
Individualized and Biomarker-Based Prognosis of Longitudinal Cognitive Decline in Early Symptomatic Alzheimer’s Disease
Abstract
Background:
Although individualized models using demographic, MRI, and biological markers have recently been applied in mild cognitive impairment (MCI), a similar study is lacking for patients with early Alzheimer’s disease (AD) with biomarker evidence of abnormal amyloid in the brain.
Objective:
We aimed to develop prognostic models for individualized prediction of cognitive change in early AD.
Methods:
A total of 421 individuals with early AD (MCI or mild dementia due to AD) having biomarker evidence of abnormal amyloid in the brain were included in the current study. The primary cognitive outcome was the slope of change in Alzheimer’s Disease Assessment Scale-cognitive subscale-13 (ADAS-Cog-13) over a period of up to 5 years.
Results:
A model combining demographics, baseline cognition, neurodegenerative markers, and CSF AD biomarkers provided the best predictive performance, achieving an overfitting-corrected R2 of 0.59 (bootstrapping validation). A nomogram was created to enable clinicians or trialists to easily and visually estimate the individualized magnitude of cognitive change in the context of patient characteristics. Simulated clinical trials suggested that the inclusion of our nomogram into the enrichment strategy would lead to a substantial reduction of sample size in a trial of early AD.
Conclusions:
Our findings may be of great clinical relevance to identify individuals with early AD who are likely to experience fast cognitive deterioration in clinical practice and in clinical trials.
INTRODUCTION
Alzheimer’s disease (AD) imposes an immense economic and clinical burden on patients, their families, and health care systems.1 Development of disease-modifying therapies and symptomatic agents has been hindered by the multifactorial nature of AD and disease heterogeneity.2–4 There are considerable variations in the rates of cognitive decline in patients with early AD [mild cognitive impairment (MCI) or mild dementia due to AD] who have evidence of elevated amyloid burden in the brain, even though the aim of the study is to create a relatively homogeneous group at baseline.5 A consequence is that the inclusion of patients with low likelihood of decline reduces the statistical power of a clinical trial.6 Successful clinical trials of early AD would benefit from strategies to enrich for participants who are more likely to experience cognitive deterioration.7 Therefore, mathematical models that can accurately predict cognitive outcomes at the individual patient level are of utmost importance for improving the patient prognostic counselling, stratification of patients for clinical trials, and timing of initiation of symptomatic or disease-modifying therapies.
Many studies have aimed to identify prognostic factors linked with cognitive decline in older adults, both in those with unimpaired and impaired cognition.8–10 Nonetheless, there is a scarcity of prediction models capable of forecasting cognitive outcomes at an individual patient level. Recently, individualized models incorporating demographic data, MRI findings, and biological markers have emerged in MCI.11–13 A similar study is lacking for patients with early AD with biomarker evidence of abnormal amyloid in the brain (MCI due to AD and mild AD dementia), a critical disease stage that a recent breakthrough has targeted.14 Furthermore, estimating the exact trajectory and extent of cognitive change, represented by numerical values of rate of decline or ‘slopes’, may be more advantageous for clinical application than predicting categorical transitions in cognitive stages (for example, the progression from MCI to dementia), which can be challenging for clinicians to interpret and for patients to comprehend accurately. A simple instrument that helps clinicians or trialists quickly and accurately identify patients with early AD likely to experience faster cognitive decline and provides an estimate of the magnitude of cognitive change could lead to more precise and more targeted surveillance strategies and possibly to better stratification of patients for future trials.
We therefore studied the usefulness of a variety of commonly used markers (demographic, cognitive, neurodegenerative, and in vivo CSF markers) for predicting longitudinal cognitive decline in individuals from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) who met inclusion criteria similar to those used in a clinical trial of early AD.14 We aimed to construct prediction models based on neurodegenerative and CSF markers for patients with early AD, taking into account individual characteristics to obtain personalized slopes of cognitive change. We integrated the findings into a nomogram that can enable easy and manual estimation of predicted values and can be useful in clinical practice and future trials. We further investigated the extent to which the nomogram could reduce the required sample sizes for theoretical clinical trials in an early AD population.
MATERIAL AND METHODS
Participants
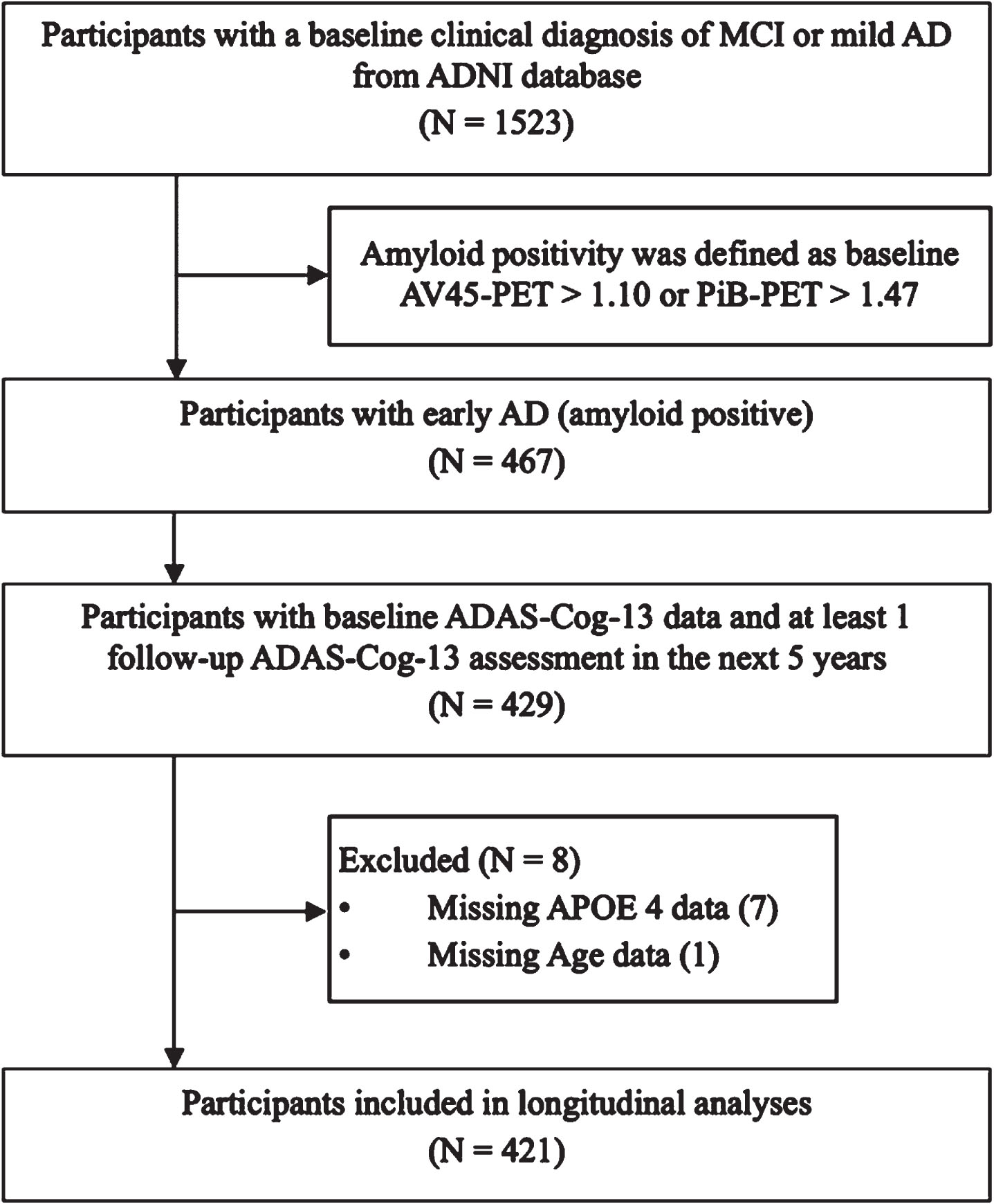
Data used in this study were obtained from the ADNI database (http://adni.loni.usc.edu). The ADNI study is a multicenter longitudinal study that aims to examine whether clinical, neuropsychological, biological, and neuroimaging makers can be integrated to track disease progression on the AD continuum.15 For the current study, data were extracted from the ADNI-1, ADNI-GO, ADNI-2, and ADNI-3 phases. We selected those participants who were diagnosed with MCI or mild AD dementia at baseline, had at least two ADAS-Cog-13 assessments within 5 years (including a baseline assessment), had elevated amyloid burden as measured by PET imaging (specific procedures and cutoff scores described below). Participants with MCI had memory complaints, objective memory impairment as evidenced by the Logical Memory II subscale (delayed paragraph recall) from the Wechsler Memory Scale-revised, Mini-Mental State Examination (MMSE)16 scores≥24, global Clinical Dementia Rating (CDR)17 scores of 0.5, essentially preserved activities of daily living, and no presence of AD or dementia. Participants with mild AD dementia were diagnosed based on the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA)18 criteria for probable AD and had MMSE scores ranging from 20 to 26 (inclusive) and global CDR scores of 0.5 or 1. In the current study, we used the inclusion criteria that were similar to those applied in a recent clinical trial targeting patients with early AD (MCI and mild AD dementia with elevated amyloid in the brain).14 Figure 1 demonstrates the sample selection procedure. The ADNI study was approved by an ethical review board and all subjects provided written informed consent at each participating study site.
