
The Causal Associations between Adipokines and Alzheimer’s Disease: A Two-Sample Mendelian Randomization Study
Abstract
Background:
Observational studies have indicated the association of alteration of adipokines with Alzheimer’s disease (AD). However, it remains unclear whether the associations are causal.
Objective:
To determine the causal associations between adipokines and AD.
Methods:
A Mendelian randomization (MR) method was applied to investigate the causal relationships of adipokines, including adiponectin and resistin, with risk of AD. Genetic proxies from genome-wide association studies (GWAS) of adiponectin and resistin were selected as instrumental variables. GWAS summary statistics for AD were extracted as outcome.
Results:
In this study, we found evidence of the causal effects of adiponectin on AD (OR: 0.850, 95% CI: 0.731-0.990, p = 0.037). However, no relationship between resistin and AD (OR: 0.936, 95% CI: 0.851-1.029, p = 0.171) was detected. In the reverse causation analysis, null associations of AD were found for adiponectin and resistin (all p > 0.05).
Conclusions:
This study provides evidence of causality between adiponectin and risk of AD. However, no genetic susceptibility of resistin was discovered for AD.
INTRODUCTION
Alzheimer’s disease (AD) is a typical neurodegenerative disease and the most common form of dementia, which accounts for nearly 80% of all kinds of dementia [1]. The earliest phase of AD starts with the accumulation of Aβ and the induction of the spread of tauopathy [2]. Several common mechanisms are responsible for AD, including imbalance of lipid metabolism, oxidative stress and inflammatory responses induced by protein accumulation [3–5]. Moreover, these pathways are also believed to be deeply involved in obesity, by participating in metabolic imbalance [6].
Obesity is a major health problem across the world. It is closely related to white matter intensity and disruption and progressive brain atrophy [7]. Some studies have demonstrated that anti-obesity therapy provides benefits for patients at risk of AD [8]. Therefore, it is important to demonstrate the potential link between obesity and AD. Studies have indicated that individuals with obesity are likely to present a chronic inflammatory status [9], with the activation of immune cells surrounding adipose tissues and the production of adipokines [10, 11]. Adipokines include a series of components mainly produced by adipose tissues. The dysregulation of adipokines has been consistently accompanied with the adverse outcomes in obesity, leading to a persistent inflammation [12]. Recent studies have indicated that the operation of adipokines could be affected during neurodegeneration and feedback to contribute to the neurodegenerative process. For instance, adiponectin interacts with blood-brain barrier [13], and regulate neurogenesis and synaptic function in hippocampus, cortex and hypothalamus [14], through its respective binding to adiponectin receptor 1 and 2 [14]. Resistin is another widely studied adipokine, associated with the integrity of brain structure. It is identified in the hypothalamus and cortex in mouse. The neuroprotective role of resistin was stated in AD mouse model by Liu et al. [15]. Therefore, it is believed that adipokines offer a window to explain the relationship between obesity and AD. However, current reports only provide observational findings concerning their associations. The causality between adipokines and AD have not been established.
Mendelian randomization (MR) approach provides a tool to estimate the causal effects of certain exposure on an outcome [16]. It uses genetic variants as instrumental variables (IVs) of exposure, and pools the effects of variants for effect estimation [16]. The approach diminishes the effects of potential confounders in observational level and overcomes the influences of reverse causation. We here used the two-sample MR analysis to determine the causal effects of adipokines on risk of AD.
MATERIALS AND METHODS
Study design
A two-sample MR analysis was done to test the causality between adipokines (adiponectin and resistin) and risk of AD, using summary data from GWAS consortia studies. Data sources were shown in Table 1. To detect the potential reverse causality, we performed an MR analysis to test the effects of AD on circulating adipokines. All participants have given informed consent in the respective center. An overview of the study assumptions was displayed in Fig. 1.
Fig. 1
The key assumptions of the MR study. First, the selected SNPs (IVs) should be significantly associated with the exposure (adipokine); second, the included SNPs for the exposure (adipokine) were not associated with any confounder; third, the selected SNPs should only affect the outcome (risk of AD) though the exposure (adipokine). AD, Alzheimer’s disease; IV, instrumental variable; MR, Mendelian randomization; SNP, single-nucleotide polymorphism.
