
Gallic Acid Alleviates Cognitive Impairment by Promoting Neurogenesis via the GSK3β-Nrf2 Signaling Pathway in an APP/PS1 Mouse Model
Abstract
Background:
Neuronal loss occurs early and is recognized as a hallmark of Alzheimer’s disease (AD). Promoting neurogenesis is an effective treatment strategy for neurodegenerative diseases. Traditional Chinese herbal medicines serve as a rich pharmaceutical source for modulating hippocampal neurogenesis.
Objective:
Gallic acid (GA), a phenolic acid extracted from herbs, possesses anti-inflammatory and antioxidant properties. Therefore, we aimed to explore whether GA can promote neurogenesis and alleviate AD symptoms.
Methods:
Memory in mice was assessed using the Morris water maze, and protein levels were examined via western blotting and immunohistochemistry. GA’s binding site in the promoter region of transcription regulator nuclear factor erythroid 2-related factor 2 (Nrf2) was calculated using AutoDock Vina and confirmed by a dual luciferase reporter assay.
Results:
We found that GA improved spatial memory by promoting neurogenesis in the hippocampal dentate gyrus zone. It also improved synaptic plasticity, reduced tau phosphorylation and amyloid-β concentration, and increased levels of synaptic proteins in APP/PS1 mice. Furthermore, GA inhibited the activity of glycogen synthase kinase-3β (GSK-3β). Bioinformatics tools revealed that GA interacts with several amino acid sites on GSK-3β. Overexpression of GSK-3β was observed to block the protective effects of GA against AD-like symptoms, while GA promoted neurogenesis via the GSK-3β-Nrf2 signaling pathway in APP/PS1 mice.
Conclusions:
Based on our collective findings, we hypothesize that GA is a potential pharmaceutical agent for alleviating the pathological symptoms of AD.
INTRODUCTION
Neuron loss is a recognized pathologically hallmark of Alzheimer’s disease (AD) [1, 2]. The phenotype manifests during preclinical stages of AD, in which neuropathological hallmarks are not yet present [3, 4]. With disease progression, neuron loss has been detected throughout the cerebral cortex, and is especially prominent in the CA1 region of the hippocampus [5, 6]. The loss of neurons contributes to a decrease in neurogenesis, which are required for normal learning processes to occur [7–9]. Amyloid-β (Aβ) plaques, another hallmark of AD, are considered the initiators of neuron loss.
A decrease in the number of neurons results in an imbalance between postnatal neurogenesis and neuronal death. Adult hippocampus neurogenesis (AHN) was first reported in the 1960 s [10]. These multipotent neural stem cells (NSCs) derived newborn neurons reside in the subgranular zone (SGZ) of the hippocampus dentate gyrus (DG) [11, 12]. Numerous studies have suggested that altered hippocampal neurogenesis contributes to neuronal loss and represents an early pathological progress in AD [13, 14]. Moreover, existing evidence suggests that repairing damaged neurogenesis could be a novel approach to treating AD. Interventions that enhance adult neurogenesis are being considered as potential therapeutic strategies for AD [15, 16].
Traditional Chinese medicine (TCM) refers to active component mixtures extracted from herbs, some of which have the potential to activate NSC proliferation and neurogenesis in neurodegenerative diseases [17]. The therapeutic effects of TCM on neurogenesis can be achieved by restoring neural function, regulating the inflammatory response, providing nutritional support, and reconstructing neural circuits and paracrine signaling involving nerve growth factors [18, 19]. GA, derived from natural plants, is a traditional Chinese medicine known for its anti-inflammatory, analgesic, and antitumor properties [20, 21]. GA can also be used to treat neurodegenerative diseases because of its antioxidant properties and oxygen free radicals eliminating ability [22]. GA could traverse the blood-brain barrier, exerting effects on both the central nervous system and peripheral tissues [23, 24]. GA is also safe and easy to take for a long time. Moreover, GA induces NSCs’ the differentiation and proliferation [25]; however, it remains unclear whether GA promotes neurogenesis in NSC.
Glycogen synthase kinase-3β (GSK-3β) is one of the dominant proteins responsible for neurogenesis [26], and this protein regulates NSC dynamics and adult neurogenesis via the Wnt/β-catenin signaling pathway [27]. Moreover, the GSK-3β/Nrf2 signaling pathway is crucial for neuroprotection and antioxidant activity [28, 29]. Nrf2 is responsible for regulating the expression of cytoprotective genes driven by binding to the antioxidant response element (ARE), and Nrf2-ARE-related pathways are implicated in the pathogenesis of AD [30, 31].
Herein, we investigated GA’s the pharmaceutical effect in an APP/PS1 mouse model and its impact on spatial memory, neuronal morphological alternation, synaptic proteins, accumulation of Aβ, and phosphorylation of tau. GA was discovered to promote neurogenesis through the GSK-3β-Nrf2 signaling pathway in APP/PS1 mice. Genetic engineering was also utilized to verify the underlying mechanisms of improving AD symptoms.
MATERIALS AND METHODS
Animals and administration
APP/PS1 mice (male, four month) were procured from the Institute of Laboratory Animals Science, Chinese Academy of Medical Sciences. Housed under a 12-h light-dark cycle at a temperature of 22±2°C, all mice were kept with ad libitum access to food and water. Twenty milligrams of GA (Sigma-Aldrich, MO, USA) were dissolved in 25 microliters of DMSO (100%) and then diluted with 12.5 milliliters of double-distilled water [32]. GA was administered to the mice via gavage (20 mg/kg) once a day and lasted for four months. All animal experiments were conducted in accordance with the “Policies on the Use of Animals and Humans in Neuroscience Research” revised and approved by the Society for Neuroscience in 1995, as well as the Institutional Animal Care and Use Committee (IACUC Number: 3190) at Tongji Medical College, Huazhong University of Science and Technology approved the study protocol.
Morris water maze
After four months of GA administration, all mice were trained in a maze for 6 consecutive days to find an underwater-hidden platform placed in a quadrant. Each day, there were three trials, with mice starting from one of the quadrants. Once the animal found and climbed on the platform (stayed for 3 s), this trail end and time would be recorded. If the animal could not find the platform within 60 s, it was guided to the underwater platform and rest on for 15–20 s. At day7, the spatial memory was tested after the underwater platform removed. The time to platform (latency), swimming path, duration in the target zone (platform zone), and swimming speed were recorded by a video camera (Techman, Chengdu, China), which was placed 2 meters from the water surface.
Stereotaxic brain injection of adeno-associated virus (AAV)
Recombinant AAV encoding s9-GSK3β (AAV2/9-CMV-s9-GSK3β-EGFP), and AAV2/9- CMV-EGFP were products of OBiO (Shanghai, China). The AAV virus titer was appropriate 5×1012 vg/ml. After anesthesia, APP mice’s heads were fixed in a RWD stereotaxic instrument (Shenzhen, China). The CA3 region of hippocampus was injected bilaterally with AAV, location AP±2.5, DV –2.0, ML –2.0, at a rate of 0.1–0.15μl per minute. The needle was left in hole for 10 min before withdrawal after injection.
Western blotting
Tissues or cultured cells were harvested on ice and homogenized with RIPA lysis buffer (1 mM PMSF, BL507A) containing protease inhibitors. The centrifuged parameter for homogenate is 12,000×g, 4°C, and 10 min. The supernatant was retained and measured for protein concentration using BCA assays. Equal amounts of protein from each sample were loaded and run on 8–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels, and then proteins on gel were electransferred to membranes. To avoid non-specific binding, the membranes without transferred proteins were blocked with 5% non-fat milk at room temperature (22–26°C) for at least 1 h, and then immersed and incubated with pre-dilution primary antibodies (1:500–1:1000) overnight at 4°C. Next day, removed primary antibodies from membranes and then incubated with horseradish peroxidase (HRP) labeled secondary antibodies (A2008 Goat-anti rabbit or A0216 Goat-anti mouse, Beyotime, Shanghai, China) for at least 1 h at RT. The blots were developed by ECL substrate (BL502A) in a luminometer (ChemiScope 6000, Clinx, China), and the protein bands on the membranes were quantified using ImageJ software (NIH, USA). The primary and secondary antibodies are listed in Table 1.
Table 1
Antibodies employed in the study
| Antibody | Host | Manufacturer | Type | Cat # | Dilution | |
| WB | IHC | |||||
| DCX | Goat | Santa Cruz | Poly | sc-8066 | 1:1000 | 1:200 |
| NeuroD | Goat | Santa Cruz | Poly | sc-1084 | 1:1000 | 1:200 |
| pS396 | Rabbit | Signalway | Poly | #11102 | 1:1000 | |
| pT231 | Rabbit | Signalway | Poly | #11110 | 1:1000 | 1:100 |
| Tau5 | Mouse | Abcam | Mono | ab80579 | 1:1000 | |
| PSD95 | Rabbit | Abcam | Poly | ab18258 | 1:2000 | |
| SYP | Mouse | Abcam | Mono | ab8049 | 1:2000 | |
| Synapsin I | Rabbit | Abcam | poly | ab64581 | 1:2000 | |
| t-GSK-3β | Rabbit | Proteintech | Poly | 22104-1-AP | 1:1000 | |
| b-catenin | Rabbit | Proteintech | Poly | 51067-2-AP | 1:1000 | |
| GAPDH | Rabbit | Abcam | Poly | ab9485 | 1:1000 | |
WB, western blotting; IHC, immunohistochemistry; SYP, synaptophysin.
