Is Vasomotion in Cerebral Arteries Impaired in Alzheimer’s Disease?
Abstract
A substantial body of evidence supports the hypothesis of a vascular component in the pathogenesis of Alzheimer’s disease (AD). Cerebral hypoperfusion and blood-brain barrier dysfunction have been indicated as key elements of this pathway. Cerebral amyloid angiopathy (CAA) is a cerebrovascular disorder, frequent in AD, characterized by the accumulation of amyloid-β (Aβ) peptide in cerebral blood vessel walls. CAA is associated with loss of vascular integrity, resulting in impaired regulation of cerebral circulation, and increased susceptibility to cerebral ischemia, microhemorrhages, and white matter damage. Vasomotion— the spontaneous rhythmic modulation of arterial diameter, typically observed in arteries/arterioles in various vascular beds including the brain— is thought to participate in tissue perfusion and oxygen delivery regulation. Vasomotion is impaired in adverse conditions such as hypoperfusion and hypoxia. The perivascular and glymphatic pathways of Aβ clearance are thought to be driven by the systolic pulse. Vasomotion produces diameter changes of comparable amplitude, however at lower rates, and could contribute to these mechanisms of Aβ clearance. In spite of potential clinical interest, studies addressing cerebral vasomotion in the context of AD/CAA are limited. This study reviews the current literature on vasomotion, and hypothesizes potential paths implicating impaired cerebral vasomotion in AD/CAA. Aβ and oxidative stress cause vascular tone dysregulation through direct effects on vascular cells, and indirect effects mediated by impaired neurovascular coupling. Vascular tone dysregulation is further aggravated by cholinergic deficit and results in depressed cerebrovascular reactivity and (possibly) impaired vasomotion, aggravating regional hypoperfusion and promoting further Aβ and oxidative stress accumulation.
INTRODUCTION
Neurovascular homeostasis in the brain is maintained through the inherent ability of vascular resistance to adapt to changes in blood pressure, thereby preserving an adequate and stable cerebral blood flow (CBF) [1]. This adaptation mechanism, termed autoregulation, is essential as the brain has a high metabolic demand.
Cerebral autoregulation is the outcome of a complex interplay of vascular, neuronal, and metabolic components, in which the vascular tonic response to increased blood pressure (known as myogenic response [2]) plays an important role in maintaining an adequate level of tissue perfusion.
Arterial resistance, especially in the arteriolar circulation, is also modulated by spontaneous rhythmic variations in the vessel lumen resulting from smooth muscle dilatation and constriction. This intrinsic phenomenon, known as vasomotion [3–5], is unrelated to cardiac rhythm, and is observed clinically through related flow variations (flowmotion) using non-invasive technologies such as laser Doppler flowmetry.
Vasomotion has been observed in vitro and in vivo in various vascular beds including, among others, skeletal muscles, cutaneous and retinal circulation, coronary and mesenteric arteries [3, 5], and cerebral arteries [6–8].
Despite research efforts spanning over 150 years, both the underlying mechanisms and the functional implications of vasomotion remain to some extent unclear.
It has been suggested that vasomotion can improve perfusion [9, 10] and local tissue oxygenation [11, 12]. Although experimental evidence is currently limited, mathematical computational models also support the role of vasomotion in oxygen (O2) delivery [13–15].
In the current view, vasomotion is the outcome of multiple and interacting systems, in which the inhibition of one may unveil the role of another [5], thus explaining the heterogeneous manifestation of vasomotion in different vascular beds within the same organism. This heterogeneous distribution may represent an adaptation mechanism to meet the dynamically changing local metabolic demand of the perfused tissues [6, 16, 17].
Impaired cerebral autoregulation in Alzheimer’s disease and cerebral amyloid angiopathy
Alzheimer’s disease
Alzheimer’s disease (AD) is the most prevalent cause of dementia in the older population accounting for 65–70% of the cases [18–20]. The formation of amyloid-β (Aβ) plaques and neurofibrillary tangles are the hallmarks of AD [21]; however, autopsy studies also show symptoms of vascular pathology in the majority of AD cases [22–25].
The mechanisms underlying AD pathogenesis are not fully understood; however, a substantial body of evidence supports the hypothesis of a vascular contribution to pathophysiology [26–29], in which cerebral hypoperfusion and blood-brain barrier dysfunction are key elements [22, 26], aggravated by impaired cerebral microcirculation [30, 31].
Cerebral autoregulation deteriorates with age [32–34], increasing neuronal vulnerability to hypoxia and ischemia [32]. In transgenic mice overexpressing amyloid-β protein precursor, impaired endothelium-dependent vasodilatation and paradoxical vasoconstriction reduce basal CBF [35], resulting in impaired autoregulation [36]. In contrast with animal models, preliminary data from AD patients show no alterations of cerebral autoregulation [37]. These reports, however, are currently few and based on very small cohorts [38–40]. Furthermore, they rely on trans-cranial Doppler (TCD) techniques for noninvasive quantification of CBF (in the middle cerebral artery). These techniques measure relative velocity instead of absolute flow, under the assumption of constant arterial diameter. On the other hand, imaging techniques such as single-photon emission computed tomography (SPECT), have provided evidence of cerebrovascular deficit with reduced regional CBF in AD patients [41–44].
Cerebral amyloid angiopathy
Cerebral amyloid angiopathy (CAA), a cerebrovascular disorder characterized by the accumulation of Aβ peptide in the tunica media and adventitia of cerebral blood vessels, is associated with vascular smooth muscle cell degeneration and loss of vessel wall integrity [45], resulting in impaired regulation of cerebral circulation [46–50], vascular insufficiency and increased susceptibility to cerebral ischemia [51], microhemorrhages [52], and white matter damage [51].
CAA is found in elderly patients without dementia; however, its prevalence significantly increases in sporadic AD [23, 49], in which it is found in more than 60% of the cases [53]. CAA has been suggested to damage the contractile apparatus of cerebral vessels, and disrupt autoregulation [31].
Focus questions
Although vasomotion is thought to play an important role in cerebral autoregulation, specific studies in the context of AD or CAA in humans are limited— possibly owing to the experimental complexity of quantitative measurements. Furthermore, while experimental techniques for in vivo measurement of vasomotion in animals are available, their application to animal models of AD or CAA remains largely unexplored.
This study reviews the current literature on vasomotion, in the attempt to address two questions:
Question I: “Is impaired vasomotion in cerebral arteries implicated in the development or progression of sporadic Alzheimer’s disease?”
This question is motivated by the current view according to which vasomotion contributes to the regulation of tissue perfusion and O2 delivery [9–12].
Vasomotion can manifest as regular, rhythmic oscillations or chaotic fluctuations [3, 54]. Chaotic patterns in vasomotion have been suggested to reflect failure of vascular tone regulation, resulting in inadequate blood supply to the tissues [17].
Increasing incidence and magnitude of vasomotion have been observed during acute conditions associated with hypoxia [55], low blood pressure and hypoperfusion [4, 56, 57], suggesting that vasomotion may play a protective role in the attempt to preserve homeostasis in acute conditions.
However, in chronic conditions such as diabetes [58–61], obesity [5, 16], and hypertension [62, 63], vasomotion appears to be depressed. Reports from longitudinal studies suggest that diabetes [64–66], untreated hypertension [67], and obesity at midlife [68–70], are associated with an increased risk of developing AD, supporting the hypothesis of an indirect link between depressed vasomotion and AD. However, the association between the above risk factors and AD remains controversial as other longitudinal studies have reported contrasting results [71–74].
Question II: “Does impaired vasomotion hinder waste product drainage in Alzheimer’s disease?”
In spite of the high metabolic rate and sensitivity of brain parenchymal cells to alterations in the extracellular environment, the brain lacks lymphatic vessels for interstitial solute and fluid clearance [75], suggesting an alternative pathway should exist.
Although the underlying mechanisms remain to some extent unclear, two hypotheses on waste product clearance in the brain have been proposed.
Observations using in vivo two-photon imaging suggest that the cerebrospinal fluid from the subarachnoid space enters the brain parenchyma following para-arterial influx routes in the Virchow-Robin space surrounding the penetrating cerebral arteries [75, 76]. The resulting interaction with the brain interstitial fluid then acts to facilitate the clearance of interstitial solutes (including Aβ) via the paravascular spaces surrounding large draining veins. This mechanism has been reported to depend on the expression of water channel aquaporin-4 in astrocytes [75]. Owing to its functional analogy to peripheral lymph vessels, this pathway has been termed “glymphatic” system [75].
A contrasting view suggests that the interstitial solutes (including Aβ) from the perivascular space enter the basement membranes of capillaries and drain along the basement membrane of the arterial tunica media toward the leptomeningeal arteries, in opposite direction to blood flow [77–80].
The driving force of this pathway would be the reflected wave of systolic pulsations, assisted by a “valve effect” of proteins in the basement membrane [77]. These proteins, however, have not been identified.
In spite of the different drainage pathways, the two above hypotheses share the common view of arterial pulsation as driving force. The question then arises, whether vasomotion could contribute to such force.
The rhythmic oscillations of vasomotion have comparable amplitude to those caused by the systolic pulsation, in spite of the lower rate (1–20 cycles/min [3, 4, 81–83]). In rat basilar arteries [82], the amplitude of vasomotion (percentage diameter change) was 19% , with a baseline diameter of 259μm.
The amplitude of systolic pulsations in the common carotid artery of the adult mouse brain is calculated to 12% according to [84, 85] (Table 1 in [84], for wild type mice; assuming a baseline diameter of 400μm [84, 85]).
This suggests that vasomotion could contribute to the driving force of perivascular drainage of extracellular fluids.
VASOMOTION
Mechanisms underlying vasomotion in cerebral arteries
The ongoing debate on the mechanisms underlying vasomotion reflects the inherent complexity of this phenomenon, which involves interacting processes that are difficult to target experimentally [86].
However, it is accepted that vasomotion is initiated when asynchronous oscillations (waves) in Ca2 + concentration ([Ca2 +]i) within vascular smooth muscle cells become synchronized along the vascular wall[5, 6].
A substantial body of evidence has consistently shown in vitro that vasomotion is associated with oscillations in the membrane potential of vascular smooth muscle cells [86–91].
The voltage-dependent cell membrane oscillator
Experimental data from rat basilar arteries support the hypothesis of a voltage-dependent membrane oscillator [6]. According to this model, the smooth muscle cell membrane undergoes cyclic depolarization and hyperpolarization stages, associated with opening and closing of Ca2 +-dependent chloride channels (ClCa) and intermediate conductance potassium channels (IKCa), respectively.
Phase I: Vascular smooth muscle cell depolarization
Inositol 1,4,5-trisphosphate (IP3), produced by phospholipase C (PLC), leads to Ca2 + release through IP3 receptors from the sarcoplasmic reticulum (SR). Increased cytosolic Ca2 + concentration then activates Ca2 +-dependent chloride channels (ClCa), depolarizing the membrane and opening voltage-dependent Ca2 + channel to extracellular Ca2 + influx [6]. This activates ryanodine receptors resulting in further Ca2 + release into the cytosol, in a Ca2 +-induced Ca2 + release fashion [7].
Phase II: Vascular smooth muscle cell hyperpolarization
The increased [Ca2 +]i opens IKCa channels resulting in membrane hyperpolarization, and subsequent closing of voltage-dependent Ca2 + channels and vasodilatation [7].
Following the increase in phase I, [Ca2 +]i decreases again as a result of two cooperating mechanisms: a decreased Ca2 + release by the SR (possibly caused by transient refractoriness to IP3) and the active removal of Ca2 + from the cytosol, through SR and plasmalemmal Ca2 +-ATPases [86].
The loss of activation of ryanodine-sensitive stores then causes IKCa channels to close, completing the cycle [7].
Role of the endothelium in vasomotion
The oscillating membrane potential of vascular smooth muscle cells synchronizes [Ca2 +]i oscillations of adjacent cells through endothelium-mediated electrical coupling, via myoendothelial gap junctions [6–8]. As membrane potential oscillations in endothelial and smooth muscle cells were found to precede the rhythmic contraction of the vessel wall, it was suggested that the endothelium might initiate the synchronized vasomotion [7].
A substantial body of evidence from in vitro studies suggests that, in cerebral arteries, endothelial nitric oxide (NO) plays a role in the regulation of vasomotion. In particular, inhibition of NO-synthesis has been consistently reported to promote vasomotion [8, 17, 92–95].In [92], NO-synthase inhibitor Nω-nitro-L-arginine (L-NA) induced cyclic variations of cortical CBF in anesthetized rats. Consistent results were obtained in [17] by blocking basal NO release with L-NA. Exposure to NO inhibitor N-nitro-L-arginine methyl ester also evoked vasomotion [8, 93, 94].
