
Neuroprotective Effects of a Multi-Herbal Extract on Axonal and Synaptic Disruption in Vitro and Cognitive Impairment in Vivo
Abstract
Background:
Alzheimer’s disease (AD) is a multifactorial disorder characterized by cognitive decline. Current available therapeutics for AD have limited clinical benefit. Therefore, preventive therapies for interrupting the development of AD are critically needed. Molecules targeting multifunction to interact with various pathlogical components have been considered to improve the therapeutic efficiency of AD. In particular, herbal medicines with multiplicity of actions produce cognitive benefits on AD. Bugu-M is a multi-herbal extract composed of Ganoderma lucidum (Antler form), Nelumbo nucifera Gaertn., Ziziphus jujuba Mill., and Dimocarpus longan, with the ability of its various components to confer resilience to cognitive deficits.
Objective:
To evaluate the potential of Bugu-M on amyloid-β (Aβ) toxicity and its in vitro mechanisms and on in vivo cognitive function.
Methods:
We illustrated the effect of Bugu-M on Aβ25–35-evoked toxicity as well as its possible mechanisms to diminish the pathogenesis of AD in rat cortical neurons. For cognitive function studies, 2-month-old female 3×Tg-AD mice were administered 400 mg/kg Bugu-M for 30 days. Behavioral tests were performed to assess the efficacy of Bugu-M on cognitive impairment.
Results:
In primary cortical neuronal cultures, Bugu-M mitigated Aβ-evoked toxicity by reducing cytoskeletal aberrations and axonal disruption, restoring presynaptic and postsynaptic protein expression, suppressing mitochondrial damage and apoptotic signaling, and reserving neurogenic and neurotrophic factors. Importantly, 30-day administration of Bugu-M effectively prevented development of cognitive impairment in 3-month-old female 3×Tg-AD mice.
Conclusion:
Bugu-M might be beneficial in delaying the progression of AD, and thus warrants consideration for its preventive potential for AD.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive, neurodegenerative disorder with multiple factors and a continuum of gradual changes developing over approximately 25 years from disease onset to death [1]. Pathologically, AD is marked by extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles formed from the microtubule-binding tau protein, which results in atrophy and neuron death [2]. Despite FDA-approved drugs are currently available, all these drugs have limited clinical benefit [3]. No therapeutic has convincingly terminated or slowed AD progression. Therefore, preventive strategies that hinder the progression of AD prior to the disease onset or when clinical manifestations are emerging are increasingly pressing [3].
Application of Aβ peptide to neuronal cultures caused cell death, axonal and synaptic dysfunction, mitochondrial damage, neuroinflammation, oxidative stress, and ultimately neurodegeneration [4]. Tau provides stability to the distal labile regions of axonal microtubules that are the conduits for trafficking cellular constituents along axonal extensions via the process of axonal transport [5, 6]. Axon demise attributes to microtubule deficits along with calcium signaling imbalance, mitochondrial dysfunction, and increased oxidative stress, leading to a dying-back pattern of neuronal degeneration that is featured in AD and Tg mouse models [7–9]. Multiple lines of evidence has shown damaged axonal transport and exacerbation of amyloid pathology in AD might be attributed to Aβ-induced microtubule disruption in presynaptic dystrophic neurites surrounding plaques [8]. Similar to the axonopathy that occurs early in AD and correlates strongly with cognitive decline [10, 11], synaptic loss is found to precede neurodegeneration in patients with symptomatic AD [12, 13].