Immunohistochemistry
Mouse brain sections were incubated in 0.1% Triton (BS084, Biosharp, Beijing, China) in phosphate buffer solution for 30 min. After being rinsed, 3% BSA (BS114) was utilized to avoid non-specific reaction on mouse sections at RT for 30 min. A pre-dilution primary antibody (1:50–1:150) was dropped onto the brain sections and kept at 4°C overnight. Next day, all the brain sections were washed with PBS and incubated with second antibody (peroxidase-labbed IgG) at 37°C water bath for 1 h. A mixture of 0.05% 3,30-diaminobenzidine (DAB) and 0.01% H2O2 was dropped onto the brain sections for color reactions. At least 5 fields of the section were selected and analyzed for quantitative assessment.
Golgi-cox staining
The Golgi-cox staining kit was a product of FD Neuro Technologies, and the staining procedure was performed according to the provided protocol. Briefly, after anesthetized by 2% pentobarbital sodium, the mice brains were removed and fixed in a mix solution (Solution A: B = 1:1) for at least two weeks in dark environment at RT. The detailed process has been described previously [31].
ELISA
GSK-3β (RX202270M, Ruixin Biotech, China), Aβ1-40/1-42 (E-EL-M3009/E-EL-M3010, Elabscience, Wuhan, China) in the rat brain were measured using ELISA kit and, and 6-7 hippocampus per group were used for assay. To assess the concentration of Aβ, hippocampus was homogenized in buffer PBS (1% PMSF), and centrifuged at 12,000 g for 10 min. Then the supernatant was collected and stored until analyze, GSK-3β and Aβ1-40/1-42 concentrations were determined by comparison with standard curves.
Electrophysiological analysis
Electrophysiological activity was analyzed according to previous literature [33]. Briefly, the brains of mice were horizontally sectioned into 300μm thick slices using a vibration microtome. Then the sections were transferred to an interface chamber and incubated within artificial cerebrospinal fluid for 30 min. After incubation, the slices were put over planar microelectrodes (8×8 array) for examination. MED64 System (Alpha MED Sciences) was used for recording voltage signals in response to stimulation, and in this study, field excitatory postsynaptic potentials (fEPSPs). To evoke fEPSP amplitudes, adjustment of the stimulation intensity is need. Additionally, the induction of long-term potentiation (LTP) was stimulated by theta-burst stimulation.
Cell counting kit (CCK8)
The cells with density of 5×103 cells per well were plated in a 96-well plate. After 24 h of culture, the cells were treated with or treated without GA at concentrations of 10, 20, 40, 80, 160, 240, 320, 480, or 640μM for another 24 h. Following this, CCK-8 solution (10μl) was added into each well, and 5-6 wells per group. The absorbance was measured at 450 nm using an EnSpire Multimode plate reader (PerkinElmer, Inc., Waltham, MA, USA). The CCK8 kit (C0038) was purchased from Beyotime (Shanghai, China).
Reverse transcription and real-time quantitative PCR
After isolation, RNA was reverse transcript and analyzed by real-time quantitative PCR (RT-PCR) according to the manufacturer’s instructions (Takara, Japan). The RT-PCR system consisted of SYBR Green PCR master mixes (10μl), MgCl2 (3 mM), forward and reverse primers (0.5μM, respectively), and cDNA template (1μl). ABI Step One Plus QPCR system (Thermo Fisher, USA) was applied to analyze RT-PCR mixture. The targeted genes expression level was normalized by the glyceraldehyde-3-phosphate dehydrogenase (GAPDH, housekeeping gene). PCR primers are as follows: doublecortin (DCX) forward and reverse primers, 5’-ACAAGGCACACGGCTTTCTT-3’ and 5’-TGGAACCACAGCAACTTTTCCAA-3’; NeuroD forward and reverse primers, 5’-AGGAATTCGCCCACGCAG-3’ and 5’-GGTCATGTTTCCACTTCCTGTTG-3’; GAPDH forward and reverse primers, 5’- CAAGCTCATTTCCTGGTATGACA-3’ and 5’-TATGGGGGTCTGGGATGGAA-3’.
Dual Luciferase reporter assay
In short, HEK293T cells were transfected with the psicheck2.0-DCX or NEUROD1-luc reporter constructs by helping with a DNA transfection reagent (Neofect, Beijing). Then, β-Amyloid (Aβ1-42, obtained from ChinaPeptides Co., Ltd. (QYAOBIO)) and GA (120μM) were added into the medium. After 24-h treatment, the cells were collected and rinsed using pre-cool PBS three times, followed by lysis with detection buffer (100μl firefly luciferase). A dual-Lumi dual luciferase reporter (DLR) gene assay kit (RG027) was applied for the luciferase activity assay of the cell extracts, which was purchased from Beyotime (Shanghai, China). The predicted ARE fragments (NRF2 binding site) on the promoter region of the gene DCX and NEUROD1 were cloned into the psicheck2.0 luciferase reporter vector (Promega, WI, USA). And the mutation site of ARE in luciferase plasmids was designed and constructed using the Gene Tailor system (Invitrogen, USA). Ahead of treatment with GA and Aβ, the cells were cultured in plates and transfected with constructs [31].
Molecular docking
AutoDock Vina and the standardized docking data was analyzed by molecular docking [34]. The 3D structure of GSK-3β was retrieved from the Protein Data Bank (PDB) database (https://www.rcsb.org/search). The grid box center in present study was set to (22.22, 1.76, 20.53) Å for (center_x, center_y, center_z). And the grid box the size was defined as (20, 20, 20) Å for (size_x, size_y, size_z). The 3D structure of the grid box for GSK3β in Fig. 4C. Figure 4D displays the structure of GA. To get more different binding modes, the num-modes was set to 9, and the energy range was set to 7 kcal/mol.
Statistical analysis
All data in present study are showed as the mean±SEM and were analyzed by other researchers in a blinded manner. Prism 8.0 (GraphPad Software) was applied to analyze and plot data. Unpaired Student’s t-test for two-group comparisons, two-way analysis of variance (ANOVA) followed by Bonferroni’s post-hoc test was applied for statistical analysis. Statistical significance was considered if a p value less than 0.05 for all experiments.
RESULTS
GA ameliorates cognitive deficits and promotes neurogenesis in APP/PS1 mice
GA is typically functioned as an inflammatory scavenging agent in the treatment of neurodegenerative diseases. To explore its capacity to enhance neurogenesis, 4-month-old APP/PS1 mice were administered GA for four months, and their cognitive capacity was assessed using the MWM. During six consecutive days of training, APP/PS1 mice treated with GA showed markedly lower latency than vehicle-treated mice from days three to six (Fig. 1A, B). During the spatial memory test (day 7) after the underwater platform was removed, escape latency decreased, while platform crossing times (Fig. 1C) and duration in the target quadrant (Fig. 1D) decreased in comparison between the APP/PS1-GA group and APP/PS1-V group. There is no significant difference in swimming speed among the groups (data not shown). Furthermore, DCX and NeuroD, two markers of neurogenesis, were markedly upregulated after the administration of GA in APP/PS1 mice, compared to vehicle-treated mice, as detected by western blotting and qPCR (Fig. 1E–H). Immunohistochemical staining also confirmed the upregulation of DCX and NeuroD in the DG subregion of the hippocampus in APP/PS1 mice (Fig. 1I, J). These results indicate that GA promotes neurogenesis and improves cognitive ability in APP/PS1 mice.
Fig. 1
GA improved spatial memory and promoted neurogenesis. A) The learning ability of APP/PS1 mice was analyzed by 6 consecutive days of training with the MWM test after 4-month administration with GA. Two-way ANOVA, F (5, 90) = 3.124, p = 0.012, n = 10 in each group. B-D) Memory was assessed at day 7, and escape latency (B), times of platform crossing (C), and time in target quadrant (D) were recorded. [B] Unpaired t-test, t = 3.90 df = 18, p = 0.012; [C] Unpaired t-test, t = 3.24 df = 18, p = 0.021; [D] Unpaired t-test, t = 4.34 df = 18, p = 0.036, n = 10 in each group. E-H) Immature neurons generated by neurogenesis were examined by western blotting using antibodies against DCX and NeuroD (E, F) and qPCR (G, H) in the brains of APP/PS1 mice with subsequent quantitative analysis. [NeuroD] Unpaired t-test, t = 3.051 df = 8, p = 0.023; [DCX] Unpaired t-test, t = 3.019 df = 8, p = 0.036; [G] Unpaired t-test, t = 4.039 df = 8, p = 0.017; [H] Unpaired t-test, t = 3.359 df = 8, p = 0.013, n = 5 in each group. I, L) Immature neurons generated from neurogenesis were examined by immunochemistry using antibodies DCX and NeuroD in the DG of hippocampus of APP/PS1 mice with quantitative analysis. [NeuroD] Unpaired t-test, t = 3.783 df = 8, p = 0.017; [DCX] Unpaired t-test, t = 4.032 df = 8, p = 0.026, n = 5 in each group. Bar = 20μm, *p < 0.05. Data are presented as mean±SD.