In non-cerebral vascular beds, the role of the endothelium is less clear, with different and sometimes conflicting results. For example, removal of the endothelium or blockade of NO production prevented vasomotion in the hamster aorta [96], rat mesenteric arteries [88, 90, 97], rabbit mesenteric [98, 99], and coronary arteries [99], and also in the human cutaneous circulation [100], whereas vasomotion was initiated upon inhibition of endothelial NO in the hamster cheek pouch [101], and rat mesenteric artery [102]. Other studies have reported the absence of any endothelium-mediated effects on vasomotion in the rat thoracic aorta [103], rabbit mesenteric arteries [98], and pig coronary arteries [104].
Neurovascular coupling in vascular tone regulation and vasomotion
Astrocytes and the regulation of vascular tone
Mounting evidence from in vitro studies on animal brain slices suggest that astrocytes may participate in cerebral vascular tone regulation [105].
Astrocytes have indeed been shown to synthesize and release vasoactive agents such as NO, prostaglandins, epoxyeicosatrienoic acids (EETs), glutamate, adenosine and adenosine triphosphate (ATP) [106, 107], which are potential mediators of vascular tone regulation. The hypothesis of astrocyte involvement is further supported by the anatomy, as astrocytes are ideally situated in apposition to cerebral arterioles and capillaries, which would facilitate the relay of vasoactive signals [105].
Although studies on brain slices are limited by the inherent absence of cerebral perfusion, which implies the lack of myogenic and shear stress-induced modulation of vascular tone [108, 109], they provide evidence that Ca2 +-related events in perivascular astrocytes can influence vascular tone.
Conflicting results were reported by the early studies regarding the vascular response to astrocytic Ca2 + elevations such as induced by neuronal stimulation, metabotropic glutamate receptor (mGluR) stimulation, or direct Ca2 + uncaging— a technique based on local release of Ca2 + from an inert molecule exposed to UV photons [110, 111]. In [112], vascular constrictions were observed when uncaging-evoked Ca2 + waves propagated to the astrocyte end-feet and caused large increases in [Ca2 +]i. The authors postulated that increased [Ca2 +]i in the astrocyte end-feet could activate phospholipase A2 and increase arachidonic acid (AA) formation. AA would then diffuse to smooth muscle cells where it would be converted to 20-hydroxyeicosatetraenoic acid by CYP4A, a cytochrome P450 (CYP450) enzyme subtype, causing vasoconstriction. Contrasting results, however, have been reported by other studies in which vasodilatation rather than vasoconstriction was observed [113–115], suggesting that the build-up of AA in astrocytes could increase the production of prostaglandins and EETs, causing parenchymal arterioles to dilate [116].
Subsequent studies have reconciled these conflicting findings, suggesting that the polarity of vascular responses (vasoconstriction versus vasodilatation) could depend on basal conditions such as resting arteriolar tone [117], O2 concentration levels [118], and NO availability [119]. The level of astrocytic increase of [Ca2 +]i following stimulation has also been suggested to play a role [120].
In [117], astrocytic stimulation by mGluR agonist trans-1-aminocyclopentane-1,3-dicarboxylic acid (t-ACPD) induced vasoconstriction in arterioles with moderate basal tone (diameter greater than 70% of maximum), and vasodilatation in arterioles with higher basal tone. In [118], t-ACPD stimulation caused arteriolar dilatation in conditions of low O2 concentration (20% ) and arteriolar constriction at high O2 concentration (95% ). Vasodilatation under low O2 was shown to depend on enhanced lactate release by activated astrocytes and increased extracellular levels of the vasodilating agent prostaglandin E2. The authors also observed increased extracellular levels of adenosine in low O2 and suggested this might act upon A2A receptors in smooth muscle cells to reduce Ca2 + channel activity and inhibit contraction.
In [119], astrocyte-induced vasoconstriction was attributed to NO interactions with AA metabolites due to NO-mediated inhibition of CYP450 and the subsequent decrease in the formation of vasodilating EET.
In [120], the level of astrocyte end-feet Ca2 + determined the polarity of arteriolar response, with increasing values shifting the vascular tone response from vasodilatation to vasoconstriction. The response was attributed to the activation of Ca2 +-activated large conductance K + channels (BK) by the increased Ca2 + concentration, which caused extracellular K + increase. This, in turn caused vasodilatation at physiological concentration levels (3 mM), and vasoconstriction at higher concentrations (8 mM).
Astrocytes mediate functional hyperemia and suppression of vasomotion
Astrocytes have been proposed to mediate vasodilatation of parenchymal arterioles in response to increased neuronal activity. This mechanism, termed ‘functional hyperemia’, increases O2 and glucose supply to the active neurons in a timely and spatially localized manner [106, 116].
In [106], increased neuronal activity by electrical stimulation, increased astrocytic [Ca2 +]i, and evoked Ca2 + waves travelling from the soma towards the end-foot. The increased Ca2 + in astrocytes was rapidly signaled to the surrounding parenchymal arterioles, which had been pre-constricted with thromboxane A2 (TXA2) agonist U46619, where it suppressed [Ca2 +]i oscillations and vasomotion. Consistent with the above, the same study also found that the activation of astrocytic glutamate receptors, which is known to increase [Ca2 +]i in cortical astrocytes [121], had similar effects. Consistent findings have been reported also in [122] and [123] using similarpreparations.
Furthermore, in [123] epoxygenase inhibitor miconazole caused arteriolar vasoconstriction and increased vasomotion oscillation frequency, suggesting that EETs may act as mediators of neuronal-activity-related dilatation of parenchymal arterioles and dampening of vasomotion.
CAA and vasomotion
In CAA, Aβ interacts predominantly with vascular smooth muscle cells in the tunica media causing structural alterations such as the disruption of actin in the cytoskeleton, and apoptosis [124–126]. The loss of vascular smooth muscle cells with disease progression weakens the vascular wall [127–129], increasing the susceptibility to abnormal vasodilatation and hemorrhage [126, 130, 131].
Aβ deposition also affects the luminal area of arterial vessels. However, experimental data are conflicting as both increase [127, 129] and decrease [128] of the internal lumen have been reported. In the former case, which is also supported by transgenic mouse studies [130], vessel dilation could reduce the vascular reactivity to vasodilatory stimuli such as ischemia [126, 132] or hypercapnia [133] (see section on cerebrovascular reactivity). In the latter case, vasoconstriction could result in hypoperfusion downstream of the narrowing of the vascular lumen [126].
However, while the experimental evidence described above can be seen as the long-term outcome, in the short-term other processes could take place, with detrimental effects on vasomotion at an early stage.
Indeed, vascular smooth muscle cells treated with Aβ1 - 40— the most frequent form of Aβ in sporadic CAA— exhibit higher inflammatory response to interleukin-1β [134]. This cytokine is expressed by endothelial cells exposed to Aβ1 - 40 [135]. As endothelial cells are adjacent to smooth muscle cells, the above finding suggests that Aβ could mediate the inflammatory response of smooth muscle cells, leading to the loss of contractile function through the disruption of α-actin in the cytoskeleton [134].
Findings from in vitro studies on neuronal cells suggest that the detrimental effects of Aβ could also impair intracellular Ca2 + dynamics as well as the membrane potential [136, 137], resulting in the loss of synchronization of cytosolic Ca2 + waves, a pre-requisite of vasomotion according to the membrane oscillator model [6].
The effect of oxidative stress
Oxidative stress has been implicated in AD and CAA from the early stages of the disease [24, 49, 138–140]. As this condition is associated with mitochondrial dysfunction [138, 141, 142], which results in ATP deficiency, it seems plausible that in AD/CAA, ATP-dependent transport might be impaired not only in neurons but also in vascular smooth muscle cells, resulting in altered Ca2 + homeostasis and disruption of vasomotion.
Vascular tortuosity and vasomotion
The long path of arterioles supplying the deep white matter tends to become tortuous with aging [33, 143, 144]. Tortuosity manifests in the form of turns and curls requiring increased perfusion pressure to maintain stable CBF [33]. It has been suggested that decreased pulsations in tortuous vessels may impede interstitial fluid flow, reducing Aβ clearance in perivascular spaces [143]. Although this suggests a potential implication of increased tortuosity in AD, existing reports are inconclusive [144, 145].
In tortuous arterial vessels, vasomotion could act as a protective mechanism by cooperating with the systolic pulse in the attempt to compensate the dispersion of kinetic energy of blood flow caused by turns and curls, and preserve adequate CBF.
Disturbed blood flow such as occurring at turns and loops of tortuous vessels is known to reduce the wall shear stress exerted by blood flow [146–148]. On the other hand, reduced wall shear stress is associated with reduced endothelial NO production [149], a condition which has been shown in vitro to promote vasomotion [8, 17, 92–95]. This further supports the hypothesis of a potential protective role of vasomotion in the presence of tortuous arteriolar paths. However, given the intrinsic difficulty in measuring vasomotion in conditions of tortuous blood flow in vivo, this hypothesis has not been tested.
Temporal variability of vasomotion
The changes in vessel diameter observed in vasomotion are identified as “rhythms”, whereas the magnitude of the diameter oscillations is referred to as “amplitude” of vasomotion.
The rhythms of vasomotion exhibit different characteristics in different tissues of the body, and are characterized by different temporal patterns.
The rhythms in vasomotion can be highly regular— nearly following a periodic sinusoidal wave (see for example [6, 8, 86, 150])— or characterized by higher complexity [3, 54, 151]. With increasing complexity, these rhythms tend to lose their periodic nature and approach a chaotic behavior, which is thought to reflect the limit beyond which vasomotionfails [17].
In [152], vasomotion was studied as a function of arteriolar diameter. Arterioles with baseline diameter in the range 50–100μm showed rhythmic oscillations at rates of 2–3 cycles/min, and magnitude of 10–20% . Rate and magnitude were found to increase with decreasing vessel diameter, exhibiting oscillation rates of 10–25 cycles/min in terminal arterioles.
Vasomotion is also dependent on the intravascular pressure. In both isolated pressurized rat cerebral arteries and in vivo measurements from rat basilar artery, the amplitude of vasomotion has been shown to decrease (and frequency to increase) with increasing pressure [62, 82]. This finding supports the idea of vasomotion as a protective mechanism, which potentiates cerebral autoregulation in the lower blood pressure range, a condition in which maintaining adequate cerebral perfusion becomes critical [5].
A neurogenic component of vasomotion, manifesting at low frequencies (between 20 and 40–60 mHz, approximately corresponding to 1–4 cycles/min) has also been reported [16, 153].
Cerebrovascular reactivity and vasomotion
In normal conditions, resistance vessels in the vascular system dilate or constrict in response to exogenous stimuli. This can be seen as the ability of blood flow regulation to respond to hemodynamic challenges beyond the resting conditions. This phenomenon is distinct from the vascular response to changes in blood pressure (autoregulation) [154].
Hypercapnic stimuli such as breath holding [155], increased inspiratory concentration of carbon dioxide [156], and acetazolamide administration [157], induce a hyperemic response which causes resistance vessels to dilate [158]. This response is thought to be mediated by increased pH, activation of K + channels endothelial hyperpolarization and subsequent [Ca2 +]i decrease in vascular smooth muscle cells, and increased NO synthesis by endothelial cells or neurons [154].
When cerebral vessels are already dilated to compensate for a pathological reduction in cerebral perfusion pressure, the above vasodilating stimuli may become ineffective. The ability of blood vessels to dilate further in response to a dilatory stimulus (or, conversely, constrict in response to vasoconstrictor stimuli) is referred to as cerebrovascular “reactivity” (CVR) or “reserve” [157, 154] (also termed “cerebral vasomotor reactivity”). The vasodilatory reserve has been studied more extensively than the vasoconstrictor reserve, as noninvasive capnic stimuli evoke the former with greater magnitude [154].
Measuring CVR can help identify patients with reduced cerebral perfusion pressure, such as caused by occlusive lesions of the brain-supplying arteries [157, 159, 160]. CVR in middle cerebral arteries has been reported to predict cerebral ischemia (stroke and transient ischemic attack) in patients with carotid occlusion [160–162] and asymptomatic carotid stenosis [160]. Reduced CVR has also been observed in AD patients [163, 164] and has been associated with cognitive decline [165].