Fig. 1
Flowchart of the sample selection procedure. MCI, mild cognitive impairment; AD, Alzheimer’s disease; ADAS-Cog-13, 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale; APOE, Apolipoprotein E; AV45, Florbetapir; PiB, Pittsburgh compound B; PET, positron emission tomography.
Cognitive outcome
The primary cognitive outcome was the 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-Cog-13)19 because it has been commonly used for tracking clinical progression and evaluating treatment effects in AD clinical trials. The ADAS-Cog-13 score used for the current study was comprised of 13 subtests that mainly capture episodic memory, language, and praxis domains. The comprehensive 13 subtests of ADAS-Cog-13 included word recall (0-10), commands (0-5), constructional praxis (0-5), delayed word recall (0-10), object and finger naming (0-5), ideational praxis (0-5), orientation (0-8), word recognition (0-12), remembering test instructions (0-5), spoken language ability (0-5), word-finding difficulty (0-5), comprehension (0-5), and digit cancellation (0-5). Adding the scores of all 13 subtests results in a total score ranging from 0 to 85, with higher scores indicating more impaired cognition. To estimate individual ADAS-Cog-13 slopes in the current study, we included participants with a baseline ADAS-Cog-13 assessment and at least one follow-up ADAS-Cog-13 assessment within the next 5 years (specific statistical procedure described below).
Biological markers
Amyloid burden in the brain was measured by Pittsburgh compound B (PiB) or Florbetapir AV-45 PET imaging (summary data were extracted from the ADNI Laboratory of Neuroimaging database: ida.loni.usc.edu). Amyloid positivity was determined according to standardized uptake values ratio (SUVR) of average uptake in four cerebral regions (cingulate, frontal, temporal, and parietal cortices) normalized to the uptake of cerebellum, using previously established cutoff scores (>1.47 for PiB and >1.10 for Florbetapir-PET).20 The FDG-PET SUVR was calculated based on the average of the temporal, angular, and bilateral cingulate regions.21 Hippocampal volume was extracted from MRI scans and was normalized using the intracranial volume (ICV) to adjust sex difference in head size. Adjusted hippocampal volume (aHV) was calculated based on the following equation: aHV = hippocampal volume (mm3)/ICV (mm3)×103. Levels of CSF Aβ1-42 (Aβ42) and phosphorylated tau (p-tau) were examined in CSF samples by the Elecsys β-amyloid (1-42) CSF and the Elecsys phospho-tau (181P) CSF immunoassays at the Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, USA. These methods were described in detail in a previous study.22 Of note, in the current analysis, we capped the values of individuals with CSF Aβ42 > 1700 pg/ml to 1700 pg/ml, as these results exceeded the upper technical limit (n = 8). The APOE4 genotype was categorized into three groups based on the number of copies of the APOE4 allele (0, 1, and 2). Demographic information included participant age at baseline, sex, and education in years.
Statistical analysis
All statistical work was performed using R version 4.1.2 (R Foundation for Statistical Computing).23 Statistical significance was set at two-tailed p < 0.05.
First, for statistical description, continuous variables were summarized as mean (standard deviation) and categorical variables as number of observations (percentage). Number of missing values in biological markers was reported.
Second, we derived participant-specific ADAS-Cog-13 slopes using a linear mixed-effects model (using the lme4 R package24) with ADAS-Cog-13 scores (at least two timepoints) as the outcome and time (years since baseline) as the independent variable. The model included random intercepts and slopes.
Third, these participant-specific slopes were subsequently treated as outcomes in a set of multivariable linear regression models with a combination of demographic variables and biological markers as predictors. Specifically, four linear regression models were constructed: the base model included participant age at baseline, sex, education in years, and APOE4 genotype as predictors; the cognition model expanded upon the base model by adding the baseline ADAS-Cog-13 score; the neurodegeneration model additionally included FDG-PET and aHV; the CSF model additionally included CSF Aβ42 and p-tau. To allow for non-linear associations between predictors and the outcome, restricted cubic splines with 3 knots (using the rms R package) were applied to baseline ADAS-Cog-13, FDG-PET, aHV, CSF Aβ42, and p-tau. To maximize the available data used for constructing the linear models, missing values in several predictors (aHV, FDG-PET, CSF Aβ42, and p-tau) were handled using simple imputation, where the missing values were replaced with the median values of their respective variables. Models were compared using R2 and Akaike Information Criterion (AIC).25 The likelihood ratio test was performed to assess the statistical significance of the four nested models.
Fourth, for model calibration, we used bootstrapping with 10,000 repetitions to obtain overfitting-corrected estimates of the predicted versus observed ADAS-Cog-13 slopes within the context of the best-fitting CSF model identified during the model development stage as stated above. The smooth nonparametric calibration estimator (loess) was applied. For model validation, we employed bootstrapping with 10,000 repetitions to validate the best-fitting CSF model and to examine whether it was overfitting the data. The optimism in a variety of measures of model predictive accuracy, including R2, mean squared error (MSE), the g-index, and the intercept and slope of an overall calibration, was estimated using the resampling technique. To predict the ADAS-Cog-13 slopes of individual patients, we constructed a nomogram that allows for manual and visual calculation of the predicted slopes on the basis of clinical and biological characteristics.
Fifth, simulations of hypothetical clinical trials were performed using the longpower R package, and sample size estimations for linear mixed-effects models were based on the equation due to Diggle.26 Clinical trials were simulated with 1 : 1 allocation of placebo and treatment groups, assuming a statistical power of 80%, a significant level of 0.05, a 20% treatment effect on the rate of cognitive change over time, a trial duration of 18 months, and cognitive assessment every 3 months (a total of 7 cognitive assessments). The bootstrap with 500 repetitions was employed to generate 500 simulations. The comparison was made between the sample size required to detect cognitive change when applying an inclusion parameter and the sample size when including all available participants.
Finally, secondary analyses were performed with (1) Clinical Dementia Rating-Sum of Boxes (CDR-SB)27 as the cognitive outcome rather than ADAS-Cog-13, and (2) MMSE as the cognitive outcome.
RESULTS
Study population characteristics
Baseline characteristics of the study population are shown in Table 1. Of the 421 patients with early AD, 183 (43%) were female, and 134 (32%), 209 (50%), and 78 (19%) had 0, 1, and 2 APOE4 alleles, respectively. The mean (SD) age was 73 (7) years, the mean (SD) education in years was 16 (3), and the mean (SD) follow-up duration was 2.55 (1.55) years. Data were missing for 43 patients for aHV, 8 for FDG-PET, 74 for CSF Aβ42, and 74 for CSF p-tau. Baseline characteristics of the CDR-SB and MMSE cohorts are provided in Supplementary Tables 1 and 2.
Table 1
Characteristics of the study sample
| Characteristic | N = 421 |
| Age, y | 73 (7) |
| Education, y | 16 (3) |
| Sex | |
| Male | 238 (57%) |
| Female | 183 (43%) |
| Numbers of APOE4 allele | |
| 0 | 134 (32%) |
| 1 | 209 (50%) |
| 2 | 78 (19%) |
| Follow-up durations, y | 2.55 (1.55) |
| Baseline ADAS-Cog-13 | 21 (10) |
| AHV | 4.34 (0.75) |
| Missing, n | 43 |
| FDG-PET | 1.15 (0.17) |
| Missing, n | 8 |
| CSF Aβ42, pg/ml | 726 (282) |
| Missing, n | 74 |
| CSF p-tau, pg/ml | 35 (16) |
| Missing, n | 74 |
| Slopes of change in ADAS-Cog-13 | 3.3 (3.1) |
Continuous variables and categorical variables are presented as Mean (SD) and n (%), respectively. APOE, Apolipoprotein E; ADAS-Cog-13, 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale; aHV, adjusted hippocampal volume; FDG-PET, fluorodeoxyglucose-positron emission tomography; CSF, cerebrospinal fluid; Aβ42, amyloid-β1-42; p-tau, phosphorylated tau.