In order to find more effective drugs for the treatment of AD, natural products and their compounds have been broadly studied because of their roles as one of the major sources of drug discovery [14]. Indeed, two cholinesterase inhibitors for treating AD, galantamine and rivastigmine, are derived from natural product with the former a natural product and the latter a derivative of physostigminea [15, 16]. Given the complexity of AD pathophysiology, multifaceted natural products either from mixtures or from extracts with neuroprotective effects may be beneficial for AD compared to single compound [17]. Also, herbs, herbal formulations and mixtures from other natural sources may offer cognitive benefits to patients with AD [14, 18, 19]. As a step in this direction, we established a multi-component agent (denoted as Bugu-M) with mixed extracts of the antler form of Ganoderma lucidum (Lingzhi), Ziziphus jujuba Mill. (red date), Dimocarpus longan (longan), and Nelumbo nucifera Gaertn. (lotus seeds) based on their neuroprotective effects in Traditional Chinese Medicine as well as in previous studies. First, Ganoderma lucidum extract and its components improve learning and alleviate cognitive deficits in APP/PS1 transgenic AD mice [20–22] and animals treated with scopolamine [23], d-galactose [24, 25], streptozotocin [26, 27], hypobaric hypoxia [28], or chronic cerebral hypoperfusion [29]. These effects are mediated through regulating fibroblast growth factor receptor 1 (FGFR1)/ERK/Akt [20], ROCK [21], CREB/p-CREB/BDNF expression [28], DNA methylation [22], Foxp3+ Treg cells [29], the Th17/Tregs axis [24], the brain-liver axis [25], acetylcholinesterase activities [23], oxidative stress, or mitochondrial dysfunction [27]. Second, Ziziphus jujuba Mill. extract possesses antiamnesic and neuroprotective effects that are mediated in part through antioxidant, anti-inflammatory, and antiapoptotic activities in Aβ25–35-induced AD mice [30], scopolamine-treated mice [31], or D-galactose-treated rats [32]. Semen Zizyphi spinosae, the seed of Ziziphus jujuba Mill, has also been applied to patients with sleep disorders to further prevent dementia [33]. Third, Longan (Dimocarpus longan) improves the cognitive ability and reduces the pathologic impairment in D-gal/aluminium chloride-treated mice, which might be partly mediated via inhibition of RAS/MEK/ERK signaling pathway [34]. In combination with Gastrodia elata, Salvia miltiorrhiza and Liriope platyphylla, Dimocarpus longan ingredients reverse Aβ-elicited memory decline and oxidative stress in mice [35]. Fourth, several compositions derived from or associated with Nelumbo nucifera Gaertn. ameliorate memory and cognitive dysfunction in aluminium chloride- or scopolamine-treated mice [36–39], and in db/db diabetic mice [40]. The antiamnesic effect of neferine may be mediated via antioxidant and anti-inflammatory capacities [37, 40] or inhibition of ChEs and BACE1 [37], while allantoin contained in Nelumbo nucifera partly mediates PI3K-Akt-GSK-3β signaling pathway [38].
The axo-and synapto-protective effect of above-mentioned composition as single agent and the potential efficacy in the early state of AD has not been previously examined. Hence, the current study investigates Bugu-M efficacy on Aβ-induced toxicity in rat cortical neurons and cognitive impairment in female 3×Tg-AD mice, and assesses potential mechanisms underlying its action.
MATERIALS AND METHODS
Primary cortical neuronal cultures
The Institutional Animal Care and Use Committee of the National Tsing Hua University approved all experiments involving animals for primary neuronal cultures (NTHU-10912H036). Primary cortical neurons were prepared from fetal brain of Sprague Dawley rats (BioLASCO Taiwan, Taipei, Taiwan) at embryonic day 18 (E18) as described previously [41]. Briefly, cortical tissues cut in Hank’s balanced salt solution (HBSS) were digested with papain (10 U/mL) at 37°C for 10 min. After digestion, tissues were incubated with DNase I (Sigma-Aldrich, St. Louis, MO, USA), followed by mechanically dispersion with a Pasteur pipette. After a low speed centrifugation, cells in the pellet were resuspended in plating medium (MEM containing 10% (v/v) horse serum, 100 U/mL penicillin, and 100 μg/mL streptomycin), followed by filtration through nylon mesh (Greiner Bio-One, Frickenhausen, Germany). Cells were then seeded onto plates or dishes coated with poly-L-lysine-coated at a density of 2.5×104 cells/cm2 for 12–16 h. The plating medium was changed with neurobasal medium containing 0.5 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin, and 2% (v/v) B27 (all from Thermo Fisher Scientific, Waltham, MA, USA). Half of the medium was replaced with fresh neurobasal medium every 3-4 days, and cells were cultured for at least 8 days for experiments. The quality of neuronal cultures was assessed by immunostaining with an anti-β-III tubulin antibody.
