
Clinical Evidence for GLP-1 Receptor Agonists in Alzheimer’s Disease: A Systematic Review
Abstract
Background:
Alzheimer’s disease (AD) is the most common cause of dementia. While preclinical studies have shown benefits of glucagon-like peptide 1 receptor agonists (GLP-1 RA) in targeting core AD pathology, clinical studies are limited.
Objective:
A systematic review was performed to evaluate GLP-1 RAs in AD for their potential to target core AD pathology and improve cognition.
Methods:
Searches were conducted via three different databases (PubMed, Embase, and Cochrane Library). Search terms included Medical Subject Headings (MeSH) terms: ‘glucagon-like peptide 1 receptor agonist’ and ‘Alzheimer’s disease’, as well as entry terms ‘GLP-1 RA’, ‘AD’, and three types of GLP-1 RA: ‘liraglutide’, ‘exenatide’, and ‘lixisenatide’.
Results:
A total of 1,444 studies were screened. Six articles that met criteria were included (four randomized control trials [RCTs] and two protocol studies). Two RCTs with amyloid-β and tau biomarker endpoints did not observe an end of treatment difference between the placebo and treated groups. In three RCTs with cognitive endpoints, there was no end of treatment difference between placebo and treated groups. GLP-1 RA showed metabolic benefits, such as lower body mass index and improved glucose levels on oral glucose tolerance tests in treated groups. GLP-1 RA may mitigate the decline in cerebral glucose metabolism and show enhanced blood-brain glucose transport capacity using 18F-FDG PET, however, more data is needed.
Conclusions:
GLP-1 RA therapy did not alter amyloid-β and tau biomarkers nor show improvements in cognition but showed potential metabolic and neuroprotective benefits.
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia and is recognized as the leading cause of dependence, disability, and mortality [1]. The deposition of amyloid-β (Aβ) plaques and tau neurofibrillary tangles are recognized as hallmarks of AD. Other contributors to the pathogenesis of AD include synaptic dysfunction, neurotransmitter imbalance, neuroinflammation, insulin resistance, metabolic derangement, oxidative stress, mitochondrial dysfunction, and impaired autophagy [2]. Currently approved treatments for AD include cholinesterase enzyme inhibitors and N-methyl D-aspartate receptor antagonists, which are symptomatic treatments [3]. Most recently, the US Food and Drug Administration (FDA) has approved lecanemab, an anti-Aβ therapy, which is the first disease targeting therapy for AD to receive full approval [4]. The effects on cognitive and functional outcomes are modest [5]. Development of disease-modifying treatment for AD is a priority.
There are several common pathological processes in AD and type 2 diabetes mellitus (T2DM), including impaired glucose metabolism, insulin resistance, mitochondrial dysfunction, increased oxidative stress, and increased neuroinflammation [6]. T2DM is associated with a risk for the development of AD [7]. Emerging evidence suggests that glucagon-like peptide 1 receptor agonist (GLP-1 RA), a class of anti-diabetic drug, may be a potential treatment for AD [8]. GLP-1 is an incretin hormone that decreases postprandial glycaemia via several pathways including glucagon inhibition, insulin secretion, delayed gastric emptying, appetite suppression, reduced intestinal nutrient absorption and promoting pancreatic beta-cell efficiency [9]. GLP-1 RA mimic the incretin hormones through induction of GLP-1 receptor activity. The currently approved GLP-1 RAs include exenatide, lixisenatide, liraglutide, dulaglutide, and semaglutide [2]. Of these, exenatide, lixisenatide and liraglutide are reported to cross the blood-brain barrier [2]. Aside from the hypoglycemic effect, they may improve cognitive dysfunction in individuals with diabetes [10]. Possible molecular mechanisms by which GLP-1 RA improve cognitive function include reduction of oxidative stress and neuroinflammation, inhibition of neuroapoptosis, facilitating insulin signal, reduction or prevention of Aβ and tau aggregation, and decreasing or suppressing neurotoxicity [9]. Evidence for these potential mechanisms of GLP-1 RA in AD predominantly stems from pre-clinical studies [9]. This systematic review focused on clinical evidence and aimed to evaluate the potential benefits of GLP-1 RA in AD, to evaluate their potential to modify underlying disease processes such as Aβ and tau accumulation, and to examine their impact on cognition. The specific questions that this review sought to address included: 1) can GLP-1 RA lower or clear soluble and aggregated Aβ and tau in AD; and 2) do GLP-1 RA improve cognitive function in AD.
MATERIALS AND METHODS
Search terms and databases
This review adheres to the PRISMA guideline and a PRISMA checklist is included in Supplementary Table 1. Searches were conducted via three databases including PubMed, Embase, and Cochrane Library. Searches were started on May 14, 2023 and completed on May 29, 2023. Search terms included Medical Subject Headings (MeSH) terms ‘glucagon-like peptide 1 receptor agonist’ and ‘Alzheimer’s disease’, as well as entry terms such as ‘GLP-1 RA’ and ‘AD’. Three types of GLP-1 agonist receptors were also used: ‘exenatide’, ‘liraglutide’, and ‘lixisenatide’. Duplicate records were removed by EndNote X9 before screening.
Study selection criteria
Inclusion criteria: i) Individuals who were diagnosed with AD, or those at risk of developing AD; ii) Studies that were original research articles, including randomized controlled trials (RCTs) and protocol studies; iii) Studies that evaluated the impact of GLP-1 RA on the core hallmarks (Aβ or tau) or other disease processes associated with AD; iv) Studies that included relevant clinical outcomes related to AD, such as changes in cognitive function.
