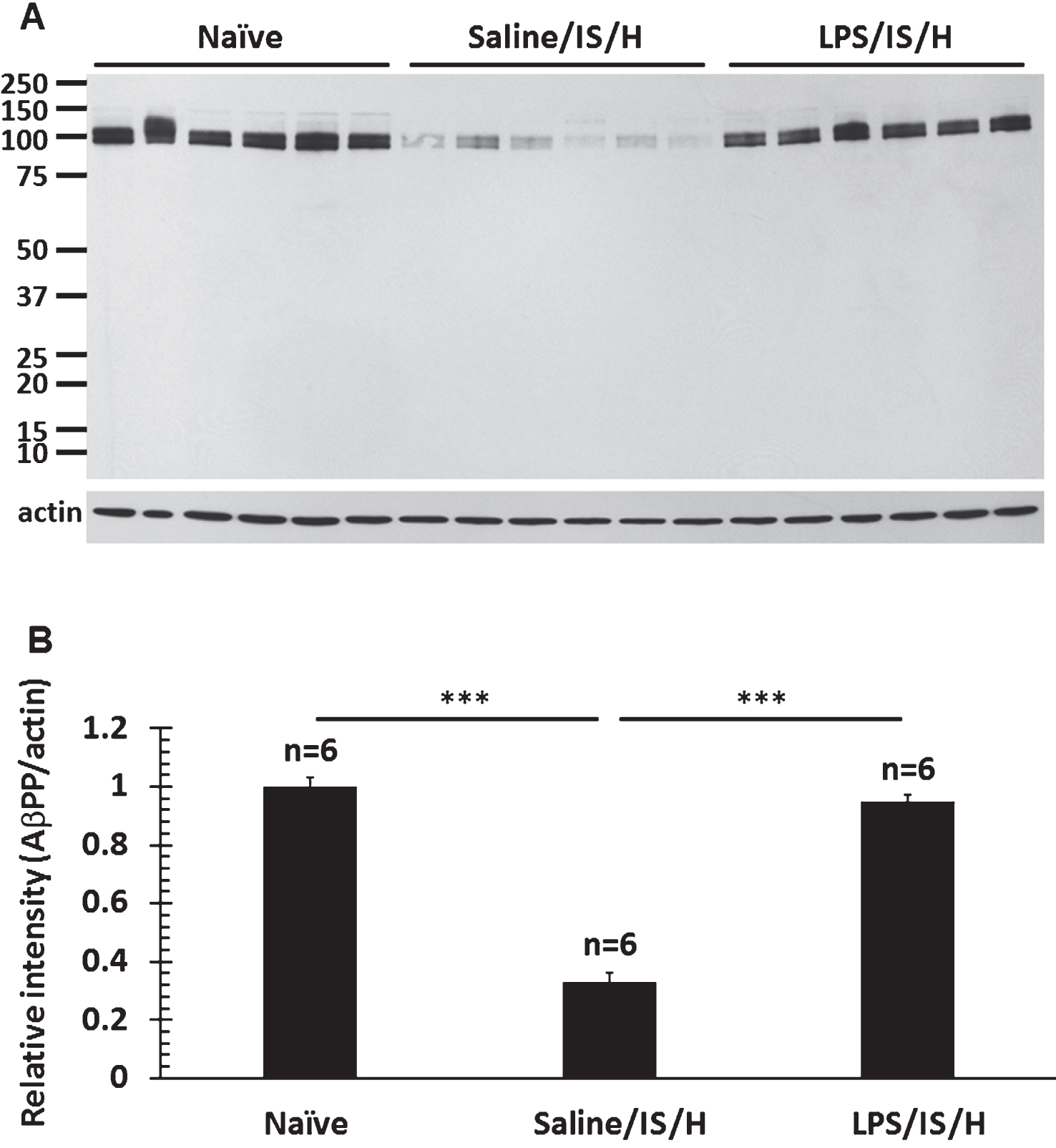
Fig.9
Co-localization of myelin aggregates with rodent Aβ and Aβ1 - 42 expression in the hemisphere ipsilateral to ischemia at 12 weeks following LPS/IS/H. A) Myelin aggregates stained with myelin basic protein (MBP) co-localized with rodent Aβ (R Aβ) deposits. Most of the MBP stained foci co-localized with rodent Aβ (white arrows, Merge) whereas some did not (yellow arrows). R Aβ likely represents AβPP. B) Myelin associated glycoprotein (MAG) co-localized with Aβ1 - 42 deposits. MAG immunostained foci in cortex co-localized with Aβ1 - 42 (B, Merge). Bars = 50 μm. C) Western blot analysis for Aβ1 - 42 at 12 weeks following saline/IS/H and LPS/IS/H. Two bands of ∼150 kDa and ∼140 kDa were detected on Western blots using the Aβ1 - 42 antibody which showed marked induction of both bands following LPS/IS/H compared to saline/IS/H. D) Quantification of the expression of Aβ1 - 42 showed that it markedly increased following LPS/IS/H compared to that following saline/IS/H. **p < 0.01.