Preparation of Aβ fibrils
Aβ25–35 (Sigma-Aldrich) was reconstituted in 1 mL of distilled water, followed by incubation at 37°C for 7 days. Formation of Aβ fibril was examined by visualizing under transmission electron microscopy.
Transmission electron microscopy
A carbon-coated copper grids (Ted Pella Inc., Redding, CA, USA) was put on a drop of sample containing Aβ25–35 for 5 min. Sample were then negatively stained with 1% (w/v) uranyl acetate (Electron Microscopy Sciences, Hatfield, PA, USA) before examination under a transmission electron microscope (HT7700, Hitachi High-Technologies, Tokyo, Japan) with an accelerating voltage of 100 kV. High-resolution images were acquired by a CCD (charge-coupled device) camera and processed further in the Adobe Photoshop CC (Adobe, San Jose, CA, USA).
Preparation of Bugu-M extract
The antler-shaped fruiting bodies of Ganoderma lucidum (purchased from Tianmei Farm, Pingtung, Taiwan), fruit of Ziziphus jujuba Mill. (purchased from Bohai Jujube Garden, Miaoli, Taiwan), fruit of Dimocarpus longan (purchased from Dongshan District Farmers Association, Tainan, Taiwan), and seed of Nelumbo nucifera Gaertn. (purchased from Jin Baoan Pharmacy, Miaoli, Taiwan) were obtained following United States Pharmacopeia and Taiwan Herbal Pharmacopeia standard. Also, molecular authentication based on the Internal transcribed spacer (ITS) sequences for raw materials were conducted. The names of botanical materials in Bugu-M can be found on https://www.theplantlist.org and https://www.mycobank.org. The Bugu-M extract evaluated in this study was prepared according to the process in US20210290710A1 [42]. The weight ratio of Ganoderma lucidum, Ziziphus jujuba Mill., Dimocarpus longan and Nelumbo nucifera Gaertn. was 3:1:1:1. First, the respective material was extracted with water under reflux for 2 h to afford respective extract. Next, the extracts were mixed and freeze-dried to obtain Bugu-M. Then, Bugu-M was stored at –20°C for in vitro and in vivo experiments.
Cell viability assay
Cell viability was assessed by 3-(4,5-dimethylthi-azol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Thermo Fisher Scientific) and the cells were incubated with MTT solution at 0.5 mg/mL for 4 h. Then dimethyl sulfoxide (DMSO) was added to solubilze MTT-formazan on an orbital shaker for 5 min. The optical density at 570 nm was evaluated by using SpectraMax® M5 Microplate Reader (Molecular Devices, Sunnyvale, CA, USA).
LDH cytotoxicity assay
Primary rat cortical neurons were treated with Bugu-M prior to Aβ25–35 exposure at 37°C in a 96-well plate for 72 h. Lactate dehydrogenase (LDH) release was determined with the cytotoxicity Detection Kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions with the absorbance measured at 490 nm using SpectraMax M5 Microplate Reader (Molecular Devices, USA).