Impaired cerebral vasoreactivity, namely the inability of dilated vessels to dilate further in response to vasodilatory stimuli, could also reduce or suppress vasomotion. Indeed, in conditions of pronounced vasodilatation such as observed in functional hyperemia [106] or the administration of nitroglycerin [82], vasomotion is suppressed. Furthermore, depressed CVR occurring in conditions of chronically low perfusion pressure may be accompanied by impaired vasomotion as an indirect effect of the insufficient O2 supply which alters the metabolism of vascular cells, impairing intracellular Ca2 + dynamics [166]. In light of the above, the observation of impaired vasoreactivity in CAA [133, 167, 168] suggests a potential indirect implication of CAA in vasomotion disruption.
Evaluation of cerebrovascular reactivity in AD by Doppler ultrasonography
CVR can be indirectly and noninvasively assessed by TCD. This method is used to measure changes in blood flow velocity in selected cerebral arteries (e.g., the middle or the posterior cerebral artery) as surrogate of the changes in CBF [157, 160]. Assuming that the vessel diameter remains nearly constant during the administration of the hypercapnic stimulus, the change in blood flow velocity measured by TCD reflects the change in CBF in response to the stimulus. Using this technique, observations of impaired autoregulation and vascular reactivity in transgenic mouse models of CAA have been confirmed in AD patients [158, 163–165, 169] and extended to vascular dementia [170]. Bär and colleagues [158] have shown that a possible cause of CVR impairment in AD might be cholinergic deficiency (a condition which has been reported in AD [171–173]), as the administration of galantamine (a substance with acetylcholinesterase inhibitor properties) increased CVR in their cohort. As vasomotion has been suggested to contain a neurogenic component, cholinergic deficiency might also affect (dampen) vasomotion, due to the cholinergic intrinsic innervation of parenchymal cerebral arteries.
A reduced CVR has also been shown to predict a worse Mini-Mental State Examination score at 12 months follow-up in AD patients with severe internal carotid artery stenosis [174], suggesting that impaired vasoregulation may be indicative of faster progression of cognitive decline in AD. Buratti and colleagues [175] found CVR depression in AD patients to be associated with the presence of obstructive sleep apnea.
CVR has also been reported to be reduced in CAA patients with a history of CAA-related hemorrhages [169].
Evaluation of cerebrovascular reactivity in AD by SPECT
Imaging of regional CBF with SPECT has been used in the quantitative assessment of cerebrovascular deficits [154] in various conditions including AD [41–44, 176–180]. Owing to the associated costs and complexity, this technique has been used in a limited number of studies compared to TCD, in the assessment of CVR. Vascular activation in response to acetazolamide (hypercapnic stimulus) has been investigated in AD patients [177–179]. However, in [177] and [179], direct comparisons between AD patients and a control group were not reported, preventing the inference of CVR impairment in AD. In [178], CVR was reduced in AD patients compared with controls, and CVR correlated with Mini-Mental State Examination score in the AD group. Pavics and colleagues [180] assessed the ability of CVR response to acetazolamide in discriminating AD from vascular dementia by SPECT analysis of regional CBF. The majority (73% ) of AD patients (n = 33) showed no CVR alteration compared to pre-injection baseline. However, comparative results with respect to the control group were not reported.
In summary, SPECT studies have not shown substantial evidence of CVR impairment in AD [154].
Experimental approaches for measuring vasomotion: Challenges and future perspective
Due to the intrinsic difficulty in measuring vasomotion, most of the data are based either on in vitro experiments of isolated arteries, or in vivo experiments on anesthetized animals (typically rat, hamster, orrabbit).
While in vitro approaches provide for the most precise measurement of the arteriole diameter changes which underpin vasomotion, insights are limited because of the lack of natural perfusion and non-physiological neurochemical environment. The use of anesthesia within in vivo studies is also problematic because of the complex and varied effects of anesthetic agents upon physiological, cerebrovascular, and neurovascular function [181].
Furthermore, as the measurement of vasomotion in the strict sense (i.e., modulation of the vessel diameter) requires complex experimental settings, many studies rely on flowmotion (i.e., modulation of blood flow velocity) as a surrogate measure. Although the two quantities are related, they are distinct as blood flow velocity can change without significant changes in vessel diameter.
The advances in optical techniques have now introduced a new perspective for noninvasive hemodynamic measurements in vivo, and in awake state [181, 182]. Advanced spectroscopic techniques (the reader is referred to [183, 184] for review) provide quantitative measurements of dynamic changes not only of vessel diameter, but also of blood volume, blood flow, and tissue oxygenation. Multi-photon microscopy methods can additionally be used to probe the specific cellular drivers of vasomotion and the mechanisms by which structural or biochemical changes can impact upon vasomotion as well as other aspects of cerebrovascular or neurovascular function [182, 185]. Important advances in the understanding of the role of vasomotion may now arise as these techniques are applied to study vasomotion in the increasingly wide range of animal models of AD and/or CAA.
In light of the literature reviewed in this study, some specific hypotheses could be tested using these models and in vivo quantitative imaging techniques:
H1) Vasomotion is present in tortuous vascular beds. The magnitude of vasomotion oscillations exhibits an inverse-U shaped relationship with respect to the degree of tortuosity.
H2) Vasomotion oscillations are driven differentially according to cell type. Cellular resolution in-vivo microscopy techniques could be used to better delineate the endothelial, neurogenic or myogenic contributors to concurrently measured vasomotion oscillations.
H3) Depressed cerebrovascular reactivity is associated with reduced vasomotion (both magnitude and frequency of oscillations). Cerebrovascular reactivity could be measured (and manipulated) in vivo and relationships to specific sources of vasomotion characterized.
H4) Vasomotion is depressed in diabetes and in hypertension. Multimodal imaging methods (such as optical spectroscopy and multiphoton imaging) could be applied to investigate vasomotion oscillations in animal models of these conditions.
IMPAIRED CEREBRAL VASOMOTION IN AD
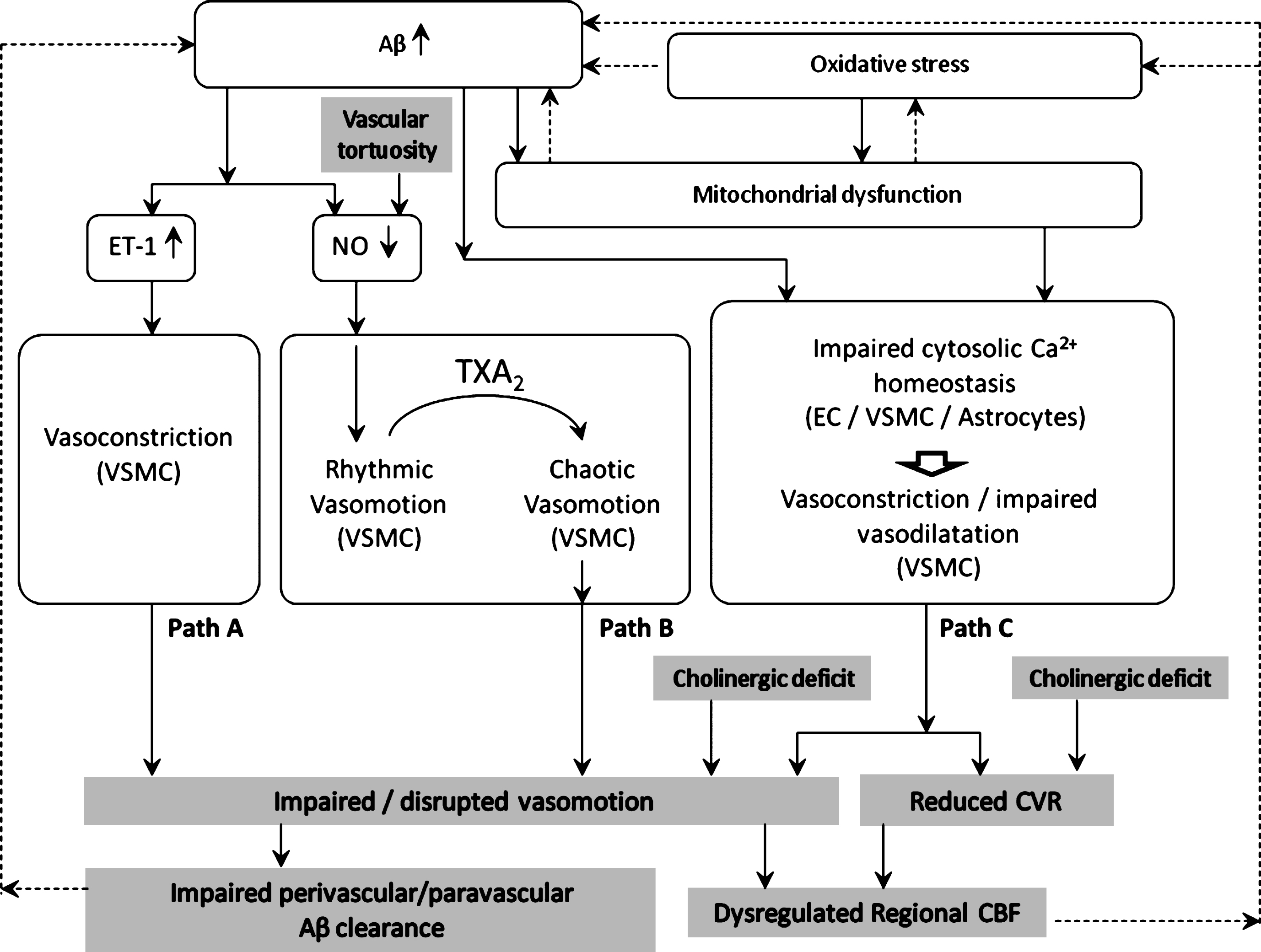
Possible pathways affecting vasomotion in AD/CAA (Question 1)
The available data suggest that impaired vasomotion may be implicated in the development or progression of AD/CAA. Different pathways can be hypothesized, as illustrated in Fig. 1.
Path A: ET-1-induced vasoconstriction depresses vasomotion
Endothelin-1 (ET-1) is a potent vasoconstrictor produced predominantly by vascular endothelial cells [186]. ET-1 is generated from its inactive precursor through cleavage by endothelin converting enzyme (ECE)-1 or ECE-2 [187]. The former is expressed in endothelial cells, in neurons and astrocytes [188], and in smooth muscle cells [189].
Substantial evidence suggests that the upregulation of ET-1 observed in AD, both in the cerebral cortex [186, 190] and in cerebral blood vessels [191, 192], is caused by the accumulation of Aβ [186, 187, 192–194].
ET-1 causes contraction of vascular smooth muscle cells by multiple signaling pathways [195], including, among others, the PLC/IP3 cascade. IP3 in turn stimulates endoplasmic reticulum (ER) release of Ca2 +, and protein kinase C activation, ultimately resulting in myosin light chain phosphatase inhibition and consequent cell contraction.
The vasoconstriction caused by upregulated ET-1 could attenuate the amplitude of vasomotion [196] or inhibit it. This is consistent with a mathematical model of vasomotion [197] in which smooth muscle cells exhibited synchronous [Ca2 +]i oscillations and vasomotion at intermediate [Ca2 +]i, shifting to tonic contractions and vasomotion suppression at greater [Ca2 +]i.
Path B: Reduced endothelial NO production may promote rhythmic vasomotion (protective) or chaotic vasomotion (detrimental)
Aβ-endothelium interaction causes decreased NO production [198, 199]. According to most experimental evidence, NO inhibition promotes vasomotion [8, 17, 92–95], which could be interpreted as a protective effect potentiating cerebral perfusion and O2 delivery.
However, administration of the thromboxane TXA2 analog U46619 has been shown to elicit irregular/chaotic vasomotion in middle cerebral arteries pre-conditioned with L-NA-induced NO blockade [17]. As TXA2 production (by activated platelets) is enhanced in AD [200], and L-NA mimics the effect of Aβ-induced endothelial NO synthesis inhibition, the above report suggests a plausible pathway for vasomotion disruption in AD.
Endothelial NO synthesis could also be reduced in tortuous vessels as a consequence of reduced shear-stress [149] caused by disturbed blood flow [146–148].
Path C: Altered Ca2 + homeostasis dysregulates vasomotion
Multiple factors can influence and impair intracellular Ca2 + homeostasis in the vascular wall and astrocytes. Aβ accumulation, mitochondrial dysfunction and oxidative stress have both individual and combined effects on Ca2 + homeostasis.
Aβ has been shown to interact with the ER in the endothelial cytosol. Incubation of rat brain endothelial cells with Aβ1 - 40 increased ER stress-induced unfolded protein response, transfer of Ca2 + from ER to mitochondria [201], and affected cytosolic Ca2 + homeostasis [202].