Exclusion criteria: i) Individuals who were diagnosed with T2DM; ii) Review articles, animal studies, or in vitro/cell experiments; iii) Studies published in languages other than English; iv) Where the full-text article was not accessible.
Studies were screened on title and abstract and then by full text by a single reviewer (Y.L).
Data extraction
Information relating to cohort demographic characteristics, intervention, primary and secondary outcomes (in particular, cognitive and biomarker outcomes), as well as treatment-emergent adverse events (including grade) were extracted by a single reviewer (Y.L.).
Quality assessment
The included studies were evaluated by the Cochrane Risk of Bias Assessment Tool (RoB 2.0): 1) Bias arising from the randomization process; 2) Bias due to deviations from the intended interventions; 3) Bias due to missing outcome data; 4) Bias in measurement of the outcome; and 5) Bias in selection of the reported result.
RESULTS
Search results and study characteristics
Based on the search strategy, 1,444 articles were identified: PubMed – 483 articles, Embase 908 articles, Cochrane – 53 articles. 163 duplicate articles were removed by EndNote X9. After browsing the title and reading the abstract, 1,262 articles were excluded, including 660 review articles or meta-analyses, 511 animal or cell studies, and 86 articles which did not include relevant outcomes. 5 articles were excluded due to inability to access the abstract or full-text article. 9 papers were assessed for eligibility and 3 papers were excluded due to undetected duplication from the previous step. The remaining 6 papers were included in this review(Fig. 1).
Fig. 1
PRISMA flowchart.
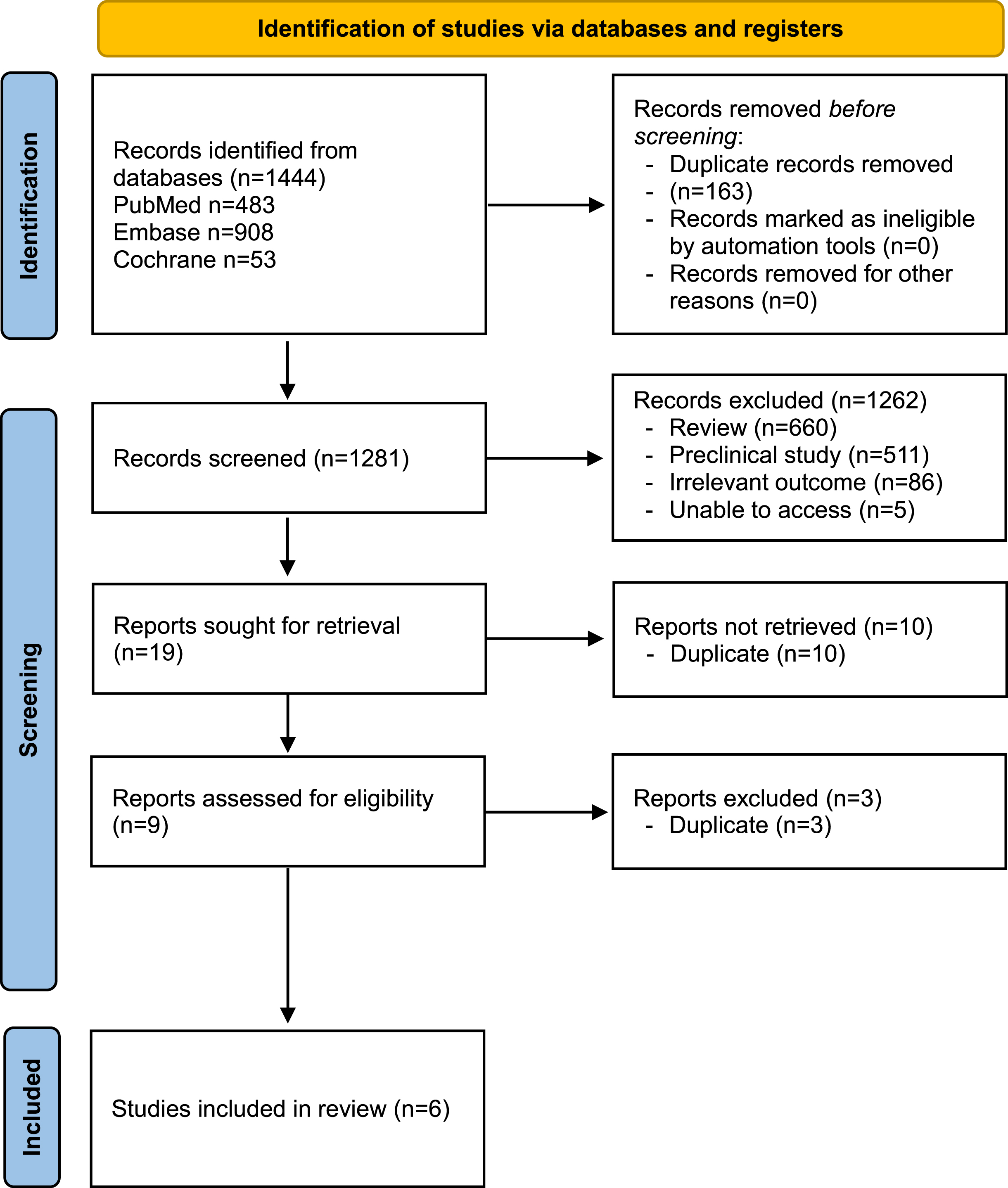
Table 1 provides a summary of the participant characteristics from the four RCTs identified. In all RCTs, the placebo and treatment groups had similar baseline demographic characteristics, including age and gender. Three RCTs included participants with a clinical diagnosis of AD [11–13], while one RCT included participants with subjective cognitive complaints and Mini-Mental State Examination (MMSE) scores >27 [14] (Table 1). In 3 of the studies, patients were treated with liraglutide [11, 12, 14], whereas exenatide was used in one study [13]. Of note, 2 studies shared an identical cohort and adhered to the same drug administration protocol [11, 12]. In this cohort, disease duration was reported to be longer in the liraglutide group (mean 29.5 versus 15 months, p = 0.015, Cohen’s d = 0.94) [11, 12]. However, it is unclear how disease duration was defined.