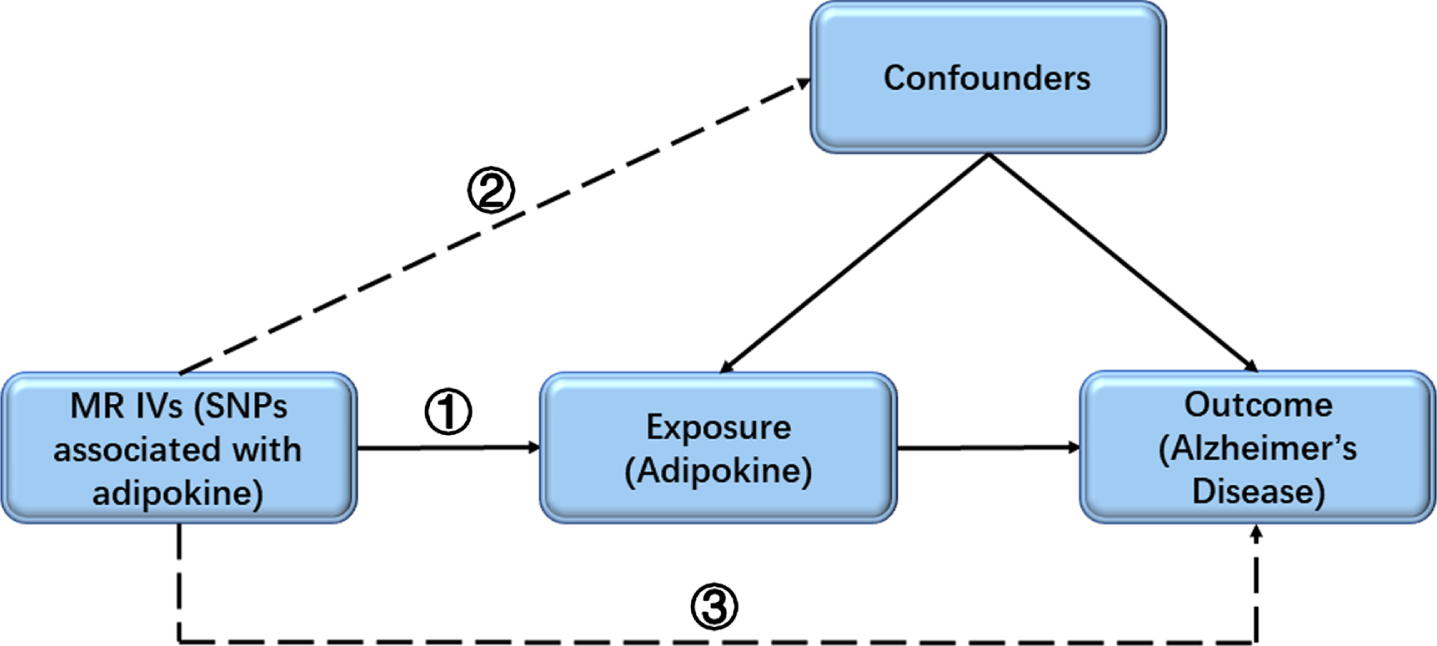
Table 1
Characteristics of the data sources in the study
| Variables | Data source | Authors | Sample size | Ancestry |
| adiponectin | ADIPOGen | Dastani et al. [17] | 39,883 | European |
| resistin | INTERVAL | Sun et al. [18] | 3,301 | European |
| AD | IGAP | Kunkle et al. [19] | 63,926 | European |
AD, Alzheimer’s disease; IGAP, International Genomics of Alzheimer’s Project.
Selection of instrumental variables
Genome-wide significant SNPs strongly (p < 5×10-8) associated with plasma adipokines, including adiponectin were selected from the study by Dastani et al. [17]. In that study, the authors performed a meta-analysis of GWAS in 39,883 individuals of European ancestry to identify genes associated with metabolic disease. SNPs, that were associated (p < 5×10-8) with plasma resistin, were screened in the GWAS dataset from the INTERVAL study [18]. In the study, Sun and colleagues measured 3,622 proteins in 3,301 healthy European blood donors. In addition, variants were further clumped for independence using the TwoSampleMR tool. A threashold of R2≤0.001 and a window of 10,000 kb were used to screen for linkage disequilibrium (LD). F statistics above 10 was required to limit the bias caused by weak instrumental variables. R2 of each SNP was defined by calculating with the formula: 2*EAF*(1-EAF)*β2, where β is the effect estimate of exposure. F statistics were calculated by the formula: R2*(N-2)/(1-R2), where N means the sample size. Details of the SNPs associated with adiponectin and resistin were shown in Supplementary Table 1.
Outcome sources
GWAS statistics for AD were obtained from a dataset released by International Genomics of Alzheimer’s Project (IGAP) (21,982 cases and 41,944 controls) [19], which consists of data from the Alzheimer Disease Genetics Consortium (ADGC), Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE), the European Alzheimer’s Disease Initiative (EADI), and Genetic and Environmental Risk in AD/Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s Disease Consortium (GERAD/PERADES). The SNPs associated with AD were shown in Supplementary Table 2.
Table 2
Causation analysis of the effects of adiponectin, resistin on AD
| Exposure | Outcome | MR method | No. of SNPs | Beta | SE | OR | 95% CI | p |
| adiponectin | AD | MR Egger | 11 | –0.160 | 0.113 | 0.852 | 0.683–1.064 | 0.191 |
| Weighted median | 11 | –0.058 | 0.100 | 0.944 | 0.776–1.148 | 0.561 | ||
| Inverse variance weighted | 11 | –0.162 | 0.077 | 0.850 | 0.731–0.990 | 0.037* | ||
| Simple mode | 11 | –0.135 | 0.158 | 0.873 | 0.641–1.189 | 0.409 | ||
| Weighted mode | 11 | –0.088 | 0.111 | 0.915 | 0.736–1.139 | 0.445 | ||
| resistin | AD | MR Egger | 4 | –0.036 | 0.100 | 0.965 | 0.793–1.174 | 0.754 |
| Weighted median | 4 | –0.056 | 0.056 | 0.946 | 0.848–1.056 | 0.321 | ||
| Inverse variance weighted | 4 | –0.066 | 0.049 | 0.936 | 0.851–1.029 | 0.171 | ||
| Simple mode | 4 | –0.039 | 0.082 | 0.962 | 0.819–1.129 | 0.666 | ||
| Weighted mode | 4 | –0.043 | 0.079 | 0.958 | 0.821–1.119 | 0.626 |
AD, Alzheimer’s disease; CI, confidence interval; MR, Mendelian randomization; OR, odds ratio; SE, standard error; SNP, single-nucleotide polymorphism.