Mitochondrial dysfunction
Intracellular Ca2 + dynamics are also susceptible to mitochondrial dysfunction and damage, a condition that has been consistently reported in regions of cerebrovascular lesions of the AD brain [142, 203–207], in endothelial and parenchymal cells. Mitochondrial dysfunction depletes cellular ATP [208], impairing ATP-dependent transport such as the sodium (Na +/K +-ATPase) and the calcium (Ca2 +-ATPase) pump [26, 209], and ATP-binding cassette (ABC) transporters [26]. Of note, the latter include Aβ transporters which are responsible for Aβ clearance from the brain parenchyma [210, 211], in particular multidrug resistance-associated protein 1 (ABCC1) [212, 213] and ABC subfamily B member 1 (ABCB1) [214–216].
As mitochondrial dysfunction may itself result from Aβ cytotoxic effects [141, 217–220], a detrimental bidirectional link exists between Aβ accumulation and mitochondrial dysfunction/damage.
Mitochondrial dysfunction is further exacerbated by oxidative stress subsequent to chronic cerebral hypoperfusion [142, 207, 221].
Membrane potential-related dysregulation of vascular tone
Impaired ATPase pump function in mitochondrial dysfunction results in dysregulation of membrane potential and consequent further alteration of intracellular Ca2 + dynamics, which in turn affects endothelial NO synthase activity [222], and NO-mediated vasodilatation.
A further element of membrane potential dysregulation is the effect of oxidative stress on K + channels in vascular smooth muscle cells. These channels play an important role in the regulation of the membrane potential and the contractile tone of arterial smooth muscle cells [223]. Among the different types of K + channels, the ATP-dependent K + channels (KATP) have consistently been found to mediate vasodilatation [224–227], and their function has been shown to decrease in pathological conditions such as diabetes [228] and ischemia [229].
A substantial body of evidence suggests that oxidative stress modulates the vascular vasomotor function through K + channel activity [230]. In vascular smooth muscle cells of the brain, the activity of KATP appears to be selectively modulated by different reactive oxygen species, such as superoxide, hydrogen peroxide, and peroxynitrite [230]. As oxidative stress manifests early in the development of AD/CAA [24, 49, 138–140], it is plausible that K + channel activity and vasodilatation are also impaired from the early stages, potentially altering the rhythmic contractions of vasomotion.
Astrocyte-related dysregulation of vascular tone
Astrocytes are thought to participate in the regulation of vascular tone. Aβ has been shown to alter Ca2 + homeostasis and signaling in astrocytes [231–238], potentially affecting neurovascular coupling and vascular tone regulation, in AD/CAA.
Exposure to exogenous Aβ has been reported to affect astrocytic Ca2 + dynamics giving rise to fast [Ca2 +]i transients and oscillations [239]. Aβ also increased the expression of key components of Ca2 + signaling such as the glutamate receptor mGluR5 and IP3 receptor-1 [240] leading to increased [Ca2 +]i. Aβ-induced [Ca2 +]i elevation has also been attributed to Ca2 +-influx pathways [234, 235] and ER stress [233]. Consistent results showing Ca2 + elevation in astrocytes exposed to Aβ have been reported also in other studies [232, 237, 238].
Other cytotoxic effects of Aβ have been observed in astrocytes in vitro, which could disrupt neurovascular coupling. Exposure of cultured rat hippocampal astrocytes to oligomeric Aβ1 - 42, reduced the production of the potent vasodilator EET [241]. Aβ also induced mitochondrial dysfunction and oxidative stress in a Ca2 +-dependent manner [235, 236]. Injection of Aβ1 - 42 into mouse neocortical pyramidal cells blocked BK channels [242]. In cultured hippocampal astrocytes, exogenous Aβ1 - 40 enhanced the expression of the non-selective cation channel TRPV4, resulting in increased [Ca2 +]i [243].
Taken together these data suggest that the cytotoxic effects of Aβ may locally de-couple neuronal activity (metabolic demand) from CBF (metabolic supply), with detrimental effects on neuronal function.
Indeed, Aβ may induce astrocyte-mediated dysregulation of local CBF by impairing vasodilatation (due to reduced EET production) and/or enhancing vasoconstriction (due to increased production of reactive oxygen species, or inhibition of astrocytic BK channels). These indirect effects on vascular tone regulation could add to the direct effects of Aβ on vascular smooth muscle cells [125, 134] and endothelial cells [140, 186, 192, 207], resulting in the alteration of Ca2 + oscillations in smooth muscle cells, which are essential to vasomotion.
Impaired CVR and vasomotion mediate cholinergic deficit-induced dysregulation of regional CBF
Cholinergic deficit has been shown in AD [154, 171–173] and implicated in CVR impairment. As the frequency spectrum of vasomotion has been suggested to contain a neurogenic component in the low frequency range [16, 153], it is plausible that the cholinergic deficit depresses not only vascular reactivity but also vasomotion (in the slow oscillations range). Both effects would then aggravate regional disruption of CBF.
Impaired vasomotion as a potential mediator of detrimental vicious circles precipitating neurovascular damage
The dysregulation of ABC transporters caused by mitochondrial dysfunction impairs Aβ clearance from the perivascular space [26, 244], resulting in Aβ accumulation [245]. This in turn causes mitochondrial dysfunction [141, 217–220] in a detrimental vicious circle. A similar bidirectional relationship exists between oxidative stress and mitochondrial dysfunction [141], which further sustains this loop.
Hypoperfusion is yet another recognized cause of oxidative stress [221] and further Aβ accumulation [22]. We postulate that impaired vasomotion (either low amplitude or chaotic) might contribute to this vicious exacerbation of neurovascular damage, by two independent pathways: by reducing peripheral perfusion, and by reducing the driving force to perivascular drainage, thus facilitating further parenchymal and perivascular accumulation of Aβ.
Vasomotion and the perivascular/glymphatic clearance of Aβ (Question 2)
In the current view, vasomotion in cerebral arteries manifests in the form of synchronous contractions of the vessel wall [6–8]. However, some evidence from in vivo studies on vasomotion in rat cerebral arteries [82] and hamster skin fold [246], suggests that vasomotion may also propagate longitudinally along the blood vessel wall, following either directions with respect to blood flow (either upstream or downstream).
The perivascular pathway proposed by Weller and colleagues [77–79] suggests that interstitial solutes (including Aβ) enter the basement membranes of blood-brain barrier capillaries and drain along the basement membrane of the arterial tunica media, in opposite direction to blood flow.
Based on the evidence that vasomotion can propagate upstream along the vessel wall, the rhythmic oscillations of vasomotion could contribute to the driving force of the perivascular drainage in the path described by Weller and colleagues.
Furthermore, the rhythmic oscillations of the arterial wall can also be viewed as a source of pressure waves exerted on the surrounding fluid in the paravascular space. According to the power-law attenuation of longitudinally propagating waves [247], higher frequency waves reach shorter distances than lower frequencies waves. Thus, the oscillations produced by vasomotion, which are lower in frequency than those induced by the systolic pulse, could facilitate the propulsion of cerebrospinal fluid from the perivascular space into the brain parenchyma, allowing deeper penetration into the latter.
In this view, the impairment of vasomotion would decrease the efficiency of both the perivascular and glymphatic clearance of Aβ, facilitating the accumulation of the toxic peptide. This, in turn, would cause further endothelial dysfunction [198], oxidative stress [248], and decreased NO production [198]. These conditions could further dampen vasomotion [17] (Fig. 1), in a detrimental vicious circle which would accelerate neurovascular damage.
CONCLUDING REMARKS
This study has reviewed the current literature on cerebral vasomotion and identified potential pathways of vasomotion impairment in AD/CAA (Fig. 1).
Aβ cytotoxicity affects Ca2 + homeostasis in the neurovascular unit, resulting in vascular tone dysregulation. Aβ also reduces endothelial NO synthesis and upregulates the potent vasoconstrictor ET-1. Impaired vasodilatation and paradoxical vasoconstriction may cooperate in the disruption of vasomotion, resulting in weakened autoregulation, chronic hypoperfusion, and reduced driving force to the perivascular/glymphatic clearance of Aβ. These effects in turn facilitate Aβ accumulation in a detrimental vicious circle.
As vascular dysfunction is thought to occur years or even decades ahead of the clinical manifestation of AD, quantitative measurements of cerebral vasomotion could predict microvascular dysfunction at an early stage of the disease, and contribute to assess the efficiency of therapeutic interventions. Detailed physiological measurements in animal models as well as advances in computational modeling approaches will be important components of future research.
ACKNOWLEDGMENTS
The present study was conducted as part of the Project VPH-DARE@IT funded by the European Union Seventh Framework Programme (FP7/2007 - 2013) under grant agreement no. 601055.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/14-2976r1).
REFERENCES
1 | Paulson OB, Strandgaard S, Edvinsson L(1990) Cerebral autoregulationCerebrovasc Brain Metab Rev2: 161192 |
2 | Johansson B, Mellander S(1975) Static and dynamic components in the vascular myogenic response to passive changes in length as revealed by electrical and mechanical recordings from the rat portal veinCirc Res36: 7683 |
3 | Pradhan RK, Chakravarthy VS(2011) Informational dynamics of vasomotion in microvascular networks: A reviewActa Physiol (Oxf)201: 193218 |
4 | Intaglietta M(1991) Arteriolar vasomotion: Implications for tissue ischemiaBlood Vessels28: Suppl 117 |
5 | Aalkjær C, Boedtkjer D, Matchkov V(2011) Vasomotion - what is currently thought?Acta Physiol (Oxf)202: 253269 |
6 | Haddock RE, Hill CE(2005) Rhythmicity in arterial smooth muscleJ Physiol566: 645656 |
7 | Haddock RE, Hill CE(2002) Differential activation of ion channels by inositol 1,4,5-trisphosphate (IP3)- and ryanodine-sensitive calcium stores in rat basilar artery vasomotionJ Physiol545: 615627 |
8 | Haddock RE, Grayson TH, Brackenbury TD, Meaney KR, Neylon CB, Sandow SL, Hill CE(2006) Endothelial coordination of cerebral vasomotion via myoendothelial gap junctions containing connexins 37 and 40Am J Physiol Heart Circ Physiol291: H2047H2056 |
9 | Sakurai T, Terui N(2006) Effects of sympathetically induced vasomotion on tissue-capillary fluid exchangeAm J Physiol Heart Circ Physiol291: H1761H1767 |
10 | Rücker M, Strobel O, Vollmar B, Roesken F, Menger MD(2000) Vasomotion in critically perfused muscle protects adjacent tissues from capillary perfusion failureAm J Physiol Heart Circ Physiol279: H550H558 |
