
Use and Reuse of Animal Behavioral, Molecular, and Biochemical Data in Alzheimer’s Disease Research: Focus on 3Rs and Saving People’s Tax Dollars
Abstract
Several decades of research on cell and animal models contributed tremendously to understanding human diseases. Particularly, research on rodents and non-human primates revealed that animal research is a major and important component in biomedical research in learning complex pathophysiological processes. Further, animal research helped us to understand human diseases, such as Alzheimer’s disease. In addition, animal research has also helped us to test hundreds of drugs and develop treatments for human use. Researchers can gain a better understanding of key biological and physiological processes in humans by comparing them to laboratory animals. Based on their relevance and resemblance to people, or even usual living conditions, scientists rationalize the use of particular animal models in their studies. It is suggested that in the National Institutes of Health and other agencies-funded research, animal models should be carefully selected to study the biology and pathophysiology of human health and diseases such as Alzheimer’s disease and other dementias. However, it is critical to use a minimum number of animals for human research. Further, it is also noted that the use and reuse of behavioral, molecular, and biochemical data from wild-type (WT) control mice with mutant lines of disease models, as long as the genetic background is the same in both WT and disease mice. On the other hand, anonymous readers have challenged the use and reuse of WT mice data for comparison. In the current article, we discuss the minimum utility of animals, covering the 3Rs, Replacement, Reduction, and Refinement, and also discuss the use and reuse of behavioral, molecular, and biochemical data.
INTRODUCTION
In biomedical research, studies of cell cultures and animal models are important and provide significant information to understand the biology, and pathophysiology of disease in human diseases, including cancer, neurodegeneration, diabetes, obesity, hypertension, and other gender and age-related conditions.
Cell cultures are the major tools used in cellular, molecular biology, and protein chemistry, providing excellent model systems for studying the physiology and biochemistry of cells in healthy and disease states in relation to age and gender, particularly the effects of toxicity and protective aspects of therapeutic agents and drugs. For the last 3 decades, researchers have been working on cell models of human diseases, simply, overexpressing and/or silencing healthy and disease genes to develop therapeutic strategies using high-throughput techniques and tools. The outcome of cell culture studies, significantly improved our understanding of the disease process. However, we also learned the limitations of cell culture studies include cell-line misidentification and contamination with microorganisms. Most importantly, studying behavioral changes such as cognition, cardiovascular, and stroke-related conditions is not possible with cell culture studies. Alternative to cell culture studies is animal models, rodents, nonhuman primates, and primates. Studying these mammalian and other higher organisms will provide information related to cognition, cardiovascular, and stroke conditions close to humans.
Animal models are important tools that mimic human diseases, and greatly help us to improve the biology and pathophysiology of diseases. For example, using animal models enables researchers to carefully modify environmental elements to learn how they affect behavior, development, and disease profile. In the field of behavioral science, particularly cognitive function, and epigenetics, meaning environmental interaction with genes, leading to changes in behavior and health. Working with laboratory mice, truly helps us to understand prenatal immune challenges, environmental toxicants, diet, and early-life stress and diagnosis. Further, mouse models of human diseases, such as Alzheimer’s, Huntington’s, and Parkinson’s, will provide new information to develop therapeutic strategies and prevent and/or delay the disease process.
The purpose of our article is to discuss the issues with the use of animals for biomedical research to improve the quality of health. Further, we also discussed the animal models of Alzheimer’s disease and other human diseases and highlighted the importance of the three Rs (3Rs), Replacement, Reduction, and Refinement, of animals in biomedical research. In addition, we also highlighted the careful use of NIH and other funding agencies’ support and the use and reuse of behavioral, molecular, and biochemical data, after following the Institutional Animal Care and Use Committee.
ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) is a heterogeneous neurodegenerative ailment that worsens with time and results in dementia. The hallmark of AD is memory loss, cognitive function impairment, and changes in behavioral and personality traits.1–3 In addition, several conditions are linked to AD, including inflammation, extracellular neuritic plaques, intracellular neurofibrillary tangles (NFTs), loss of synapses, microRNA (miRNA) dysregulation, and deficiencies in the structure and function of the mitochondria.2–7 While late-onset sporadic AD is brought on by aging and other lifestyle factors, early-onset familial AD is mostly caused by genetic abnormalities.8–10 Mutations in amyloid precursor protein (APP), presenilin 1 (PS1), presenilin 2 (PS2), apolipoprotein E4 (APOE4), sortilin-related receptor 1, clusterin, complement component receptor 1, CD2 Associated Protein (CD2AP), Aph-1 Homolog A (APHA1), and membrane-spanning 4-domains subfamily A (MS4A4/MS4A6E) genes have been found in different AD patients.11
MOUSE MODELS OF ALZHEIMER’S DISEASE
Tremendous progress has been made in understanding the molecular basis of AD through the development and use of a large number of mouse models of AD and AD-related diseases.12–18 These models include mice with modified, as well as models with a combination of multiple AD mutations, such as APP-PS1,19 APP-PS1-Tau,20 and 5XFAD21 mice. Researchers have also developed knockin (physiological expression) mouse models with mutations in APP, PS1, and Tau.22–26 Finally, other groups have created knockout lines of mice to better understand the functions of APP, PS1, PS2, Tau, and ApoE4.27
NFT aggregation and Aβ accumulation are the primary biomarkers of AD, making them crucial for creating the right AD mice models. Sophisticated technologies like CRISPR/Cas9, Cre-loxP, and other techniques of genetic manipulation allow targeted insertion/deletion/point mutation and have enabled researchers to build animal models that better mimic the origin and depict the manifestation of AD-like features in mice. The initial model, PDAPP, carried the V717F Indiana mutation of the human APP gene using the platelet-derived growth factor B (PDGF) promoter through a minigene construct. In critical areas of the brain such as the entorhinal cortex, cingulate cortex, and hippocampus, these mice had thick and diffuse plaques exhibiting synaptic loss, microgliosis, and astrocytosis.13 Another widely used mouse model was the Tg2576 line, with an overexpression of Swedish mutation (K670M/N671L) within the 695 isoforms of human APP gene induced by a Prion Protein (PrP) promoter into B6; SJL F2 mice.12 This mouse model has become popular since there is a progressive development of plaques and successive decline in memory. On the other hand, mice solely overexpressing PSEN1 mutations did not exhibit plaques or other AD symptoms. That’s why scientists crossed the PSEN1 transgenic line with APP overexpression lines yielding a significant increase in amyloid pathology. Plaque deposition developed at 8–10 weeks when Tg2576 mated with mice carrying the human PSEN1–M146L mutant, as opposed to 8–12 months for single APP-mutated animals.28,29 To study the p-tau pathology of AD, the JNPL3 line was introduced expressing the 4R0N isoform of human hMAPT (microtubule-associated protein tau) with P301L mutation, which showed tau pathology neurodegeneration and memory loss.30 Later, 3xTg mice (bearing the Swedish mutation in APP, M146v in PSEN1, and P301L in MAPT),20 and 5xFAD mice (containing 5 familial AD mutations (APP KM670/671NL (Swedish), APP I716V (Florida), APP V717I (London), PSEN1 M146L, PSEN1 L286V)21 were developed to study more elaborated pathological developments of AD.
Although most of these models mimic some aspects of AD pathology, none have a phenotype that mimics the trajectory of late-onset AD cases, which are not driven by mutations in APP, PS1, or PS2. ApoE2/3/4 mouse models have been generated, but again, the biology underlying these models is not representative of the vast majority of late-onset AD patients.