Table 1
Characteristics of the included studies
| Reference | Clinical trial identification number | Total population and randomization | Participant characteristics | Participant ethnicity | Type of GLP-1 RA, dosage and duration | Sex (M/F) | Recruitment location | Age (M±SD) |
| [11] | NCT01469351 | Total n = 38 (1 : 1 randomization) Liraglutide n = 18 Placebo n = 20 | Clinically diagnosed AD (MMSE 18–21). If MMSE>22, diagnosis was made based on CSF results. | Caucasian | s.c. liraglutide for 26 weeks 0.6 mg (1 wk)⟶1.2 mg (1 wk)⟶1.8 mg (thereafter) | Total 21/13 Placebo 15/5 Liraglutide 6/8 | Dementia clinics in Central Denmark | Placebo 66.6±1.8 Liraglutide 63.1±1.3 |
| [12] | NCT01469351 | Total n = 38 (1 : 1 randomization) Liraglutide n = 18 Placebo n = 20 | Clinically diagnosed AD (MMSE 18–21). If MMSE>22, diagnosis was made based on CSF results. | Caucasian | s.c. liraglutide for 26 weeks 0.6 mg (1 wk)⟶1.2 mg (1 wk)⟶1.8 mg (thereafter) | Total 21/13 Placebo 15/5 Liraglutide 6/8 | Dementia clinics in Central Denmark | Placebo 66.6±1.8 Liraglutide 63.1±1.3 |
| [13] | NCT01255163 | Total n = 27 randomized Exenatide n = 14 Placebo n = 13 | Clinical diagnosis of amnestic MCI/probable AD with supportive CSF biomarkers (low Aβ42, high total tau±pTau181) | N/A | s.c. exenatide for 78 weeks 5 mcg⟶10 mcg | Total 11/10 Placebo 4/6 Exenatide 7/4 | NIA Clinical Research Unit (Baltimore, MD) | Placebo 74.0±6.4 Exenatide 71.7±6.9 |
| [14] | N/A | Total n = 41 (1 : 1 randomization) Liraglutide n = 25 Placebo n = 16 | Cognitively normal participants aged 45–70 with subjective cognitive complaints and MMSE>27 (50% with first-degree relative(s) with AD) | N/A | s.c. liraglutide for 12 weeks 0.6 mg (1 wk)⟶1.2 mg (1 wk)⟶1.8 mg (thereafter) | Total 16/25 Placebo 5/11 Liraglutide 11/14 | N/A | Placebo 59.56±5.69 Exenatide 60.78±5.79 |
AD, Alzheimer’s disease; F, female; M, male; M±SD, mean±standard deviation; MCI, mild cognitive impairment; MMSE, Mini-Mental State Examination; N/A, not available; s.c., subcutaneous; wk, week.
Study quality
The quality of selected RCTs were evaluated by the Cochrane Risk of Bias Assessment Tool (RoB 2.0). A summary of risk of bias for all selected RCTs is shown in Fig. 2. Overall, the risk of bias for the selected RCTs was low.
Fig. 2
Risk of bias assessment.

Randomized controlled trials
Table 2 provides a summary of the primary and secondary outcomes of the four RCTs identified. Limited data was available for most of the outcome measures, particularly for biomarker outcomes, which were largely restricted to findings from single studies.
Table 2
Summary of the randomized controlled trials. ↑=increase; ↓decrease; — =no change
| Reference | Primary outcomes | Secondary outcomes | AEs (dropout rate due to AEs) |
| [11] | Liraglutide versus placebo —in Aβ burden measured with 11C-PiB PET global and regional binding potential ratios (scan 2/ scan 1) (p≥0.38) Baseline versus end-of-treatment (within groups) ↑ in PiB BP in temporal lobe (p = 0.04) in placebo group ↑ in PiB BP in temporal and occipital lobes (both p = 0.04) in liraglutide group | Biomarker, Cognitive and Metabolic outcomes Liraglutide versus placebo ↓ fasting glucose level (p = 0.004, d = 1.21) in liraglutide group —in total cognitive score (WMS-IV) (p = 0.50) ↓session2/session1 ratios of CMRglc (cingulate p = 0.04, occipital p = 0.04) in placebo group Baseline versus end-of-treatment (within groups) ↓CMRglc measured with 18F-FDG PET (precuneus p = 0.009; parietal, p = 0.04; temporal, p = 0.046; occipital, p = 0.009; cerebellum, p = 0.04) in the placebo group ↑ CMRglc measured with 18F-FDG PET (parietal, temporal, occipital, cerebellum; not statistically significant) in the liraglutide group —in total cognitive score (WMS-IV) in both placebo and liraglutide groups | nausea, anorexia (n = 1) |
| [12] | Liraglutide versus placebo —Tmax at 6-months Baseline versus end-of-treatment (within groups) —Tmax at 6 months (p = 0.24) in placebo groups ↑ Tmax at 6 months (p < 0.0001) in liraglutide group | N/A | |