11 | Nilsson H, Aalkjaer C(2003) Vasomotion: Mechanisms and physiological importanceMol Interv3: 7989 |
12 | Tsai AG, Intaglietta M(1993) Evidence of flowmotion induced changes in local tissue oxygenationInt J Microcirc Clin Exp12: 7588 |
13 | Goldman D, Popel AS(2001) A computational study of the effect of vasomotion on oxygen transport from capillary networksJ Theor Biol209: 189199 |
14 | Pradhan RK, Chakravarthy VS(2007) A computational model that links non-periodic vasomotion to enhanced oxygenation in skeletal muscleMath Biosci209: 486499 |
15 | Hapuarachchi T, Park CS, Payne S(2010) Quantification of the effects of vasomotion on mass transport to tissue from axisymmetric blood vesselsJ Theor Biol264: 553559 |
16 | De Boer MP, Wijnstok NJ, Serné EH, Eringa EC, Stehouwer CDA, Flyvbjerg A, Hoekstra T, Heymans MW, Meijer RI, Twisk JW, Smulders YM(2014) Body mass index is related to microvascular vasomotion, this is partly explained by adiponectinEur J Clin Invest44: 660667 |
17 | Lacza Z, Hermán P, Görlach C, Hortobágyi T, Sándor P, Wahl M, Benyó Z(2001) NO synthase blockade induces chaotic cerebral vasomotion via activation of thromboxane receptorsStroke32: 26092614 |
18 | Plassman BL, Langa KM, Fisher GG, Heeringa SG, Weir DR, Ofstedal MB, Burke JR, Hurd MD, Potter GG, Rodgers WL, Steffens DC, Willis RJ, Wallace RB(2007) Prevalence of dementia in the United States: The aging, demographics, and memory studyNeuroepidemiology29: 125132 |
19 | Whitehouse PJ, Sciulli CG, Mason RM(1997) Dementia drug development: Use of information systems to harmonize global drug developmentPsychopharmacol Bull33: 129133 |
20 | Wilson RS, Weir DR, Leurgans SE, Evans DA, Hebert LE, Langa KM, Plassman BL, Small BJ, Bennett DA(2011) Sources of variability in estimates of the prevalence of Alzheimer’s disease in the United StatesAlzheimers Dement7: 7479 |
21 | Hardy J, Selkoe DJ(2002) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeuticsScience297: 353356 |
22 | Kelleher RJ, Soiza RL(2013) Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’s a vascular disorder?Am J Cardiovasc Dis3: 197226 |
23 | Jellinger KA, Attems J(2005) Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer diseaseJ Neurol Sci229-230: 3741 |
24 | de la Torre JC(2002) Alzheimer’s disease: How does it start?J Alzheimers Dis4: 497512 |
25 | Kalaria RN(2000) The role of cerebral ischemia in Alzheimer’s diseaseNeurobiol Aging21: 321330 |
26 | Zlokovic BV(2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disordersNat Rev Neurosci12: 723738 |
27 | de la Torre JC(2004) Personal view: Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialecticsLancet Neurol3: 184190 |
28 | Cordonnier C, van der Flier WM(2011) Brain microbleeds and Alzheimer’s disease: Innocent observation or key player?Brain134: 335344 |
29 | Van Norden AGW, van Dijk EJ, de Laat KF, Scheltens P, Olderikkert MGM, de Leeuw FE(2012) Dementia: Alzheimer pathology and vascular factors: From mutually exclusive to interactionBiochim Biophys Acta1822: 340349 |
30 | de la Torre JC, Mussivand T(1993) Can disturbed brain microcirculation cause Alzheimer’s disease?Neurol Res15: 146153 |
31 | Stopa EG, Butala P, Salloway S, Johanson CE, Gonzalez L, Tavares R, Hovanesian V, Hulette CM, Vitek MP, Cohen RA(2008) Cerebral cortical arteriolar angiopathy, vascular beta-amyloid, smooth muscle actin, Braak stage, and APOE genotypeStroke39: 814821 |
32 | Popa-Wagner A, Buga A-M, Popescu B, Muresanu D(2013) Vascular cognitive impairment, dementia, aging and energy demand. A vicious cycleJ Neural Transm10.1007/s00702-013-1129-3 |
33 | Brown WR, Thore CR(2011) Review: Cerebral microvascular pathology in ageing and neurodegenerationNeuropathol Appl Neurobiol37: 5674 |
34 | Van Beek AH, Claassen JA, Rikkert MGO, Jansen RW(2008) Cerebral autoregulation: An overview of current concepts and methodology with special focus on the elderlyJ Cereb Blood Flow Metab28: 10711085 |
35 | Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA(1999) SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor proteinNat Neurosci2: 157161 |
36 | Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C(2002) Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor proteinAm J Physiol Heart Circ Physiol283: H315H323 |
37 | Claassen JAHR, Zhang R(2011) Cerebral autoregulation in Alzheimer’s diseaseJ Cereb Blood Flow Metab31: 15721577 |
38 | Claassen JAHR, Diaz-Arrastia R, Martin-Cook K, Levine BD, Zhang R(2009) Altered cerebral hemodynamics in early Alzheimer disease: A pilot study using transcranial DopplerJ Alzheimers Dis17: 621629 |
39 | Tarumi T, Dunsky DI, Khan MA, Liu J, Hill C, Armstrong K, Martin-Cook K, Cullum CM, Zhang R(2014) Dynamic cerebral autoregulation and tissue oxygenation in amnestic mild cognitive impairmentJ Alzheimers Dis41: 765778 |
40 | Gommer ED, Martens EGHJ, Aalten P, Shijaku E, Verhey FRJ, Mess WH, Ramakers IHGB, Reulen JPH(2012) Dynamic cerebral autoregulation in subjects with Alzheimer’s disease, mild cognitive impairment, and controls: Evidence for increased peripheral vascular resistance with possible predictive valueJ Alzheimers Dis30: 805813 |
41 | Habert MO, Spampinato U, Mas JL, Piketty ML, Bourdel MC, de Recondo J, Rondot P, Askienazy S(1991) A comparative technetium 99m hexamethylpropylene amine oxime SPET study in different types of dementiaEur J Nucl Med18: 311 |
42 | Perani D, Di Piero V, Vallar G, Cappa S, Messa C, Bottini G, Berti A, Passafiume D, Scarlato G, Gerundini P(1988) Technetium-99m HM-PAO-SPECT study of regional cerebral perfusion in early Alzheimer’s diseaseJ Nucl Med29: 15071514 |
43 | Yoshikawa T, Murase K, Oku N, Imaizumi M, Takasawa M, Rishu P, Kimura Y, Ikejiri Y, Kitagawa K, Hori M, Hatazawa J(2003) Heterogeneity of cerebral blood flow in Alzheimer disease and vascular dementiaAJNR Am J Neuroradiol24: 13411347 |
44 | Staffen W, Schönauer U, Zauner H, Spindler I, Mair A, Iglseder B, Bernroider G, Ladurner G(2006) Brain perfusion SPECT in patients with mild cognitive impairment and Alzheimer’s disease: Comparison of a semiquantitative and a visual evaluationJ Neural Transm113: 195203 |
45 | Blaise R, Mateo V, Rouxel C, Zaccarini F, Glorian M, Béréziat G, Golubkov VS, Limon I(2012) Wild-type amyloid beta 1-40 peptide induces vascular smooth muscle cell death independently from matrix metalloprotease activityAging Cell11: 384393 |
46 | Maier FC, Wehrl HF, Schmid AM, Mannheim JG, Wiehr S, Lerdkrai C, Calaminus C, Stahlschmidt A, Ye L, Burnet M, Stiller D, Sabri O, Reischl G, Staufenbiel M, Garaschuk O, Jucker M, Pichler BJ(2014) Longitudinal PET-MRI reveals β-amyloid deposition and rCBF dynamics and connects vascular amyloidosis to quantitative loss of perfusionNat Med20: 14851492 |
47 | Li H, Guo Q, Inoue T, Polito VA, Tabuchi K, Hammer RE, Pautler RG, Taffet GE, Zheng H(2014) Vascular and parenchymal amyloid pathology in an Alzheimer disease knock-in mouse model: Interplay with cerebral blood flowMol Neurodegener9: 28 |
48 | Kara F, van Dongen ES, Schliebs R, van Buchem MA, de Groot HJM, Alia A(2012) Monitoring blood flow alterations in the Tg2576 mouse model of Alzheimer’s disease by in vivo magnetic resonance angiography at 17.6 TNeuroimage60: 958966 |
49 | Park L, Koizumi K, El Jamal S, Zhou P, Previti M Lou, Van Nostrand WE, Carlson G, Iadecola C(2014) Age-dependent neurovascular dysfunction and damage in a mouse model of cerebral amyloid angiopathyStroke45: 18151821 |
50 | Thal DR, Griffin WST, de Vos RAI, Ghebremedhin E(2008) Cerebral amyloid angiopathy and its relationship to Alzheimer’s diseaseActa Neuropathol115: 599609 |
51 | Maia LF, Mackenzie IRA, Feldman HH(2007) Clinical phenotypes of cerebral amyloid angiopathyJ Neurol Sci257: 2330 |
52 | Chen H, Zhang JH(2011) Cerebral amyloid angiopathy-related microhemorrhages in Alzheimer’s disease: A review of investigative animal modelsActa Neurochir Suppl111: 1517 |
53 | Kalaria RN(1999) The blood-brain barrier and cerebrovascular pathology in Alzheimer’s diseaseAnn N Y Acad Sci893: 113125 |
54 | Griffith TM, Edwards DH(1994) Fractal analysis of role of smooth muscle Ca2+ fluxes in genesis of chaotic arterial pressure oscillationsAm J Physiol266: H1801H1811 |
55 | Mayevsky A, Ziv I(1991) Oscillations of cortical oxidative metabolism and microcirculation in the ischaemic brainNeurol Res13: 3947 |
56 | Gustafsson U, Wårdell K, Nilsson GE, Lewis DH(1991) Vasomotion in rat skeletal muscle induced by hemorrhage as recorded by laser-Doppler flowmetryMicrovasc Res42: 224228 |
57 | Vollmar B, Preissler G, Menger MD(1994) Hemorrhagic hypotension induces arteriolar vasomotion and intermittent capillary perfusion in rat pancreasAm J Physiol267: H1936H1940 |
58 | Bek T, Jeppesen P, Kanters JK(2013) Spontaneous high frequency diameter oscillations of larger retinal arterioles are reduced in type 2 diabetes mellitusInvest Ophthalmol Vis Sci54: 636640 |
59 | Benbow SJ, Pryce DW, Noblett K, MacFarlane IA, Friedmann PS, Williams G(1995) Flow motion in peripheral diabetic neuropathyClin Sci (Lond)88: 191196 |
60 | Stansberry KB, Shapiro SA, Hill MA, McNitt PM, Meyer MD, Vinik AI(1996) Impaired peripheral vasomotion in diabetesDiabetes Care19: 715721 |
61 | Schmiedel O, Schroeter ML, Harvey JN(2007) Microalbuminuria in Type 2 diabetes indicates impaired microvascular vasomotion and perfusionAm J Physiol Heart Circ Physiol293: H3424H3431 |
62 | Osol G, Halpern W(1988) Spontaneous vasomotion in pressurized cerebral arteries from genetically hypertensive ratsAm J Physiol254: H28H33 |
63 | Lefer DJ, Lynch CD, Lapinski KC, Hutchins PM(1990) Enhanced vasomotion of cerebral arterioles in spontaneously hypertensive ratsMicrovasc Res39: 129139 |
64 | Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, Sulkava R, Kivipelto M(2010) Diabetes, Alzheimer disease, and vascular dementia: A population-based neuropathologic studyNeurology75: 11951202 |
65 | Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA(2004) Diabetes mellitus and risk of Alzheimer disease and decline in cognitive functionArch Neurol61: 661666 |
66 | Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM(1999) Diabetes mellitus and the risk of dementia: The Rotterdam StudyNeurology53: 19371942 |
67 | Bermejo-Pareja F, Benito-León J, Louis ED, Trincado R, Carro E, Villarejo A, de la Cámara AG(2010) Risk of incident dementia in drug-untreated arterial hypertension: A population-based studyJ Alzheimers Dis22: 949958 |
68 | Beydoun MA, Lhotsky A, Wang Y, Dal Forno G, An Y, Metter EJ, Ferrucci L, O’Brien R, Zonderman AB(2008) Association of adiposity status and changes in early to mid-adulthood with incidence of Alzheimer’s diseaseAm J Epidemiol168: 11791189 |
69 | Fitzpatrick AL, Kuller LH, Lopez OL, Diehr P, O’Meara ES, Longstreth WT, Luchsinger JA(2009) Midlife and late-life obesity and the risk of dementia: Cardiovascular health studyArch Neurol66: 336342 |
70 | Hassing LB, Dahl AK, Thorvaldsson V, Berg S, Gatz M, Pedersen NL, Johansson B(2009) Overweight in midlife and risk of dementia: A 40-year follow-up studyInt J Obes (Lond)33: 893898 |
71 | Morris MC, Scherr PA, Hebert LE, Glynn RJ, Bennett DA, Evans DA(2001) Association of incident Alzheimer disease and blood pressure measured from 13 years before to 2 years after diagnosis in a large community studyArch Neurol58: 16401646 |