Immunofluorescence microscopy
Cells were processed for double-label immunofluorescence microscopy as previously described [41]. Briefly, cells were washed with phosphate-buffered saline (PBS), fixed in 4% (w/v) paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) in PBS, and then permeabilized with 0.2% (v/v) Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA) in PBS. After being washed with PBS, cells were blocked with 10% (w/v) normal goat serum (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) in PBS, followed by incubation with primary antibodies (Table 1) for 1 h. After several washes with PBS, cells were incubated with secondary antibodies conjugated with Alexa Fluor 488 (1:400) or Alexa Fluor 594 (Thermo Fisher Scientific, Waltham, MA, USA), and were counterstained with 4′,6- diamino- 2- phenylindole (DAPI). After immunostaining, cells were examined by a Zeiss LSM800 laser scanning confocal microscope (Carl Zeiss, Jena, Germany) using 20×(0.7 NA) Plan-Neofluar or 40×(1.3 NA) Apochromat objective lens. Images were acquired by the Zen software (Ver. 2.3) and processed for figures using Adobe Photoshop CC (Adobe Systems, San Jose, CA, USA).
Table 1
Summary of antibodies used for Western blotting and immunofluorescence analysis
| Antibody | RRID | Host | Supplier |
| β-III tubulin | AB_2313773 | Mouse | BioLegend |
| MAP2 | AB_2722660 | Rabbit | Cell Signaling Technology |
| Tau | AB_2536235 | Mouse | Thermo Fisher Scientific |
| β-actin | AB_787885 | Mouse | Novus Biologicals |
| Synaptophysin | AB_10126406 | Rabbit | Novus Biologicals |
| PSD-95 | AB_325399 | Mouse | Thermo Fisher Scientific |
| Neurogranin | AB_447526 | Rabbit | Abcam |
| SV2A | AB_778192 | Rabbit | Abcam |
| SNAP25 | AB_2716878 | Rabbit | Thermo Fisher Scientific |
| VAMP2 | AB_2798240 | Rabbit | Cell Signaling Technology |
| Synaptogyrin-3 | AB_11004965 | Rabbit | Novus Biologicals |
| GAP43 | AB_10860076 | Rabbit | Cell Signaling Technology |
| GAPDH | AB_10622025 | Rabbit | Cell Signaling Technology |
| Phospho-PKC (pan) (βII Ser660) | AB_2168219 | Rabbit | Cell Signaling Technology |
| mGluR1 | AB_2797953 | Rabbit | Cell Signaling Technology |
| mGluR2 | AB_2799878 | Rabbit | Cell Signaling Technology |
| NMDAR1 | AB_10862307 | Rabbit | Abcam |
| AMPAR1 (GluA1) | AB_2732897 | Rabbit | Cell Signaling Technology |
| CamKII (pan) | AB_2067938 | Rabbit | Cell Signaling Technology |
| Phospho-CamKII (Thr286) | AB_2713889 | Rabbit | Cell Signaling Technology |
| Phospho-CREB (Ser133) | AB_2561044 | Rabbit | Cell Signaling Technology |
| Akt | AB_1147620 | Mouse | Cell Signaling Technology |
| Phospho-Akt (Ser473) | AB_2224726 | Rabbit | Cell Signaling Technology |
| Phospho-PI3Kp85α (Tyr607) | AB_2834667 | Rabbit | Affinity Biosciences |
| PI3K (p85) | AB_329869 | Rabbit | Cell Signaling Technology |
| Phospho-GSK-3β (Ser9) | AB_2115196 | Rabbit | Cell Signaling Technology |
| GSK-3α/β | AB_10547140 | Rabbit | Cell Signaling Technology |
| BDNF | AB_10862052 | Rabbit | Abcam |
| Doublecortin | AB_561007 | Rabbit | Cell Signaling Technology |
| Neurofilament-L | AB_823575 | Rabbit | Cell Signaling Technology |
| Cleaved caspase-3 | AB_2070042 | Rabbit | Cell Signaling Technology |
| α-tubulin cleaved by caspase-6 (TubΔCasp6) | Not available | Rabbit | MyBioSource |
| TDP-43 | AB_2200505 | Rabbit | Proteintech |
| Phospho-TDP-43 (Ser409/410) | AB_2564934 | Rat | Biolegend |
| HSP90 | AB_2233307 | Rabbit | Cell Signaling Technology |
| HSP40 | AB_2094571 | Rabbit | Cell Signaling Technology |
| HSP60 | AB_2636980 | Rabbit | Cell Signaling Technology |
Neurite outgrowth assay
Neurite length obtained from the aforementioned immunofluorescence microscopy analysis of β-III tubulin was measured by using Image J for loading images followed by quantification with the Neuron J plug-in [41]. For individual neuron, the sum of neurites was analyzed after tracing of each neurite.