Statistical analyses
To detect possible SNPs that were related to AD, SNPs associated with adiponectin and resistin were searched in the Phenoscanner (http://www.phenoscanner.medschl.cam.ac.uk/) [20]. The online web tool mRnd [21] (http://shiny.cnsgenomics.com/mRnd/) was used to calculate the statistical power of the MR analysis. Inverse variance weighted (IVW), MR-Egger, weighted median, simple mode and weighted mode methods were used in the study, with effect estimates (β) and 95% confidence interval (CI). IVW estimates were considered as the primary MR outcomes, to provide the highest precision of combined effects of adipokines with the assumption of balanced pleiotrophy. Weighted median method gave a consistent estimated effect, even when half of the results were generated from the analysis of invalid or weak instruments. MR-Egger regression analysis was performed to explore the presence of horizontal pleiotrophy by evaluating the intercept. Simple mode and weighted mode methods were also done to assess the robustness of the results. An MR-PRESSO analysis was applied to detect and adjust for horizontal pleiotrophy through removing outliers. A leave-one-out analysis and Cochrane Q statistic were performed to check for any pleiotrophy affected by a single SNP. Statistical analyses were done using the TwoSampleMR and MRPRESSO package in R (version 4.1.3). Statistical significance was considered when p value was less than 0.05.
RESULTS
Genetic instruments for adiponectin and resistin
A total of 14 and 4 variants were initially identified as IVs for adiponectin and resistin, respectively. The F statistics for the SNPs were all above 10, except for rs1597466 of adiponectin (F statistics = 9.354). For adiponectin, 11 SNPs were employed in the main analysis of AD after further excluding 2 SNPs (rs2980879 and rs7964945) for being palindromic with intermediate allele frequencies. For resistin, the F statistics for the SNPs were above 10, indicating that the weak IV bias did not exist. Therefore, all SNPs associated with resistin were included for MR analysis. Phenoscanner was searched, and no SNPs were excluded. Details of the selected SNPs were shown in Supplementary Table 1.
Causal effects of adiponectin on AD
The analysis results of the relationship between adiponectin and AD were displayed in Table 2. IVW mode indicated a detrimental effect of the decreased level of circulating adiponectin on risk of AD (OR: 0.850; 95% CI: 0.731-0.990, p = 0.037). However, such trend was not confirmed in the estimates in MR-Egger, weighted median, simple mode and weighted mode methods. The statistical power to detect the causal effects of adiponectin on AD was 100%.
Cochrane’s Q test and MR-PRESSO global test did not find obvious pleiotrophic effects (Table 3). The MR-Egger intercept analysis did not find significances (p = 0.982), indicating no evidence of potential pleiotrophy (Table 3).
Table 3
Results of the Cochrane’s Q test, MR-Egger intercept test and MRPRESSO Global test in the MR study
| Cochrane’s Q test | MR-Egger intercept test | MR-PRESSO Global test | |||
| Q-value | P-Q test | intercept | P-intercept | P-Global | |
| Adiponectin versus AD | |||||
| 8.502 | 0.580 | –0.0002 | 0.982 | 0.376 | |
| Resistin versus AD | |||||
| 2.010 | 0.570 | –0.008 | 0.761 | 0.606 | |
AD, Alzheimer’s disease; MR, Mendelian randomization. *p < 0.05.