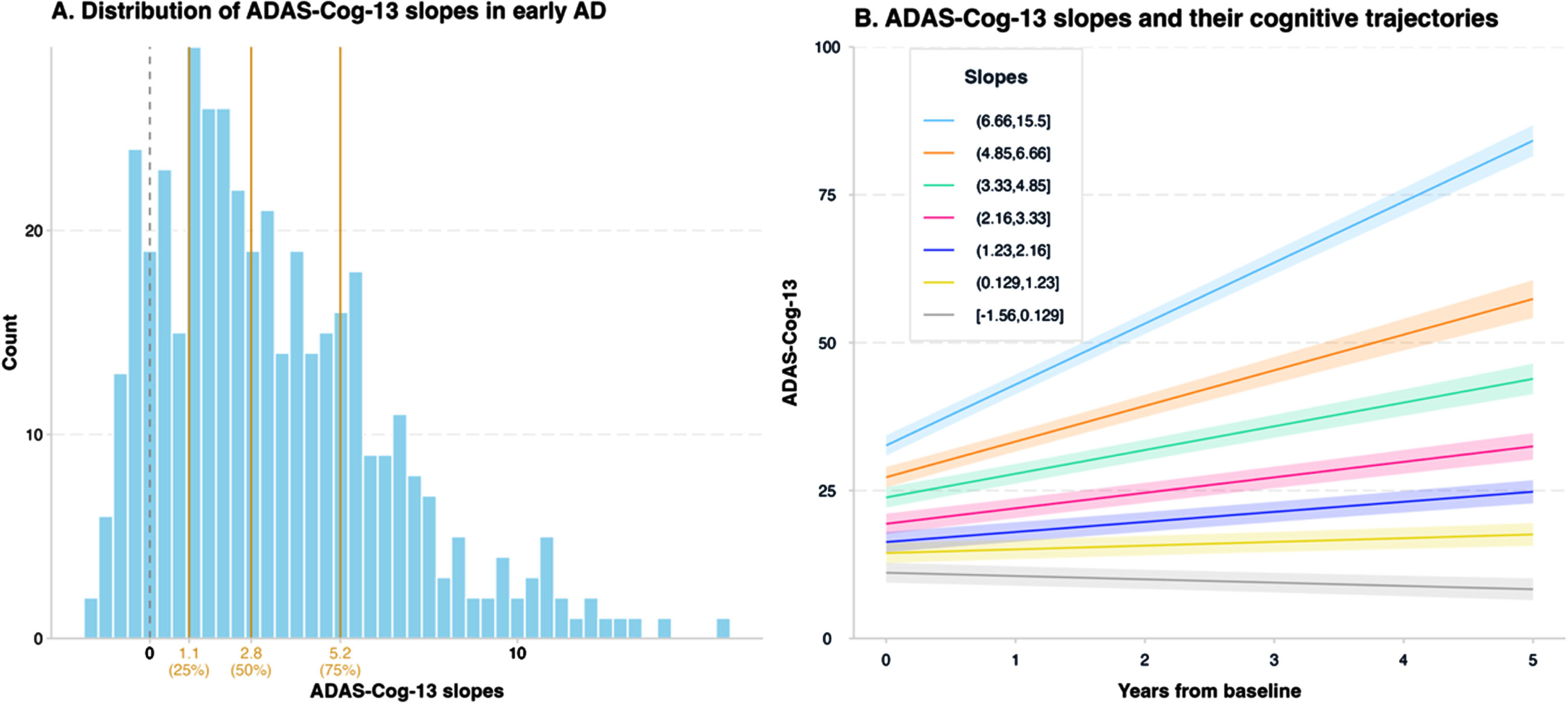
Slopes of cognitive decline in early AD
The histogram in Fig. 2A shows the distribution of participant-specific ADAS-Cog-13 slopes in patients with early AD and illustrates that slopes vary substantially, ranging from –1.557 to 15.36 points annually (average change per year: +3.34 points; 95% CIs based on the Wald method: 2.93 to 3.75; p < 0.001). The quartiles of ADAS-Cog-13 slopes were 1.1 (Q1), 2.8 (Q2), and 5.2 (Q3), respectively (Fig. 2A). For illustrative purposes, individuals were divided into seven groups with approximately equal sample size based on slopes, and Fig. 2B demonstrates the mean cognitive change of individuals with different slopes. Participant-specific slopes on the CDR-SB ranged from –0.38 to 5.84 points annually (average change per year: +1.1 points; 95% CIs: 0.97 to 1.24; p < 0.001) (Supplementary Figure 1), and participant slopes on the MMSE ranged from –9.1 to 0.59 points annually (average change per year: –1.6 points; 95% CIs: –1.8 to –1.4; p < 0.001) (Supplementary Figure 2).
Fig. 2
Participant-specific slopes on the ADAS-Cog-13. Panel A depicts the distribution of participant-specific ADAS-Cog-13 slopes in patients with early AD. The vertical gray dashed line represents a slope of 0, and the three vertical orange solid lines represent the quartiles (25% = 1.1, 50% = 2.8, and 75% = 5.2) of ADAS-Cog-13 slopes. Panel B demonstrates the mean cognitive change in participants with different slopes in the next 5 years since baseline. Participants were classified into seven groups with approximately equal numbers of observations based on slopes. The mean cognitive change of participants with slopes ranging from –1.56 (inclusive) to 0.129 (inclusive), 0.129 to 1.23 (inclusive), 1.23 to 2.16 (inclusive), 2.16 to 3.33 (inclusive), 3.33 to 4.85 (inclusive), 4.85 to 6.66 (inclusive), 6.66 to 15.5 (inclusive) are represented by the gray, golden, purple, pink, green, orange, and blue solid lines, respectively. The shaded areas represent the 95% confidence intervals for the regression lines at the group level. ADAS-Cog-13, 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale; AD, Alzheimer’s disease.
Modeling longitudinal cognitive decline in early AD
We constructed a set of multivariable linear regression models to predict ADAS-Cog-13 slopes in patients with early AD. The predictors included in each model (base model, cognition model, neurodegeneration model, and CSF model) and their corresponding regression coefficients [95% CIs] are shown in Table 2. The cognition model that additionally included baseline ADAS-Cog-13 score as a predictor (R2 = 0.542; AIC = 1840.9) fit the data significantly better than the base model that included age, sex, education, and APOE4 status (R2 = 0.025; AIC = 2154.8; model comparison between the base model and cognition model: p < 0.001 based on the likelihood ratio test). In terms of R2, the base model only captured 2.5% of the variability in the ADAS-Cog-13 slopes, while adding baseline ADAS-Cog-13 information to the base model explained an additional 51.7% of the total variability in the ADAS-Cog-13 slopes. Subsequently, we constructed the neurodegeneration model to assess whether adding variables representing neurodegeneration (aHV and FDG-PET) to the cognition model can improve model performance. The neurodegeneration model (R2 = 0.602; AIC = 1789.7) demonstrated a significantly better fit to the data compared to the cognition model (p < 0.001 based on the likelihood ratio test). This means that the neurodegenerative markers explained an additional 6% of the total variability in the ADAS-Cog-13 slopes after accounting for age, sex, education, APOE4 status, and baseline ADAS-Cog-13 score. Finally, to test whether adding CSF AD biological markers (Aβ42 and p-tau) can further improve prediction of ADAS-Cog-13 change compared to the neurodegeneration model, we constructed the CSF model, which combines demographic, cognitive, neurodegenerative, and CSF AD biological variables. The CSF model (R2 = 0.618; AIC = 1780.3) showed a significantly better fit to the data than the neurodegeneration model (p = 0.002 based on the likelihood ratio test) and explained an additional 1.6% of the total variability in the ADAS-Cog-13 slopes after accounting for predictors included in the neurodegeneration model. Hence, the CSF model was chosen as the best-fitting model for prediction of ADAS-Cog-13 slopes based on the R2, AIC, and likelihood ratio test.