Subcellular fractionation
Primary neurons were extracted with RIPA buffer, and the resulting total, supernatant, and pellet fractions were prepared as described previously [41]. Membrane-enriched fractions were prepared according to a published protocol [43]. Briefly, primary neurons were homogenized with homogenization buffer (7.5 mM sodium phosphate (pH 7.0), 0.25 M sucrose, 5 mM EDTA, and 5 mM EGTA) containing a cocktail of protease and phosphatase inhibitors. Homogenates were centrifuged at 1,000×g in a bench top centrifuge (Eppendorf, 5415R), and the resulting supernatants were further centrifuged at 100,000×g for 30 min at 4°C. The final pellet was taken as the membrane-enriched fraction, and the supernatant as cytosolic fraction. After concentration determination by bicinchoninic acid assay (BCA), protein samples were equalized by mixing with appropriate volumes of sample buffer (25 mM Tris-HCl, pH 6.8, 10% (v/v) glycerol, 1% (w/v) SDS, and 5% (v/v) β-mercaptoethanol) prior to Western blot analysis.
Western blot analysis
Protein samples were analyzed by SDS-PAGE followed by Western blotting as described previously [41]. The nitrocellulose membranes (Pall Life Sciences, Port Washington, NY, USA) were washed with Tris-buffered saline (TBS, 150 mM NaCl, 20 mM Tris-HCl, pH 7.4), and then blocked with 3% (w/v) BSA in TBS containing 0.1% (v/v) Tween 20 (TBST) for at least 1 h at room temperature. After several washes with TBST, membranes were incubated with primary antibody (Table 1) at 4°C overnight, followed by incubation with anti-mouse, anti-rabbit or anti-rat secondary antibodies conjugated with horseradish peroxidase (Jackson ImmunoResearch Laboratories, West Glove, PA, USA) for at least 2 h. Chemiluminescent signal was developed by Enhanced Chemiluminescence Substrate (Western Lightning, PerkinElmer Life Sciences, Waltham, MA, USA) and visualized using a LAS 4000 (GE Healthcare, Chicago, IL, USA) imaging system. Signal intensities of immunoblots were quantified using the ImageQuant TL 7.0 software (IQTL ver 7.0, GE Healthcare, Chicago, IL, USA) and adjusted for protein loading by normalizing to GAPDH or β-actin.
Detection of extracellular tau levels
Following the stimulation of the cells with Aβ, cell culture supernatants were collected and assayed for tau using High Sensitive ELISA Kit for Microtubule Associated Protein Tau (Taipei, Taiclone) according to the manufacturer’s protocol.
Animals for behavioral testing
The Institutional Animal Care and Use Committee at Industrial Technology Research Institute (ITRI) approved all animal procedures for behavioral testing under protocol ITRI-IACUC-2021-037V1. Animals were maintained in an AAALAC approved ITRI animal facility under a 12 h light/12 h dark cycle (lights on at 7:30am, 23±2°C). Animals were provided ad libitum water and Standard Rodent Diet (altromin 1326, Altromin, Lage, Germany) until time of sacrifice, aged approximately 3 months for 3×Tg-AD [44] and NonTg mice. 3×Tg-AD and NonTg C57BL/6 background female mice were obtained from Jackson Laboratory (stock 004807) and BioLASCO (Taipei City, Taiwan), respectively.