72 | Rönnemaa E, Zethelius B, Lannfelt L, Kilander L(2011) Vascular risk factors and dementia: 40-year follow-up of a population-based cohortDement Geriatr Cogn Disord31: 460466 |
73 | Atti AR, Palmer K, Volpato S, Winblad B, De Ronchi D, Fratiglioni L(2008) Late-life body mass index and dementia incidence: Nine-year follow-up data from the Kungsholmen ProjectJ Am Geriatr Soc56: 111116 |
74 | Hassing LB, Johansson B, Nilsson SE, Berg S, Pedersen NL, Gatz M, McClearn G(2002) Diabetes mellitus is a risk factor for vascular dementia, but not for Alzheimer’s disease: A population-based study of the oldest oldInt Psychogeriatr14: 239248 |
75 | Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M(2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid βSci Transl Med4: 147ra111 |
76 | Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M(2013) Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brainJ Neurosci33: 1819018199 |
77 | Hawkes CA, Jayakody N, Johnston DA, Bechmann I, Carare RO(2014) Failure of perivascular drainage of β-amyloid in cerebral amyloid angiopathyBrain Pathol24: 396403 |
78 | Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JAR, Perry VH, Weller RO(2008) Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: Significance for cerebral amyloid angiopathy and neuroimmunologyNeuropathol Appl Neurobiol34: 131144 |
79 | Weller RO, Subash M, Preston SD, Mazanti I, Carare RO(2008) Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s diseaseBrain Pathol18: 253266 |
80 | Schley D, Carare-Nnadi R, Please CP, Perry VH, Weller RO(2006) Mechanisms to explain the reverse perivascular transport of solutes out of the brainJ Theor Biol238: 962974 |
81 | Diehl RR, Diehl B, Sitzer M, Hennerici M(1991) Spontaneous oscillations in cerebral blood flow velocity in normal humans and in patients with carotid artery diseaseNeurosci Lett127: 58 |
82 | Fujii K, Heistad DD, Faraci FM(1990) Vasomotion of basilar arteries in vivoAm J Physiol258: H1829H1834 |
83 | Funk W, Endrich B, Messmer K, Intaglietta M(1983) Spontaneous arteriolar vasomotion as a determinant of peripheral vascular resistanceInt J Microcirc Clin Exp2: 1125 |
84 | Hartley CJ, Reddy AK, Madala S, Entman ML, Michael LH, Taffet GE(2004) Noninvasive ultrasonic measurement of arterial wall motion in miceAm J Physiol Heart Circ Physiol287: H1426H1432 |
85 | Hartley CJ, Reddy AK, Madala S, Entman ML, Taffet GE(2010) Feasibility of dual Doppler velocity measurements to estimate volume pulsations of an arterial segmentUltrasound Med Biol36: 11691175 |
86 | Aalkjaer C, Nilsson H(2005) Vasomotion: Cellular background for the oscillator and for the synchronization of smooth muscle cellsBr J Pharmacol144: 605616 |
87 | Segal SS, Bény JL(1992) Intracellular recording and dye transfer in arterioles during blood flow controlAm J Physiol263: H1H7 |
88 | Gustafsson H(1993) Vasomotion and underlying mechanisms in small arteries. An in vitro study of rat blood vesselsActa Physiol Scand Suppl614: 144 |
89 | Gokina NI, Bevan RD, Walters CL, Bevan JA(1996) Electrical activity underlying rhythmic contraction in human pial arteriesCirc Res78: 148153 |
90 | Peng H, Matchkov V, Ivarsen A, Aalkjaer C, Nilsson H(2001) Hypothesis for the initiation of vasomotionCirc Res88: 810815 |
91 | Oishi H, Schuster A, Lamboley M, Stergiopulos N, Meister J-J, Bény J-L(2002) Role of membrane potential in vasomotion of isolated pressurized rat arteriesLife Sci71: 22392248 |
92 | Dirnagl U, Lindauer U, Villringer A(1993) Nitric oxide synthase blockade enhances vasomotion in the cerebral microcirculation of anesthetized ratsMicrovasc Res45: 318323 |
93 | Yuill KH, McNeish AJ, Kansui Y, Garland CJ, Dora KA(2010) Nitric oxide suppresses cerebral vasomotion by sGC-independent effects on ryanodine receptors and voltage-gated calcium channelsJ Vasc Res47: 93107 |
94 | Thomsen ABK, Kim S, Aalbaek F, Aalkjaer C, Boedtkjer E(2014) Intracellular acidification alters myogenic responsiveness and vasomotion of mouse middle cerebral arteriesJ Cereb Blood Flow Metab34: 161168 |
95 | Hempelmann RG, Pradel RH, Mehdorn HM, Ziegler A(1998) Contractions induced by NO synthase inhibition in isolated rat basilar artery: Role of the endothelium and endogenous vasoconstrictorsNeurol Res20: 6372 |
96 | Jackson WF(1988) Oscillations in active tension in hamster aortas: Role of the endotheliumBlood Vessels25: 144156 |
97 | Okazaki K, Seki S, Kanaya N, Hattori J-I, Tohse N, Namiki A(2003) Role of endothelium-derived hyperpolarizing factor in phenylephrine-induced oscillatory vasomotion in rat small mesenteric arteryAnesthesiology98: 11641171 |
98 | Omote M, Mizusawa H(1993) The role of sarcoplasmic reticulum in endothelium-dependent and endothelium-independent rhythmic contractions in the rabbit mesenteric arteryActa Physiol Scand149: 1521 |
99 | Akata T, Kodama K, Takahashi S(1995) Role of endothelium in oscillatory contractile responses to various receptor agonists in isolated small mesenteric and epicardial coronary arteriesJpn J Pharmacol68: 331343 |
100 | Kvandal P, Stefanovska A, Veber M, Kvernmo HD, Kvermmo HD, Kirkeb>øen KA(2003) Regulation of human cutaneous circulation evaluated by laser Doppler flowmetry, iontophoresis, and spectral analysis: Importance of nitric oxide and prostaglandinesMicrovasc Res65: 160171 |
101 | Bertuglia S, Colantuoni A, Intaglietta M(1995) Capillary reperfusion after L-arginine, L-NMMA, and L-NNA treatment in cheek pouch microvasculatureMicrovasc Res50: 162174 |
102 | Sell M, Boldt W, Markwardt F(2002) Desynchronising effect of the endothelium on intracellular Ca2+ concentration dynamics in vascular smooth muscle cells of rat mesenteric arteriesCell Calcium32: 105120 |
103 | Chemtob S, Inayatulla A, Varma DR(1992) Eicosanoid-dependent and endothelium-independent oscillations of rat aortaJ Vasc Res29: 270280 |
104 | Von der Weid PY, Bény JL(1993) Simultaneous oscillations in the membrane potential of pig coronary artery endothelial and smooth muscle cellsJ Physiol471: 1324 |
105 | Filosa JA, Iddings JA(2013) Astrocyte regulation of cerebral vascular tone.H609-HAm J Physiol Heart Circ Physiol305: 619 |
106 | Filosa JA, Bonev AD, Nelson MT(2004) Calcium dynamics in cortical astrocytes and arterioles during neurovascular couplingCirc Res95: e73e81 |
107 | Carmignoto G, Gómez-Gonzalo M(2010) The contribution of astrocyte signalling to neurovascular couplingBrain Res Rev63: 138148 |
108 | Girouard H, Iadecola C(2006) Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer diseaseJ Appl Physiol100: 328335 |
109 | Iadecola C(2004) Neurovascular regulation in the normal brain and in Alzheimer’s diseaseNat Rev Neurosci5: 347360 |
110 | Brown EB, Shear JB, Adams SR, Tsien RY, Webb WW(1999) Photolysis of caged calcium in femtoliter volumes using two-photon excitationBiophys J76: 489499 |
111 | Soeller C, Cannell MB(1999) Two-photon microscopy: Imaging in scattering samples and three-dimensionally resolved flash photolysisMicrosc Res Tech47: 182195 |
112 | Mulligan SJ, MacVicar BA(2004) Calcium transients in astrocyte endfeet cause cerebrovascular constrictionsNature431: 195199 |
113 | Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann K-A, Pozzan T, Carmignoto G(2003) Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculationNat Neurosci6: 4350 |
114 | Peng X, Carhuapoma JR, Bhardwaj A, Alkayed NJ, Falck JR, Harder DR, Traystman RJ, Koehler RC(2002) Suppression of cortical functional hyperemia to vibrissal stimulation in the rat by epoxygenase inhibitorsAm J Physiol Heart Circ Physiol283: H2029H2037 |
115 | Peng X, Zhang C, Alkayed NJ, Harder DR, Koehler RC(2004) Dependency of cortical functional hyperemia to forepaw stimulation on epoxygenase and nitric oxide synthase activities in ratsJ Cereb Blood Flow Metab24: 509517 |
116 | Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA(2010) Glial and neuronal control of brain blood flowNature468: 232243 |
117 | Blanco VM, Stern JE, Filosa JA(2008) Tone-dependent vascular responses to astrocyte-derived signalsAm J Physiol Heart Circ Physiol294: H2855H2863 |
118 | Gordon GRJ, Choi HB, Rungta RL, Ellis-Davies GCR, MacVicar BA(2008) Brain metabolism dictates the polarity of astrocyte control over arteriolesNature456: 745749 |
119 | Metea MR, Newman EA(2006) Glial cells dilate and constrict blood vessels: A mechanism of neurovascular couplingJ Neurosci26: 28622870 |
120 | Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT(2010) Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constrictionProc Natl Acad Sci U S A107: 38113816 |
121 | Porter JT, McCarthy KD(1996) Hippocampal astrocytes in situ respond to glutamate released from synaptic terminalsJ Neurosci16: 50735081 |
122 | Brown LA, Key BJ, Lovick TA(2002) Inhibition of vasomotion in hippocampal cerebral arterioles during increases in neuronal activityAuton Neurosci95: 137140 |
123 | Lovick TA, Brown LA, Key BJ(2005) Neuronal activity-related coupling in cortical arterioles: Involvement of astrocyte-derived factorsExp Physiol90: 131140 |
124 | Ruzali WAW, Kehoe PG, Love S(2013) Influence of LRP-1 and apolipoprotein E on amyloid-β uptake and toxicity to cerebrovascular smooth muscle cellsJ Alzheimers Dis33: 95110 |
125 | Stukas S, Robert J, Wellington CL(2014) High-density lipoproteins and cerebrovascular integrity in Alzheimer’s diseaseCell Metab19: 574591 |
126 | Attems J, Jellinger K, Thal DR, Van Nostrand W(2011) Review: Sporadic cerebral amyloid angiopathyNeuropathol Appl Neurobiol37: 7593 |
127 | Imaoka K, Kobayashi S, Fujihara S, Shimode K, Nagasaki M(1999) Leukoencephalopathy with cerebral amyloid angiopathy: A semiquantitative and morphometric studyJ Neurol246: 661666 |
128 | Tian J, Shi J, Smallman R, Iwatsubo T, Mann DMA(2006) Relationships in Alzheimer’s disease between the extent of Abeta deposition in cerebral blood vessel walls, as cerebral amyloid angiopathy, and the amount of cerebrovascular smooth muscle cells and collagenNeuropathol Appl Neurobiol32: 332340 |
129 | Zekry D, Duyckaerts C, Belmin J, Geoffre C, Moulias R, Hauw J-J(2003) Cerebral amyloid angiopathy in the elderly: Vessel walls changes and relationship with dementiaActa Neuropathol106: 367373 |
130 | Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, Jucker M(2001) Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathyJ Neurosci21: 16191627 |
131 | Christie R, Yamada M, Moskowitz M, Hyman B(2001) Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathyAm J Pathol158: 10651071 |
132 | Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C(1997) Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor proteinJ Neurosci17: 76557661 |
133 | Princz-Kranz FL, Mueggler T, Knobloch M, Nitsch RM, Rudin M(2010) Vascular response to acetazolamide decreases as a function of age in the arcA beta mouse model of cerebral amyloidosisNeurobiol Dis40: 284292 |
134 | Vromman A, Trabelsi N, Rouxel C, Béréziat G, Limon I, Blaise R(2013) β-Amyloid context intensifies vascular smooth muscle cells induced inflammatory response and de-differentiationAging Cell12: 358369 |
135 | Grammas P(2011) Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s diseaseJ Neuroinflammation8: 26 |