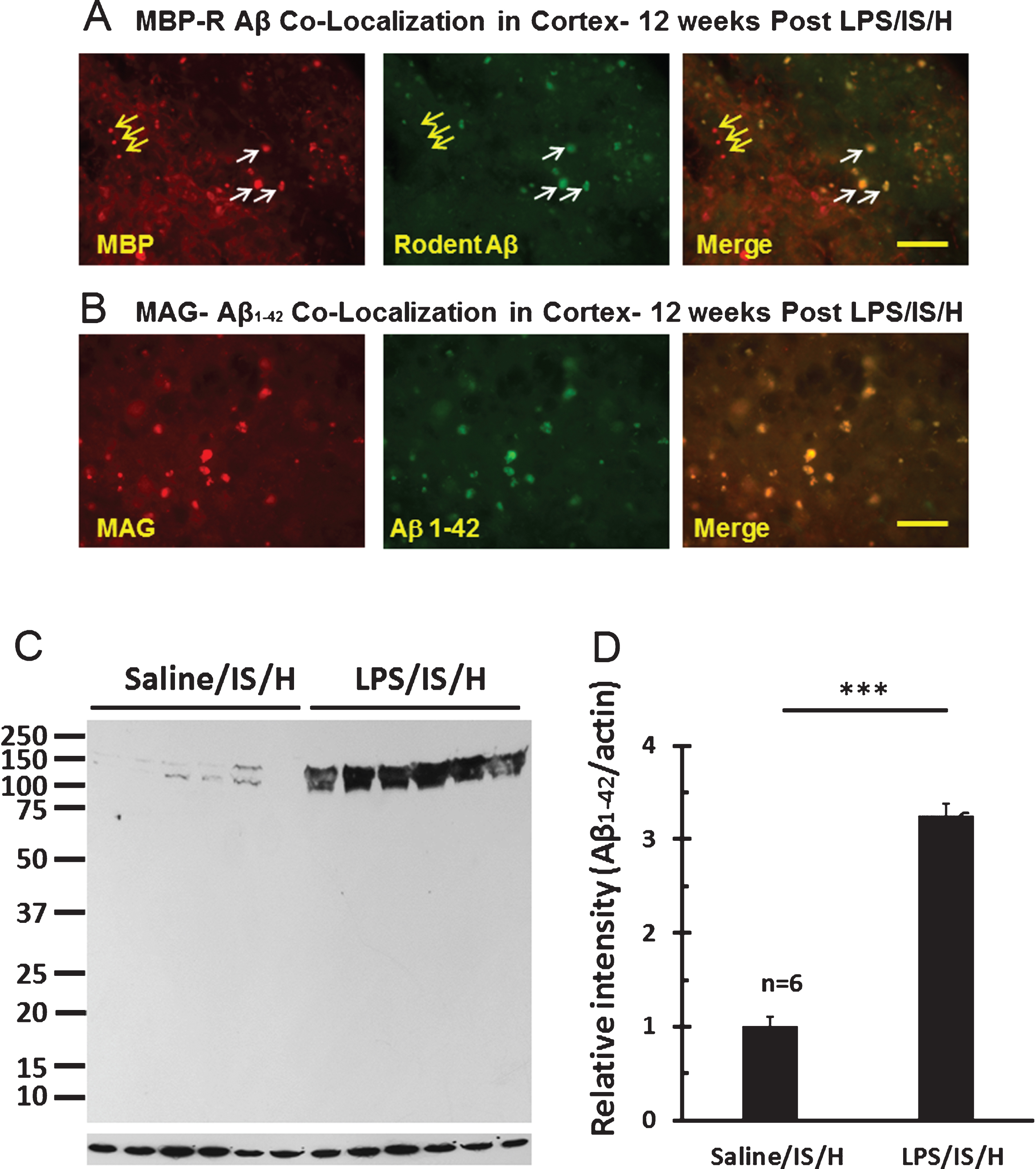
Fig.10
Western blot analysis for Aβ1 - 40/42 in the hemisphere ipsilateral to ischemia at 12 weeks following LPS/IS/H and saline/IS/H compared to naïve control. A) A protein with molecular weight as 150 kDa was detected on Western blot analysis for Aβ1 - 40/42 either following saline/IS/H or following LPS/IS/H and in naïve control. B) The expression of Aβ1 - 40/42 decreased following saline/IS/H compared to naïve, and Aβ1 - 40/42 increased following LPS/IS/H compared to saline/IS/H. The expression of Aβ1 - 40/42 following LPS/IS/H was not significantly different from naïve controls. β-actin was used as the loading control. ***p < 0.001.