ALZHEIMER’S MOUSE MODELS STUDIED IN REDDY LABORATORY
The Reddy laboratory has been working on multiple lines of mice in the last 25 years, briefly described below: Currently, our lab extensively conducted experiments using early-onset AD mice, APP transgenic mice (with Swedish mutation 670/671, Tg2576 strain), 5XFAD, APPxPS1, and late-onset AD mice,31–42 human amyloid beta knockin (AbetaKI),43 and mutant Tau mice37–39,44 in different stages of disease progression. More recently, we studied naturally occurring diabetes in mice, referred to as TallyHo.45–48 In addition, we generated microRNA-455-3p Tg and KO lines of mice 49, microRNA stroke Tg mice (Vijayan et al unpublished), and also Rlip Tg mice (Reddy et al unpublished) (Table 1).
Table 1
Summarizing Alzheimer’s disease mice models with their genotype and phenotype investigated in the Reddy’s laboratory
| Sl | Mice | Mutation | AD Onset | Pathology | Cognitive Behavior | Synaptic features | Mitochondrial dysfunction | Reference |
| 1 | APP TG | Swedish mutation (KM670/671NL) | Early-onset | Amyloid-β | Impaired spatial learning and working memory | Dendritic spine loss, decline in LTP, synaptic damage | Mitophagy | 31 |
| 2 | hAbetaKI | Humanized Abeta sequence | Late-onset | Amyloid-β | Impairments in spatial learning and memory. | Reduced dendritic spines, decline in synaptic proteins. | Revealed increased mt fragmentation and reduced mt length | 43 |
| 3 | Mutant Tau | P301L | Early-onset | p-tau/NFT | Impaired hippocampal learning and memory. | Dendritic spines are significantly reduced | Increased Mitochondrial biogenesis | 37 |
| 4 | Rlip KO (Rlip +/-) | Conditional knockout | Late-onset | Amyloid-β | Reduced spatial and learning memory, | Reduced synaptophysin expression | Alterations in mitochondrial dynamics. | 53 |
| 5 | Drp1 KO (Drp 1 +/-) | Reduced expression of Drp1 | N/A | Amyloid-β /p-tau | Provided protection against impaired cognition in APP mice | Increased levels of mRNA and proteins of synaptic genes | Mitochondrial function was increased | 35 |
| 6 | VDAC1+/- | Reduced expression of VDAC1 | N/A | Amyloid-β /p-tau | Provided protection against impaired cognitive activities in a transgenic Tau mouse | Enhanced synaptic activity | Upregulated mitochondrial function | 59 |
| 7 | miR-455-3p KO | Knock out the miR-455-3p | Early-onset | Amyloid-β | Decline in cognitive behavior, spatial learning and memory | Alteration in synaptic activities | Reduced mitochondrial biogenesis and dynamics | 49 |
| 8 | TallyHo mice (TH) | Polygenic inbred model for type 2 diabetes | N/A | N/A | Cognitive impairments | mRNA levels of synaptic genes were significantly reduced | Mitochondrial dysfunction | 45 |
APP Tg mice (Tg2576)
One of the most popular and thoroughly studied mouse models of AD is the Tg2576 model due to its therapeutic relevance to the disease. It overexpresses a mutant form of APP (isoform 695) with the Swedish mutation (KM670/671NL), resulting in elevated levels of Aβ and ultimately amyloid plaques. Karen Hsiao Ashe and colleagues created the Tg2576 model, which is currently offered by Taconic and Charles River. Mice that are hemizygous developed widespread amyloid pathology and experienced cognitive impairments and deficits in spatial learning, working memory, and contextual fear conditioning at 6 months of age.12 31 Dendritic spine loss in the hippocampal CA1 area, alterations in synaptic plasticity, and decline in LTP in the dentate gyrus following perforant route stimulation have also been reported.50 Our laboratory has extensively studied for gene expression analysis,40 molecular and cellular links amyloid beta/mutant APP with mitochondria and mitochondrial proteins such as VDAC1, Drp1,33,36,42 and mitophagy/autophagy and synaptic damage.31
Humanized amyloid beta knockin mice (hAbetaKI)
To understand the mechanisms and pathophysiology of late-onset AD, recently a humanized Abeta knockin (hAb-KI) mouse model was developed. These hAbeta-loxP-KI mice express endogenous mouse APP protein with a humanized Aβ peptide sequence. Early characterization work has found that this model recapitulates many biological features that are consistent with the vast majority of late-onset AD cases.51 There are several advantages of this AD mouse model (hAb-KI). First, it avoids APP overexpression, which was a limitation of earlier AD mouse models. Second, this mouse model has been longitudinally characterized in several behavioral tests.51 Further, we recently characterized the model for early cellular, molecular, and behavioral changes43 where 7-month-old hAbKI mice showed deficits in memory and motor coordination compared to age-matched control. Alteration in mitochondrial biogenesis was revealed by increased mt fragmentation and reduced mt length in both hippocampal and cortical tissues in hAbKI mice.43 These observations offer defined criteria to evaluate the effects of exercise and a healthy diet at both preventive and curative stages of AD progression.
Tau mice
The mutant Tau mouse model (P301L mouse line) was generated with a human Tau P301L mutation.30 These mice develop age-dependent p-tau and NFTs in the neocortex, hippocampus, and cortex, as do humans with AD. Cognitive impairments have been observed at 4.5 months of age in homozygous Tau mice and at 6 months in hemizygous Tau mice.37 Cognitive impairments are similar to those developed by humans with AD. The P301L line of mice is a well-studied model of p-tau/NFT pathologies and will be important for studies into the effects of a healthy diet and regular exercise on p-tau formation, quality of mt, mitophagy, synaptic health, and miRNA and mRNA expression.37
Recently our lab studied Tau mice for several aspects of mitochondria and synaptic activities.37 Tau mice showed deficits in learning, and memory in our MWM test and a decline in motor coordination in our rotarod tests compared to WT mice. Irregular mitochondrial dynamics were reported in tau mice where mitochondrial fission and fusion proteins were altered compared to WT control mice. As expected, total and phosphorylated tau proteins were increased in tau mice. A significant reduction in the number of dendritic spines in tau was observed in a Golgi-Cox staining whereas a considerable increment in mitochondrial numbers and decline in mitochondrial length in tau mice were revealed in our transmission electron microscopy analysis. Taken together, aggregation of phosphorylated tau in the hippocampus generates mitochondrial dysfunction, and alteration in dendritic and synaptic morphogenesis leading to learning and memory impairments in tau mice.37
Rlip mice
Heterozygous Rlip knockout mice (Rlip+/–), were generated with the help of Biocytogen LLC (Wakefield, MA).52 We designed a conditional knockout of exon 4 of the Ralbp1 gene on chromosome 17 using a Cre-loxP system. The intron 3-4 and intron 4-5 are big and insertion of loxP elements will not interfere with mRNA splicing. Both of the loxP sites were inserted into non-conserved regions. These mice are maintained in our animal facility by us and are in use. We recently reported our initial investigations into the phenotypic behavior, oxidative stress/mitochondrial dysfunction, and synaptic activities in these mice.53 Our laboratory recently generated Rlip transgenic RlipTg/Tg) mice using CRISPR/Cas9-based extreme genome editing (EGE) technology in collaboration with Cyagen Biosciences (Santa Clara, CA, USA). Single sgRNAs were designed to insert a CAG promoter-mouse Ralbp1 coding sequence-WPRE-polyA cassette into intron 1-2 of the mouse Rosa26 gene. The mouse Ralbp1 gene, driven by the CAG promoter, is over-expressed in all tissues.
miR-455-3p Tg and miR-455-3p KO mouse models
To understand the molecular causes of AD and create treatment approaches for the disease, mouse models are proven to be the most crucial research instruments. Our team recently created a miR-455-3p-Tg (TG) mouse model on a C57BL6 background by pronuclear injecting the miR-455-3p expression vector (pRP[Exp]-U6 > hsa- miR-455-3p-CAG-EGFP), with the support from Cyagen Biosciences, Santa Clara, CA, USA.49 In addition, a miR-455-3p knockout (KO) mouse model on a C57BL6 background was also generated to study the effects of depleted miR-455-3p in AD development. We used CRISPR /Cas9 technology to knock out the endogenous miR-455-3p. MiR-455-KO pups were genotyped and their sequencing analysis confirmed the depletion of miR-455 locus of 82 bp total size. Further, miR-455-3p-KO mice did not exhibit any deviation in their physical features and reproductive activities. The miR-455-3p TG mice showed increased neuronal expression and reduced levels of microglia and astrocytes; on the other hand, KO miR-455-3p mice showed the opposite, meaning reduced neuronal expression and enhanced glia and astrocytes.