Table 2
Summary of regression models
| Base model | Cognition model | Neurodegeneration model | CSF model | |
| Intercept | 3.707 [–0.353, 7.766] | 1.018 [–1.857, 3.893] | 9.020 [4.272, 13.769] | 9.333 [4.503, 14.163] |
| p value: 0.073 | p value: 0.487 | p value: <0.001 | p value: <0.001 | |
| Age | 0.002 [–0.041, 0.046] | –0.032 [–0.063, –0.002] | –0.028 [–0.059, 0.002] | –0.031 [–0.062, –0.001] |
| p value: 0.922 | p value: 0.038 | p value: 0.071 | p value: 0.042 | |
| Education | –0.070 [–0.183, 0.043] | 0.010 [–0.068, 0.088] | –0.018 [–0.092, 0.056] | –0.015 [–0.088, 0.058] |
| p value: 0.222 | p value: 0.799 | p value: 0.638 | p value: 0.686 | |
| Presence of 1 APOE4 allele | 0.710 [0.034, 1.386] | 0.292 [–0.175, 0.759] | 0.290 [–0.150, 0.729] | 0.116 [–0.328, 0.560] |
| p value: 0.040 | p value: 0.219 | p value: 0.196 | p value: 0.608 | |
| Presence of 2 APOE4 alleles | 1.295 [0.398, 2.192] | 0.686 [0.063, 1.310] | 0.557 [–0.029, 1.143] | 0.175 [–0.437, 0.787] |
| p value: 0.005 | p value: 0.031 | p value: 0.062 | p value: 0.574 | |
| Female sex | –0.005 [–0.627, 0.618] | 0.131 [–0.298, 0.561] | 0.270 [–0.137, 0.676] | 0.162 [–0.250, 0.573] |
| p value: 0.989 | p value: 0.548 | p value: 0.193 | p value: 0.440 | |
| ADAS-Cog-13 | 0.176 [0.118, 0.234] | 0.086 [0.024, 0.147] | 0.062 [0.001, 0.124] | |
| p value: <0.001 | p value: 0.007 | p value: 0.047 | ||
| ADAS-Cog-13’ | 0.075 [0.004, 0.147] | 0.107 [0.036, 0.179] | 0.121 [0.050, 0.192] | |
| p value: 0.040 | p value: 0.003 | p value: <0.001 | ||
| FDG-PET | –6.133 [–8.893, –3.372] | –6.084 [–8.806, –3.363] | ||
| p value: <0.001 | p value: <0.001 | |||
| FDG-PET’ | 1.296 [–1.688, 4.280] | 1.881 [–1.079, 4.841] | ||
| p value: 0.394 | p value: 0.212 | |||
| aHV | 0.254 [–0.390, 0.898] | 0.170 [–0.476, 0.816] | ||
| p value: 0.438 | p value: 0.605 | |||
| aHV’ | –0.747 [–1.485, –0.009] | –0.638 [–1.375, 0.100] | ||
| p value: 0.047 | p value: 0.090 | |||
| CSF Aβ42 | –0.001 [–0.003, 0.001] | |||
| p value: 0.239 | ||||
| CSF Aβ42’ | 0.000 [–0.002, 0.003] | |||
| p value: 0.771 | ||||
| CSF p-tau | 0.054 [0.013, 0.095] | |||
| p value: 0.010 | ||||
| CSF p-tau’ | –0.042 [–0.095, 0.011] | |||
| p value: 0.121 | ||||
| Number of Observations | 421 | 421 | 421 | 421 |
| R2 | 0.025 | 0.542 | 0.602 | 0.618 |
| AIC | 2154.8 | 1840.9 | 1789.7 | 1780.3 |
This table presents the results of four linear regression models: the base model included participant age at baseline, sex, education in years, and APOE4 genotype as predictors; the cognition model expanded upon the base model by adding the baseline ADAS-Cog-13 score; the neurodegeneration model additionally included FDG-PET and aHV; the CSF model additionally included CSF Aβ42 and p-tau. The proportion of variance explained by these models, quantified as R-squared values, are respectively 0.025, 0.542, 0.602, and 0.618. Meanwhile, the Akaike Information Criteria (AIC) for these models are reported as 2154.8, 1840.9, 1789.7, and 1780.3, respectively. Effect sizes are presented as unstandardized βs [95% CIs]. Two tailed t-tests on regression coefficients were used throughout. APOE, Apolipoprotein E; ADAS-Cog-13 : 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale; aHV: Adjusted hippocampal volume; FDG-PET: Fluorodeoxyglucose-Positron Emission Tomography; CSF: Cerebrospinal fluid; Aβ42, amyloid-β1-42; p-tau: Phosphorylated tau; AIC: Akaike Information Criterion.
We next examined the partial effects of predictors in the CSF model (Fig. 3). For demographic variables, the Wald tests suggested that ADAS-Cog-13 slopes were significantly associated with participant age at baseline (F statistics = 4.18, p = 0.042; Fig. 3A), but not with education (F statistics = 0.16, p = 0.69; Fig. 3B), sex (F statistics = 0.6, p = 0.44; Fig. 3C), or APOE4 status (F statistics = 0.19, p = 0.82; Fig. 3D). To relax the linearity assumption of linear models, restricted cubic splines with 3 knots were applied to several predictors, including baseline ADAS-Cog-13 score, FDG-PET, aHV, CSF Aβ42, and p-tau. The associations between slopes of cognitive change and baseline ADAS-Cog-13 score (p for overall < 0.001; p for non-linearity<0.001), FDG-PET (p for overall < 0.001; p for non-linearity = 0.212), aHV (p for overall = 0.041; p for non-linearity = 0.09), CSF Aβ42 (p for overall = 0.06; p for non-linearity = 0.77), and p-tau (p for overall = 0.0017; p for non-linearity = 0.121) are presented in Fig. 3E-I. Subsequently, the chi-square statistic minus the degrees of freedom (χ2 – df) was used as a measure for predictor importance (higher values represent greater importance).28 As shown in Fig. 3J, baseline ADAS-Cog-13 score was the strongest predictor for the model, followed by FDG-PET and CSF p-tau. Finally, level plots based on the CSF model were created to facilitate easy and visual calculation of predicted ADAS-Cog-13 slopes according to the top 5 important predictors (baseline ADAS-Cog-13, FDG-PET, CSF p-tau, aHV, and CSF Aβ42) (Fig. 4).
Fig. 3
Associations of predictors with the ADAS-Cog-13 slopes in the CSF model and ranking of importance of predictors. The panels A-I show the partial effects of predictors in the CSF model. The partial effect of a predictor was examined when holding all other variables constant [median age = 73.9 years; median education = 16 years; APOE4 genotype = 1; gender = male; baseline median ADAS-Cog-13 = 20; median FDG SUVRs = 1.158; median aHV = 4.28; median CSF Aβ= 685.7 pg/ml; median p-tau = 31.45 pg/ml]. For instance, panel F illustrates the partial effect of FDG SUVRs on the ADAS-Cog-13 slopes, assuming that all other variables in the model remain constant ([median age = 73.9 years; median education = 16 years; APOE4 = 1 (range, 0–2 APOE4 alleles); gender = male; baseline median ADAS-Cog-13 = 20; median aHV = 4.28; median CSF Aβ= 685.7 pg/ml; median p-tau = 31.45 pg/ml]). For the categorical predictors (gender and APOE4 genotype), solid dots represent means of predicted slopes and error bars represent their corresponding 95% confidence intervals. For the continuous predictors, solid lines represent estimated mean regression lines and shaded regions represent their 95% confidence intervals. In this model, we employed restricted cubic spline transformations with 3 knots to relax the linearity assumption of the linear model. Specifically, we applied these transformations to the baseline ADAS-Cog-13 score, aHV, FDG-PET, CSF Aβ42, and p-tau variables. Knots were placed at the quantiles (0.1, 0.5, and 0.9) of the respective predictors. Panel J shows the ranking of predictor importance based on χ2 – df. APOE, Apolipoprotein E; ADAS-Cog-13, 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale; aHV, adjusted hippocampal volume; FDG-PET, fluorodeoxyglucose-positron emission tomography; CSF, cerebrospinal fluid; Aβ42, amyloid-β1-42; p-tau, phosphorylated tau.