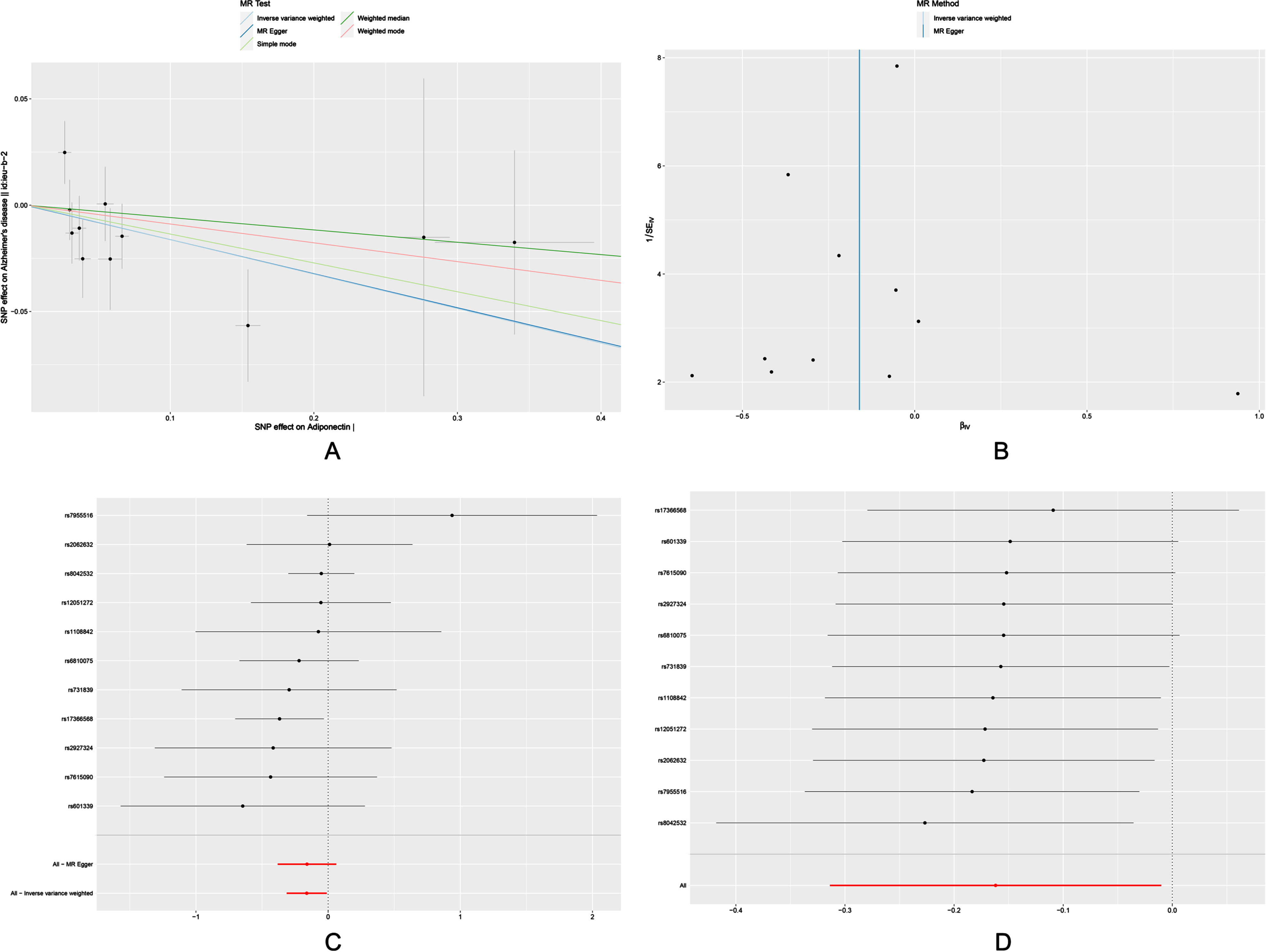
Scatter plots, funnels plots, forest plots and leave-one-out analysis of the MR analysis were shown in Fig. 2. rs17366568 was discovered to be a possible driving factor for the significant association of adiponectin with risk of AD. Therefore, we re-analyzed by excluding rs17366568 (Supplementary Table 3). It indicated no significant associations between adiponectin and AD in the IVW model.
Fig. 2
Scatter plot (A), funnel plot (B), forest plot (C), and leave-one-out sensitivity analysis (D) of the associations of adiponectin with risk of AD. AD, Alzheimer’s disease.
Causal effects of resistin on AD
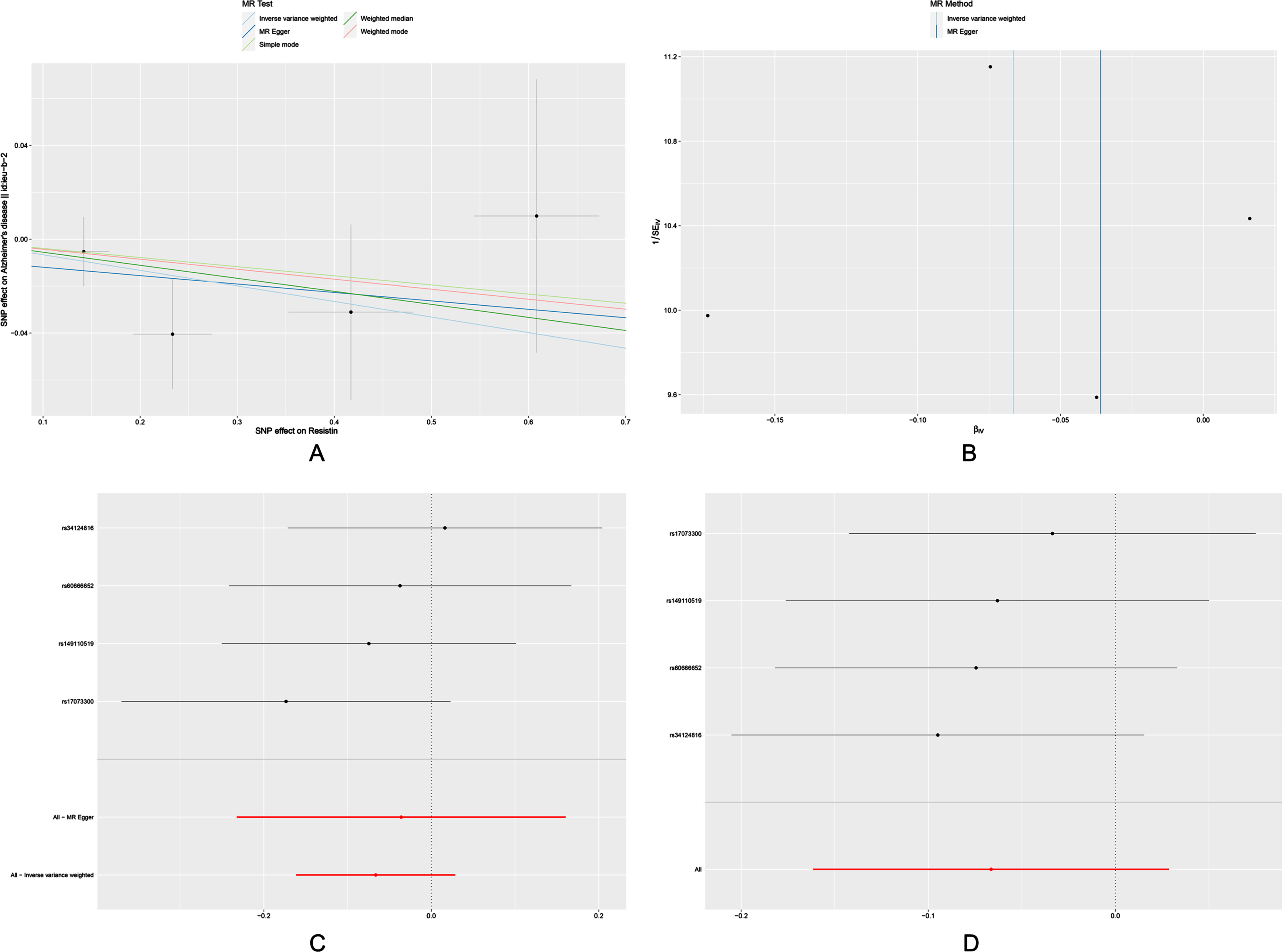
The OR for the associations of resistin with AD was 0.936 (95% CI: 0.851-1.029, p = 0.171) (Table 2). Null associations were also found in the MR-Egger, weighted median, simple mode and weighted mode methods. Moreover, we did not find significant results in Cochrane’s Q test and MR-PRESSO global test (Table 3). And the MR-Egger intercept test did not reveal pleiotrophy. Power analysis using the mRnd yielded 100% power to detect the effects of resistin on AD.
There was no significant change in the estimates of risk of diseases by genetically determined resistin level in the leave-one-out analysis, demonstrating that no specific SNP would change the results. Scatter plots, forest plots, funnels plots and leave-one-out sensitivity analysis were shown in Fig. 3.
Fig. 3
Scatter plot (A), funnel plot (B), forest plot (C), and leave-one-out sensitivity analysis (D) of the associations of resistin with risk of AD. AD, Alzheimer’s disease.
Reverse causation between AD and adiponectin, resistin
All models in reverse analysis did not suggest the genetic prediction of the levels of adiponectin and resistin by AD (Supplementary Table 4). Moreover, no horizontal pleiotrophy and heterogeneity were observed using MR-Egger intercept test, Cochrane Q test and MR-PRESSO global test (Supplementary Table 5). Sensitivity analysis using the leave-one-out approach confirmed that the inverse causation was not substantially driven by any individual SNP. In addition, scatter plots, funnels plots, forest plots and leave one out sensitivity analysis of the reverse causation analysis were shown in Supplementary Figure 1 and 2.