| [13] | Exenatide versus placebo Safety, tolerability, and AEs: ↑ Nausea (Grade 1 or 2) (38% versus 0%, p = 0.016) in exenatide group ↑ upper GI infection symptoms (62% versus 7%, p = 0.004) in exenatide group ↑ loss of appetite/weight loss (31% versus 0%, p = 0.041) in exenatide group | Biomarker, Cognitive and Metabolic outcomes Exenatide versus placebo —in cognition measured by cognitive tests* at 18 monthsImprovement in digit-span forward task (total and maximum scores) at 6-months in exenatide group —in Aβ42 and tau (total Tau, pTau181) in CSF —plasma Aβ40 and Aβ42 —plasma EV (pTau181, IRS-1, Aβ40) ↓ in EV Aβ42 (F[1,16.3] = 4.71, p = 0.045) at 18 months in exenatide group —in the progression of grey matter atrophy at 18 months ↓ glucose level on 3-hour OGTT (comparing follow-up to baseline) (p < 0.001) at 18 months in exenatide group Baseline versus end-of-treatment (within groups) ↑ progression of grey matter atrophy in precuneus and medial-temporo-parietal regions and cortical thinning in placebo and exenatide groups ↓ BMI at 18-months (F[3,52.9] = 3.203, p = 0.031) in exenatide group ↓ glucose level on 3-h OGTT (p < 0.001) in exenatide group | As per primary outcomes |
| [14] | Liraglutide versus placebo ↑ associations between bilateral hippocampus and 3 clusters (left middle frontal gyrus, bilateral posterior cingulate and precuneus, and left lateral occipital cortex) on fMRI in liraglutide group | Cognitive and Metabolic outcomes Liraglutide versus placebo —in total cognitive score measured by cognitive testing battery** (all p>0.05) Baseline versus end-of-treatment (within groups) ↓ glucose level on OGTT at 120 minutes in liraglutide group at 12 weeks (p = 0.06, d = 0.05) | Total n = 5 GI side-effects n = 3 concern about potential SEs, n = 2 |
11C-PiB, 11C-Pittsburgh Compound B; 18F-FDG, 18F-fluoro-2-deoxyglucose; AD, Alzheimer’s disease; AEs, adverse effects; Aβ, amyloid-β; BP, binding potential; CMRglc, cerebral metabolic rate of glucose; CSF, cerebrospinal fluid; DMN, default mode network; EV, extracellular vesicle; OGTT, oral glucose tolerance tests; PET, positron emission tomography; SE, side effect; Tmax, maximum blood-brain glucose transfer capacity; WMS, Wechsler Memory Scale. *Cognitive tests: Clinical Dementia Rating Sum of Boxes (CDR-SOB), Mini-Mental State Exam (MMSE) and Alzheimer’s Disease Assessment Scale cognitive subscale (ADAS-Cog), the Frontal Assessment Battery (FAB), Verbal Fluency, the California Verbal Learning Task (CVLT), Boston Naming Test, Wechsler Memory Scale Logical Memory Subtest, Trail-making Test Parts A & B, American National Adult Reading Test (ANART), Clock drawing, Wechsler Adult Intelligence Scale Digit-Symbol and Digit Span Forward & Backward Subtests, the Benton Visual Retention Task (BVRT) and the University of Pennsylvania Smell Identification Test (UPSIT). **cognitive testing battery: Benton Visual Retention Test-Fifth Edition (BVRT), California Verbal Learning Test-Second Edition (CVLT-II), selected subtests of the Delis-Kaplan Executive Functioning System (D-KEFS, Rey Complex Figure Test (RCFT), Purdue Pegboard, selected subtests of the Wechsler Adult Intelligence Scale-3rd Edition (WAIS-III) and 2 subtests of the Wechsler Abbreviated Scale of Intelligence (WASI).
Biomarker outcome measures
CSF and plasma biomarkers
Cerebrospinal fluid (CSF) and plasma biomarkers were assessed in a pilot study, which observed no significant effect of treatment with exenatide on CSF (Aβ42, total Tau, p181-Tau), plasma (Aβ42, Aβ40) biomarkers, and plasma neuronal extracellular vesicle (EVs) markers (p181-tau, IRS-1, Aβ40), with the exception of a decrease in plasma-based EV Aβ42 in the exenatide group compared to the placebo group at 18 months [13].
Aβ positron emission tomography
Aβ plaque burden was assessed in one RCT [11], which was evaluated using 11C-Pittsburgh Compound B (11C-PiB) positron emission tomography (PET). Baseline PiB retention was higher in the liraglutide group compared to placebo across all brain regions examined. However, statistical comparison and effect sizes were not provided. There was a significant mean increase in PiB binding potential (BP) at 26 weeks compared to baseline in the temporal lobes in the placebo group, and the temporal and occipital lobes in the liraglutide group [11]. The two groups did not differ in terms of the global and regional BP ratios (scan 2/ scan 1) [11].
Cerebral glucose metabolism
Cerebral glucose metabolic rate with 18F-fluoro-2-deoxyglucose (18F-FDG) PET was observed in one RCT, which suggested a non-significant increase in CMRglc in all regions aside from the precuneus in the liraglutide group at the end of 6-month treatment period compared to baseline, whilst a decrease was seen in the precuneus, parietal, temporal, occipital lobe, cerebellum, and cingulate cortex in the placebo group at the end of treatment period compared to baseline [11]. A group comparison revealed a significant decline in the BP ratios (scan 2/scan 1) in the placebo group in the cingulate and occipital lobe compared to the treated group [11]. However, as quantitative values were not reported, it was not possible to estimate effect sizes.