136 | Mark RJ, Hensley K, Butterfield DA, Mattson MP(1995) Amyloid beta-peptide impairs ion-motive ATPase activities: Evidence for a role in loss of neuronal Ca2+ homeostasis and cell deathJ Neurosci15: 62396249 |
137 | Hermes M, Eichhoff G, Garaschuk O(2010) Intracellular calcium signalling in Alzheimer’s diseaseJ Cell Mol Med14: 3041 |
138 | Aliev G, Li Y, Palacios HH, Obrenovich ME(2011) Oxidative stress induced mitochondrial DNA deletion as a hallmark for the drug development in the context of the cerebrovascular diseasesRecent Pat Cardiovasc Drug Discov6: 222241 |
139 | Zhu X, Smith MA, Honda K, Aliev G, Moreira PI, Nunomura A, Casadesus G, Harris PLR, Siedlak SL, Perry G(2007) Vascular oxidative stress in Alzheimer diseaseJ Neurol Sci257: 240246 |
140 | Aliev G, Smith MA, Seyidov D, Neal ML, Lamb BT, Nunomura A, Gasimov EK, Vinters HV, Perry G, LaManna JC, Friedland RP(2002) The role of oxidative stress in the pathophysiology of cerebrovascular lesions in Alzheimer’s diseaseBrain Pathol12: 2135 |
141 | Müller WE, Eckert A, Kurz C, Eckert GP, Leuner K(2010) Mitochondrial dysfunction: Common final pathway in brain aging and Alzheimer’s disease–therapeutic aspectsMol Neurobiol41: 159171 |
142 | Aliev G, Priyadarshini M, Reddy VP, Grieg NH, Kaminsky Y, Cacabelos R, Ashraf GM, Jabir NR, Kamal MA, Nikolenko VN, Zamyatnin AAJ, Benberin VV, Bachurin SO(2014) Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer diseaseCurr Med Chem21: 22082217 |
143 | Black S, Gao F, Bilbao J(2009) Understanding white matter disease: Imaging-pathological correlations in vascular cognitive impairmentStroke40: S48S52 |
144 | Thore CR, Anstrom JA, Moody DM, Challa VR, Marion MC, Brown WR(2007) Morphometric analysis of arteriolar tortuosity in human cerebral white matter of preterm, young, and aged subjectsJ Neuropathol Exp Neurol66: 337345 |
145 | Moody DM, Brown WR, Challa VR, Ghazi-Birry HS, Reboussin DM(1997) Cerebral microvascular alterations in aging, leukoaraiosis, and Alzheimer’s diseaseAnn N Y Acad Sci826: 103116 |
146 | Cuhlmann S, Van der Heiden K, Saliba D, Tremoleda JL, Khalil M, Zakkar M, Chaudhury H, Luong LA, Mason JC, Udalova I, Gsell W, Jones H, Haskard DO, Krams R, Evans PC(2011) Disturbed blood flow induces RelA expression via c-Jun N-terminal kinase 1: A novel mode of NF-κB regulation that promotes arterial inflammationCirc Res108: 950959 |
147 | Heo K-S, Fujiwara K, Abe J-I(2014) Shear stress and atherosclerosisMol Cells37: 435440 |
148 | Chiu J-J, Chien S(2011) Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectivesPhysiol Rev91: 327387 |
149 | Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM(1999) Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylationNature399: 601605 |
150 | Mayhew JE, Askew S, Zheng Y, Porrill J, Westby GW, Redgrave P, Rector DM, Harper RM(1996) Cerebral vasomotion: A 0.1-Hz oscillation in reflected light imaging of neural activityNeuroimage4: 183193 |
151 | Griffith TM(1996) Temporal chaos in the microcirculationCardiovasc Res31: 342358 |
152 | Intaglietta M(1990) Vasomotion and flowmotion: Physiological mechanisms and clinical evidenceVasc Med1: 101112 |
153 | Zhang Z, Khatami R(2014) Predominant endothelial vasomotor activity during human sleep: A near-infrared spectroscopy studyEur J Neurosci40: 33963404 |
154 | Glodzik L, Randall C, Rusinek H, de Leon MJ(2013) Cerebrovascular reactivity to carbon dioxide in Alzheimer’s diseaseJ Alzheimers Dis35: 427440 |
155 | Balestrini S, Perozzi C, Altamura C, Vernieri F, Luzzi S, Bartolini M, Provinciali L, Silvestrini M(2013) Severe carotid stenosis and impaired cerebral hemodynamics can influence cognitive deteriorationNeurology80: 21452150 |
156 | Ringelstein EB, Sievers C, Ecker S, Schneider PA, Otis SM(1988) Noninvasive assessment of CO2-induced cerebral vasomotor response in normal individuals and patients with internal carotid artery occlusionsStroke19: 963969 |
157 | Ringelstein EB, Van Eyck S, Mertens I(1992) Evaluation of cerebral vasomotor reactivity by various vasodilating stimuli: Comparison of CO2 to acetazolamideJ Cereb Blood Flow Metab12: 162168 |
158 | Bär K-J, Boettger MK, Seidler N, Mentzel HJ, Terborg C, Sauer H(2007) Influence of galantamine on vasomotor reactivity in Alzheimer’s disease and vascular dementia due to cerebral microangiopathyStroke38: 31863192 |
159 | Bullock R, Mendelow AD, Bone I, Patterson J, Macleod WN, Allardice G(1985) Cerebral blood flow and CO2 responsiveness as an indicator of collateral reserve capacity in patients with carotid arterial diseaseBr J Surg72: 348351 |
160 | Markus H, Cullinane M(2001) Severely impaired cerebrovascular reactivity predicts stroke and TIA risk in patients with carotid artery stenosis and occlusionBrain124: 457467 |
161 | Vernieri F, Pasqualetti P, Passarelli F, Rossini PM, Silvestrini M(1999) Outcome of carotid artery occlusion is predicted by cerebrovascular reactivityStroke30: 593598 |
162 | Vernieri F, Pasqualetti P, Matteis M, Passarelli F, Troisi E, Rossini PM, Caltagirone C, Silvestrini M(2001) Effect of collateral blood flow and cerebral vasomotor reactivity on the outcome of carotid artery occlusionStroke32: 15521558 |
163 | Vicenzini E, Ricciardi MC, Altieri M, Puccinelli F, Bonaffini N, Di Piero V, Lenzi GL(2007) Cerebrovascular reactivity in degenerative and vascular dementia: A transcranial Doppler studyEur Neurol58: 8489 |
164 | Den Abeelen ASSM, Lagro J, van Beek AHEA, Claassen JAHR(2014) Impaired cerebral autoregulation and vasomotor reactivity in sporadic Alzheimer’s diseaseCurr Alzheimer Res11: 1117 |
165 | Silvestrini M, Pasqualetti P, Baruffaldi R, Bartolini M, Handouk Y, Matteis M, Moffa F, Provinciali L, Vernieri F(2006) Cerebrovascular reactivity and cognitive decline in patients with Alzheimer diseaseStroke37: 10101015 |
166 | de la Torre JC(2012) Cerebral hemodynamics and vascular risk factors: Setting the stage for Alzheimer’s diseaseJ Alzheimers Dis32: 553567 |
167 | Han BH, Zhou M-L, Abousaleh F, Brendza RP, Dietrich HH, Koenigsknecht-Talboo J, Cirrito JR, Milner E, Holtzman DM, Zipfel GJ(2008) Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: Contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibitionJ Neurosci28: 1354213550 |
168 | Dorr A, Sahota B, Chinta LV, Brown ME, Lai AY, Ma K, Hawkes CA, McLaurin J, Stefanovic B(2012) Amyloid-β-dependent compromise of microvascular structure and function in a model of Alzheimer’s diseaseBrain135: 30393050 |
169 | Menendez-Gonzalez M, Garcia-Garcia J, Calleja S, Rojo J, Ribacoba R(2011) Vasomotor reactivity is similarly impaired in patients with Alzheimer’s disease and patients with amyloid hemorrhageJ Neuroimaging21: e83e85 |
170 | Tsivgoulis G, Katsanos AH, Papageorgiou SG, Dardiotis E, Voumvourakis K, Giannopoulos S(2014) The role of neurosonology in the diagnosis of vascular dementiaJ Alzheimers Dis42: Suppl 3S251S257 |
171 | Farkas E, Luiten PG(2001) Cerebral microvascular pathology in aging and Alzheimer’s diseaseProg Neurobiol64: 575611 |
172 | Claassen JAHR, Jansen RWMM(2006) Cholinergically mediated augmentation of cerebral perfusion in Alzheimer’s disease and related cognitive disorders: The cholinergic-vascular hypothesisJ Gerontol A Biol Sci Med Sci61: 267271 |
173 | Van Beek AHEA, Claassen JAHR(2011) The cerebrovascular role of the cholinergic neural system in Alzheimer’s diseaseBehav Brain Res221: 537542 |
174 | Silvestrini M, Viticchi G, Falsetti L, Balucani C, Vernieri F, Cerqua R, Luzzi S, Bartolini M, Provinciali L(2011) The role of carotid atherosclerosis in Alzheimer’s disease progressionJ Alzheimers Dis25: 719726 |
175 | Buratti L, Viticchi G, Falsetti L, Cagnetti C, Luzzi S, Bartolini M, Provinciali L, Silvestrini M(2014) Vascular impairment in Alzheimer’s disease: The role of obstructive sleep apneaJ Alzheimers Dis38: 445453 |
176 | Zaknun JJ, Leblhuber F, Schücktanz H(2008) Value of cerebral blood flow quantification in the diagnosis of dementiaNucl Med Commun29: 260269 |
177 | Knapp WH, Dannenberg C, Marschall B, Zedlick D, Löschmann K, Bettin S, Barthel H, Seese A(1996) Changes in local cerebral blood flow by neuroactivation andvasoactivation in patients with impaired cognitive functionEur J Nucl Med23: 878888 |
178 | Stoppe G, Schütze R, Kögler A, Staedt J, Munz DL, Emrich D, Rüther ECerebrovascular reactivity to acetazolamide in (senile) dementia of Alzheimer’s type: Relationship to disease severityDementia6: 7382 |
179 | Bonte FJ, Devous MD, Reisch JS, Ajmani AK, Weiner MF, Hom J, Tintner R(1989) The effect of acetazolamide on regional cerebral blood flow in patients with Alzheimer’s disease or stroke as measured by single-photon emission computed tomographyInvest Radiol24: 99103 |
180 | Pávics L, Grünwald F, Reichmann K, Séra T, Ambrus E, Horn R, Hartmann A, Menzel C, Csernay L, Biersack HJ(1998) rCBF SPECT and the acetazolamide test in the evaluation of dementiaNucl Med Rev Cent East Eur1: 1319 |
181 | Martin C, Martindale J, Berwick J, Mayhew J(2006) Investigating neural-hemodynamic coupling and the hemodynamic response function in the awake ratNeuroimage32: 3348 |
182 | Summers P, Taylor Z, Shih A(2014) Two-Photon imaging of cerebral vasodynamics in awake mice during health and diseaseAdvances in Intravital MicroscopyWeigert R2543SpringerNetherlands |
183 | Devor A, Sakadžić S, Srinivasan VJ, Yaseen MA, Nizar K, Saisan PA, Tian P, Dale AM, Vinogradov SA, Franceschini MA, Boas DA(2012) Frontiers in optical imaging of cerebral blood flow and metabolismJ Cereb Blood Flow Metab32: 12591276 |
184 | Martin C(2014) Contributions and complexities from the use of in vivo animal models to improve understanding of human neuroimaging signalsFront Neurosci8: 211 |
185 | Shih A, Drew P, Kleinfeld D(2014) Imaging vasodynamics in the awake mouse brain with two-photon microscopyNeurovascular Coupling MethodsZhao M, Ma H, Schwartz TH5573New YorkSpringer |
186 | Palmer JC, Barker R, Kehoe PG, Love S(2012) Endothelin-1 is elevated in Alzheimer’s disease and upregulated by amyloid-βJ Alzheimers Dis29: 853861 |
187 | Miners JS, Palmer JC, Tayler H, Palmer LE, Ashby E, Kehoe PG, Love S(2014) Aβ degradation or cerebral perfusion? Divergent effects of multifunctional enzymesFront Aging Neurosci6: 238 |
188 | Palmer JC, Kehoe PG, Love S(2010) Endothelin-converting enzyme-1 in Alzheimer’s disease and vascular dementiaNeuropathol Appl Neurobiol36: 487497 |
189 | Maguire JJ(2002) Endothelin-converting enzyme activity in vascular smooth muscle preparations in vitroMethods Mol Biol206: 165177 |
190 | Minami M, Kimura M, Iwamoto N, Arai H(1995) Endothelin-1-like immunoreactivity in cerebral cortex of Alzheimer-type dementiaProg Neuropsychopharmacol Biol Psychiatry19: 509513 |
191 | Luo J, Grammas P(2010) Endothelin-1 is elevated in Alzheimer’s disease brain microvessels and is neuroprotectiveJ Alzheimers Dis21: 887896 |
192 | Palmer JC, Tayler HM, Love S(2013) Endothelin-converting enzyme-1 activity, endothelin-1 production, and free radical-dependent vasoconstriction in Alzheimer’s diseaseJ Alzheimers Dis36: 577587 |
193 | Palmer JC, Baig S, Kehoe PG, Love S(2009) Endothelin-converting enzyme-2 is increased in Alzheimer’s disease and up-regulated by AbetaAm J Pathol175: 262270 |
194 | Paris D, Humphrey J, Quadros A, Patel N, Crescentini R, Crawford F, Mullan M(2003) Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer’s disease: Role of inflammationNeurol Res25: 642651 |