![GA improved spatial memory and promoted neurogenesis. A) The learning ability of APP/PS1 mice was analyzed by 6 consecutive days of training with the MWM test after 4-month administration with GA. Two-way ANOVA, F (5, 90) = 3.124, p = 0.012, n = 10 in each group. B-D) Memory was assessed at day 7, and escape latency (B), times of platform crossing (C), and time in target quadrant (D) were recorded. [B] Unpaired t-test, t = 3.90 df = 18, p = 0.012; [C] Unpaired t-test, t = 3.24 df = 18, p = 0.021; [D] Unpaired t-test, t = 4.34 df = 18, p = 0.036, n = 10 in each group. E-H) Immature neurons generated by neurogenesis were examined by western blotting using antibodies against DCX and NeuroD (E, F) and qPCR (G, H) in the brains of APP/PS1 mice with subsequent quantitative analysis. [NeuroD] Unpaired t-test, t = 3.051 df = 8, p = 0.023; [DCX] Unpaired t-test, t = 3.019 df = 8, p = 0.036; [G] Unpaired t-test, t = 4.039 df = 8, p = 0.017; [H] Unpaired t-test, t = 3.359 df = 8, p = 0.013, n = 5 in each group. I, L) Immature neurons generated from neurogenesis were examined by immunochemistry using antibodies DCX and NeuroD in the DG of hippocampus of APP/PS1 mice with quantitative analysis. [NeuroD] Unpaired t-test, t = 3.783 df = 8, p = 0.017; [DCX] Unpaired t-test, t = 4.032 df = 8, p = 0.026, n = 5 in each group. Bar = 20μm, *p < 0.05. Data are presented as mean±SD.](https://ip.ios.semcs.net:443/media/adr/2024/8-1/adr-8-1-adr230171/adr-8-adr230171-g001.jpg)
GA reduces Aβ protein levels and tau hyperphosphorylation
Aβ level and tau hyperphosphorylation are closely related to reduced neurogenesis, impairments of LTP, and synaptic plasticity [35]. We detected tau phosphorylation levels in the mouse brain after GA treatment and observed that the concentrations of Aβ1 - 40 and Aβ1 - 42 were markedly reduced, compared to their respective levels in vehicle-treated mice (Fig. 2A). The levels of hyperphosphorylated tau at pS396 and pT231 were also reduced after GA treatment (Fig. 2B, C). Immunohistochemical staining revealed reduced pT231 expression in the cortex and hippocampus following GA treatment (Fig. 2D, E).
Fig. 2
GA decreased the concentration of Aβ and phosphorylation of tau in APP/PS1 mouse brains. A) The concentrations of Aβ1 - 40 and Aβ1 - 42 in the mouse brains of APP/PS1mice were detected by ELISA after 4-month administration of GA. [NeuroD] Unpaired t-test, t = 3.954 df = 18, p = 0.013; [DCX] Unpaired t-test, t = 3.287 df = 18, p = 0.019, n = 10 in each group. B, C) The phosphorylation of tau at pS396 and pT231 was assayed by western blotting in the mouse brains of APP/PS1 after GA administration with subsequent quantitative analysis. [pS396] Unpaired t-test, t = 5.56 df = 8, p = 0.015; [pT231] Unpaired t-test, t = 4.2351 df = 8, p = 0.013, n = 5 in each group. D, E) The protein level of tau at pT231 was assayed by immunohistochemical staining in the mouse brains of APP/PS1 after GA administration with quantitative analysis. [Cortex] Unpaired t-test, t = 3.051 df = 8, p = 0.023; [DCX] Unpaired t-test, t = 3.019 df = 8, p = 0.036, n = 5 in each group. Bar = 20μm. Unpaired t-test, *p < 0.05. Data are presented as mean±SD.
![GA decreased the concentration of Aβ and phosphorylation of tau in APP/PS1 mouse brains. A) The concentrations of Aβ1 - 40 and Aβ1 - 42 in the mouse brains of APP/PS1mice were detected by ELISA after 4-month administration of GA. [NeuroD] Unpaired t-test, t = 3.954 df = 18, p = 0.013; [DCX] Unpaired t-test, t = 3.287 df = 18, p = 0.019, n = 10 in each group. B, C) The phosphorylation of tau at pS396 and pT231 was assayed by western blotting in the mouse brains of APP/PS1 after GA administration with subsequent quantitative analysis. [pS396] Unpaired t-test, t = 5.56 df = 8, p = 0.015; [pT231] Unpaired t-test, t = 4.2351 df = 8, p = 0.013, n = 5 in each group. D, E) The protein level of tau at pT231 was assayed by immunohistochemical staining in the mouse brains of APP/PS1 after GA administration with quantitative analysis. [Cortex] Unpaired t-test, t = 3.051 df = 8, p = 0.023; [DCX] Unpaired t-test, t = 3.019 df = 8, p = 0.036, n = 5 in each group. Bar = 20μm. Unpaired t-test, *p < 0.05. Data are presented as mean±SD.](https://ip.ios.semcs.net:443/media/adr/2024/8-1/adr-8-1-adr230171/adr-8-adr230171-g002.jpg)
GA improves synaptic proteins, synaptic plasticity, and neuron morphological complexity in APP/PS1 mice
Neurogenesis refers to the formation of large numbers of mature neurons. We examined the synaptic proteins in the brains of APP/PS1 mice. As expected, GA increased the levels of synaptic proteins (PSD95, Syn I, and synaptophysin) in APP/PS1 mice, as examined by western blotting (Fig. 3A, B). Spine density (Fig. 2C, D) and mature spines (mushrooms) (Fig. 3E) increased after GA administration. Moreover, synaptic plasticity, as measured by LTP (Fig. 3F, G), was ameliorated by GA administration in APP/PS1 mice.
Fig. 3
GA ameliorated synaptic impairment and improved synaptic plasticity. A, B) The protein levels of synapse were assayed by western blotting in the mouse brains of APP/PS1 after GA administration with quantitative analysis. [PSD95] Unpaired t-test, t = 3.221 df = 8, p = 0.0218; [SYP] Unpaired t-test, t = 3.904 df = 8, p = 0.0101; [Synapsin I] Unpaired t-test, t = 3.352 df = 8, p = 0.0213, n = 5 in each group. C-E) The spine number (D) and shape (E) were examined by Golgi-cox staining in the mouse brains of APP/PS1 mice after GA administration with quantitative analysis. [D] Unpaired t-test, t = 2.700 df = 8, p = 0.0356; [E] Unpaired t-test, t = 2.904 df = 8, p = 0.0204, n = 5 in each group. F, G) Synaptic plasticity was detected in the brain slice of APP/PS1 after GA administration. Unpaired t-test, t = 4.353 df = 8, p = 0.0102, n = 5 in each group. *p < 0.05. Data are presented as mean±SD.
![GA ameliorated synaptic impairment and improved synaptic plasticity. A, B) The protein levels of synapse were assayed by western blotting in the mouse brains of APP/PS1 after GA administration with quantitative analysis. [PSD95] Unpaired t-test, t = 3.221 df = 8, p = 0.0218; [SYP] Unpaired t-test, t = 3.904 df = 8, p = 0.0101; [Synapsin I] Unpaired t-test, t = 3.352 df = 8, p = 0.0213, n = 5 in each group. C-E) The spine number (D) and shape (E) were examined by Golgi-cox staining in the mouse brains of APP/PS1 mice after GA administration with quantitative analysis. [D] Unpaired t-test, t = 2.700 df = 8, p = 0.0356; [E] Unpaired t-test, t = 2.904 df = 8, p = 0.0204, n = 5 in each group. F, G) Synaptic plasticity was detected in the brain slice of APP/PS1 after GA administration. Unpaired t-test, t = 4.353 df = 8, p = 0.0102, n = 5 in each group. *p < 0.05. Data are presented as mean±SD.](https://ip.ios.semcs.net:443/media/adr/2024/8-1/adr-8-1-adr230171/adr-8-adr230171-g003.jpg)
GA directly interacts with and inhibits the activity of GSK-3β: The mechanism of the GSK3β-NRF2 signaling pathway
Overactivation of GSK-3β in transgenic mice (Tet/GSK3β) depletes the neurogenic niche in adults [36]. Moreover, tau phosphorylation and synaptic toxicity closely relate to GSK3β activity [37, 38]; thus, we explored its alterations after treatment with GA. As shown below, we observed an increase in GSK-3β Ser9 (the unactive form of GSK-3β) after four-month treatment with GA (Fig. 4A, B). Furthermore, the activity of GSK-3β, as measured by ELISA, indicated that GA administration greatly inhibited GSK-3β activity in the mouse brain (Fig. 4C), which suggests that GA exerts its protective effect via GSK-3β. To verify this hypothesis, we performed a bioinformatic analysis to investigate interaction between GSK-3β and GA. VINA 1.1.2 software was applied for docking studies between proteins and compounds. The binding energy for this docking was –7.2 kcal/mol, indicating that GA could spontaneously bind strongly to the protein GSK-3β. Subsequently, PYMOL and Discovery Studio were used for analyses, and GA was able to stably bind to the cavity of the GSK-3β and interact with its surrounding amino acids. As shown in Fig. 4D and 4E, GA interacted with GSK-3β mainly through hydrogen bonds and salt bridges. The functional groups in the compounds were able to form hydrogen bonds with amino acids GLN265 and ASP260 in the A chain of GSK-3β and with amino acids ARG220, ARG223, ASP260, GLY262, and GLN265 in the B chain of GSK-3β. The carboxyl functional group of the compound formed a salt-bridge interaction with amino acid ARG223 in the protein A chain, and these were the main forces contributing to the binding of the compound to the active site.