A second study using the same cohort hypothesized a loss of normal glucose transporters at the blood-brain barrier in AD and proposed that native GLP-1 (and thus its analogues) may increase the maximum blood-brain glucose transfer capacity (Tmax) as estimated from 18F-FDG PET. This measure was used as a surrogate measure for the density and activity of glucose transporters [12]. As mentioned, disease duration was reported to be longer in the liraglutide group. At baseline, the estimated average Tmax was significantly lower in the liraglutide group compared to placebo (p = 0.02). There was no change in the cortical Tmax estimate in the placebo group (p = 0.24) from baseline to 6 months. However, the average Tmax estimate in liraglutide group increased significantly (p < 0.0001) in the cerebral cortex at 6 months. The estimates of Tmax increased significantly more in the liraglutide group than in the placebo group (p = 0.002). There was no difference in Tmax between the two groups at 6 months. The liraglutide group had comparable Tmax to estimates derived from a healthy control group, as reported in the prior literature. However, as quantitative values were not reported, it was not possible to estimate effect sizes [12].
AD-specific brain atrophy
Progression of grey matter atrophy was assessed in one RCT, which observed no differences or trends in the progression of grey matter atrophy on anatomical MRI between the two groups (exenatide versus placebo) [13]. Both groups were observed to have progressive grey matter atrophy in the precuneus and medial-temporo-parietal regions and significant cortical thinning over the 18-month study period [13].
Functional connectivity
Seed-based resting state functional connectivity (RSFC) was assessed in one RCT, as evaluated by functional MRI (fMRI) in a cohort of participants with subjective cognitive complaints and MMSE>27 [14]. AD-specific biomarkers (for Aβ and tau) were not reported for these participants. The association between the bilateral hippocampus and 3 clusters (left middle frontal gyrus, bilateral posterior cingulate and precuneus, and left lateral occipital cortex) was enhanced in the liraglutide group by the end of the 12-week treatment period [14].
Cognitive outcomes
Cognitive outcomes were assessed in 3 RCTs, which were evaluated either by the Brief Cognitive Examination from the Wechsler Memory Scale [11] or a cognitive testing battery [13, 14] (Table 2). The cognitive testing battery in Mullins et al. included Clinical Dementia Rating Sum of Boxes, MMSE and Alzheimer’s Disease Assessment Scale cognitive subscale, the Frontal Assessment Battery, Verbal Fluency, the California Verbal Learning Task, Boston Naming Test, Wechsler Memory Scale Logical Memory Subtest, Trail-making Test Parts A & B, American National Adult Reading Test, Clock drawing, Wechsler Adult Intelligence Scale Digit-Symbol and Digit Span Forward & Backward Subtests, the Benton Visual Retention Task, and the University of Pennsylvania Smell Identification Test [13]. Similarly, Watson et al included Benton Visual Retention Task and California Verbal Learning Task in their cognitive battery test, as well as selected subtests of the Delis-Kaplan Executive Functioning System, Rey Complex Figure Test, Purdue Pegboard, selected subtests of the Wechsler Adult Intelligence Scale-3rd Edition, and 2 subtests of the Wechsler Abbreviated Scale of Intelligence [14]. In all three RCTs, no significant differences were observed at the end of the treatment period between placebo and treated groups. Of these, one RCT suggested better performance in the digit-span forward task (total and maximum scores) in the exenatide-treated group compared to placebo, observed only at the 6-month timepoint [13].
Metabolic measures
Glucose levels from oral glucose tolerance tests (OGTT) were evaluated in two RCTs [13, 14]. Both RCTs reported a decrease in glucose level on OGTT in GLP-1 RA treated groups at the end of treatment period compared to baseline [13, 14]. One RCT reported a significant group difference (exenatide versus placebo) in glucose level on OGTT by the end of treatment period [13]. In addition, there was a significant group difference (liraglutide versus placebo) in fasting glucose levels at the end of the treatment period, with comparable baseline levels in one RCT [11].
One RCT assessed the effect of GLP-1 RA on body mass index (BMI) and suggested a significant decrease in BMI over 18 months of the study in the treatment group compared to baseline [13].
Treatment-emergent adverse events
Three RCTs discussed gastrointestinal-related symptoms as the main treatment-related adverse events (AEs) [11, 13, 14]. While one RCT noted that the rate of AEs was significantly higher in the GLP-1 RA-treated group, there were no related serious AEs or AEs Grade 3 or above in both groups [13] (Table 2). Dropout rates due to AEs were relatively low as mentioned in two RCTs [11, 14].
Protocol studies
Table 3 provides a summary of the participant characteristics for two prospective randomized placebo-controlled studies identified. Both studies are multi-centered. Participants in one proposed study will be treated with subcutaneous liraglutide [15] whereas in the other study participants will be treated with oral semaglutide [16] (Table 4). The rationale for the ELAD study is based on proposed shared pathophysiological mechanisms between T2DM and AD, and hypothesizes a potential for GLP-1 RA to influence AD pathology through multiple mechanisms, including neuroprotection, reducing Aβ plaque formation and neuroinflammation, with supportive evidence from mouse models and a pilot study in AD participants [15]. EVOKE and EVOKE+studies hypothesize a neuroprotective disease-modifying effect of semaglutide in early AD, based on evidence from animal studies showing potential benefits of GLP-1 RA in modifying underlying disease processes including reduction of neuroinflammation and tau phosphorylation and improved memory function, and studies in individuals with T2DM showing an association with lower risk of all-cause dementia and clinically diagnosed AD dementia [16].