Drp1 mouse models
Recent Drp1 knockout studies revealed that Drp1 homozygous knockouts are embryonic-lethal.54,55 The longevity, fertility, and viability of Drp1+/– mice are normal, and their phenotype is not different from that of WT mice. Our lab investigated and characterized the synaptic, dendritic, and mt protein levels in Drp1+/– and WT mice. The Drp1+/– animals showed much lower levels of hydrogen peroxide and lipid peroxidation, while their mitochondrial and synaptic functioning and GTPase Drp1 enzymatic activity appeared normal suggesting that a partial reduction in Drp1 is beneficial against oxidative stress. Next, we conducted cross-breeding of Drp1+/– mice with APP mice to explore the role of Drp1 in Aβ pathogenesis. 35 In the double mutant lines APP X Drp1+/–, we observed decreased mt fission and enhanced mt fusion, mt biogenesis, and synaptic proteins inferring that a reduction in free radicals may protect Aβ in AD neurons.
VDAC1 mouse model (VDAC1+/–)
VDAC, ubiquitously located in the outer membrane of mitochondria, is generally thought to be the primary means by which metabolites diffuse in and out of mitochondria.56 In mammalian mitochondria, three VDAC isoforms have been identified: VDAC1, VDAC2, and VDAC3. VDAC1 is the most extensively expressed of the three isoforms in the skeletal muscles, liver, heart, and brain. VDAC proteins have key roles in synaptic plasticity, maintaining mitochondrial biogenesis, and regulating apoptosis.
Researchers designed VDAC1 and VDAC3 heterozygote embryonic stem (ES) cells to obtain heterozygote and homozygote knockout mice where the VDAC heterozygote knockout (VDAC1+/– and VDAC3+/–) mice had no problem with fertility and longevity.57,58 Our laboratory focused on VDAC1 in our studies because VDAC2 and VDAC3 expression levels are low in mammalian brains. In our lab, while characterizing the role of VDAC1 in AD progression, we found a reduction in mRNA levels in the AD-related genes, including APP, Tau, PS1, PS2, and BACE1. In addition, we found that the expression of mitochondrial fusion genes Mfn1, and Mfn2 were upregulated and the level of fission genes Drp1 and Fis1 were decreased suggesting an enhanced mitochondrial activity.59 Henceforth, it could be inferred that partially reduced VDAC1 expression in the VDAC1+/– mice generated a protective role against AD-related bioburden in mice brains. It is unclear whether and how partially reduced VDAC1 affects the mitochondrial phenotype and synaptic activity in AD transgenic mice expressing AβPP and mutant Tau.
The TallyHo mice
An inbred polygenic model for type 2 diabetes (T2D), the TALLYHO/Jng (TH) mouse is typified by elevated body and fat pad weights, hyperleptinemia, hyperinsulinemia, and hyperlipidemia.60 Our lab examined potential mitochondrial changes in the pancreas and liver brought on by hyperglycemia in diabetes using strains of non-diabetic SWR/J mice and diabetic TallyHO/JngJ (TH) mice.45,46 Intraperitoneal glucose tolerance tests at different time intervals were conducted in both diabetic and non-diabetic animals. Mitochondrial dynamics, biogenesis, and function were studied in our experiment on three groups of mice; controls, diabetic TH mice, and diabetic TH mice treated with SS31 peptide intraperitoneally. After 24 weeks of age, the diabetic mice displayed overt pancreatic injury, as evidenced by reduced islet size, vacuolization, and β cell disintegration and atrophy. TH mice showed signs of mitochondrial dysfunction, including markedly increased H2O2 generation, lipid peroxidation, and decreased ATP synthesis. In addition, diabetic mice showed a significant alteration in mitochondrial dynamics gene immunoblot analysis and mRNA expression when compared to control groups. On the other hand, SS31 therapy decreased mitochondrial dysfunction and nearly normalized most mitochondrial functions and mitochondrial dynamics processes in TH mice. In summary, SS31 therapy may protect against the mitochondrial changes brought on by hyperglycemia.
Conducting experiments with mouse models of Alzheimer’s disease expressing human mutation with APP, PS1, PS2, Tau, ApoE4 to understand the impact of early-onset and late-onset disease process. Further other mouse models of mitochondrial proteins such as Voltage-dependent anion channel protein 1 (VDAC1), dynamin-related protein 1 (Drp1), and others are equally important to understand aging, age-related mitochondrial changes in AD.
In mouse model studies concerning AD and other neurological diseases such as multiple sclerosis, Huntington’s disease, and Parkinson’s disease, age-matched WT mice comparisons are critical to understanding the disease process. When we compare WT mice’s behavioral and biochemical data, it is important to maintain the disease mutation in the same/similar genetic background of mutant lines of mice (Fig. 1). In other words, it is important to keep transgene and/or knock-in negative animals as WT for behavioral and biochemical data. Keeping these things in the real sense, mouse models provide valuable information, in terms of cognitive function, cell and molecular biology, electrophysiology, and synaptic/mitochondrial function with disease progression in relation to gender.
Fig. 1
A diagram shows cross-breeding. WT mice is relevant as a control for the offspring mice. So, Use and reuse of behavioral and biochemical data can be applied to other phenotypes as well.