Fig. 4
GA directly binds to the GSK-3β amino acid sequence. The mechanism of GSK-3β-Nrf2 signaling pathway. A, B) Phosphorylation of GSK-3β at Ser9 was assayed by western blotting in the mouse brains of APP/PS1 mice after GA administration with quantitative analysis. Two-way ANOVA, F (2, 8) = 18.983, p = 0.0335, n = 5 in each group. C) GSK-3β activity was assayed by ELISA in the mouse brains of APP/PS1 mice after GA administration with quantitative analysis. Two-way ANOVA, F (2, 8) = 23.143, p = 0.0244, n = 5 in each group. D) The triadic interaction of GA with GSK-3β was estimated based on their 3D-structures in molecular DOCK Server. The key binding residues of GSK-3β are labeled. E) Molecular structure of GA. F, G) The protein level of Nrf2 was assayed by western blotting in the mouse brains of APP/PS1 mice after GA administration and the overexpression of GSK-3β was assessed by quantitative analysis. Two-way ANOVA, F (2, 8) = 13.534, p = 0.0401, n = 5 in each group. H) CCK8 analyzed cell viability after GA treatment with different concentration of GA in 293T cell. I-L) Diagrams showing the predicted anti-oxidative response element (ARE) for Nrf2 binding in the promoter (–2000±200 bp) of DCX (I) and NEUROD1 (K) genes. The AREs or the mutated (MUT) AREs plasmids of DCX (J) and NEUROD1 (L) genes were co-treated with Abeta, with or without GA into HEK293 cells for 24 h, and then the luciferase activity was measured. [J] Unpaired t-test, [DCX] t = 5.322 df = 4, p = 0.0204; [ARE1] t = 4.784 df = 4, p = 0.0174. [L] Unpaired t-test, [NEUROD1] t = 4.532 df = 4, p = 0.0308; [ARE1] t = 5.523 df = 4, p = 0.0294. *p < 0.05 versus APP/PS1-V, #p < 0.05 versus APP/PS1-GA. Data are presented as mean±SD.
![GA directly binds to the GSK-3β amino acid sequence. The mechanism of GSK-3β-Nrf2 signaling pathway. A, B) Phosphorylation of GSK-3β at Ser9 was assayed by western blotting in the mouse brains of APP/PS1 mice after GA administration with quantitative analysis. Two-way ANOVA, F (2, 8) = 18.983, p = 0.0335, n = 5 in each group. C) GSK-3β activity was assayed by ELISA in the mouse brains of APP/PS1 mice after GA administration with quantitative analysis. Two-way ANOVA, F (2, 8) = 23.143, p = 0.0244, n = 5 in each group. D) The triadic interaction of GA with GSK-3β was estimated based on their 3D-structures in molecular DOCK Server. The key binding residues of GSK-3β are labeled. E) Molecular structure of GA. F, G) The protein level of Nrf2 was assayed by western blotting in the mouse brains of APP/PS1 mice after GA administration and the overexpression of GSK-3β was assessed by quantitative analysis. Two-way ANOVA, F (2, 8) = 13.534, p = 0.0401, n = 5 in each group. H) CCK8 analyzed cell viability after GA treatment with different concentration of GA in 293T cell. I-L) Diagrams showing the predicted anti-oxidative response element (ARE) for Nrf2 binding in the promoter (–2000±200 bp) of DCX (I) and NEUROD1 (K) genes. The AREs or the mutated (MUT) AREs plasmids of DCX (J) and NEUROD1 (L) genes were co-treated with Abeta, with or without GA into HEK293 cells for 24 h, and then the luciferase activity was measured. [J] Unpaired t-test, [DCX] t = 5.322 df = 4, p = 0.0204; [ARE1] t = 4.784 df = 4, p = 0.0174. [L] Unpaired t-test, [NEUROD1] t = 4.532 df = 4, p = 0.0308; [ARE1] t = 5.523 df = 4, p = 0.0294. *p < 0.05 versus APP/PS1-V, #p < 0.05 versus APP/PS1-GA. Data are presented as mean±SD.](https://ip.ios.semcs.net:443/media/adr/2024/8-1/adr-8-1-adr230171/adr-8-adr230171-g004.jpg)
The results showed that GA enhanced Nrf2 protein level, and the overexpression of GSK-3β attenuated the nuclear translation of Nrf2 (Fig. 4F, G). These results indicate that GSK-3β influence Nrf2 protein level during GA treatment. The relative viability of HEK293T cells was then evaluated using CCK8 after treatment with a concentration gradient of GA (Fig. 4 H).
To verify this assumption that Nrf2 transcription function, a DLR assay was performed using constructs with specific ARE promoter elements of neurogenesis-related proteins. After transfecting DCX/NEUROD1-ARE-plasmid constructs, a significant increase in luciferase activity in the GA group was observed compared to the control group (Fig. 6I–L). And this increase was completely block when the cells were transfected with the mutated ARE plasmid (Fig. 6I–L). Taken together, these data suggest that in response to GA, Nrf2 binds to specific ARE promoter elements in DCX/NEUROND1 and increases genes transcription.
Reactivation of GSK-3β blocks GA’s protective effects on spatial memory and neurogenesis
To further determine whether the activity of GSK-3β mediates GA’s protective effects against Aβ in APP/PS1 mice, we explored whether reactivating GSK-3β could block the protective effects of GA. As revealed by the spatial memory test, the six-day training in the MWM test indicated that APP/PS1 mice that received GA and GSK-3β had a longer latency in finding the platform on days three through five (Fig. 5A). On day seven, when the platform was removed, APP/PS1 mice with GSK-3β overexpression took more time to reach the platform location, spend less time crossing the platform location, and spent less time in the target quadrant (Fig. 5B-D). Western botting and qPCR analysis indicated that both DCX and NeuroD decreased after overexpressing GSK-3β (Fig. 5E-H). Immunohistochemical staining confirmed that the DCX and NeuroD levels were markedly reduced in the DG of the hippocampus (Fig. 5I, J). Moreover, β-catenin also markedly decreased after overexpressing GSK-3β (Fig. 5K, L), indicating an impairment of neurogenesis.
Fig. 5
Overexpressing GSK-3β blocked GA’s improvement on spatial memory and neurogenesis. A) The learning ability of APP/PS1 mice was analyzed by 6 consecutive days of training with the MWM test after overexpressing GSK-3β along with GA. Two-way ANOVA, F (10, 135) = 2.542, p = 0.0324, n = 10 in each group. B-D) Memory was assessed at day 7, and escape latency (B), times of platform crossing (C), and time in target quadrant (D) were recorded. [B] Two-way ANOVA, F (2, 18) = 33.423, p = 0.0104; [C] Two-way ANOVA, F (2, 18) = 45.294, p = 0.0110; [D] Two-way ANOVA, F (2, 18) = 49.423, p = 0.0201, n = 10 in each group. E-H) Immature neurons generated from neurogenesis were examined by western blot using antibodies DCX and NeuroD (E, F) and qPCR (G, H) in the brains of APP/PS1 mice after overexpressing GSK-3β and GA administration with quantitative analysis. [DCX] Two-way ANOVA, F (2, 8) = 16.675, p = 0.0132; [NeuroD] Two-way ANOVA, F (2, 8) = 15.313, p = 0.0138; [G] Two-way ANOVA, F (2, 8) = 12.324, p = 0.0129; [H] Two-way ANOVA, F (2, 8) = 18.973, p = 0.0164, n = 5 in each group. I, J) Immature neurons generated from neurogenesis were examined by immunochemistry using antibodies DCX and NeuroD in the DG of hippocampus of APP/PS1 mice overexpressing GSK-3β and GA administration with quantitative analysis. [DCX] Two-way ANOVA, F (2, 18) = 35.535, p = 0.0104; [NeuroD] Two-way ANOVA, F (2, 18) = 32.546, p = 0.0104, n = 10 in each group. K, L) β-catenin also markedly decreased after overexpressing GSK-3β and GA administration with quantitative analysis. Two-way ANOVA, F (2, 8) = 35.113, p = 0.001, n = 5 in each group. Bar = 20μm. *p < 0.05 versus APP/PS1-V, #p < 0.05 versus APP/PS1-GA. Data are presented as mean±SD.