Animal treatment
The dose of 400 mg/kg/day Bugu-M was selected from our unpublished data that showed 28-day treatment of Bugu-M rescues cognitive deficits in rodents injected with Aβ1-42 or Aβ1–40 peptide intracerebroventricularly. From 2 months of age Bugu-M was administered via oral gavage. Mice were divided to three groups of 10 mice each at 2 months of age: 1) NonTg with water treatment; 2) 3×Tg-AD mice with water treatment; 3) 3×Tg-AD mice with 400 mg/kg of Bugu-M treatment. Bugu-M and water was administered by gavage once daily for 30 days. Throughout the experimental process, evaluation of body weight was continuously monitored until animal sacrifice. Behavioral tests were conducted by the same trained experimenter one week before animal sacrifice. We performed behavioral test during the light phase (between 12:00–16:00) of the animal’s sleep-wake cycle. One hour prior to testing, mice were allowed to acclimate to the testing arena and returned to individual cages. At 3 months of age animals were sacrificed by CO2 asphyxiation.
T-maze test
T-maze analysis was performed as described by Wenk [45] with slight modifications. The middle arm was starting arm, while the other two arms were assigned as novel and familiar arms. The novel arm was blocked to form an L-shape arena in the acquisition phase and kept open as T-shape in the test phase. After placed into the starting arm, mice were allowed to explore freely for 5 min in both acquisition and test phases. Between these 2 phases, mice were removed from arena to a non-home cage, and stayed for 2 min as inter-trial interval. In the test phase, total distance moved in novel arm was calculated. In fully 5 min of test phase, spontaneous alternation behavior (%Alternations) was determined as (100×number of alternation) / (total arm entries – 2), while the alternation was defined as a set of consecutive entries into all 3 different arms, and the individual with total arm entries less than 3 was excluded. All tests were recorded with Logitech Capture software using Logitech C922 camera that was fixed on the top of the testing arena. The recorded videos were then analyzed using the EthoVision XT software v13 (Noldus Information Technology, Wageningen, Netherlands).
Morris water maze test
We performed the Morris water maze (MWM) as reported by Morris [46] with slight modifications. A water tank pool of 115 cm in diameter with a 40 cm wall was divided into quadrants with opaque water of 22°C±1°C by adding skimmed milk powder. During the training session, a platform of 10 cm in diameter was constantly placed one cm in the middle of one quadrant beneath the water surface with two stickers as visual cues on the walls of the pool. Mice were randomly placed in one of the quadrants and allowed to swim for 60 sec in training trials for 3 days (four training trials per day). The latency time for mice to find the platform was assessed. A probe trial was conducted without a platform by the same protocol as the training session. The time for the mice to remain at the previous location of the platform was evaluated. All tests were recorded with Logitech Capture software using Logitech C922 camera that was fixed on the top of the testing arena. The recorded videos were then analyzed using the EthoVision XT software v13 (Noldus Information Technology, Wageningen, Netherlands). The swimming paths of each mouse were analysed by its navigational strategies classified as non-goal-directed or goal-directed search stragtegies as described by Baeta-Corral and Giménez-Llort [47].
Open field test
The open field arena was made of white acrylic and measured 50×50×30 cm. The central 25×25 cm square was defined as the center area. Each mouse was placed in the corner of the box and allowed to freely move for 5 min. All tests were recorded with Logitech Capture software using Logitech C922 camera that was fixed on the top of the testing arena. The recorded videos were then analyzed using the EthoVision XT software v13 (Noldus Information Technology, Wageningen, Netherlands). The total distance traveled, time spent in the periphery, and the frequency of freezing were measured.