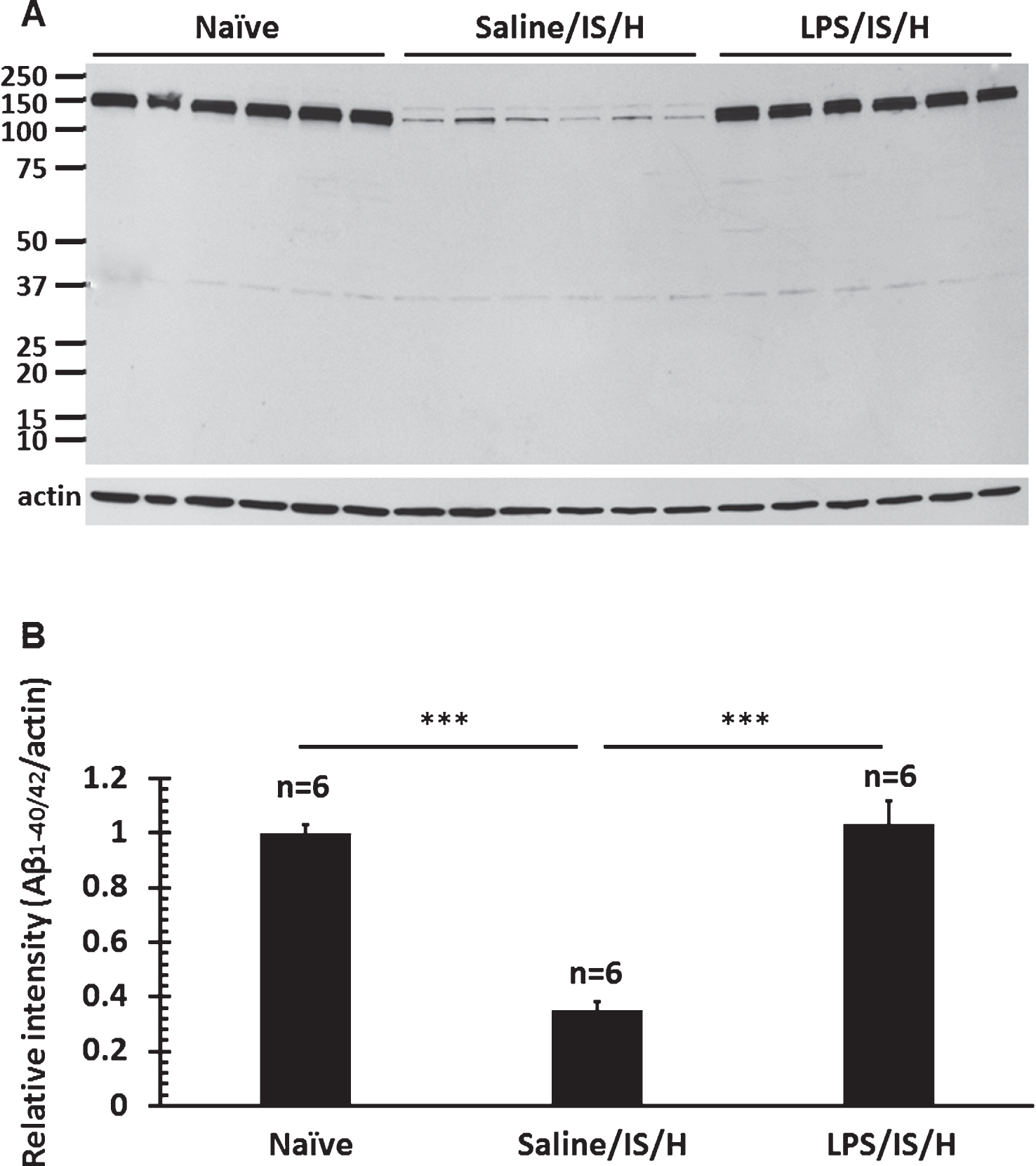
Fig.11
Double staining of myelin basic protein (MBP) and (E, E)-1-fluoro-2,5-bis (3-hydroxycarbonyl-4-hydroxy) styrylbenzene (FSB) in the amyloid plaques of 5XFAD mice. MBP positive myelin aggregates were detected in cortex of 8 month (8M, left panel) old (A) and 10 month (10M, left panel) old (b) 5XFAD mice. These MBP myelin aggregates co-localized (Merge) with FSB stained amyloid plaques in the cortex of 8 month old (8M, middle upper panel) and 10 month (10M, middle lower panel) old 5XFAD mice. MBP aggregates at 10 months (10M) seemed somewhat fragmented (B). The FSB stained plaques co-localized with MBP staining in 8M (upper right panel) and 10M (lower right panel) old animals (Merge). Bar = 50 μm.