![Overexpressing GSK-3β blocked GA’s improvement on spatial memory and neurogenesis. A) The learning ability of APP/PS1 mice was analyzed by 6 consecutive days of training with the MWM test after overexpressing GSK-3β along with GA. Two-way ANOVA, F (10, 135) = 2.542, p = 0.0324, n = 10 in each group. B-D) Memory was assessed at day 7, and escape latency (B), times of platform crossing (C), and time in target quadrant (D) were recorded. [B] Two-way ANOVA, F (2, 18) = 33.423, p = 0.0104; [C] Two-way ANOVA, F (2, 18) = 45.294, p = 0.0110; [D] Two-way ANOVA, F (2, 18) = 49.423, p = 0.0201, n = 10 in each group. E-H) Immature neurons generated from neurogenesis were examined by western blot using antibodies DCX and NeuroD (E, F) and qPCR (G, H) in the brains of APP/PS1 mice after overexpressing GSK-3β and GA administration with quantitative analysis. [DCX] Two-way ANOVA, F (2, 8) = 16.675, p = 0.0132; [NeuroD] Two-way ANOVA, F (2, 8) = 15.313, p = 0.0138; [G] Two-way ANOVA, F (2, 8) = 12.324, p = 0.0129; [H] Two-way ANOVA, F (2, 8) = 18.973, p = 0.0164, n = 5 in each group. I, J) Immature neurons generated from neurogenesis were examined by immunochemistry using antibodies DCX and NeuroD in the DG of hippocampus of APP/PS1 mice overexpressing GSK-3β and GA administration with quantitative analysis. [DCX] Two-way ANOVA, F (2, 18) = 35.535, p = 0.0104; [NeuroD] Two-way ANOVA, F (2, 18) = 32.546, p = 0.0104, n = 10 in each group. K, L) β-catenin also markedly decreased after overexpressing GSK-3β and GA administration with quantitative analysis. Two-way ANOVA, F (2, 8) = 35.113, p = 0.001, n = 5 in each group. Bar = 20μm. *p < 0.05 versus APP/PS1-V, #p < 0.05 versus APP/PS1-GA. Data are presented as mean±SD.](https://ip.ios.semcs.net:443/media/adr/2024/8-1/adr-8-1-adr230171/adr-8-adr230171-g005.jpg)
Reactivation of GSK-3β blocked GA’s protective effects on the production of Aβ, tau phosphorylation, and synaptic toxicity
Furthermore, we explored the effect of overexpressing GSK-3β on the production of Aβ, tau hyperphosphorylation, and synaptic toxicity in APP/PS1 mice. We observed that overexpressing GSK-3β increased the concentration of Aβ1 - 40/1 - 42 in APP/PS1 mice with GA, compared to those treated with only GA (Fig. 6A). Moreover, overexpressing GSK-3β also increased tau hyperphosphorylated at pT231 and pS396 (Fig. 6B, C). The synaptic protein (PSD95, SYP, and synapsin I) levels also decreased (Fig. 6D, E) after overexpressing GSK-3β. Golgi-cox staining indicated a marked reduction in spine density (Fig. 6F-H) along with impaired LTP after overexpressing GSK3β (Fig. 6I, J). The above results indicates that GA protects against the toxicity induced by Aβ via the deactivation of GSK-3β.
Fig. 6
GSK-3β overexpression increases the concentration Aβ, tau phosphorylation, and impaired synaptic protein. A) The concentration of Aβ1 - 40 and Aβ1 - 42 was detected by ELISA after overexpressing GSK-3β and GA treatment in the mouse brains of APP/PS1 mice. [Aβ1-40] Two-way ANOVA, F (2, 18) = 43.435, p = 0.0123; [Aβ1-42] Two-way ANOVA, F (2, 18) = 46.342, p = 0.0108, n = 10 in each group. B, C) The phosphorylation of tau at pS396 and pT231 was assayed by western blotting in the mouse brains of APP/PS1 mice after overexpressing GSK-3β and GA administration with quantitative analysis. [pS396] Two-way ANOVA, F (2, 8) = 25.944, p = 0.0156; [pT231] Two-way ANOVA, F (2, 8) = 24.535, p = 0.0236, n = 5 in each group. D, E) The protein levels of synapse were assayed by western blotting in the mouse brains of APP/PS1 mice with quantitative analysis. [PSD95] Two-way ANOVA, F (2, 8) = 32.255, p = 0.0108; [SYP] Two-way ANOVA, F (2, 8) = 26.435, p = 0.0208, [Synapsin I] Two-way ANOVA, F (2, 8) = 32.542, p = 0.0194, n = 5 in each group. F-H) The spine number and shape were examined by Golgi-cox staining in the mouse brains of APP/PS1 mice, with quantitative analysis. Two-way ANOVA, F (2, 8) = 29.4235, p = 0.0324, n = 5 in each group. I, J) Synaptic plasticity was detected in brain slices of APP/PS1 mice. Two-way ANOVA, F (2, 8) = 19.432, p = 0.0321, n = 5 in each group. *p < 0.05 versus APP/PS1-V, #p < 0.05 versus APP/PS1-GA. Data are presented as mean±SD.
![GSK-3β overexpression increases the concentration Aβ, tau phosphorylation, and impaired synaptic protein. A) The concentration of Aβ1 - 40 and Aβ1 - 42 was detected by ELISA after overexpressing GSK-3β and GA treatment in the mouse brains of APP/PS1 mice. [Aβ1-40] Two-way ANOVA, F (2, 18) = 43.435, p = 0.0123; [Aβ1-42] Two-way ANOVA, F (2, 18) = 46.342, p = 0.0108, n = 10 in each group. B, C) The phosphorylation of tau at pS396 and pT231 was assayed by western blotting in the mouse brains of APP/PS1 mice after overexpressing GSK-3β and GA administration with quantitative analysis. [pS396] Two-way ANOVA, F (2, 8) = 25.944, p = 0.0156; [pT231] Two-way ANOVA, F (2, 8) = 24.535, p = 0.0236, n = 5 in each group. D, E) The protein levels of synapse were assayed by western blotting in the mouse brains of APP/PS1 mice with quantitative analysis. [PSD95] Two-way ANOVA, F (2, 8) = 32.255, p = 0.0108; [SYP] Two-way ANOVA, F (2, 8) = 26.435, p = 0.0208, [Synapsin I] Two-way ANOVA, F (2, 8) = 32.542, p = 0.0194, n = 5 in each group. F-H) The spine number and shape were examined by Golgi-cox staining in the mouse brains of APP/PS1 mice, with quantitative analysis. Two-way ANOVA, F (2, 8) = 29.4235, p = 0.0324, n = 5 in each group. I, J) Synaptic plasticity was detected in brain slices of APP/PS1 mice. Two-way ANOVA, F (2, 8) = 19.432, p = 0.0321, n = 5 in each group. *p < 0.05 versus APP/PS1-V, #p < 0.05 versus APP/PS1-GA. Data are presented as mean±SD.](https://ip.ios.semcs.net:443/media/adr/2024/8-1/adr-8-1-adr230171/adr-8-adr230171-g006.jpg)
DISCUSSION
Numerous studies have provided experimental evidence supporting the antioxidation, anti-inflammatory, and antimicrobial activities of GA [39, 40]. In addition to the neuronal compartment, the brain’s immunological system is closely related to AD pathogenesis [41], and neuronal loss contributes to the pathogenesis of AD [42]. In this study, GA was proved to effectively promote neurogenesis and synaptic proteins and reduce tau hyperphosphorylation and the concentration of Aβ, thus improving spatial memory. Molecular docking analysis revealed that GA directly binds to GSK-3β and inhibits its activity. Furthermore, overexpressing GSK-3β blocked GA’s protective effect against Aβ accumulation, increased tau phosphorylation, impaired synaptic protein expression, and impaired spatial memory. Therefore, GA appears to exert protective effects against AD progression. GA also alleviates cognitive impairment by promoting neurogenesis via the GSK3β-Nrf2 signaling pathway in APP/PS1 mice. The mechanisms are illustrated in Fig. 7.
Fig. 7
Proposed mechanisms of GA in promoting neurogenesis and alleviating AD symptoms. GA binds to and inhibits GSK-3β activity, reducing the phosphorylation level of Nrf2 and degrading its ubiquitination. Nrf2 transcriptionally regulates the expression of NEUROD1 and DCX genes in the nucleus, promoting an increase in the number of neural synapses in the dentate gyrus of the hippocampus and improving spatial memory in APP/PS1 mice.