DISCUSSION
This MR study reported causal associations between genetically determined adiponectin and AD. Besides, the genetic susceptibility of resistin was not responsible for the development of AD.
It is believed that there is a connection between being overweight and AD [22]. Unwanted weight loss usually occurs among individuals with AD before the presence of clinical symptoms, even with unchanged eating patterns [23]. These suggest the alteration of adipose tissue metabolism along neurodegeneration [24]. Studies have indicated that the metabolic status is regulated by endocrine, paracrine and autocrine “adipokines” [25]. The agents can interact with blood brain barrier and participate in CNS homeostasis through their control of energy metabolism and immune responses [13]. Therefore, adipokines seem to be an intermediate player between obesity and AD.
Adiponectin is secreted by adipose tissues and participates in multiple cellular functions, including insulin responses, antioxidation and suppression of inflammation [14]. Accumulating evidence has suggested the presence of hypoadiponectinemia in metabolic disorders. Further, plasma adiponectin concentration appears to be inversely associated with the severity of adiposity and type 2 diabetes mellitus [26, 27]. Moreover, low level of adiponectin is found in AD dementia [28], which represents metabolic dysfunction in brain [29]. The inverse association between adiponectin and AD accords with our findings using IVW model that a descending adiponectin was associated with a higher risk of AD. Our findings indicated a neuroprotective potential of adiponectin against Alzheimer’s pathology. Interestingly, our findings were inconsistent with a previous MR study [30] revealing no causal effects of adiponectin on AD, using the same GWAS datasets with our study. In that study, all 14 SNPs significantly associated with adiponectin were included. However, we excluded 3 SNPs rs1597466, rs2980879 and rs7964945 for their unsatisfactory performances. Therefore, a relatively strict analysis process ensured the reliability of results. This may help find the potential role of promising markers in disorders.