Table 3
Characteristics of the proposed prospective studies
| Reference | Clinical trial identification number | Total population and randomization | Participant characteristics | Participant ethnicity | Type of GLP-1 RA, dosage and duration | Sex (M/F) | Recruitment location | Age |
| [15] | NCT01843075 | Total n = 206 (1 : 1 randomization) Liraglutide n = 103 Placebo n = 103 | Participants with a clinical diagnosis of mild AD dementia | N/A | s.c. liraglutide for 52 weeks (4-week dose escalation from starting dose of 0.6 mg) →1.8 mg | N/A | Local Memory Clinics and national databases across the UK | 50+ |
| [16] | NCT04777396 | Evoke: Total n = 1840 (1 : 1 randomization) Semaglutide n = 920 Placebo n = 920 | Amyloid positive participants with MCI due to AD (CDR = 0.5) or mild AD dementia (CDR = 1.0) | N/A | Oral semaglutide for 156 weeks (104 weeks plus 52-week extension phase) (8-week dose escalation) →14 mg | N/A | Argentina, Australia, Austria, Belgium, Brazil, Bulgaria, Canada, Czechia, Denmark, Finland, France, Germany, Greece, Hungary, Ireland, Israel, Italy, Japan, Korea, Republic of Netherlands, Norway, Poland, Portugal, Romania, Russian Federation, Serbia, Slovakia, Slovenia, South Africa, Spain, Sweden, Switzerland, Turkey, Ukraine, United Kingdom, United States | 55-85 |
| [16] | NCT04777409 | Evoke+: Total n = 1840 Semaglutide n=920 Placebo n = 920 | Amyloid positive participants with MCI due to AD (CDR = 0.5) or mild AD dementia (CDR = 1.0), with≥20% of participants with small-vessel co-pathology | N/A | Oral semaglutide for 156 weeks (104 weeks plus 52-week extension phase) (8-week dose escalation) →14 mg | N/A | Argentina, Australia, Austria, Belgium, Brazil, Bulgaria, Canada, China, Croatia, Czechia, Denmark, Finland, France, Germany, Greece, Hungary, Ireland, Israel, Italy, Japan, Korea, Republic of Netherlands, Poland, Portugal, Romania, Russian Federation, Serbia, Slovakia, Slovenia, South Africa, Spain, Sweden, Switzerland, Taiwan, Turkey, Ukraine, United Kingdom, United States | 55–85 |
AD, Alzheimer’s disease; CDR, Clinical Dementia Rating; s.c., subcutaneous.
Table 4
Proposed outcome measures of the prospective studies
| Reference | Primary outcomes | Secondary outcomes |
| [15] | Change in cerebral glucose metabolic rate in the hippocampus, medial temporal lobe, and posterior cingulate as measured by 18F-FDG PET. | Change in MRI (entorhinal cortex and hippocampal volumes; diffusion tensor parameters). Changes in microglial activation –as measured by TSPO PET. Changes in plasma markers of neuroinflammation (IL-6, TNF-α, IL-8, IL-10, IL-12, CRP, TGF-β). Change in Z-scores for the CDR-SOB, ADAS Exec, ADCS-ADL.incidence and severity of treatment-emergent adverse events. |
| [16] | Change from baseline to week 104 in CDR-SB score. | Confirmatory secondary endpoints: Time to progression to dementia (CDR global≥1.0) for participants with MCI at baseline (from baseline to week 104). Change from baseline to week 104 in ADCS-ADL-MCI score. Exploratory endpoints: Change from baseline to week 104 for plasma markers of neuroinflammation and neurodegeneration (NfL, GFAP, and other validated biomarkers). Change from baseline to week 104 in plasma p-tau181 |
18F-FDG, 18F-fluoro-2-deoxyglucose; ADAS Exec, Alzheimer’s Disease Assessment Scale-Cognitive Subscale and Executive domain scores; ADCS-ADL, Alzheimer’s Disease Cooperative Study-Activities Of Daily Living; CDR-SOB, Clinical Dementia Rating Sum of Boxes; CRP, C-reactive protein; IL, interleukin; MCI, mild cognitive impairment; PET, positron emission tomography; s.c., subcutaneous; TGF-β, transforming growth factor-beta; TNF-α, tumor necrosis factor-alpha; TSPO, translocator protein.
DISCUSSION
In Australia, the currently approved treatments for AD remain symptomatic and emerging preclinical evidence suggests that GLP-1 RA could be a potential disease-modifying treatment for AD. However, clinical studies on GLP-1 RA in AD are very limited. Hence, this systematic review evaluated the available evidence from clinical studies to determine whether GLP-1 RAs alter core AD pathology and improve cognition.
Aβ plaque and tau neurofibrillary tangle formation and deposition are recognized as the hallmarks of AD. Several animal studies suggest GLP-1 RA may decrease Aβ and tau deposition [17], whilst in this review, two RCTs suggested no difference in Aβ plaque and tau deposition between placebo and GLP-1 RA-treated groups. Interestingly, one RCT reported a decrease in plasma-based EV biomarkers (Aβ42) in the pilot study of exenatide actions in AD. However, given other biomarkers remained unchanged and this was the only positive finding in this pilot study, this finding should not be overinterpreted and future studies are required to draw a definite conclusion.
Studies have reported an association between GLP-1 RA and improved memory and other cognitive functions in animal models [17]. Potential cognitive benefits also have been observed in individuals with T2DM, treated with GLP-1 RA [18]. For example, a post-hoc analysis of an RCT with longitudinal follow-up of more than 4 years observed a potential cognitive benefit associated with a type of GLP-1 RA, dulaglutide, in individuals with T2DM [19]. In contrast, in this review, three out of the four RCTs suggested no difference in cognitive outcomes between placebo and GLP-1 RA intervention groups, except for improvement in digit-span forward task with exenatide treatment at the 6-month point only. However, this test is a measure of verbal and short-term memory and is not specific to dementia.