![Associations of predictors with the ADAS-Cog-13 slopes in the CSF model and ranking of importance of predictors. The panels A-I show the partial effects of predictors in the CSF model. The partial effect of a predictor was examined when holding all other variables constant [median age = 73.9 years; median education = 16 years; APOE4 genotype = 1; gender = male; baseline median ADAS-Cog-13 = 20; median FDG SUVRs = 1.158; median aHV = 4.28; median CSF Aβ= 685.7 pg/ml; median p-tau = 31.45 pg/ml]. For instance, panel F illustrates the partial effect of FDG SUVRs on the ADAS-Cog-13 slopes, assuming that all other variables in the model remain constant ([median age = 73.9 years; median education = 16 years; APOE4 = 1 (range, 0–2 APOE4 alleles); gender = male; baseline median ADAS-Cog-13 = 20; median aHV = 4.28; median CSF Aβ= 685.7 pg/ml; median p-tau = 31.45 pg/ml]). For the categorical predictors (gender and APOE4 genotype), solid dots represent means of predicted slopes and error bars represent their corresponding 95% confidence intervals. For the continuous predictors, solid lines represent estimated mean regression lines and shaded regions represent their 95% confidence intervals. In this model, we employed restricted cubic spline transformations with 3 knots to relax the linearity assumption of the linear model. Specifically, we applied these transformations to the baseline ADAS-Cog-13 score, aHV, FDG-PET, CSF Aβ42, and p-tau variables. Knots were placed at the quantiles (0.1, 0.5, and 0.9) of the respective predictors. Panel J shows the ranking of predictor importance based on χ2 – df. APOE, Apolipoprotein E; ADAS-Cog-13, 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale; aHV, adjusted hippocampal volume; FDG-PET, fluorodeoxyglucose-positron emission tomography; CSF, cerebrospinal fluid; Aβ42, amyloid-β1-42; p-tau, phosphorylated tau.](https://ip.ios.semcs.net:443/media/adr/2024/8-1/adr-8-1-adr240049/adr-8-adr240049-g003.jpg)
Fig. 4
Level plots for the prediction of ADAS-Cog-13 slopes. Predicted ADAS-Cog-13 slopes can be visually calculated based on baseline ADAS-Cog-13 score in combination with FDG-PET (A), aHV (B), CSF Aβ42 (C), and p-tau (D). The model from which the level plots are based on was adjusted for other predictors [median age = 73.9 years; median education = 16 years; APOE4 genotype = 1; gender = male; baseline median ADAS-Cog-13 = 20; median FDG SUVRs = 1.158; median aHV = 4.28; median CSF Aβ= 685.7 pg/ml; median p-tau = 31.45 pg/ml]. For example, the level plot in panel A is based on the model that was adjusted for several other predictors [median age = 73.9 years; median education = 16 years; APOE4 genotype = 1; gender = male; median aHV = 4.28; median CSF Aβ= 685.7 pg/ml; median p-tau = 31.45 pg/ml]. APOE, Apolipoprotein E; ADAS-Cog-13, 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale; aHV, adjusted hippocampal volume; FDG-PET, fluorodeoxyglucose-positron emission tomography; CSF, cerebrospinal fluid; Aβ42, amyloid-β1-42; p-tau, phosphorylated tau.
![Level plots for the prediction of ADAS-Cog-13 slopes. Predicted ADAS-Cog-13 slopes can be visually calculated based on baseline ADAS-Cog-13 score in combination with FDG-PET (A), aHV (B), CSF Aβ42 (C), and p-tau (D). The model from which the level plots are based on was adjusted for other predictors [median age = 73.9 years; median education = 16 years; APOE4 genotype = 1; gender = male; baseline median ADAS-Cog-13 = 20; median FDG SUVRs = 1.158; median aHV = 4.28; median CSF Aβ= 685.7 pg/ml; median p-tau = 31.45 pg/ml]. For example, the level plot in panel A is based on the model that was adjusted for several other predictors [median age = 73.9 years; median education = 16 years; APOE4 genotype = 1; gender = male; median aHV = 4.28; median CSF Aβ= 685.7 pg/ml; median p-tau = 31.45 pg/ml]. APOE, Apolipoprotein E; ADAS-Cog-13, 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale; aHV, adjusted hippocampal volume; FDG-PET, fluorodeoxyglucose-positron emission tomography; CSF, cerebrospinal fluid; Aβ42, amyloid-β1-42; p-tau, phosphorylated tau.](https://ip.ios.semcs.net:443/media/adr/2024/8-1/adr-8-1-adr240049/adr-8-adr240049-g004.jpg)
Subsequently, we repeated the analyses with CDR-SB and MMSE slopes as the cognitive outcomes. For the CSR-SB cohort, the model summary is listed in Supplementary Table 3, and the neurodegeneration model (R2 = 0.544; AIC = 1031.9) was selected as the best-fitting model according to the likelihood ratio test (p = 0.177 based on the comparison between the neurodegeneration and CSF model). Next, the partial effects of predictors in the neurodegeneration model were investigated (Supplementary Figure 3), and predictor importance was examined based on χ2 – df (Supplementary Figure 4). For the MMSE cohort, summary of regression models is shown in Supplementary Table 4, and the CSF model (R2 = 0.603; AIC = 1311.5) was chosen as the best-fitting model according to the likelihood ratio test (p = 0.012 based on the comparison between the neurodegeneration and CSF model). Next, the partial effects of predictors in the CSF model were investigated (Supplementary Figure 5), and predictor importance was examined based on χ2 – df (Supplementary Figure 6).
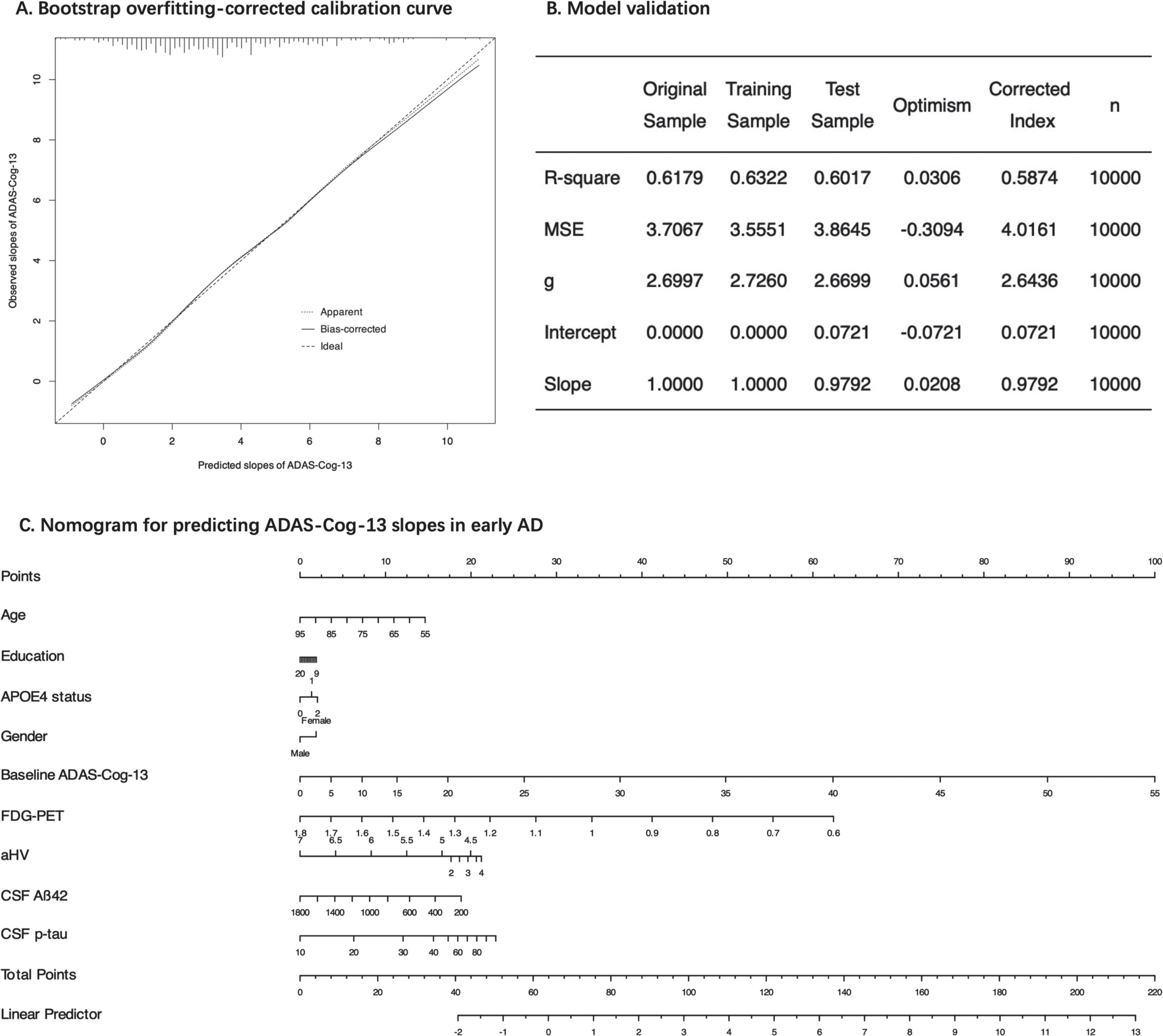
Calibration, validation, and nomogram
Model calibration (or reliability) performance (based on the best-fitting model) was evaluated by a 10,000-iteration bootstrap overfitting-corrected calibration curve (Fig. 5A). Based on visual inspection, the excellent calibration is illustrated by the close alignment of the calibration curve with the 45° line (Fig. 5A). The calibration errors were quantified using several metrics, including mean absolute error (0.072), mean squared error (MSE, 0.0081), and the 0.9 quantile of absolute error (0.134). Internal model validation was assessed by a 10,000-iteration bootstrap approach. Estimates of the optimism in several metrics of model performance, including R2, MSE, the g-index, and the intercept and slope of an overall calibration, were obtained. The optimism-corrected measures of model performance are summarized in Fig. 5B. For instance, the estimated R2 across the 10,000 training samples was 0.6322, while the average R2 was 0.6017 in the testing samples, resulting in an optimism of 0.03057. To obtain the optimism-corrected R2, we subtracted the estimated optimism (0.03057) from the original R2 (0.6179) obtained using our original data (n = 421), resulting in a corrected R2 value of 0.587. Finally, a nomogram was created to facilitate easy and visual calculation of the individualized slopes of cognitive change (Fig. 5C). Using an example for our nomogram, a 75-year-old female patient with early AD who has 15 years of education, one APOE4 allele, an ADAS-Cog-13 score of 25, with an aHV of 3 and FDG SUVRs of 1.0, and with CSF Aβ42 of 400 pg/ml and CSF p-tau of 30 pg/ml has a predicted slope of 4.7 points per year (for the calculation process, please refer to Supplementary Figure 7).