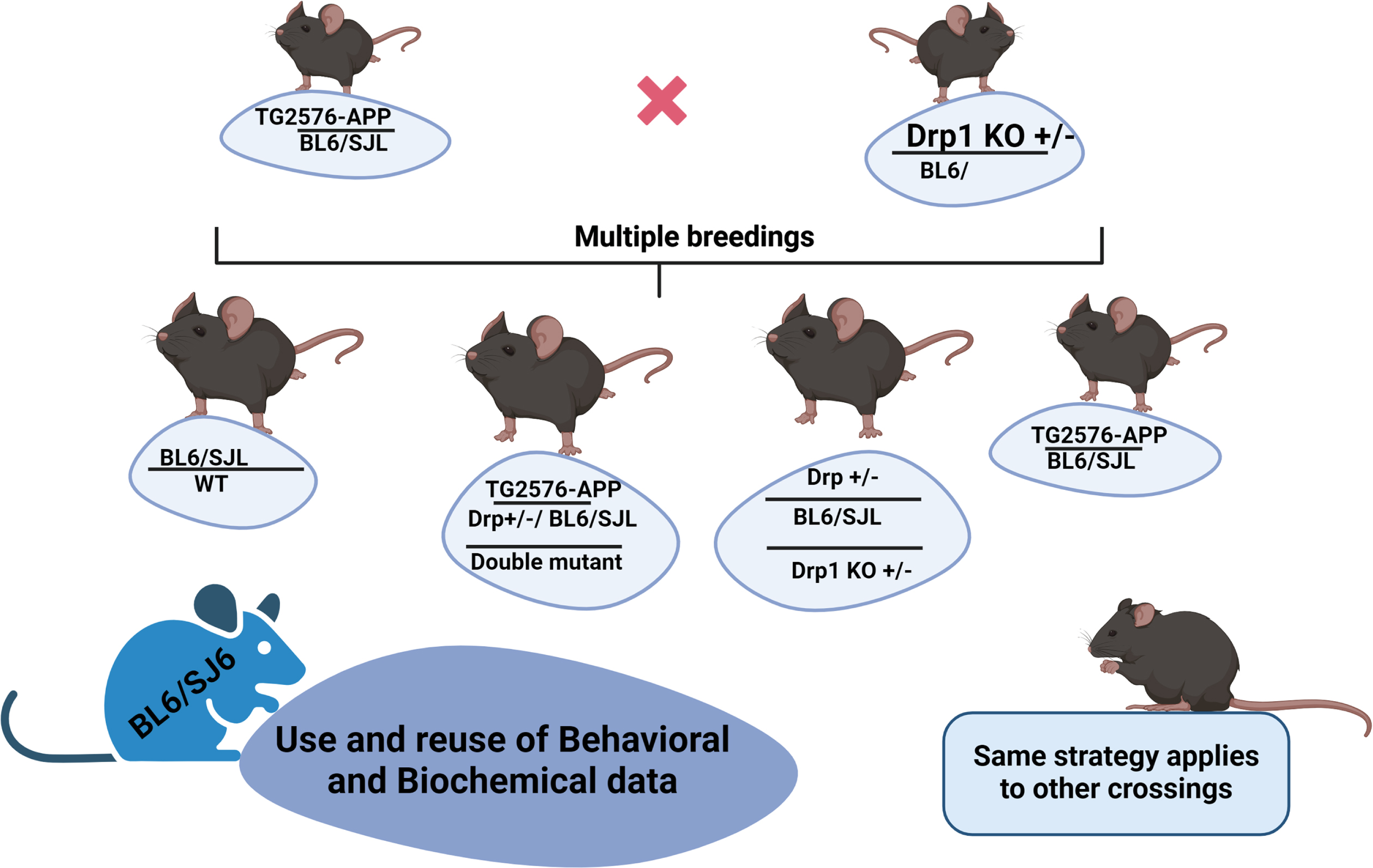
We maintained WT mice genetic background similar/same to several mutant lines of mice, including Drp1+/–, APP (Tg2576), Tau (P301L), TallyHo (naturally occurring diabetic mutation/trait, similar to human Type 2 Diabetes), and others. Behavioral/biochemical data of WT mice can be used for Drp1+/–, APP (Tg2576), Tau (P301L), and TallyHo mice because they all maintained C57BL backgrounds. This is important to NIH dollars. Studying these lines of mice can be time-consuming, labor-intensive, and expensive.
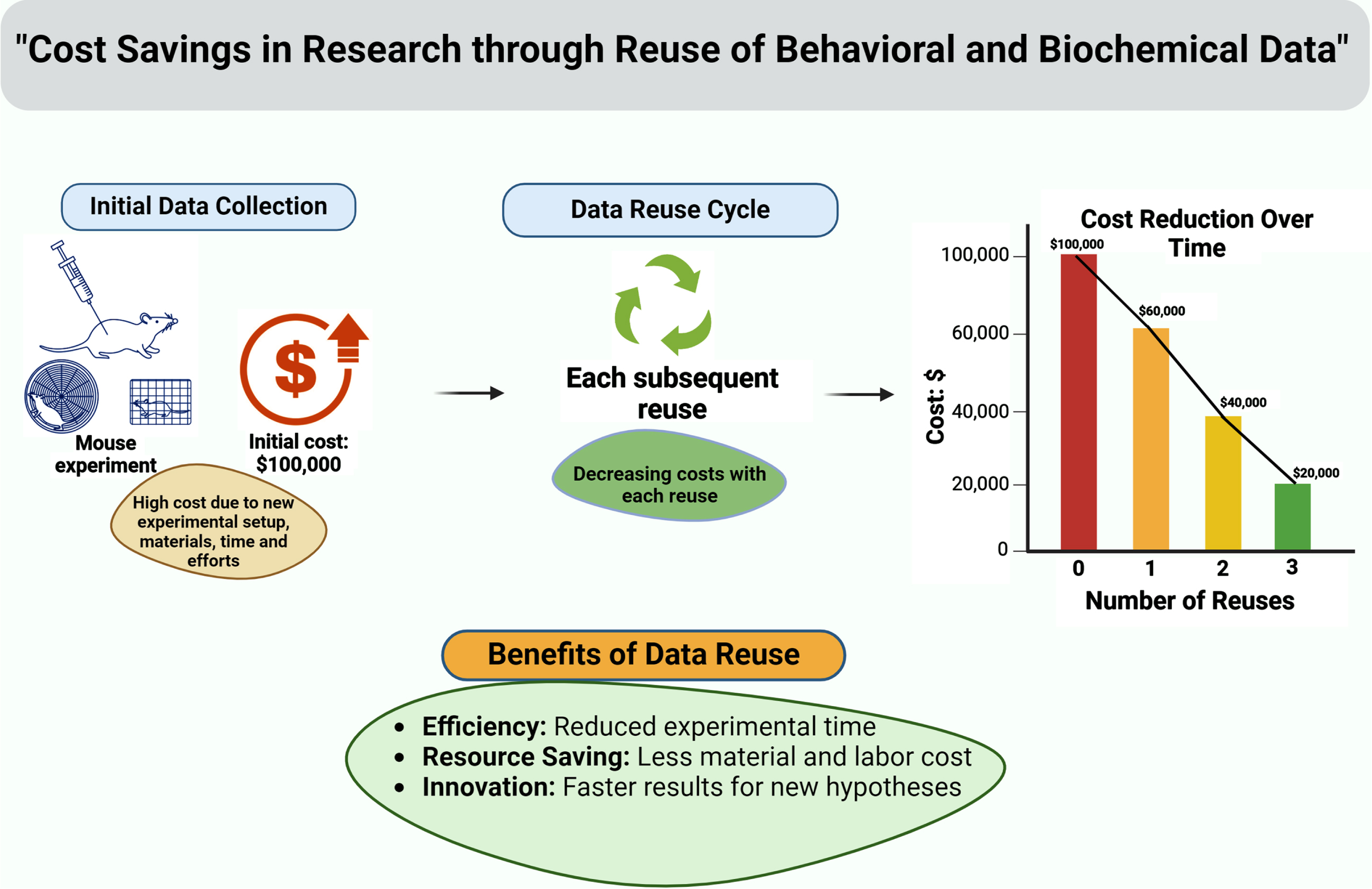
Reusing existing behavioral/biochemical data of WT mice, if the background strain of WT mice is the same as mutant mice within the same research group, researchers can optimize with maximum resource utilization and minimize animal usage (Fig. 2). This is particularly important from an ethical standpoint, as it reduces the number of animals needed for research purposes, while still generating valuable scientific knowledge.
Fig. 2
A graphical depiction shows financial advantages of data reuse in animal research.