Some studies have referred to the effects of adiponectin against amyloid pathology. A study used the SH-SY5YswAPP cell line to mimic amyloid burden in neuronal cells. And it found that adiponectin could improve cellular survival [31]. Mouse with adiponectin deficiency showed impaired memory and learning abilities, as well as increased Aβ depositions in brain, along with the over-production of pro-inflammatory cytokines [32]. Therefore, we proposed that adiponectin functioned against AD through its suppression of amyloid pathology and related inflammatory responses.
However, some studies have provided varied findings concerning adiponectin. Increase [33], or no change [34, 35] of adiponectin level has been found among AD individuals. Some factors seem to be responsible for the inconsistence. It is believed that increased weight is associated with decreased level of adiponectin [36]. The weight loss commonly seen in AD patients may induce the upregulation of adiponectin. However, the findings from the Framing Heart Study indicated elevated serum adiponectin was still associated with increased risk of AD in women, when adjusting for age, weight change and BMI [37]. Most studies evaluating adiponectin in AD patients did not take treatment details into consideration. A previous study reported increased adiponectin following donepezil application [38]. These may also explain the null association between adiponectin and AD without rs17366568 taking into analysis, for the sake that the findings may be interfered by underlying agents. Further exploration of the effects of adiponectin in AD should be done in the future.
Resistin is another adipokine protein derived from adipose tissue. It is involved in the production of pro-inflammatory cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) [12, 39]. It plays an important role in metabolic, inflammatory, and autoimmune conditions [39, 40]. The effects of resistin on AD has been reported in previous studies. Marcinnò et al. [41] found lower resistin concentrations in AD patients. Mooldijk et al. [42] proved that resistin level was inversely associated with the risk of AD. Resistin could suppress the production of oxidative factors in Neuro2a cells transfected with the Swedish amyloid precursor protein (swAPP) mutant and Presenilin exon 9 deletion mutant [43], indicating an anti-amyloidosis effect. Therefore, it is believed that resistin should also protect against AD, like adiponectin. However, our findings did not reveal causal effects of resistin on AD. This echoes a previous study that higher resistin is more likely to be associated with dementia with a vascular cause, but not with AD dementia [44]. Further studies indicated that resistin is involved in the regulation of mitochondrial function but not in APP metabolism [43].
This study explored the bidirectional relationship between circulating adipokines and risk of AD. GWAS datasets were used, and we identified association between adiponectin and AD. Although MR method provides a reliable tool to estimate the causal link between an exposure and an outcome using genetic variants, some additional caveats and limitations should be listed. First, the varied data sources of SNP-exposure, SNP-disease should be non-negligible. As seen in Table 1, data used in the study were from population of a European ancestry. Our findings should be carefully when applied to other population. Second, the participants with AD may not be accordant in advancing stages and pathological indications. This might bias the MR estimates and cause the discrepancies between our findings and previous reports. Third, adiponectin and resistin perfumed their intrinsic abilities via the binding with their own receptors. Future efforts could be paid to explore the interactive mode between receptors and ligands. Fourth, not all factors in adipokine family were analyzed in the study, for the lack of adequate GWAS data sources. Further exploration for future datasets may help solve it.
Conclusions
The MR study provided evidence of causal associations between reduced adiponectin level and an increased risk of AD, indicating a potential protective effect of adiponectin on AD. Moreover, no causal associations of resistin were indicated for the development of AD.
ACKNOWLEDGMENTS
Data for the genetic association with adiponectin, resistin and Alzheimer’s disease were obtained from GWAS respectively by Dastani et al., Sun et al., and Kunkle et al. We thank all researchers for sharing the genome-wide summary statistics.
FUNDING
This study was supported by grants from National Natural Science Foundation of China (Grant No. 81971877) and Basic and Applied Basic Research Foundation of Guangdong Province (Grant No. 2019A1515110087).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
All data used in the current study were extracted from public genome-wide association study summary statistics described above.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-230110.