In terms of metabolic effect, a decrease in glucose level on the OGTT was reported in two RCTs. In addition, a decrease in fasting glucose level and BMI were reported. These findings suggest a potential role for GLP-1 RA in addressing metabolic dysregulation even in individuals without diabetes. Notably, the investigations by Geji et al. showed GLP-1 RA prevents the expected decline in cerebral glucose metabolism and increases a surrogated measure of the number of glucose transporters at the blood-brain barrier, although no benefit in cognition was observed [11, 12]. In addition, the authors reported a negative correlation between AD progression and blood-brain glucose transfer and cerebral glucose metabolism [12]. Another study by Waston et al. using fMRI suggested that GLP-1 RA enhanced RSFC associations within the studied region [14]. Hence, even though GLP-1 RA did not alter biomarkers of Aβ plaques and tau neurofibrillary tangles in these studies, they show some potential metabolic benefits, which supports the purported neuroprotective mechanisms of this class of drugs. While a narrative review has identified sex differences in the use of GLP-1 RAs for treatment of T2DM and obesity, with females demonstrating superior weight loss outcomes [20], the small sample size and lack of disaggregation by sex in the present studies preclude evaluation of the sex differences in the use of GLP-1 RAs for AD.
In terms of drug safety, three RCTs reported side effects (SEs) of GLP-1 RA and the most common SE was gastrointestinal. Two RCTs reported participant withdrawal due to SEs; however, the dropout rate was relatively low. Overall, this class of drugs appears relatively safe and well-tolerated.
There are some limitations to this systematic review. First, due to the strict inclusion and exclusion criteria, only six papers were included in this review, four of which report original data. Secondly, the sample size in all RCTs was relatively small. This may be addressed by forthcoming studies with larger sample sizes. Additionally, the proposed Evoke/Evoke+ study involves the investigation of an oral GLP-RA drug, which may facilitate trial recruitment. Evoke+ also includes a proposed subset of participants with vascular co-pathology, which will enhance the representativeness of the study population. Thirdly, the study duration for most of the RCTs was relatively short. This limitation could possibly pose challenges in detecting differences, even if they exist. This issue may also be addressed by the upcoming studies, with a longer study duration outlined in the protocol papers. Fourthly, in some studies, participants’ baseline characteristics differed. For example, in both studies by Geji et al, disease duration was longer in the liraglutide group [11, 12]. These differences may have had an impact on the primary and secondary outcomes. Finally, the screening process and data extraction were performed by a single reviewer, which could potentially introduce bias.
Nonetheless, given that there is limited clinical evidence on GLP-1 RA in AD, this review adds to the existing literature and complements the studies that have been reported. Aside from cognitive outcomes and Aβ plaques/tau biomarker outcomes, this study reported on a range of other measures associated with AD pathogenesis and metabolic factors, which adds to the understanding of GLP-1 RA’s potential neuroprotective effects and provides direction for future study.
Despite the lack of evidence for direct Aβ or tau clearance/lowering, the observed metabolic benefits of GLP-1, both peripheral and central, indicate a promising direction for further exploration. In addition, GLP-1 RA’s potential to mitigate vascular contributions to cognitive impairment warrants further investigation. Given its potential benefit, GLP-1 RA may be a suitable adjunctive therapy for therapies targeting Aβ and tau. To draw a definite conclusion, further studies with larger and more representative populations are required.
Conclusions
In conclusion, this systematic review summarized the clinical studies on GLP-1 RA in AD and found that GLP-1 RA show no benefit in lowering/clearing Aβ plaques and tau neurofibrillary tangles deposition or improving cognition, but highlights the metabolic and neuroprotective potential for this class of drugs. Due to the limitations of the included studies, further studies with large sample sizes are needed to draw more definite conclusions.
AUTHOR CONTRIBUTIONS
Yulin Liang (Writing – original draft; Writing – review & editing); Vincent Doré (Writing – review & editing); Christopher C. Rowe (Writing – review & editing); Natasha Krishnadas (Supervision; Writing – review & editing).
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICTS OF INTEREST
Christopher C. Rowe has received research grants from NHMRC, Enigma Australia, Biogen, Eisai and Abbvie. He is on the scientific advisory board for Cerveau Technologies and has consulted for Prothena, Eisai, Roche, and Biogen Australia.
All other authors have no conflict of interest to report.
DATA AVAILABILITY
Data sharing is not applicable to this article, as no datasets were generated or analyzed during this study.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-230181.