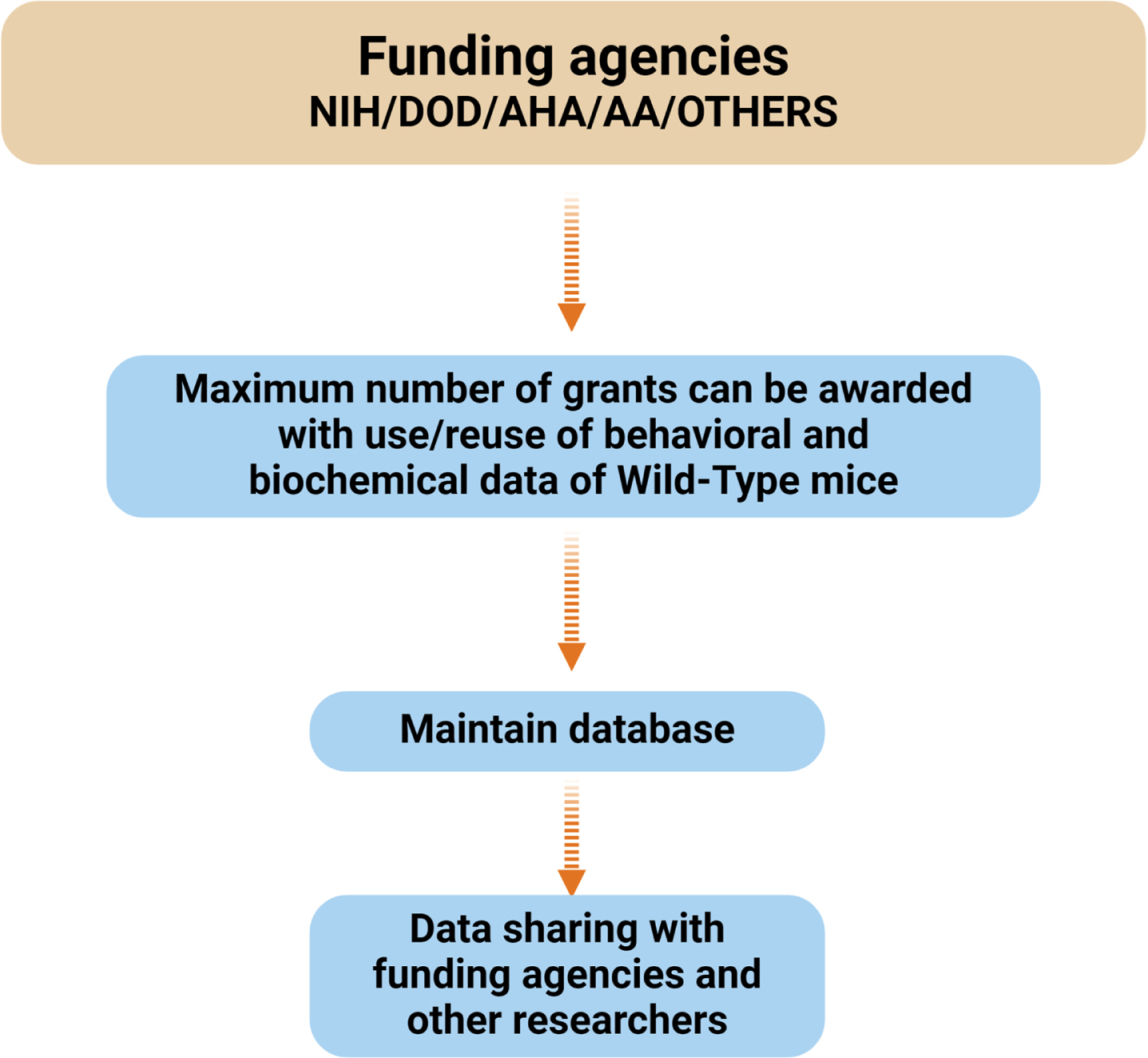
The funding agencies, such as NIH and DoD, VA, and Institutional Animal Care and Use Committees (IACUC) recommended using the minimum number of mice for biomedical research (Fig. 3), after following three RRRs (3Rs) rules/guidelines.
Fig. 3
Flowchart illustrating the relationship between funding agencies (NIH, DOD, AHA, AA) and the management of behavioral and biochemical data. The maximum number of grants can be awarded by saving and reusing behavioral and biochemical data. This data is maintained in a database, which facilitates data sharing with the funding agencies and researchers. NIH, National Institute of Health; AHA, American Heart Association; DOD, Department of Defense; AA, American Alzheimer’sAssociation.
NIH GUIDELINES FOR ANIMAL RESEARCH
Researchers should consider several essential factors when planning studies involving animals and designing research proposals for NIH grant applications. First, researchers need to be very explicit about why using animals is essential to their work. Second, throughout the study process, they should select the bare minimum required to guarantee thorough and reproducible investigations. Third, scientists ought to take into account certain animal models that are relevant to public health and suitable for the questions being posed in the research. Fourth, every surgery that will be carried out on the animals should be meticulously recorded, taking into account the least amount of possible pain, discomfort, harm, or distress that the animals might go through. Finally, but just as importantly, researchers must explore alternative, acceptable methods that could take the place of animal models in studies, their results, and their possible advantages.
THE 3Rs: REPLACEMENT, REFINEMENT, AND REDUCTION
The “3Rs” refer to the fundamental principles of responsible animal use in research, which the NIH mandates researchers to consider when designing their studies. These principles include Reduction, Replacement, and Refinement. Reduction involves designing experiments with appropriate statistical analyses, appropriate timelines, and suitable comparison groups to ensure the minimum number of animals is used.61 Replacement encourages the use of non-animal models whenever possible and appropriate for the scientific objectives. Refinement focuses on selecting experimental procedures that minimize pain or distress for the animals involved (Fig. 4).
Fig. 4
Schematic representation of 3Rs. Researchers must follow the strict guidelines of replacement, reduction, and refinement in practicing animal studies.
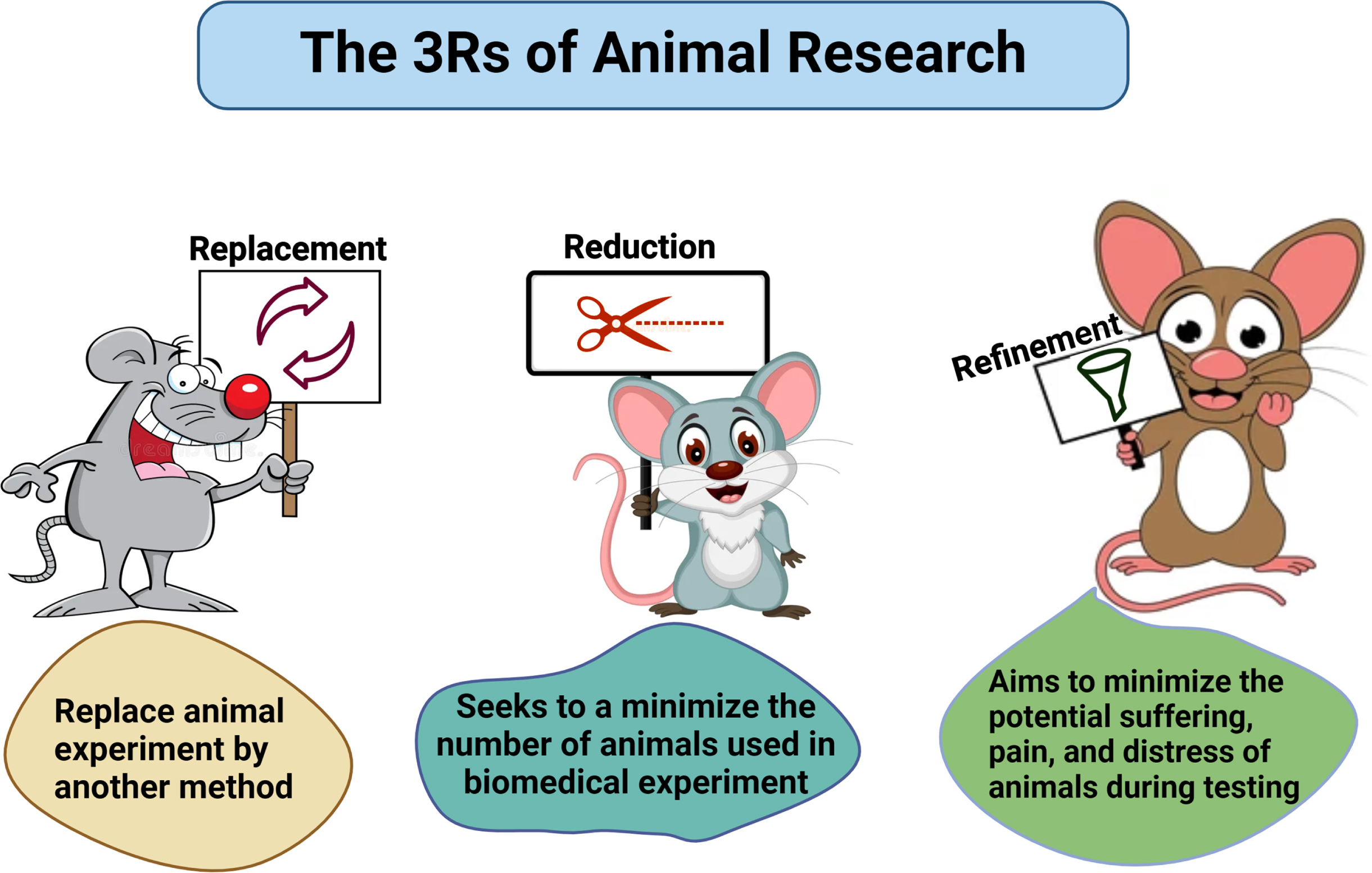
The principles of Replacement, Reduction, and Refinement are crucial for all animal studies and are a prerequisite for successful animal experiments. The significance of humanity and its intimate connection with efficiency in experimentation should be carefully considered. Russell and Burch define “efficiency” as producing the greatest amount of scientific or medical outcomes from the best use of resources—including money, animals, facilities, and staff. They contend that when animals endure needless suffering, these resources are frequently squandered or fail to produce the greatest outcomes. Our objective aligns with the efficiency approach and we opted to use the fewest animals possible while achieving the maximum outcome. Developed over 50 years ago, the 3Rs framework was designed to promote more humane animal research.61 These principles have since been incorporated into national and international laws and regulations governing the use of animals in scientific procedures, as well as into the policies of organizations that fund or conduct animal research. The use of a minimum number of animals is important for funding agencies and researchers across the globe, who are involved with animal research. The general attitudes consistently show that support for animal research is conditional on the 3Rs being put into practice. The strategy to advance the 3Rs focuses on their scientific impacts and benefits. Recently, the definitions of the 3Rs have been updated to better align with contemporary scientific practices and developments. Originally, the 3Rs were introduced by Russell and Burch.62 Scientists follow a hierarchical arrangement of the Rs, wherein Replacement—which can completely prevent harm—should come first, followed by Reduction—which lowers the number of animals harmed—and Refinement—which comes last.
Replacement
Where animals would typically be used in research, it is imperative to avoid using them or find alternatives. In many cases, important research issues can be investigated without using animals by applying the most recent scientific and technological advancements in selecting reliable, predictive models and tools. The term “replacement” describes methods or technology that circumvent using animals in research when it would not have been required to do so.62–65 Research animals have long been used to help address important scientific issues, especially those pertaining to human health. However, using animal models can be expensive, time-consuming, and have limitations for science, particularly when studying non-human primates. Alternative models can mitigate some of these issues. In recent years, advancements in science and technology have created realistic opportunities to replace animal use. Replacement can be categorized into full and partial replacement. Full replacement methods completely avoid using animals in research and testing, including human volunteers, tissues, cells, mathematical and computer models, and established cell lines—collectively known as non-animal technologies or methods. Partial replacement involves using animals that are not considered capable of experiencing suffering based on current scientific understanding, such as invertebrates.