Fig. 5
Model calibration, internal validation, and nomogram. Panel A shows a bootstrap overfitting-corrected calibration curve. Panel B shows various metrics of model performance based on resampling technique with 10,000 iterations. Panel C displays a nomogram for prediction of ADAS-Cog-13 slopes based on 9 predictors in the model. ADAS-Cog-13, 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale; MSE, mean squared error; g, the g-index; APOE, Apolipoprotein E; aHV, adjusted hippocampal volume; FDG-PET, fluorodeoxyglucose-positron emission tomography; CSF, cerebrospinal fluid; Aβ42, amyloid-β1-42; p-tau, phosphorylated tau.
Subsequently, we repeated the analyses with CDR-SB and MMSE slopes as the cognitive outcomes. For the CDR-SB cohort, calibration performance based on the best-fitting model (the neurodegeneration model) was assessed by a bootstrap overfitting-corrected calibration curve (Supplementary Figure 8). The calibration errors were quantified using several metrics, including mean absolute error (0.03), mean squared error (0.0014), and the 0.9 quantile of absolute error (0.07). Supplementary Table 5 illustrates the optimism-corrected measures of model performance in relation to internal model validation. A nomogram was created to manually calculate the individualized CDR-SB slopes (Supplementary Figure 9). For the MMSE cohort, calibration performance based on the best-fitting model (the CSF model) was examined by a bootstrap overfitting-corrected calibration curve (Supplementary Figure 10). The calibration errors were quantified using several metrics, including mean absolute error (0.049), mean squared error (0.004), and the 0.9 quantile of absolute error (0.11). Supplementary Table 6 illustrates the optimism-corrected measures of model performance in relation to internal model validation. A nomogram was created to manually calculate the individualized MMSE slopes (Supplementary Figure 11).
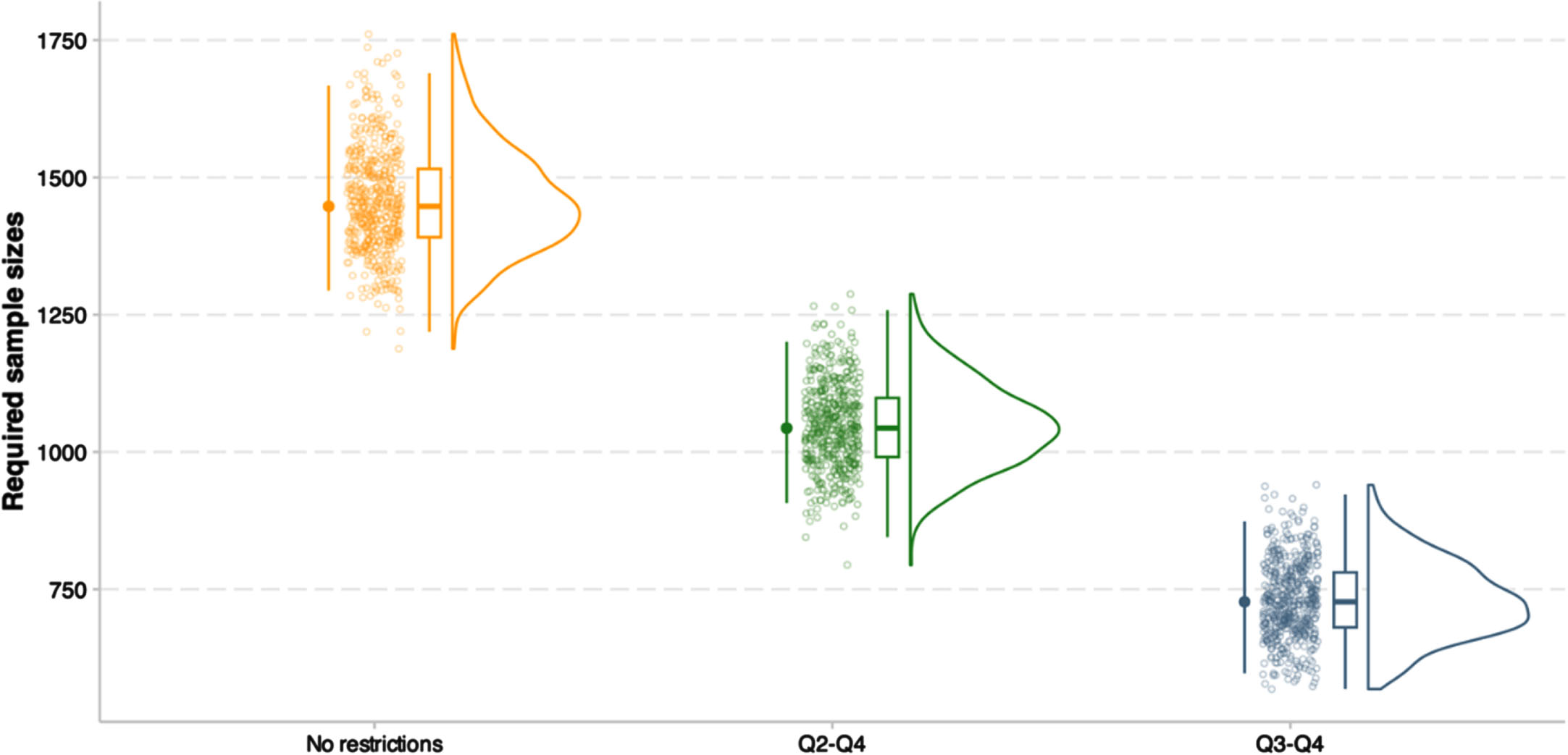
Clinical trials simulations
We investigated whether utilizing the nomogram for participant screening would lead to the reduction of the required sample size of hypothetical clinical trials. In clinical trial simulations using ADAS-Cog-13 slopes as the cognitive outcome with inclusion of all study participants (no restrictions applied), the required sample sizes were 1447.29 (i.e., approximately 724 participants in the treatment group and 724 participants in the placebo group; 95% CIs: 1294.1 to 1667.0; Fig. 6). When including the greatest 3 quantiles (Q2-Q4) of predicted ADAS-Cog-13 slopes using the nomogram, the required sample sizes were reduced to 1043.4 (95% CIs: 907.1 to 1200.6; Fig. 6). When including the greatest 2 quantiles (Q3-Q4) of predicted ADAS-Cog-13 slopes, the required sample sizes were reduced to 727.1 (95% CIs: 597 to 873.7; Fig. 6).