195 | Ivey ME, Osman N, Little PJ(2008) Endothelin-1 signalling in vascular smooth muscle: Pathways controlling cellular functions associated with atherosclerosisAtherosclerosis199: 237247 |
196 | Delgado E, Marques-Neves C, Rocha I, Sales-Luís J, Silva-Carvalho L(2010) Endothelin-1 effects on spontaneous oscillations in choroidal arteriolesActa Ophthalmol88: 742747 |
197 | Koenigsberger M, Sauser R, Bény J-L, Meister J-J(2005) Role of the endothelium on arterial vasomotionBiophys J88: 38453854 |
198 | Gentile MT, Vecchione C, Maffei A, Aretini A, Marino G, Poulet R, Capobianco L, Selvetella G, Lembo G(2004) Mechanisms of soluble beta-amyloid impairment of endothelial functionJ Biol Chem279: 4813548142 |
199 | Toda N, Okamura T(2012) Cerebral blood flow regulation by nitric oxide in Alzheimer’s diseaseJ Alzheimers Dis32: 569578 |
200 | Ciabattoni G, Porreca E, Di Febbo C, Di Iorio A, Paganelli R, Bucciarelli T, Pescara L, Del Re L, Giusti C, Falco A, Sau A, Patrono C, Davì G(2007) Determinants of platelet activation in Alzheimer’s diseaseNeurobiol Aging28: 336342 |
201 | Fonseca ACRG, Ferreiro E, Oliveira CR, Cardoso SM, Pereira CF(2013) Activation of the endoplasmic reticulum stress response by the amyloid-beta 1-40 peptide in brain endothelial cellsBiochim Biophys Acta1832: 21912203 |
202 | Fonseca ACRG, Moreira PI, Oliveira CR, Cardoso SM, Pinton P, Pereira CF(2014) Amyloid-beta disrupts calcium and redox homeostasis in brain endothelial cellsMol Neurobiol10.1007/s12035-014-8740-7 |
203 | Aliev G, Palacios HH, Walrafen B, Lipsitt AE, Obrenovich ME, Morales L(2009) Brain mitochondria as a primary target in the development of treatment strategies for Alzheimer diseaseInt J Biochem Cell Biol41: 19892004 |
204 | Claudio L(1996) Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer’s disease patientsActa Neuropathol91: 614 |
205 | Stewart PA, Hayakawa K, Akers MA, Vinters HV(1992) A morphometric study of the blood-brain barrier in Alzheimer’s diseaseLab Invest67: 734742 |
206 | Aliev G, Seyidova D, Neal ML, Shi J, Lamb BT, Siedlak SL, Vinters HV, Head E, Perry G, Lamanna JC, Friedland RP, Cotman CW(2002) Atherosclerotic lesions and mitochondria DNA deletions in brain microvessels as a central target for the development of human AD and AD-like pathology in aged transgenic miceAnn N Y Acad Sci977: 4564 |
207 | Aliev G, Smith MA, Obrenovich ME, de la Torre JC, Perry G(2003) Role of vascular hypoperfusion-induced oxidative stress and mitochondria failure in the pathogenesis of Azheimer diseaseNeurotox Res5: 491504 |
208 | Grammas P, Martinez J, Miller B(2011) Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseasesExpert Rev Mol Med13: e19 |
209 | De Bock M, Wang N, Decrock E, Bol M, Gadicherla AK, Culot M, Cecchelli R, Bultynck G, Leybaert L(2013) Endothelial calcium dynamics, connexin channels and blood-brain barrier functionProg Neurobiol108: 120 |
210 | ElAli A, Hermann DM(2011) ATP-binding cassette transporters and their roles in protecting the brainNeuroscientist17: 423436 |
211 | Ueno M, Nakagawa T, Wu B, Onodera M, Huang C-L, Kusaka T, Araki N, Sakamoto H(2010) Transporters in the brain endothelial barrierCurr Med Chem17: 11251138 |
212 | Abuznait AH, Kaddoumi A(2012) Role of ABC transporters in the pathogenesis of Alzheimer’s diseaseACS Chem Neurosci3: 820831 |
213 | Krohn M, Lange C, Hofrichter J, Scheffler K, Stenzel J, Steffen J, Schumacher T, Brüning T, Plath A-S, Alfen F, Schmidt A, Winter F, Rateitschak K, Wree A, Gsponer J, Walker LC, Pahnke J(2011) Cerebral amyloid-β proteostasis is regulated by the membrane transport protein ABCC1 in miceJ Clin Invest121: 39243931 |
214 | Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM(2005) P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse modelJ Clin Invest115: 32853290 |
215 | Silverberg GD, Messier AA, Miller MC, Machan JT, Majmudar SS, Stopa EG, Donahue JE, Johanson CE(2010) Amyloid efflux transporter expression at the blood-brain barrier declines in normal agingJ Neuropathol Exp Neurol69: 10341043 |
216 | Van Assema DME, Lubberink M, Bauer M, van der Flier WM, Schuit RC, Windhorst AD, Comans EFI, Hoetjes NJ, Tolboom N, Langer O, Müller M, Scheltens P, Lammertsma AA, van Berckel BNM(2012) Blood-brain barrier P-glycoprotein function in Alzheimer’s diseaseBrain135: 181189 |
217 | Suhara T, Magrané J, Rosen K, Christensen R, Kim H-S, Zheng B, McPhie DL, Walsh K, Querfurth H. Abeta42 generation is toxic to endothelial cells and inhibits eNOS function through an Akt/GSK-3beta signaling-dependent mechanismNeurobiol Aging24: 437451 |
218 | Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li Q-X, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA(2005) Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42J Neurosci25: 672679 |
219 | Xu J, Chen S, Ku G, Ahmed SH, Chen H, Hsu CY(2001) Amyloid beta peptide-induced cerebral endothelial cell death involves mitochondrial dysfunction and caspase activationJ Cereb Blood Flow Metab21: 702710 |
220 | Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH(2006) Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progressionHum Mol Genet15: 14371449 |
221 | Aliev G, Palacios HH, Lipsitt AE, Fischbach K, Lamb BT, Obrenovich ME, Morales L, Gasimov E, Bragin V(2009) Nitric oxide as an initiator of brain lesions during the development of Alzheimer diseaseNeurotox Res16: 293305 |
222 | Busse R, Mülsch A(1990) Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulinFEBS Lett265: 133136 |
223 | Park WS, Han J, Earm YE(2008) Physiological role of inward rectifier K(+) channels in vascular smooth muscle cellsPflugers Arch457: 137147 |
224 | Rodrigo GC, Standen NB(2005) ATP-sensitive potassium channelsCurr Pharm Des11: 19151940 |
225 | Rosenblum WI, Wei EP, Kontos HA(2004) Vasodilation of brain surface arterioles by blockade of Na-H+ antiport and its inhibition by inhibitors of KATP channel openersBrain Res1005: 7783 |
226 | Janigro D, Nguyen TS, Meno J, West GA, Winn HR(1997) Endothelium-dependent regulation of cerebrovascular tone by extracellular and intracellular ATP.H878-HAm J Physiol273: 885 |
227 | Brian JE, Faraci FM, Heistad DD(1996) Recent insights into the regulation of cerebral circulationClin Exp Pharmacol Physiol23: 449457 |
228 | Mayhan WG(1994) Effect of diabetes mellitus on response of the basilar artery to activation of ATP-sensitive potassium channelsBrain Res636: 3539 |
229 | Bari F, Louis TM, Meng W, Busija DW(1996) Global ischemia impairs ATP-sensitive K+ channel function in cerebral arterioles in piglets.1874-1880; discussionStroke27: 18801881 |
230 | Liu Y, Gutterman DD(2002) Oxidative stress and potassium channel functionClin Exp Pharmacol Physiol29: 305311 |
231 | Simpson JE, Ince PG, Shaw PJ, Heath PR, Raman R, Garwood CJ, Gelsthorpe C, Baxter L, Forster G, Matthews FE, Brayne C, Wharton SB (2011) Neurobiol Aging 32: 1795 1807 Microarray analysis of the astrocyte transcriptome in the aging braRelationshito Alzheimer’s pathology and APOE genotype |
232 | Casley CS, Lakics V, Lee H, Broad LM, Day TA, Cluett T, Smith MA, O’Neill MJ, Kingston AE(2009) Up-regulation of astrocyte metabotropic glutamate receptor 5 by amyloid-β peptideBrain Res1260: 6575 |
233 | Alberdi E, Wyssenbach A, Alberdi M, Sánchez-Gómez MV, Cavaliere F, Rodríguez JJ, Verkhratsky A, Matute C(2013) Ca(2+) -dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease.292-302Aging Cell12: |
234 | Chow S-K, Yu D, Macdonald CL, Buibas M, Silva GA(2010) Amyloid β-peptide directly induces spontaneous calcium transients, delayed intercellular calcium waves and gliosis in rat cortical astrocytes.eASN Neuro2: 00026 |
235 | Abramov AY, Canevari L, Duchen MR(2003) Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity.5088-5095J Neurosci23: |
236 | Abramov AY, Canevari L, Duchen MR(2004) Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture.81-87Biochim Biophys Acta1742: |
237 | Haughey NJ, Mattson MP(2003) Alzheimer’s amyloid beta-peptide enhances ATP/gap junction-mediated calcium-wave propagation in astrocytes.173-180Neuromolecular Med3: |
238 | Lim D, Iyer A, Ronco V, Grolla AA, Canonico PL, Aronica E, Genazzani AA(2013) Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kBGlia61: 11341145 |
239 | Lim D, Ronco V, Grolla AA, Verkhratsky A, Genazzani AA(2014) Glial calcium signalling in Alzheimer’s diseaseRev Physiol Biochem Pharmacol167: 4565 |
240 | Grolla AA, Fakhfouri G, Balzaretti G, Marcello E, Gardoni F, Canonico PL, DiLuca M, Genazzani AA, Lim D(2013) Aβ leads to Ca2+ signaling alterations and transcriptional changes in glial cellsNeurobiol Aging34: 511522 |
241 | Sarkar P, Narayanan J, Harder DR(2011) Differential effect of amyloid β on the cytochrome P450 epoxygenase activity in rat brainNeuroscience194: 241249 |
242 | Yamamoto K, Ueta Y, Wang L, Yamamoto R, Inoue N, Inokuchi K, Aiba A, Yonekura H, Kato N(2011) Suppression of a neocortical potassium channel activity by intracellular amyloid-β and its rescue with Homer1aJ Neurosci31: 1110011109 |
243 | Bai J-Z, Lipski J(2014) Involvement of TRPV4 channels in Aβ(40)-induced hippocampal cell death and astrocytic Ca(2+) signallingNeurotoxicology41: 6472 |
244 | Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE(1998) Cerebral amyloid angiopathy: Amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s diseaseAm J Pathol153: 725733 |
245 | Weller RO, Nicoll JA(2003) Cerebral amyloid angiopathy: Pathogenesis and effects on the ageing and Alzheimer brainNeurol Res25: 611616 |
246 | Colantuoni A, Bertuglia S, Intaglietta M(1985) Variations of rhythmic diameter changes at the arterial microvascular bifurcationsPflugers Arch403: 289295 |
247 | Szabo T, Wu J(2000) A model for longitudinal and shear wave propagation in viscoelastic mediaJ Acoust Soc Am107: 24372446 |
248 | Bamji-Mirza M, Callaghan D, Najem D, Shen S, Hasim MS, Yang Z, Zhang W(2014) Stimulation of insulin signaling and inhibition of JNK-AP1 activation protect cells from amyloid-β-induced signaling dysregulation and inflammatory responseJ Alzheimers Dis40: 105122 |
Figures and Tables
Fig.1
Hypothetic pathways of impaired vasomotion Path A: Aβ upregulates ET-1 causing vasoconstriction and cessation of vasomotion. Path B: Aβ reduces endothelial NO synthesis. According to most studies, this promotes vasomotion (protective). However, exposure to the vasoconstrictor thromboxane (TXA2) could increase vascular susceptibility to chaotic vasomotion. Path C: Aβ induces ER stress in endothelial cells and astrocytes, resulting in impaired calcium (Ca2 +) homeostasis. Aβ also interacts with vascular smooth muscle cells dysregulating vascular tone. Mitochondrial dysfunction disrupts Ca2 + homeostasis by reducing ATP production and impairing ATP-mediated transport. Cholinergic deficit aggravates CVR depression and vasomotion, resulting in regional CBF dysregulation, which in turn leads to hypoperfusion and subsequent oxidative stress and Aβ accumulation. Dotted lines indicate “feed-back” loops triggering vicious circles. Gray (white) rectangles indicate status (process). CBF, cerebral blood flow; CVR, cerebrovascular reactivity; EC, endothelial cell; ER, endoplasmic reticulum; ET-1, endothelin-1; NO, nitric oxide; VSMC, vascular smooth muscle cells.