Reduction
Minimizing the number of animals used in research while maintaining scientific integrity is crucial. Properly designed and analyzed animal experiments should be robust, reproducible, and contribute significantly to the existing knowledge base. Reduction refers to methods that minimize the number of animals used in each experiment or study, aligning with scientific objectives. This includes designing and analyzing studies in a way that ensures robust and reproducible results, as well as maximizing the data obtained from each animal.62–65 For example, imaging modalities that allow for longitudinal measurements in the same animal or micro-sampling of blood for repeated sampling can reduce the need for additional animals. It’s important to balance reducing animal numbers with avoiding additional suffering caused by repeated use. Sharing data and resources, such as animals, tissues, and equipment, between research groups and organizations also contributes to reduction. Additionally, reusing behavioral and biochemical data with appropriate cross-referencing and acknowledgment in research reports is essential.
Refinement
Minimizing the pain, suffering, distress, and lasting harm that research animals might experience is essential for advancing laboratory animal welfare. The use of the Animal Welfare Act, which mandates that investigators take into account the use of alternatives to painful or distressing procedures, is also emphasized by the US Department of Agriculture Animal Plant and Health Inspection Service.66 Because refinement focuses on the actual conduct of research and the treatment of sentient study animals, it is promoted as a unique approach to eliminating inhumanity. By utilizing the latest in vivo technologies, we can reduce pain and distress while improving our understanding of how welfare impacts scientific outcomes. Refinement involves methods that minimize these negative experiences and enhance animal welfare. This covers every facet of employing animals, including scientific research housing, and husbandry. Examples include providing housing that allows for species-specific behaviors, using appropriate anesthesia and analgesia to minimize pain, and training animals to cooperate with procedures to reduce distress. Evidence shows that pain and suffering can alter an animal’s behavior, physiology, and immunology, leading to variations in experimental results that affect the reliability and repeatability of studies. The welfare of the animal is another important factor in this situation, as it allows for uniformity and the usage of fewer animals by eliminating discomfort, even minor distress. This eliminates wasteful repetition and allows for the regular practice of a number that yields substantial results.
Before animal research is approved, the Institutional Animal Care and Use Committee (IACUC) is tasked with evaluating the projects’ ethical viability guided by a set of laws and regulations inspired by the 3Rs. 67 Our lab followed NIH, TTUHSC, and IACUC guidelines (The Institutional Animal Care and Use Committee (IACUC) for the three RRRs: the principles of Replacement, Reduction, and Refinement for all animal (mice) studies and our goal was to use a minimum number of animals and achieve/obtain the maximum outcome. We mostly focus on minimizing the use of a number of animals from the same genetic background in similar types of experiments which reduces the amount of study animals that may be in distress, hence mitigating their suffering. In instances where scientists are acquainted with the anticipated fluctuation in the bioassay, it allows them to predict the right quantity of animals in advance based on their experience. Moreover, improvements in statistical and experimental procedures should lead to continuous revision and further reduction in the number of animals used in experiments.65 To provide a scientifically sound and statistically feasible experiment, we recommend selecting the ideal number of animals from separate experiment groups. Another important factor is carefully planning the experiment to optimize the amount of data obtained from a specific number of animals, allowing researchers to save labor, time, and the need for additional animals.
The core principles and guidelines for the utilization of animals in laboratory research are governed by the Animal Welfare Act and Regulations issued by the United States Department of Agriculture. This policy directs the IACUC’s operational policies in any institution. To guarantee that animals are used in research facilities properly, the IACUC has regular reporting requirements and conducts unexpected inspections.68 Animal Research Advisory Committee (ARAC) Guidelines from the Office of Animal Care and Use, NIH also provide specific guidelines for animal-based experiments following the 3Rs principle, and all NIH-funded research involving live vertebrate animals is managed by the Office of Laboratory Animal Welfare (OLAW).69 In addition, the researcher should follow the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments) which constitutes the minimum requirement for sample size of experimental animals, experimental procedures, and statistical analysis of research outcome.70 Taken together, arbitrary use of research animals is strictly prohibited, and rational reduction of animal use in research is highly prescribed. Please find below some examples from our published article on how to use/reuse the behavioral, molecular, and biochemical data generated from laboratory animals in our laboratory.