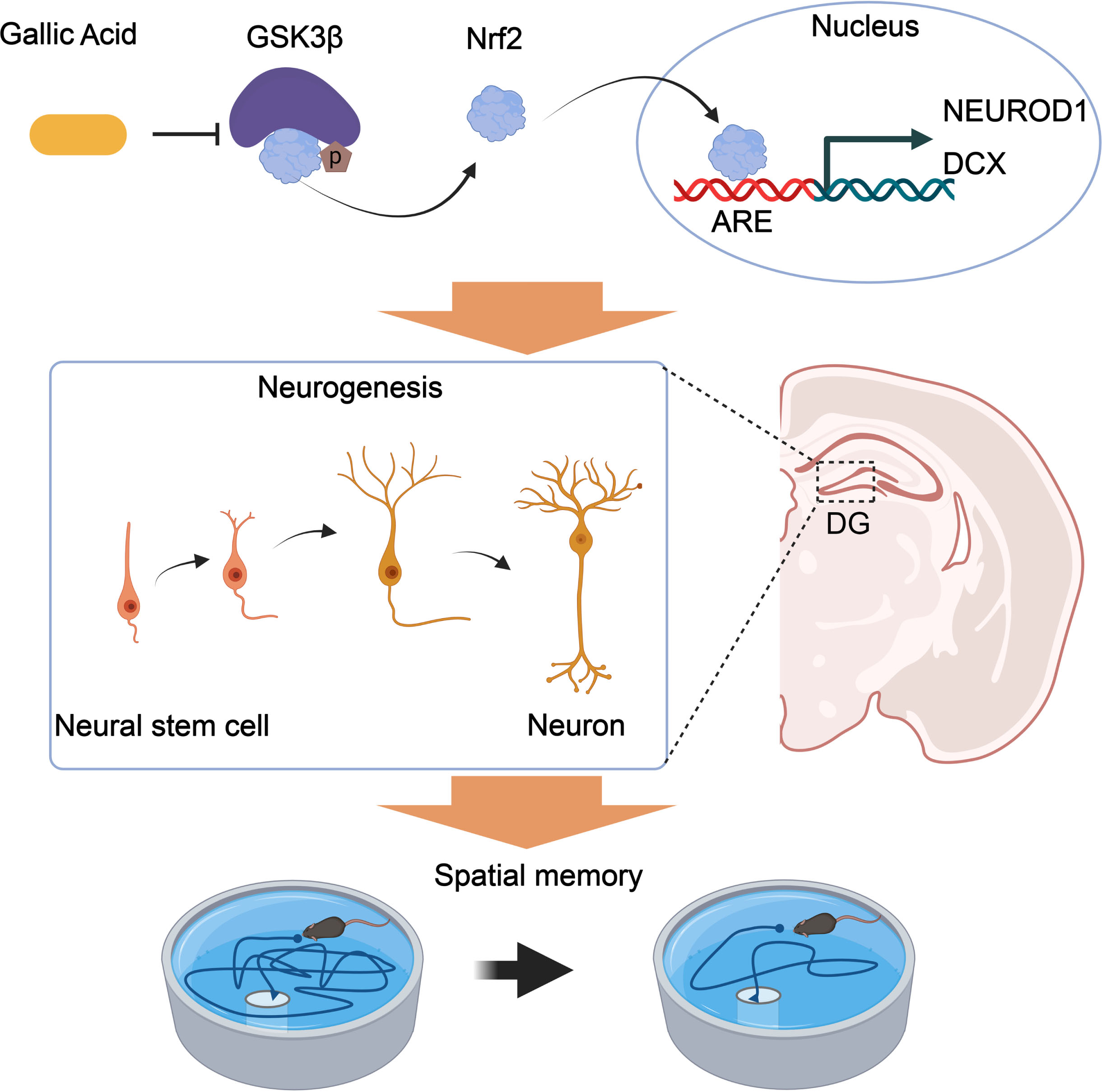
GA, a member of the hydrolyzable tannin family, has been studied in several animal models where it displays anti-anxiety, antidepressant, and anti-dementia effects [43–46]. By the enhancement of mitogen-activated protein kinase/extracellular-regulated kinase (MAPK/ERK) pathway, NSCs proliferation could be promoted by GA and differentiate into immature neurons [25]. AHN occurs in the SGZ of the hippocampus and contributes to various hippocampus-related work, i.e., learning and memory [47, 48]. Mature granular cells in the SGZ undergo four developmental stages. In stage one, they originate from NSCs and express nestin, Sox2, and glial fibrillary acidic protein. Stage two mainly comprises intermediate progenitor cells that express DCX. Stage three comprises neuroblasts that express NeuroD, DCX, and PSA-NCAM. Axonal and dendritic targeting (Stage four) is characterized by mature neurons that express NeuroN, and synaptic integration (stage five) is neuron expressed calbindin [49]. Consistent with these results, we found that GA promoted neurogenesis in the DG of hippocampus, as shown by increased levels of DCX+ and NeuroD+, two markers of new-born neurons in the DG and improved spatial memory in APP/PS1 mice. Furthermore, more mushroom spines, which are considered substrates of long-term memory due to their longevity and structural and functional characteristics [50, 51], appear after GA administration. Such spinogenesis represents the basis of the GA protective effects against Aβ-induced amnesia in APP/PS1 mice. Postsynaptic proteins, such as PSD95, Syn1, and synaptophysin, were upregulated after GA administration in the synapse, indicating the presence of more mature neurons in the brains of APP/PS1 mice.
In a previous literature, a cellular cascade that involved phosphoinositide 3-kinase /mammalian target of rapamycin/GSK3β/β-catenin signaling was verified as a key contributor to neurogenesis in the hippocampus of adult brain [52–54]. Aβ oligomers accumulated in NSCs impaired the neurogenesis of Tg2576 progenitors at a presymptomatic stage of AD [55]. We therefore examined tau’s phosphorylation level and Aβ concentration in APP/PS1 mice brains. The phosphorylation level of tau at two sites (pS396 and pT231) and the concentration of Aβ1 - 40/1 -42 were greatly enhanced in APP/PS1 mice and decreased after treatment with GA, which indicates GA owns a protective effect against tau and Aβ’s toxicity.
Apart from being as a kinase involved in tau phosphorylation and the production of Aβ [56, 57], GSK-3β has a role in keeping homeostasis of neural progenitor in the rodent brains during the early developmental period [58]. In our preceding scholarly publication, the impact of GSK-3 activation or inhibition on neurogenesis was examined [59, 60]. If deletion by genetic engineering, GSK-3β causes neural progenitors’ hyperproliferation; however, GSK-3β’s activation during prenatal neurogenesis reduces neural progenitors’ number and inhibits axon formation [61, 62]. In the present study, GSK-3β in APP/PS1 mouse brain was deactivated after the administration of GA, and there was a corresponding increase in β-catenin, which promoted neurogenesis. Further analysis by molecular docking revealed that GA could directly interact with several amino acids of GSK-3β. Moreover, the reactivation of GSK-3β by overexpression blocked its protective effect, indicating that GA exerted its pharmaceutical effect on neurogenesis via deactivation of GSK-3β. However, although we hypothesize that GA directly binds to GSK-3β via molecular docking, there is no direct experimental evidence that proves the direct interaction between GSK-3β and GA.
The transcription factor Nrf2 was identified to undergo posttranscriptional modification by an upstream protein, such as KEAP1 and p62, and bond to the ARE region of phase II detoxifying and antioxidant enzymes [31, 63]. Two serines of Nrf2 are phosphorylated by GSK-3β and targeted by the E3 ligase substrate adaptor β-TrCP, which marks the protein for ubiquitination and proteasomal degradation [64]. Nrf2 transcriptionally regulates PSD93, PSD95, SYN1 [31], BACE1, and BACE1 mRNA-stabilizing antisense RNA [65]. In this study, we observed that DCX and NEUROD1 were transcriptionally regulated by Nrf2, as evidenced by a dual luciferase reporter assay. Thus, we proposed that inhibition of GSK-3β by GA reversed the transcriptional activity of Nrf2 and enhanced the protein levels of DCX and NeuroD.
In conclusion, we report that the administration of GA promotes hippocampal neurogenesis to partly remediated behavioral deficits and reduced the abundance of hyperphosphorylated tau and deposited Aβ in an APP/PS1 mouse model. Mechanically, GA treatment exerted anti-neuroinflammation and balanced oxidative stress ability via binding to GSK-3β and activation of Nrf2. Our experimental evidence suggests GA is a promising therapeutic agent for delaying AD symptoms.
AUTHOR CONTRIBUTIONS
Yu Ding (Validation; Investigation); Jinrong He (Validation; Investigation); Fanli Kong (Methodology; Formal analysis; Resources; Software; Writing - Original Draft); Dongsheng Sun (Methodology; Formal analysis; Resources); Weiqun Chen (Visualization; Supervision); Bo Luo (Visualization; Supervision); Jia Wu (Visualization; Supervision); Shaoying Zhang (Visualization; Supervision); Peiyan Zhan (Conceptualization; Funding acquisition); Caixia Peng (Conceptualization; Funding acquisition; Project administration; Writing – original draft; Writing – review & editing).
ACKNOWLEDGMENTS
We would like to thank Editage (www.editage.cn) for English language editing.