Fig. 6
Clinical trials simulations using predicted ADAS-Cog-13 slopes based on the nomogram for inclusion. This figure illustrates the reduction in required sample sizes resulting from using the nomogram for inclusion enrichment in hypothetical clinical trials aimed at slowing cognitive decline measured by ADAS-Cog-13 in patients with early AD. These clinical trial simulations had statistical power of 80% at α; = 0.05 using the R package “longpower”, assuming a 20% treatment effect on slopes of cognitive change over time, an 1 : 1 allocation of treatment, a trial duration of 18 months, and cognitive assessments every 3 months (at month 0, 3, 6, 9, 12, 15, and 18). Required samples sizes were estimated from 500 bootstrapped trials over all 421 patients with early AD. The reference model (No restrictions; representing no enrichment) included all participants in the quartile (Q) 1 through Q4 of the predicted ADAS-Cog-13 slopes based on the nomogram. In the first enrichment model (Q2-Q4), only participants in the Q2 through Q4 were included. In the second enrichment model (Q3-Q4), only participants in the Q3 through Q4 were included. The quartile limits of the predicted ADAS-Cog-13 slopes were –1.66 (inclusive) to 1.61 for quartile 1, 1.61 (inclusive) to 2.97 for quartile 2, 2.97 (inclusive) to 4.66 for quartile 3, and 4.66 to 12.18 for quartile 4. ADAS-Cog-13, 13-item version of the Alzheimer’s Disease Assessment Scale-cognitive subscale; Q, quartile.
DISCUSSION
In this study, we investigated the variations in the rates of decline over 5 years among individuals with early AD based on commonly used cognitive outcome measures in AD clinical trials. We found that individual slopes of cognitive change (ADAS-Cog-13, CDR-SB, and MMSE) differed substantially among individuals with early AD, despite applying inclusion criteria similar to those of a typical early AD trial in order to create a relatively homogeneous clinical group. Subsequently, we constructed prognostic models to estimate individualized slopes of cognitive change among patients with early AD based on patient characteristics and continuous marker values. These models were internally validated based on the bootstrap technique and showed strong model performance and calibration. We translated these biomarker-based prognostic models into nomograms, enabling a straightforward and manual calculation of personalized slopes of cognitive change. Simulated clinical trials illustrated great reductions in required sample sizes when enriching for individuals with early AD using higher predicted slopes based on the nomogram. This novel and practical instrument might improve clinical care and cognitive monitoring and could make clinical trials for early AD more precise and cost-effective.
The finding that individuals with early AD who have biomarker evidence of abnormal amyloid demonstrated substantial heterogeneity in terms of the rates of cognitive progression aligns with previous studies showing that individuals have highly variable times to progress from MCI to dementia as well as distinct magnitudes of cognitive decline.4,5,29 The variability in how individuals experience cognitive decline presents a hurdle for discerning therapeutic impacts through cognitive endpoints.30 In the context of AD clinical trials, enrolling participants with negligible or no anticipated cognitive deterioration over the course of the study could diminish the decline observed in the placebo arm, thereby complicating efforts to distinguish treatment effects from placebo responses.31 On the other hand, the diverse patterns of cognitive decline can yield substantial discrepancies in cognitive assessment results between the placebo group and the treatment group, where these differences may stem not from the therapy itself but from the inherent inconsistency in the extent of decline among individual patients.5 The average individual patient slopes of cognitive change among patients with early AD from our study were +3.3 points (SD = 3.1; range = –1.557 to 15.36 points) on the ADAS-Cog-13, +1.1 points (SD = 1.17; range = –0.38 to 5.84 points) on the CDR-SB, and –1.6 points (SD = 1.72; range = –9.1 to 0.59) on the MMSE. These results further suggested that current inclusion criteria for early AD trials are not optimal, highlighting the need for a more refined recruitment strategy. To the best of our knowledge, this is the first study to provide a useful instrument enabling an easy and manual estimation of individualized cognitive slopes of widely used cognitive outcome measures in AD trials among patients with early AD who have biomarker evidence of elevated amyloid in the brain, taking into account individual patient characteristics.
The model with both baseline cognitive information, neurodegenerative markers, and CSF measures provided the best predictive performance, which indicates the complementary nature of these biomarkers and aligns with findings in previous studies.32–34 When examining the contribution of individual predictors (Fig. 3J), baseline cognitive information was identified to be the strongest determinant of subsequent cognitive decline, which is consistent with previous findings.35 The powerful effect of baseline cognitive performance on the slopes of cognitive decline among patients with early AD may be explained by that baseline cognition serves as a snapshot that reflects the dynamics of the interaction between the patient’s initial cognitive abilities, brain reserve, and underlying neuropathophysiological changes. An individual with better cognition at baseline might be more likely to experience a slower downward cognitive trajectory compared to an individual with worse initial cognition, even though both of them have elevated levels of amyloid deposition in the brain. That is, the initial higher level of cognition may indicate greater cognitive reserve that confers an advantage enabling to counterbalance neuropathophysiological changes by, for instance, adopting alternative cognitive strategies or engaging alternative brain networks.36,37 With the progression of the disease (i.e., cognitive worsening, and neuropathophysiological advancing) and exhaustion of compensatory mechanisms, individuals with worse cognition at baseline might experience a steeper cognitive decline because of more advanced neuropathophysiological changes at that point.38,39 In support of this notion, our findings in Figs. 3E and 5C showed a non-linear association between baseline ADAS-Cog-13 and the magnitude of future cognitive decline. Specifically, when baseline ADAS-Cog-13 scores are below approximately 15 points, the relationship between the two variables remains relatively flat. However, as baseline ADAS-Cog-13 scores exceed approximately 15 points, the relationship exhibits a more pronounced steepening (Figs. 3E; 5C also shows that baseline ADAS-Cog-13 scores above 15 points have larger effects on the total points in the nomogram). In terms of other modalities, hypometabolism on FDG-PET that is supposed to reflect neuronal and synaptic dysfunction performed better than aHV based on MRI and CSF biomarkers in predicting slopes of cognitive decline, which is consistent with previous findings.40 The modest effect of CSF biomarkers, especially Aβ42, was somehow expected. Several former studies illustrated that amyloid deposition reaches a plateau and then becomes static when overt cognitive symptoms are worsening among patients with mild dementia.41 Statistically speaking, the floor effect (CSF Aβ42 were skewed towards lower ranges of values since all study participants with early AD had abnormal amyloid in the brain) may also contribute to the attenuation of the association between the two.
The simulation of clinical trials of early AD individuals showed that the use of the nomogram as an enrichment strategy significantly reduced the required sample sizes. This finding is promising because current clinical trials are increasingly shifting to earlier stages of AD.42 To conduct clinical trials in a cost-effective manner, it is crucial to enhance our ability to efficiently identify individuals most likely to experience a steeper downward cognitive trajectory. We did not include individuals in the greatest 4th quantile of predicted ADAS-Cog-13 slopes as a standalone group for enrichment due to the concern that individuals experiencing more pronounced declines in cognition may potentially have advanced underlying conditions, leading to reduced efficacy of certain treatments.
Several limitations should be kept in mind when interpreting the findings of our study. The mean (SD) follow-up duration was 2.55 (1.55) years, which was relatively short and variable among our study sample. We included individuals who underwent at least two timepoints of cognitive assessments over a period of up to 5 years. However, our study did not have sufficient power to estimate precise 5-year slopes of cognitive decline. In addition, the lack of external validation using an independent sample limits the generalizability of our models to new settings and populations, though internal validation using bootstrapping was performed to partially address this issue. Substantial variabilities across different methods and cohorts in terms of volumetric MRI measurements, PET imaging techniques, and CSF concentrations prohibit us from conducting an external validation, since the generalizability of our models is reliant on the utilization of identical methods. In the future study, we aim to validate our models using an independent sample once the standardization of different methods becomes available for these measures, similar to the case where measurements from different Aβ tracers, including florbetaben, florbetapir, and flutemetamol, were normalized into the Centiloid scale.43 Finally, our model did not include CSF Aβ40 measurements and the CSF Aβ42/Aβ40 ratio. These markers should be included in future studies due to the fact that CSF Aβ42/Aβ40 ratio is more closely related with amyloid deposition in the brain than CSF Aβ42 alone.
Our study provides a straightforward and useful statistical instrument that allows clinicians to estimate the magnitude of cognitive decline in patients with early AD, contributing to an improvement in the interpretation of biomarker results and potentially providing more targeted and precise patient management strategies. It can also inform the selection of an enriched population for early AD clinical trials and significantly reduce required sample sizes.