Manczak et al. (2016) (Drp1+/– X APP) and Kandimalla et al. (2016) (Drp1+/– X Tau) – Genetic background is the same for WT, Drp1+/–, APP, Tau, and double mutant (Drp1+/– X APP; Drp1+/– X Tau) mice. Therefore, we used WT mice behavioral data in Manczak et al. (2016) (Drp1+/– X APP), and Kandimalla et al. (2016) (Drp1+/– X Tau) studies. 38,42
In Bhatti et al. (2020), we studied the pancreas in TallyHo mice, findings were published in 2020 online, printed form in 202145; In Bhatti et al. (2021), we studied the liver in the same TallyHo mice, treated with SS31 molecule, findings were published in 2021. We assessed glucose levels in 8, 16, and 24 weeks, used the data in the pancreatic study, and upon request of reviewers from the previous article, we reused 24 weeks of glucose levels data in the liver study.
In Kshirsagar et al. (2022) 43, we studied open-field, rotarod behavioral analysis in 6-month-old humanized Abeta knockin (hAPPKI) mice. We did reuse the same behavioral (open field, rotarod) data for comparison in hAPPKI mice treated with mitophagy enhancers (Urolithin A + EGCG).71
Conducting experiments with animal models such as wild-type mice and/or other mutant mice (hAPPKI, APP, Tau, TallyHo) can be time-consuming, labor-intensive, and expensive. By reusing existing data for another paper within the same research group, researchers can optimize with maximum resource utilization and minimize animal usage. This is particularly important from an ethical standpoint, as it reduces the number of animals needed for research purposes, while still generating valuable scientific knowledge.
CONCLUSIONS AND FUTURE DIRECTIONS
Animal research has significantly advanced our understanding of complex human diseases, such as cancer, cardiovascular, kidney, diabetes/obesity, and a large number of age-related neurodegenerative diseases, particularly AD, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and inherited mitochondrial diseases by elucidating key pathophysiological processes. The use of well-cared-for animal models has facilitated the development and testing of numerous treatments, underscoring the importance of ethical practices and the 3Rs—Replacement, Reduction, and Refinement—in biomedical research. Similarly, studying cell cultures and in vitro mouse models of AD provides critical insights into the disease’s pathophysiology. Cell cultures allow for a controlled environment to examine specific cellular mechanisms and drug responses, offering the advantage of reproducibility and cost-effectiveness. However, these models lack the complexity of living organisms and do not fully replicate the multifaceted nature of AD. Mouse models address some of these limitations by providing a more comprehensive understanding of disease progression and systemic interactions, particularly behavioral phenotype and structural and functional aspects of different organs in the body. They offer the advantage of studying the effects of AD in a whole organism, which can lead to more translationally relevant findings. Nevertheless, ethical considerations arise, necessitating adherence to the 3R principles. Researchers are encouraged to design experiments that minimize animal use, employ non-animal models when possible, and choose procedures that reduce distress. Additionally, reusing existing behavioral, biochemical, and molecular data within the same research group can optimize resources, minimize animal usage, and reduce costs, making research more efficient and ethically responsible by limiting the number of animals needed for experiments. Funding agencies and institutions play a crucial role in promoting these practices, ensuring that the number of laboratory animals used is minimized, and fostering responsible and ethical research practices.
AUTHOR CONTRIBUTIONS
Md Ariful Islam (Conceptualization; Data curation; Formal analysis; Investigation; Methodology); Sudhir Kshirsagar (Conceptualization; Data curation; Formal analysis; Investigation; Methodology); Arubala P. Reddy (Data curation; Formal analysis; Investigation); Ujala Sehar (Conceptualization; Formal analysis; Investigation; Methodology); Hemachandra Reddy (Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration).
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This investigation was supported in part by NIH grant AG079264 (to PHR).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
1. | Mattson MP . Pathways towards and away from Alzheimer’s disease. Nature (2004) ; 430: : 631–639. |
2. | LaFerla FM , Green KN and Oddo S. Intracellular amyloid-β in Alzheimer’s disease. Nat Rev Neurosci (2007) ; 8: : 499–509. |
3. | Reddy PH , Manczak M , Mao P , et al. Amyloid-β and mitochondria in aging and Alzheimer’s disease: implications for synaptic damage and cognitive decline. J Alzheimers Dis (2010) ; 20: : S499–S512. |
4. | Selkoe DJ . Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloidβ-protein. J Alzheimers Dis (2001) ; 3: : 75–82. |
5. | Reddy PH , Tripathi R , Troung Q , et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta (2012) ; 1822: : 639–649. |
6. | Kumar S and Reddy PH. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim Biophys Acta (2016) ; 1862: : 1617–1627. |
7. | Pradeepkiran JA , Baig J , Islam MA , et al. Amyloid-β and phosphorylated tau are the key biomarkers and predictors of Alzheimer’s disease. Aging Dis (2024) . doi: 10.14336/AD.2024.0286 |
8. | Amakiri N , Kubosumi A , Tran J , et al. Amyloid beta and microRNAs in Alzheimer’s disease. Front Neurosci (2019) ; 13: : 430. |
9. | Rawat P , Sehar U , Bisht J , et al. Phosphorylated tau in Alzheimer’s disease and other tauopathies. Int J Mol Sci (2022) ; 23: : 12841. |
10. | Sehar U , Rawat P , Reddy AP , et al. Amyloid beta in aging and Alzheimer’s disease. Int J Mol Sci (2022) ; 23: : 12924. |
11. | Mao P and Reddy PH. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: implications for early intervention and therapeutics. Biochimica et Biophysica Acta (2011) ; 1812: : 1359–1370. |
12. | Hsiao K , Chapman P , Nilsen S , et al. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science (1996) ; 274: : 99–103. |
13. | Games D , Adams D , Alessandrini R , et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature (1995) ; 373: : 523–527. |
14. | Borchelt DR , Thinakaran G , Eckman CB , et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron (1996) ; 17: : 1005–1013. |
15. | Sturchler-Pierrat C , Abramowski D , Duke M , et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A (1997) ; 94: : 13287–13292. |
16. | Mucke L , Yu GQ , McConlogue L , et al. Astroglial expression of human alpha(1)-antichymotrypsin enhances Alzheimer-like pathology in amyloid protein precursor transgenic mice. Am J Pathol (2000) ; 157: : 2003–2010. |
17. | Chishti MA , Yang DS , Janus C , et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem (2001) ; 276: : 21562–21570. |
18. | Davis HS and Rockwood K. Conceptualization of mild cognitive impairment: a review. Int J Geriatr Psychiatry (2004) ; 19: : 313–319. |
19. | Jankowsky JL , Xu G , Fromholt D , et al. Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Neuropathol Exp Neurol (2003) ; 62: : 1220–1227. |
20. | Oddo S , Caccamo A , Shepherd JD , et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron (2003) ; 39: : 409–421. |
21. | Oakley H , Cole SL , Logan S , et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci (2006) ; 26: : 10129–10140. |
22. | Lamb BT , Bardel KA , Kulnane LS , et al. Amyloid production and deposition in mutant amyloid precursor protein and presenilin-1 yeast artificial chromosome transgenic mice. Nat Neurosci (1999) ; 2: : 695–697. |