REFERENCES
[1] | Terzo S , Amato A , Mulè F ((2021) ) From obesity to Alzheimer’s disease through insulin resistance, J Diabetes Complications 35: , 108026. |
[2] | Scheltens P , De Strooper B , Kivipelto M , Holstege H , Chételat G , Teunissen CE , Cummings J , van der Flier WM ((2021) ) Alzheimer’sdisease. Lancet 397: , 1577–1590. |
[3] | Cheignon C , Tomas M , Bonnefont-Rousselot D , Faller P , Hureau C , Collin F ((2018) ) Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol 14: , 450–464. |
[4] | Sinyor B , Mineo J , Ochner C ((2020) ) Alzheimer’s disease, inflammation, and the role of antioxidants. J Alzheimers Dis Rep 4: , 175–183. |
[5] | Yin F ((2023) ) Lipid metabolism and Alzheimer’s disease: Clinical evidence, mechanistic link and therapeutic promise. FEBS J 290: , 1420–1453. |
[6] | Fernández-Sánchez A , Madrigal-Santillán E , Bautista M , Esquivel-Soto J , Morales-González Á , Esquivel-Chirino C , Durante-Montiel I , Sánchez-Rivera G , Valadez-Vega C , Morales-González JA ((2011) ) Inflammation, oxidative stress, andobesity. Int J Mol Sci 12: , 3117–3132. |
[7] | Dekkers IA , Jansen PR , Lamb HJ ((2019) ) Obesity, brain volume, and white matter microstructure at MRI: A cross-sectional UK Biobank Study. Radiology 291: , 763–771. |
[8] | Zhuang QS , Meng L , Wang Z , Shen L , Ji HF ((2021) ) Associations between obesity and Alzheimer’s disease: Multiple bioinformatic analyses. J Alzheimers Dis 80: , 271–281. |
[9] | Nielsen FH ((2010) ) Magnesium, inflammation, and obesity in chronic disease. Nutr Rev 68: , 333–340. |
[10] | Bondia-Pons I , Ryan L , Martinez JA ((2012) ) Oxidative stress and inflammation interactions in human obesity. J Physiol Biochem 68: , 701–711. |
[11] | Ferrante AW Jr ((2013) ) The immune cells in adipose tissue. Diabetes Obes Metabol 15: , 34–38. |
[12] | Yaturu S , Daberry RP , Rains J , Jain S ((2006) ) Resistin and adiponectin levels in subjects with coronary artery disease and type 2 diabetes. Cytokine 34: , 219–223. |
[13] | Pan W , Kastin AJ ((2007) ) Adipokines and the blood-brain barrier. Peptides 28: , 1317–1330. |
[14] | Samant NP , Gupta GL ((2021) ) Adiponectin: A potential target for obesity-associated Alzheimer’s disease.. Metab Brain Dis 36: , 1565–1572. |
[15] | Liu J , Chi N , Chen H , Zhang J , Bian Y , Cui G , Xiu C ((2013) ) Resistin protection against endogenous Aβ neuronal cytotoxicity from mitochondrial pathway. Brain Res 1523: , 77–84. |
[16] | Lawlor DA , Harbord RM , Sterne JA , Timpson N , Davey Smith G ((2008) ) Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat Med 27: , 1133–1163. |
[17] | Dastani Z , Hivert M-F , Timpson N , Perry JR , Yuan X , Scott RA , Henneman P , Heid IM , Kizer JR , Lyytikäinen L-P ((2012) ) Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: A multi-ethnic meta-analysis of 45,891 individuals, PLoS Genet 8: , e1002607. |
[18] | Sun BB , Maranville JC , Peters JE , Stacey D , Staley JR , Blackshaw J , Burgess S , Jiang T , Paige E , Surendran P ((2018) ) Genomic atlas of the human plasma proteome. Nature 558: , 73–79. |
[19] | Kunkle BW , Grenier-Boley B , Sims R , Bis JC , Damotte V , Naj AC , Boland A , Vronskaya M , Van Der Lee SJ , Amlie-Wolf A ((2019) ) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 51: , 414–430. |
[20] | Kamat MA , Blackshaw JA , Young R , Surendran P , Burgess S , Danesh J , Butterworth AS , Staley JR ((2019) ) PhenoScanner V2: An expanded tool for searching human genotype–phenotype associations. Bioinformatics 35: , 4851–4853. |
[21] | Brion M-JA , Shakhbazov K , Visscher PM ((2013) ) Calculating statistical power in Mendelian randomization studies. Int J Epidemiol 42: , 1497–1501. |
[22] | Al-Kuraishy HM , Al-Gareeb AI , Alsayegh AA , Hakami ZH , Khamjan NA , Saad HM , Batiha GE , De Waard M ((2023) ) A potential link between visceral obesity and risk of Alzheimer’s disease. Neurochem Res 48: , 745–766. |
[23] | Gillette-Guyonnet S , Nourhashemi F , Andrieu S , de Glisezinski I , Ousset PJ , Riviere D , Albarede JL , Vellas B ((2000) ) Weight loss in Alzheimer disease, Am J Clin Nutr 71: , 637s–642s |
[24] | Letra L , Santana I ((2017) ) The influence of adipose tissue on brain development, cognition, and risk of neurodegenerative disorders. Adv Neurobiol 19: , 151–161. |