AUTHOR CONTRIBUTIONS
Xiwu Wang (Investigation; Methodology; Visualization; Writing – original draft); Teng Ye (Formal analysis; Investigation; Methodology; Visualization; Writing – original draft); Ziye Huang (Formal analysis; Investigation; Methodology; Visualization); Wenjun Zhou (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Supervision; Validation; Writing – review & editing); Jie Zhang, Ph.D., M.D. (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Supervision; Validation; Visualization; Writing – review & editing).
ACKNOWLEDGMENTS
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
FUNDING
This work did not receive any grant from funding agencies in the public, commercial, or not-for-profit sectors.
CONFLICT OF INTEREST
Jie Zhang serves as the Founder of Hangzhou Shansier Medical Technologies Co., Ltd., and holds shares in the company, which is dedicated to slowing down or halting cognitive decline in Alzheimer’s Disease. Wenjun Zhou is employed by Hangzhou Shansier Medical Technologies Co., Ltd. and holds shares in the company. The other authors declare that they have no conflict of interest.
DATA AVAILABILITY
Data used in the present study has been made publicly available by the ADNI in the Laboratory of Neuro Imaging (LONI) database.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/ADR-240049.
REFERENCES
1. | 2023 Alzheimer’s disease facts and figures. Alzheimers Dement (2023) ; 19: : 1598–1695. |
2. | Cummings J , Feldman HH and Scheltens P. The “rights” of precision drug development for Alzheimer’s disease. Alzheimers Res Ther (2019) ; 11: : 76. |
3. | Dong A , Toledo JB , Honnorat N , et al. Heterogeneity of neuroanatomical patterns in prodromal Alzheimer’s disease: links to cognition, progression and biomarkers. Brain (2017) ; 140: : 735–747. |
4. | Wang X , Ye T , Zhou W , et al. Uncovering heterogeneous cognitive trajectories in mild cognitive impairment: a data-driven approach. Alzheimers Res Ther (2023) ; 15: : 57. |
5. | Jutten RJ , Sikkes SAM , Van der Flier WM , et al. Finding treatment effects in Alzheimer trials in the face of disease progression heterogeneity. Neurology (2021) ; 96: : e2673–e2684. |
6. | Cummings JL . Lessons learned from Alzheimer disease: clinical trials with negative outcomes. Clin Transl Sci (2017) ; 11: : 147–152. |
7. | Wolz R , Schwarz AJ , Gray KR , et al. Enrichment of clinical trials in MCI due to AD using markers of amyloid and neurodegeneration. Neurology (2016) ; 87: : 1235–1241. |
8. | Garcia MJ , Leadley R , Ross J , et al. Prognostic and predictive factors in early Alzheimer’s disease: a systematic review. J Alzheimers Dis Rep (2024) ; 8: : 203–240. |
9. | Tam A , Laurent C , Gauthier S , et al. Prediction of cognitive decline for enrichment of Alzheimer’s disease clinical trials. J Prev Alzheimers Dis (2022) ; 9: : 400–409. |
10. | Kwon HS , Kim JY , Koh SH , et al. Predicting cognitive stage transition using p-tau181, Centiloid, and other measures. Alzheimers Dement (2023) ; 19: : 4641–4650. |
11. | van Maurik IS , Zwan MD , Tijms BM , et al. Interpreting biomarker results in individual patients with mild cognitive impairment in the Alzheimer’s Biomarkers in Daily Practice (ABIDE) Project. JAMA Neurol (2017) ; 74: : 1481–1491. |
12. | van Maurik IS , Vos SJ , Bos I , et al. Biomarker-based prognosis for people with mild cognitive impairment (ABIDE): a modelling study. Lancet Neurol (2019) ; 18: : 1034–1044. |
13. | Cullen NC , Leuzy A , Palmqvist S , et al. Individualized prognosis of cognitive decline and dementia in mild cognitive impairment based on plasma biomarker combinations. Nat Aging (2021) ; 1: : 114–123. |
14. | van Dyck CH , Swanson CJ , Aisen P , et al. Lecanemab in early Alzheimer’s disease. N Engl J Med (2023) ; 388: : 9–21. |
15. | Veitch DP , Weiner MW , Aisen PS , et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement (2019) ; 15: : 106–152. |
16. | Folstein MF , Folstein SE and McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res (1975) ; 12: : 189–198. |
17. | Morris JC . The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology (1993) ; 43: : 2412–2414. |
18. | McKhann G , Drachman D , Folstein M , et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology (1984) ; 34: : 939–944. |
19. | Rosen WG , Mohs RC and Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry (1984) ; 141 11: : 1356–1364. |
20. | Landau SM , Breault C , Joshi AD , et al. Amyloid-β imaging with Pittsburgh Compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med (2013) ; 54: : 70–77. |
21. | Landau SM , Harvey D , Madison CM , et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging (2011) ; 32: : 1207–1218. |
22. | Bittner T , Zetterberg H , Teunissen CE , et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of β-amyloid (1–42) in human cerebrospinal fluid. Alzheimers Dement (2016) ; 12: : 517–526. |
23. | R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. |
24. | Bates DM , Machler M , Bolker BM , et al. Fitting linear mixed-effects models using lme4. J Stat Softw (2014) ; 67: : 1–48. |
25. | Akaike H . A new look at the statistical model identification. IEEE Trans Automat Contr (1974) ; 19: : 716–723. |
26. | Diggle P . Analysis of Longitudinal Data. OUP Oxford, (2002) . |
27. | Williams MM , Storandt M , Roe CM , et al. Progression of Alzheimer’s disease as measured by Clinical Dementia Rating Sum of Boxes scores. Alzheimers Dement (2013) ; 9: : S39–S44. |
28. | Harrell FE . Regression modeling strategies: with applications to linear models, logistic regression, and survival analysis. Springer, (2001) . |
29. | Kim YJ , Cho SK , Kim HJ , et al. Data-driven prognostic features of cognitive trajectories in patients with amnestic mild cognitive impairments. Alzheimers Res Ther (2019) ; 11: : 10. |
30. | Duara R and Barker W. Heterogeneity in Alzheimer’s disease diagnosis and progression rates: implications for therapeutic trials. Neurotherapeutics (2022) ; 19: : 8–25. |
31. | Edmonds EC , Ard MC , Edland SD , et al. Unmasking the benefits of donepezil via psychometrically precise identification of mild cognitive impairment: A secondary analysis of the ADCS vitamin E and donepezil in MCI study. Alzheimers Dement (N Y) (2018) ; 4: : 11–18. |
32. | Davatzikos C , Bhatt P , Shaw LM , et al. Prediction of MCI to AD conversion, via MRI, CSF biomarkers, and pattern classification. Neurobiol Aging (2011) ; 32: : e2322.19–27. |
33. | Vos SJB , Rossum IAv , Burns L , et al. Test sequence of CSF and MRI biomarkers for prediction of AD in subjects with MCI. Neurobiol Aging (2012) ; 33: : 2272–2281. |
34. | Walhovd KB , Fjell AM , Brewer JB , et al. Combining MR imaging, positron-emission tomography, and CSF biomarkers in the diagnosis and prognosis of Alzheimer disease. Am J Neuroradiol (2010) ; 31: : 347–354. |
35. | Schaeverbeke JM , Gabel S , Meersmans K , et al. Baseline cognition is the best predictor of 4-year cognitive change in cognitively intact older adults. Alzheimers Res Ther (2021) ; 13: : 75. |
36. | Stern Y . What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc (2002) ; 8: : 448–460. |
37. | Stern Y , Zarahn E , Hilton J , et al. Exploring the neural basis of cognitive reserve. J Clin Exp Neuropsychol (2003) ; 25: : 691–701. |
38. | Hall CB , Derby CA , LeValley AJ , et al. Education delays accelerated decline on a memory test in persons who develop dementia. Neurology (2007) ; 69: : 1657–1664. |
39. | Stern Y , Albert S , Tang MX , et al. Rate of memory decline in AD is related to education and occupation: cognitive reserve? Neurology (1999) ; 53: : 1942–1947. |
40. | Caminiti SP , Ballarini T , Sala A , et al. FDG-PET and CSF biomarker accuracy in prediction of conversion to different dementias in a large multicentre MCI cohort. Neuroimage Clin (2018) ; 18: : 167–177. |
41. | Jack CR , Knopman DS , Jagust WJ , et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol (2013) ; 12: : 207–216. |
42. | van der Flier WM and Tijms BM. Treatments for AD: towards the right target at the right time. Nat Rev Neurol (2023) ; 19: : 581–582. |
43. | Klunk WE , Koeppe RA , Price JC , et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement (2015) ; 11: : 1–15.e11–14. |