REFERENCES
[1] | Lane CA , Hardy J , Schott JM ((2018) ) Alzheimer’s disease. Eur J Neurol 25: , 59–70. |
[2] | Du H , Meng X , Yao Y , Xu J ((2022) ) The mechanism and efficacy of GLP-1 receptor agonists in the treatment of Alzheimer’s disease. Front Endocrinol (Lausanne) 13: , 1033479. |
[3] | Breijyeh Z , Karaman R ((2020) ) Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 25: , 5789. |
[4] | Harris E ((2023) ) Alzheimer drug lecanemab gains traditional FDA approval. JAMA 330: , 495. |
[5] | van Dyck CH , Swanson CJ , Aisen P , Bateman RJ , Chen C , Gee M , Kanekiyo M , Li D , Reyderman L , Cohen S , Froelich L , Katayama S , Sabbagh M , Vellas B , Watson D , Dhadda S , Irizarry M , Kramer LD , Iwatsubo T ((2023) ) Lecanemab in early Alzheimer’s disease. N Engl J Med 388: , 9–21. |
[6] | Candeias E , Duarte AI , Carvalho C , Correia SC , Cardoso S , Santos RX , Plácido AI , Perry G , Moreira PI ((2012) ) The impairment of insulin signaling in Alzheimer’s disease. IUBMB Life 64: , 951–957. |
[7] | Li L , Cavuoto M , Biddiscombe K , Pike KE ((2020) ) Diabetes mellitus increases risk of incident dementia in APOEɛ4 carriers: a meta-analysis. J Alzheimers Dis 74: , 1295–1308. |
[8] | Reich N , Hölscher C ((2022) ) The neuroprotective effects of glucagon-like peptide 1 in Alzheimer’s and Parkinson’s disease: An in-depth review. Front Neurosci 16: , 970925. |
[9] | Yaribeygi H , Rashidy-Pour A , Atkin SL , Jamialahmadi T , Sahebkar A ((2021) ) GLP-1 mimetics and cognition. Life Sci 264: , 118645. |
[10] | Longo M , Di Meo I , Caruso P , Muscio MF , Scappaticcio L , Maio A , Maiorino MI , Bellastella G , Signoriello G , Knop FK , Rizzo MR , Esposito K ((2023) ) Circulating levels of endothelial progenitor cells are associated with better cognitive function in older adults with glucagon-like peptide 1 receptor agonist-treated type 2 diabetes. Diabetes Res Clin Pract 200: , 110688. |
[11] | Gejl M , Gjedde A , Egefjord L , Møller A , Hansen SB , Vang K , Rodell A , Brændgaard H , Gottrup H , Schacht A , Møller N , Brock B , Rungby J ((2016) ) In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front Aging Neurosci 8: , 108. |
[12] | Gejl M , Brock B , Egefjord L , Vang K , Rungby J , Gjedde A ((2017) ) Blood-brain glucose transfer in Alzheimer’s disease: effect of GLP-1 analog treatment. Sci Rep 7: , 17490. |
[13] | Mullins RJ , Mustapic M , Chia CW , Carlson O , Gulyani S , Tran J , Li Y , Mattson MP , Resnick S , Egan JM , Greig NH , Kapogiannis D ((2019) ) A pilot study of exenatide actions in Alzheimer’s disease. Curr Alzheimer Res 16: , 741–752. |
[14] | Watson KT , Wroolie TE , Tong G , Foland-Ross LC , Frangou S , Singh M , McIntyre RS , Roat-Shumway S , Myoraku A , Reiss AL , Rasgon NL ((2019) ) Neural correlates of liraglutide effects in persons at risk for Alzheimer’s disease. Behav Brain Res 356: , 271–278. |
[15] | Femminella GD , Frangou E , Love SB , Busza G , Holmes C , Ritchie C , Lawrence R , McFarlane B , Tadros G , Ridha BH , Bannister C , Walker Z , Archer H , Coulthard E , Underwood BR , Prasanna A , Koranteng P , Karim S , Junaid K , McGuinness B , Nilforooshan R , Macharouthu A , Donaldson A , Thacker S , Russell G , Malik N , Mate V , Knight L , Kshemendran S , Harrison J , Hölscher C , Brooks DJ , Passmore AP , Ballard C , Edison P ((2019) ) Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: study protocol for a randomised controlled trial (ELAD study). Trials 20: , 191. |
[16] | Atri A , Feldman HH , Hansen CT , Honore JB , Johannsen P , Knop FK , Poulsen P , Raket LL , Sano M , Soininen H , Cummings J ((2022) ) evoke and evoke+: design of two large-scale, double-blind, placebo-controlled, phase 3 studies evaluating the neuroprotective effects of semaglutide in early Alzheimer’s disease. Alzheimers Dement 18: , e062415. |
[17] | Katsenos AP , Davri AS , Simos YV , Nikas IP , Bekiari C , Paschou SA , Peschos D , Konitsiotis S , Vezyraki P , Tsamis KI ((2022) ) New treatment approaches for Alzheimer’s disease: preclinical studies and clinical trials centered on antidiabetic drugs. Expert Opin Investig Drugs 31: , 105–123. |
[18] | Tian S , Jiang J , Wang J , Zhang Z , Miao Y , Ji X , Bi Y ((2023) ) Comparison on cognitive outcomes of antidiabetic agents for type 2 diabetes: A systematic review and network meta-analysis. Diabetes Metab Res Rev 39: , e3673. |
[19] | Cukierman-Yaffe T , Gerstein HC , Colhoun HM , Diaz R , García-Pérez LE , Lakshmanan M , Bethel A , Xavier D , Probstfield J , Riddle MC , Rydén L , Atisso CM , Hall S , Rao-Melacini P , Basile J , Cushman WC , Franek E , Keltai M , Lanas F , Leiter LA , Lopez-Jaramillo P , Pirags V , Pogosova N , Raubenheimer PJ , Shaw JE , Sheu WH , Temelkova-Kurktschiev T ((2020) ) Effect of dulaglutide on cognitive impairment in type 2 diabetes: an exploratory analysis of the REWIND trial. Lancet Neurol 19: , 582–590. |
[20] | Rentzeperi E , Pegiou S , Koufakis T , Grammatiki M , Kotsa K ((2022) ) Sex differences in response to treatment with glucagon-like peptide 1 receptor agonists: opportunities for a tailored approach to diabetes and obesity care. J Pers Med 12: , 454. |