23. | Köhler C , Ebert U , Baumann K , et al. Alzheimer’s disease-like neuropathology of gene-targeted APP-SLxPS1mut mice expressing the amyloid precursor protein at endogenous levels. Neurobiol Dis (2005) ; 20: : 528–540. |
24. | Andorfer C , Kress Y , Espinoza M , et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem (2003) ; 86: : 582–590. |
25. | Chui DH , Tanahashi H , Ozawa K , et al. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med (1999) ; 5: : 560–564. |
26. | Simon T , Becquemont L , Mary-Krause M , et al. Combined glutathione-S-transferase M1 and T1 genetic polymorphism and tacrine hepatotoxicity. Clin Pharmacol Ther (2000) ; 67: : 432–437. |
27. | Jankowsky JL and Zheng H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol Neurodegener (2017) ; 12: : 89. |
28. | McGowan E , Sanders S , Iwatsubo T , et al. Amyloid phenotype characterization of transgenic mice overexpressing both mutant amyloid precursor protein and mutant presenilin 1 transgenes. Neurobiol Dis (1999) ; 6: : 231–244. |
29. | Kurt MA , Davies DC , Kidd M , et al. Neurodegenerative changes associated with β-amyloid deposition in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin-1 transgenes. Exp Neurol (2001) ; 171: : 59–71. |
30. | Lewis J , McGowan E , Rockwood J , et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet (2000) ; 25: : 402–405. |
31. | Manczak M , Kandimalla R , Yin X , et al. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet (2018) ; 27: : 1332–1342. |
32. | Manczak M , Anekonda TS , Henson E , et al. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet (2006) ; 15: : 1437–1449. |
33. | Manczak M and Reddy PH. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum Mol Genet (2012) ; 21: : 5131–5146. |
34. | Reddy PH , Manczak M , Yin X , et al. Protective effects of Indian spice curcumin against amyloid-β in Alzheimer’s disease. J Alzheimers Dis (2018) ; 61: : 843–866. |
35. | Manczak M and Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet (2012) ; 21: : 2538–2547. |
36. | Manczak M , Calkins MJ and Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet (2011) ; 20: : 2495–2509. |
37. | Kandimalla R , Manczak M , Yin X , et al. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet (2018) ; 27: : 30–40. |
38. | Kandimalla R , Manczak M , Fry D , et al. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet (2016) ; 25: : 4881–4897. |
39. | Kandimalla R , Manczak M , Pradeepkiran JA , et al. A partial reduction of Drp1 improves cognitive behavior and enhances mitophagy, autophagy and dendritic spines in a transgenic Tau mouse model of Alzheimer disease. Hum Mol Genet (2022) ; 31: : 1788–1805. |
40. | Reddy PH , McWeeney S , Park BS , et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet (2004) ; 13: : 1225–1240. |
41. | Reddy PH , Yin X , Manczak M , et al. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum Mol Genet (2018) ; 27: : 2502–2516. |
42. | Manczak M , Kandimalla R , Fry D , et al. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet (2016) ; 25: : 5148–5166. |
43. | Kshirsagar S , Alvir RV , Hindle A , et al. Early cellular, molecular, morphological and behavioral changes in the humanized amyloid-beta-knock-in mouse model of late-onset Alzheimer’s disease. Cells (2022) ; 11: : 733. |
44. | Vijayan M , George M , Bunquin LE , et al. Protective effects of a small-molecule inhibitor DDQ against tau-induced toxicities in a transgenic tau mouse model of Alzheimer’s disease. Hum Mol Genet (2022) ; 31: : 1022–1034. |
45. | Bhatti JS , Thamarai K , Kandimalla R , et al. Mitochondria-targeted small peptide, SS31 ameliorates diabetes induced mitochondrial dynamics in male TallyHO/JngJ mice. Mol Neurobiol (2021) ; 58: : 795–808. |
46. | Bhatti JS , Tamarai K , Kandimalla R , et al. Protective effects of a mitochondria-targeted small peptide SS31 against hyperglycemia-induced mitochondrial abnormalities in the liver tissues of diabetic mice, Tallyho/JngJ mice. Mitochondrion (2021) ; 58: : 49–58. |
47. | Ramasubramanian B and Reddy PH. Are TallyHo mice a true mouse model for type 2 diabetes and Alzheimer’s disease? J Alzheimers Dis (2019) ; 72: : S81–S93. |
48. | Ramasubramanian B , Griffith C , Hanson M , et al. Protective effects of Chaya against mitochondrial and synaptic toxicities in the type 2 diabetes mouse model TallyHO. Cells (2022) ; 11: : 744. |
49. | Kumar S , Morton H , Sawant N , et al. MicroRNA-455-3p improves synaptic, cognitive functions and extends lifespan: Relevance to Alzheimer’s disease. Redox Biol (2021) ; 48: : 102182. |
50. | Lanz T , Carter D and Merchant K. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis (2003) ; 13: : 246–253. |
51. | Baglietto-Vargas D , Forner S , Cai L , et al. Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer’s disease-like pathology. Nat Commun (2021) ; 12: : 2421. |
52. | Singh SP , Lee J , Bose C , et al. Haploinsufficiency interactions between RALBP1 and p53 in ERBB2 and PyVT models of mouse mammary carcinogenesis. Cancers (2021) ; 13: : 3329. |
53. | Awasthi S , Hindle A , Sawant NA , et al. RALBP1 in oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Cells (2021) ; 10: : 3113. |
54. | Wakabayashi J , Zhang Z , Wakabayashi N , et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol (2009) ; 186: : 805–816. |
55. | Ishihara N , Nomura M , Jofuku A , et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol (2009) ; 11: : 958–966. |
56. | Reddy PH . Is the mitochondrial outermembrane protein VDAC1 therapeutic target for Alzheimer’s disease? Biochim Biophys Acta (2013) ; 1832: : 67–75. |
57. | Weeber EJ , Levy M , Sampson MJ , et al. The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J Biol Chemi (2002) ; 277: : 18891–18897. |
58. | Raghavan A , Sheiko T , Graham BH , et al. Voltage-dependant anion channels: novel insights into isoform function through genetic models. Biochim Biophys Acta (2012) ; 1818: : 1477–1485. |
59. | Manczak M , Sheiko T , Craigen WJ , et al. Reduced VDAC1 protects against Alzheimer’s disease, mitochondria, and synaptic deficiencies. J Alzheimers Dis (2013) ; 37: : 679–690. |
60. | Kim JH , Sen Ś , Avery CS , et al. Genetic analysis of a new mouse model for non-insulin-dependent diabetes. Genomics (2001) ; 74: : 273–286. |
61. | Hubrecht RC and Carter E. The 3Rs and humane experimental technique: implementing change. Animals (2019) ; 9: : 754. |
62. | Russell WMS , Burch RL and Hume CW. The principles of humane experimental technique. Methuen London, (1959) . |
63. | Curzer HJ , Perry G , Wallace MC , et al. The three Rs of animal research: what they mean for the institutional animal care and use committee and why. Sci Eng Ethics (2016) ; 22: : 549–565. |
64. | Regulations AW . Animal Welfare Act. Animal Welfare Act (2013) . |
65. | Tannenbaum J and Bennett BT. Russell and Burch’s 3Rs then and now: the need for clarity in definition and purpose. J Am Assoc Lab Anim Sci (2015) ; 54: : 120–132. |
66. | Animal Welfare Act Regulations. 9 C.F.R. §2.31(d)(ii). |
67. | Curzer HJ , Perry G , Wallace MC , et al. The three Rs of animal research: what they mean for the institutional animal care and use committee and why. Sci Eng Ethics (2016) ; 22: : 549–565. |
68. | Bradfield JF , Bennett BT and Gillett CS. Chapter 2 – Oversight of research animal welfare in the United States. In: Guillén J (ed) Laboratory animals. Academic Press, (2014) , pp. 5–59. |
69. | Oversight of NIH-Supported Animal Research, https://grants.nih.gov/grants/policy/air/how-nih-ensures. |
70. | Sprague W . Arrive Guidelines 2.0. Vet Clin Pathol (2020) ; 49: : 378–379. |
71. | Kshirsagar S , Alvir RV , Pradeepkiran JA , et al. A combination therapy of urolithin A+ EGCG has stronger protective effects than single drug urolithin a in a humanized amyloid beta knockin mice for late-onset Alzheimer’s disease. Cells (2022) ; 11: : 2660. |