FUNDING
This work was supported, in whole or in part, by grants from the National Natural Science Foundation of China (grant number 81301084), the Medical Scientific Research Foundation of Wuhan Municipal Health Commission (No. WZ22C47, WX21A08, WX20Q35).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
[1] | Henstridge CM , Hyman BT , Spires-Jones TL ((2019) ) Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat Rev Neurosci 20: , 94–108. |
[2] | Jurado-Arjona J , Llorens-Martín M , Ávila J , Hernández F ((2016) ) GSK3β overexpression in dentate gyrus neural precursor cells expands the progenitor pool and enhances memory skills. J Biol Chem 291: , 8199–8213. |
[3] | Hampel H , Mesulam MM , Cuello AC , Farlow MR , Giacobini E , Grossberg GT , Khachaturian AS , Vergallo A , Cavedo E , Snyder PJ , Khachaturian ZS ((2018) ) The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141: , 1917–1933. |
[4] | Isaacson RS , Ganzer CA , Hristov H , Hackett K , Caesar E , Cohen R , Kachko R , Meléndez-Cabrero J , Rahman A , Scheyer O , Hwang MJ , Berkowitz C , Hendrix S , Mureb M , Schelke MW , Mosconi L , Seifan A , Krikorian R ((2018) ) The clinical practice of risk reduction for Alzheimer’s disease: A precision medicine approach. Alzheimers Dement 14: , 1663–1673. |
[5] | Wright AL , Zinn R , Hohensinn B , Konen LM , Beynon SB , Tan RP , Clark IA , Abdipranoto A , Vissel B ((2013) ) Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS One 8: , e59586. |
[6] | Cope EC , Gould E ((2019) ) Adult neurogenesis, glia, and the extracellular matrix. Cell Stem Cell 24: , 690–705. |
[7] | Gould E , Beylin A , Tanapat P , Reeves A , Shors TJ ((1999) ) Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci 2: , 260–265. |
[8] | Wiskott L , Rasch MJ , Kempermann G ((2006) ) A functional hypothesis for adult hippocampal neurogenesis: Avoidance of catastrophic interference in the dentate gyrus. Hippocampus 16: , 329–343. |
[9] | Duncan K , Doll BB , Daw ND , Shohamy D ((2018) ) More than the sum of its parts: A role for the hippocampus in configural reinforcement learning. Neuron 98: , 645–657.e646. |
[10] | Kozareva DA , Cryan JF , Nolan YM ((2019) ) Born this way: Hippocampal neurogenesis across the lifespan. Aging Cell 18: , e13007. |
[11] | Bonaguidi MA , Wheeler MA , Shapiro JS , Stadel RP , Sun GJ , Ming GL , Song H ((2011) ) In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 145: , 1142–1155. |
[12] | Anacker C , Luna VM , Stevens GS , Millette A , Shores R , Jimenez JC , Chen B , Hen R ((2018) ) Hippocampal neurogenesis confers stress resilience by inhibiting the ventral dentate gyrus. Nature 559: , 98–102. |
[13] | Mu Y , Gage FH ((2011) ) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6: , 85. |
[14] | Leiter O , Zhuo Z , Rust R , Wasielewska JM , Gronnert L , Kowal S , Overall RW , Adusumilli VS , Blackmore DG , Southon A , Ganio K , McDevitt CA , Rund N , Brici D , Mudiyan IA , Sykes AM , Runker AE , Zocher S , Ayton S , Bush AI , Bartlett PF , Hou ST , Kempermann G , Walker TL ((2022) ) Selenium mediates exercise-induced adult neurogenesis and reverses learning deficits induced by hippocampal injury and aging. Cell Metab 34: , 408–423.e8. |
[15] | Berger T , Lee HA , Young AH , Aarsland D , Thuret S ((2020) ) Adult hippocampal neurogenesis in major depressive disorder and Alzheimer’s disease. Trends Mol Med 26: , 803–818. |
[16] | Moreno-Jimenez EP , Flor-Garcia M , Terreros-Roncal J , Rabano A , Cafini F , Pallas-Bazarra N , Avila J , Llorens-Martin M ((2019) ) Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med 25: , 554-+. |
[17] | Wang J , Hu J , Chen X , Lei X , Feng H , Wan F , Tan L ((2021) ) Traditional Chinese Medicine monomers: Novel strategy for endogenous neural stem cells activation after stroke. Front Cell Neurosci 15: , 628115. |
[18] | Chen X , Chen H , He Y , Fu S , Liu H , Wang Q , Shen J ((2020) ) Proteomics-guided study on Buyang Huanwu decoction for its neuroprotective and neurogenic mechanisms for transient ischemic stroke: Involvements of EGFR/PI3K/Akt/Bad/14-3-3 and Jak2/Stat3/Cyclin D1 signaling cascades. Mol Neurobiol 57: , 4305–4321. |
[19] | Xu W , Jiang Y , Wang N , Bai H , Xu S , Xia T , Xin H ((2022) ) Traditional Chinese Medicine as a promising strategy for the treatment of Alzheimer’s disease complicated with osteoporosis. Front Pharmacol 13: , 842101. |
[20] | Yang RA , Li ZJ , Zou YT , Yang JJ , Li L , Xu XM , Schmalzing G , Nie H , Li GL , Liu SM , Liang SD , Xu CS ((2021) ) Gallic acid alleviates neuropathic pain behaviors in rats by inhibiting P2X7 receptor-mediated NF-kappa B/STAT3 signaling pathway. Front Pharmacol 12: , 680139. |
[21] | Javed F , Jabeen Q , Aslam N , Awan AM ((2020) ) Pharmacological evaluation of analgesic, anti-inflammatory and antipyretic activities of ethanolic extract of Indigofera argentea Burm. J Ethnopharmacol 259: , f112966. |
[22] | Chandrasekhar Y , Phani Kumar G , Ramya EM , Anilakumar KR ((2018) ) Gallic acid protects 6-OHDA induced neurotoxicity by attenuating oxidative stress in human dopaminergic cell line. Neurochem Res 43: , 1150–1160. |
[23] | Ferruzzi MG , Lobo JK , Janle EM , Cooper B , Simon JE , Wu Q-L , Welch C , Ho L , Weaver C , Pasinetti GM ((2009) ) Bioavailability of gallic acid and catechins from grape seed polyphenol extract is improved by repeated dosing in rats: Implications for treatment in Alzheimer’s disease. J Alzheimers Dis 18: , 113–124. |
[24] | Mori T , Koyama N , Yokoo T , Segawa T , Maeda M , Sawmiller D , Tan J , Town T ((2020) ) Gallic acid is a dual α/β-secretase modulator that reverses cognitive impairment and remediates pathology in Alzheimer mice. J Biol Chem 295: , 16251–16266. |
[25] | Jiang JX , Hai JT , Liu WY , Luo Y , Chen KQ , Xin YR , Pan JP , Hu Y , Gao Q , Xiao F , Luo HM ((2021) ) Gallic acid induces neural stem cell differentiation into neurons and proliferation through the MAPK/ERK pathway. J Agr Food Chem 69: , 12456–12464. |
[26] | Gupta V , Mahata T , Roy R , Gharai PK , Jana A , Garg S , Ghosh S ((2022) ) Discovery of imidazole-based GSK-3β inhibitors for transdifferentiation of human mesenchymal stem cells to neurons: A potential single-molecule neurotherapeutic foresight. Front Mol Neurosci 15: , 1002419. |
[27] | Singh S , Mishra A , Bharti S , Tiwari V , Singh J , Parul , Shukla S ((2018) ) Glycogen synthase kinase-3beta regulates equilibrium between neurogenesis and gliogenesis in rat model of Parkinson’s disease: A crosstalk with Wnt and Notch signaling. Mol Neurobiol 55: , 6500–6517. |
[28] | Duan J , Cui J , Yang Z , Guo C , Cao J , Xi M , Weng Y , Yin Y , Wang Y , Wei G , Qiao B , Wen A ((2019) ) Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3β/Nrf2 signaling. J Neuroinflammation 16: , 24. |
[29] | Guo X-H , Pang L , Gao C-Y , Meng F-L , Jin W ((2023) ) Lyoniresinol attenuates cerebral ischemic stroke injury in MCAO rat based on oxidative stress suppression via regulation of Akt/GSK-3β/Nrf2 signaling. Biomed Pharmacother 167: , 115543. |
[30] | Fão L , Mota SI , Rego AC ((2019) ) Shaping the Nrf2-ARE-related pathways in Alzheimer’s and Parkinson’s diseases. Ageing Res Rev 54: , 100942. |
[31] | Xie J-Z , Zhang Y , Li S-H , Wei H , Yu H-L , Zhou Q-Z , Wei L-Y , Ke D , Wang Q , Yang Y , Wang J-Z ((2022) ) P301S-hTau acetylates KEAP1 to trigger synaptic toxicity via inhibiting NRF2/ARE pathway: A novel mechanism underlying hTau-induced synaptic toxicities. Clin Transl Med 12: , e1003. |