[25] | Giralt M , Cereijo R , Villarroya F ((2016) ) Adipokines and the endocrine role of adipose tissues. Handb Exp Pharmacol 233: , 265–282. |
[26] | Yang WS , Lee WJ , Funahashi T , Tanaka S , Matsuzawa Y , Chao CL , Chen CL , Tai TY , Chuang LM ((2002) ) Plasma adiponectin levels in overweight and obese Asians. Obes Res 10: , 1104–1110. |
[27] | Li S , Shin HJ , Ding EL , van Dam RM ((2009) ) Adiponectin levels and risk of type 2 diabetes: A systematic review and meta-analysis. JAMA 302: , 179–188. |
[28] | Teixeira AL , Diniz BS , Campos AC , Miranda AS , Rocha NP , Talib LL , Gattaz WF , Forlenza OV ((2013) ) Decreased levels of circulating adiponectin in mild cognitive impairment and Alzheimer’s disease. Neuromol Med 15: , 115–121. |
[29] | Suzanne M , Tong M ((2014) ) Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem Pharmacol 88: , 548–559. |
[30] | Jin T , Huang W , Cao F , Yu X , Ying Z , Guo S , Cheng Y , Xu C ((2022) ) Causal association between adiponectin and the risk of Alzheimer’s disease: A Mendelian randomization study. Front Neurol 13: , 1038975. |
[31] | Chan K-H , Lam KS-L , Cheng O-Y , Kwan JS-C , Ho PW-L , Cheng KK-Y , Chung SK , Ho JW-M , Guo VY , Xu A ((2012) ) Adiponectin is protective against oxidative stress induced cytotoxicity in amyloid-beta neurotoxicity, PloS One 7: , e52354. |
[32] | Ng RC-L , Cheng O-Y , Jian M , Kwan JS-C , Ho PW-L , Cheng KK-Y , Yeung PKK , Zhou LL , Hoo RL-C , Chung SK ((2016) ) Chronic adiponectin deficiency leads to Alzheimer’s disease-like cognitive impairments and pathologies through AMPK inactivation and cerebral insulin resistance in aged mice. Mol Neurodegener 11: , 71. |
[33] | Letra L , Matafome P , Rodrigues T , Duro D , Lemos R , Baldeiras I , Patrício M , Castelo-Branco M , Caetano G , Seica R , Santana I ((2019) ) Association between adipokines and biomarkers of Alzheimer’sdisease: A cross-sectional study. J Alzheimers Dis 67: , 725–735. |
[34] | Dukic L , Simundic A-M , Martinic-Popovic I , Kackov S , Diamandis A , Begcevic I , Diamandis EP ((2016) ) The role of human kallikrein 6, clusterin and adiponectin as potential blood biomarkers of dementia. Clin Biochem 49: , 213–218. |
[35] | Warren MW , Hynan LS , Weiner MF ((2012) ) Lipids and adipokines as risk factors for Alzheimer’s disease. J Alzheimers Dis 29: , 151–157. |
[36] | Lubkowska A , Radecka A , Bryczkowska I , Rotter I , Laszczyńska M , Dudzińska W ((2015) ) Serum adiponectin and leptin concentrationsin relation to body fat distribution, hematological indices andlipid profile in humans., Int J Environ Res Public Health 12: , 11528–11548. |
[37] | Van Himbergen TM , Beiser AS , Ai M , Seshadri S , Otokozawa S , Au R , Thongtang N , Wolf PA , Schaefer EJ ((2012) ) Biomarkers for insulin resistance and inflammation and the risk for all-cause dementia and Alzheimer disease: Results from the Framingham Heart Study. Arch Neurol 69: , 594–600. |
[38] | Pákáski M , Fehér A , Juhász A , Drótos G , FazekasÖ C , Kovács J , Janka Z , Kálmán J ((2014) ) Serumadipokine levels modified by donepezil treatment in Alzheimer’sdisease, Alzheimers Dis 38: , 371–377. |
[39] | Tripathi D , Kant S , Pandey S , Ehtesham NZ ((2020) ) Resistin in metabolism, inflammation, and disease. FEBS J 287: , 3141–3149. |
[40] | Versini M , Jeandel P-Y , Rosenthal E , Shoenfeld Y ((2014) ) Obesity in autoimmune diseases: Not a passive bystander. Autoimmun Rev 13: , 981–1000. |
[41] | Marcinnò A , Gallo E , Roveta F , Boschi S , Grassini A , Rainero I , Rubino E ((2022) ) Decreased resistin plasmatic concentrations in patients with Alzheimer’s disease: A case-control study, Heliyon 8: , e11738. |
[42] | Mooldijk SS , Ikram MK , Ikram MA ((2022) ) Adiponectin, leptin, and resistin and the risk of dementia. J Gerontol A Biol Sci Med Sci 77: , 1245–1249. |
[43] | Xiaoying L , Li T , Yu S , Jiusheng J , Jilin Z , Jiayi W , Dongxin L , Wengang F , Xinyue Z , Hao Y , Yuhua C , Deshu S ((2019) ) Resistin-inhibited neural stem cell-derived astrocyte differentiation contributes to permeability destruction of the blood-brain barrier. Neurochem Res 44: , 905–916. |
[44] | Bednarska-Makaruk M , Graban A , Wiśniewska A , Łojkowska W , Bochyńska A , Gugała-Iwaniuk M , Sławińska K , Ługowska A , Ryglewicz D , Wehr H ((2017) ) Association ofadiponectin, leptin and resistin with inflammatory markers andobesity in dementia. Biogerontology 18: , 561–580. |