[32] | Lin S , Qin H-Z , Li Z-Y , Zhu H , Long L , Xu L-B ((2022) ) Gallic acid suppresses the progression of triple-negative breast cancer HCC1806 cells via modulating PI3K/AKT/EGFR and MAPK signaling pathways. Front Pharmacol 13: , 1049117. |
[33] | Li X-G , Hong X-Y , Wang Y-L , Zhang S-J , Zhang J-F , Li X-C , Liu Y-C , Sun D-S , Feng Q , Ye J-W , Gao Y , Ke D , Wang Q , Li H-L , Ye K , Liu G-P , Wang J-Z ((2019) ) Tau accumulation triggers STAT1-dependent memory deficits by suppressing NMDA receptor expression. EMBO Rep 20: , e47202. |
[34] | Forli S , Huey R , Pique ME , Sanner MF , Goodsell DS , Olson AJ ((2016) ) Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 11: , 905–919. |
[35] | Largo-Barrientos P , Apostolo N , Creemers E , Callaerts-Vegh Z , Swerts J , Davies C , McInnes J , Wierda K , De Strooper B , Spires-Jones T , de Wit J , Uytterhoeven V , Verstreken P ((2021) ) Lowering Synaptogyrin-3 expression rescues Tau-induced memory defects and synaptic loss in the presence of microglial activation. Neuron 109: , 767–777 e765. |
[36] | Eom TY , Jope RS ((2009) ) Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol Psychiatry 66: , 494–502. |
[37] | Banach E , Jaworski T , Urban-Ciecko J ((2022) ) Early synaptic deficits in GSK-3beta overexpressing mice. Neurosci Lett 784: , 136744. |
[38] | Sayas CL , Avila J ((2021) ) GSK-3 and tau: A key duet in Alzheimer’s disease. Cells 10: , 721. |
[39] | Li Y , Xie Z , Gao T , Li L , Chen Y , Xiao D , Liu W , Zou B , Lu B , Tian X , Han B , Guo Y , Zhang S , Lin L , Wang M , Li P , Liao Q ((2019) ) A holistic view of gallic acid-induced attenuation in colitis based on microbiome-metabolomics analysis. Food Funct 10: , 4046–4061. |
[40] | Sohrabi F , Dianat M , Badavi M , Radan M , Mard SA ((2021) ) Gallic acid suppresses inflammation and oxidative stress through modulating Nrf2-HO-1-NF-kappaB signaling pathways in elastase-induced emphysema in rats. Environ Sci Pollut Res Int 28: , 56822–56834. |
[41] | Heneka MT , Carson MJ , El Khoury J , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[42] | Zhang J , Wu N , Wang S , Yao Z , Xiao F , Lu J , Chen B ((2021) ) Neuronal loss and microgliosis are restricted to the core of Abeta deposits in mouse models of Alzheimer’s disease. Aging Cell 20: , e13380. |
[43] | Chhillar R , Dhingra D ((2013) ) Antidepressant-like activity of gallic acid in mice subjected to unpredictable chronic mild stress. Fundam Clin Pharmacol 27: , 409–418. |
[44] | Dhingra D , Chhillar R , Gupta A ((2012) ) Antianxiety-like activity of gallic acid in unstressed and stressed mice: Possible involvement of nitriergic system. Neurochem Res 37: , 487–494. |
[45] | Huang HL , Lin CC , Jeng KC , Yao PW , Chuang LT , Kuo SL , Hou CW ((2012) ) Fresh green tea and gallic acid ameliorate oxidative stress in kainic acid-induced status epilepticus. J Agric Food Chem 60: , 2328–2336. |
[46] | Korani MS , Farbood Y , Sarkaki A , Fathi Moghaddam H , Taghi Mansouri M ((2014) ) Protective effects of gallic acid against chronic cerebral hypoperfusion-induced cognitive deficit and brain oxidative damage in rats. Eur J Pharmacol 733: , 62–67. |
[47] | Jurkowski MP , Bettio L , E KW , Patten A , Yau SY , Gil-Mohapel J ((2020) ) Beyond the hippocampus and the SVZ: Adult neurogenesis throughout the brain. Front Cell Neurosci 14: , 576444. |
[48] | Fares J , Bou Diab Z , Nabha S , Fares Y ((2019) ) Neurogenesis in the adult hippocampus: History, regulation, and prospective roles. Int J Neurosci 129: , 598–611. |
[49] | Kumar A , Pareek V , Faiq MA , Ghosh SK , Kumari C ((2019) ) Adult neurogenesis in humans: A review of basic concepts, history, current research, and clinical implications. Innov Clin Neurosci 16: , 30–37. |
[50] | Ashby MC , Maier SR , Nishimune A , Henley JM ((2006) ) Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci 26: , 7046–7055. |
[51] | Choi JH , Sim SE , Kim JI , Choi DI , Oh J , Ye S , Lee J , Kim T , Ko HG , Lim CS , Kaang BK ((2018) ) Interregional synaptic maps among engram cells underlie memory formation. Science 360: , 430–435. |
[52] | Dioli C , Patricio P , Trindade R , Pinto LG , Silva JM , Morais M , Ferreiro E , Borges S , Mateus-Pinheiro A , Rodrigues AJ , Sousa N , Bessa JM , Pinto L ((2017) ) Tau-dependent suppression of adult neurogenesis in the stressed hippocampus. Eur Neuropsychopharm 27: , S546–S546. |
[53] | Wang Q , Huang X , Su Y , Yin G , Wang S , Yu B , Li H , Qi J , Chen H , Zeng W , Zhang K , Verkhratsky A , Niu J , Yi C ((2022) ) Activation of Wnt/beta-catenin pathway mitigates blood-brain barrier dysfunction in Alzheimer’s disease. Brain 145: , 4474–4488. |
[54] | Sun X , Peng X , Cao Y , Zhou Y , Sun Y ((2020) ) ADNP promotes neural differentiation by modulating Wnt/β-catenin signaling. Nat Commun 11: , 2984. |
[55] | Scopa C , Marrocco F , Latina V , Ruggeri F , Corvaglia V , La Regina F , Ammassari-Teule M , Middei S , Amadoro G , Meli G , Scardigli R , Cattaneo A ((2020) ) Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular A beta oligomers. Cell Death Differ 27: , 2035–2035. |
[56] | Wang X , Bian Y , Wong CTT , Lu JH , Lee SM ((2022) ) TRPV1 modulator ameliorates Alzheimer-like amyloid-beta neuropathology via Akt/Gsk3beta-mediated Nrf2 activation in the Neuro-2a/APP cell model. Oxid Med Cell Longev 2022: , 1544244. |
[57] | Zhou Q , Li S , Li M , Ke D , Wang Q , Yang Y , Liu GP , Wang XC , Liu E , Wang JZ ((2022) ) Human tau accumulation promotes glycogen synthase kinase-3beta acetylation and thus upregulates the kinase: A vicious cycle in Alzheimer neurodegeneration. EBioMedicine 78: , 103970. |
[58] | Pandey MK , DeGrado TR ((2016) ) Glycogen synthase kinase-3 (GSK-3)-targeted therapy and imaging. Theranostics 6: , 571–593. |
[59] | Hong X-P , Peng C-X , Wei W , Tian Q , Liu Y-H , Yao X-Q , Zhang Y , Cao F-Y , Wang Q , Wang J-Z ((2010) ) Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus 20: , 1339–1349. |
[60] | Hong X-P , Peng C-X , Wei W , Tian Q , Liu Y-H , Cao F-Y , Wang Q , Wang J-Z ((2011) ) Relationship of adult neurogenesis with tau phosphorylation and GSK-3β activity in subventricular zone. Neurochem Res 36: , 288–296. |
[61] | Dohare P , Kidwai A , Kaur J , Singla P , Krishna S , Klebe D , Zhang X , Hevner R , Ballabh P ((2019) ) GSK3beta inhibition restores impaired neurogenesis in preterm neonates with intraventricular hemorrhage. Cereb Cortex 29: , 3482–3495. |
[62] | Kim WY , Wang X , Wu Y , Doble BW , Patel S , Woodgett JR , Snider WD ((2009) ) GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci 12: , 1390–1397. |
[63] | Li L , Dong H , Song E , Xu X , Liu L , Song Y ((2014) ) Nrf2/ARE pathway activation, HO-1 and NQO1 induction by polychlorinated biphenyl quinone is associated with reactive oxygen species and PI3K/AKT signaling. Chem Biol Interact 209: , 56–67. |
[64] | Jiang Y , Bao H , Ge Y , Tang W , Cheng D , Luo K , Gong G , Gong R ((2015) ) Therapeutic targeting of GSK3β enhances the Nrf2 antioxidant response and confers hepatic cytoprotection in hepatitis C. Gut 64: , 168–179. |
[65] | Bahn G , Park J-S , Yun UJ , Lee YJ , Choi Y , Park JS , Baek SH , Choi BY , Cho YS , Kim HK , Han J , Sul JH , Baik S-H , Lim J , Wakabayashi N , Bae SH , Han J-W , Arumugam TV , Mattson MP , Jo D-G ((2019) ) NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc Natl Acad Sci U S A 116: , 12516–12523. |