Elevated plus maze test
The elevated plus maze (EPM) was a plus-shaped maze made of white acrylic, consisting of two open arms (30×5 cm) with 0.5 cm-high guardrail and two closed arms (30×5×15 cm). Four arms were connected to the center square (5×5 cm), which was elevated 50 cm above the floor. Each mouse was placed in the center square and allowed to freely explore for 5 min. All tests were recorded with Logitech Capture software using Logitech C922 camera that was fixed on the top of the testing arena. The recorded videos were then analyzed using the EthoVision XT software v13 (Noldus Information Technology, Wageningen, Netherlands). The total distance traveled, distance moved in the open arms, and the frequency of freezing were measured.
Statistical analysis
All quantitative data are presented as mean±standard error of the mean (SEM). Statistical significance between multiple groups were measured by One-way or Two-way analysis of variance (ANOVA) followed by Dunnett’s or Fisher’s least significant difference (LSD) comparisons test that was described in the figure legends using GraphPad Prism (version 8.3.0) (San Diego, CA, USA). Analysis of the strategies of goal- or non-goal-directed search was performed using logistic regression. Differences in the incidence were measured by Chi-square test. Identification and removal of outlier values was done using the robust regression and outlier removal test (ROUT) with Q = 1%. A p-value less than 0.05 was considered statistically significant.
RESULTS
Bugu-M rescues Aβ neurotoxicity in rat cortical neurons
To examine the effect of Bugu-M on cell viability of rat cortical neurons, we performed MTT assay. As shown in Fig. 1B, Bugu-M (12.5, 25, 50, and 100 μg/mL) was not toxic to neurons after 72 h exposure (Fig. 1B). Thus, the concentration of 100 μg/mL or below was used for further studies. Aβ accumulation is neurotoxic and pivotal for developing AD [17]. To assess whether Bugu-M enhances neuronal survival against Aβ injury, rat cortical neurons were incubated with Aβ25–35 peptide that results in neuronal shrinkage, neurite breakdown and apoptotic reactions [41]. Compared to control neurons, 72 h exposure of 20 and 40 μM Aβ25–35 significantly reduced cell viability (Fig. 1C). However, pretreatment of Bugu-M (25, 50, and 100 μg/mL) significantly reversed 20 μM Aβ-induced neuronal toxicity (p < 0.01; Fig. 1D). The release of LDH was also measured as an indicator of membrane damage induced by Aβ25–35. Neurons co-treated with Bugu-M (100 μg/mL) and Aβ (20 μM) decreased the amounts of LDH leakage relative to the Aβ alone group (p = 0.0142; Fig. 1E). Overall, the LDH assay in neurons confirmed the results of MTT assay.
Fig. 1
Bugu-M rescues Aβ-induced neurotoxicity in neurons. A) A representative image of Aβ25–35 after incubation at 37°C for 7 days. Scale bar, 200 nm. Rat cortical neurons were cultured in Bugu-M (B) or Aβ25–35 (C) at indicated concentrations for 72 h. (D) Rat cortical neurons were cultured in Aβ25–35 at 20 μM with or without Bugu-M (12.5, 25, 50, and 100 μg/mL) for 72 h. Cell viability was measured using the MTT assay (B-D). E) Rat cortical neurons were cultured in Aβ25–35 at 20 μM in the absence or presence of Bugu-M (100 μg/mL) for 72 h. Cytotoxicity was measured by LDH release assay. F) Rat cortical neurons were pretreated with 100 μg/mL Bugu-M for 1 h prior to Aβ25–35 incubation for 72 h. Neurite outgrowth was analyzed by immunofluorescent labeling of β-III tubulin. Confocal images are single optical slices, and camera and microscope setting were equivalent for comparisons between groups. Shown are representative images. Scale bar, 20 μm. G) Quantification of total length of axons and minor neurites by NeuronJ software. Data are expressed as mean±SEM (n = 3). Statistic significance between groups were analyzed by one-way ANOVA and Dunnett’s multiple comparisons test. For all tests: #p < 0.05, ##p < 0.01, and ###p < 0.001 versus the untreated control group; *p < 0.05, **p < 0.01, and ***p < 0.001 versus the Aβ group.
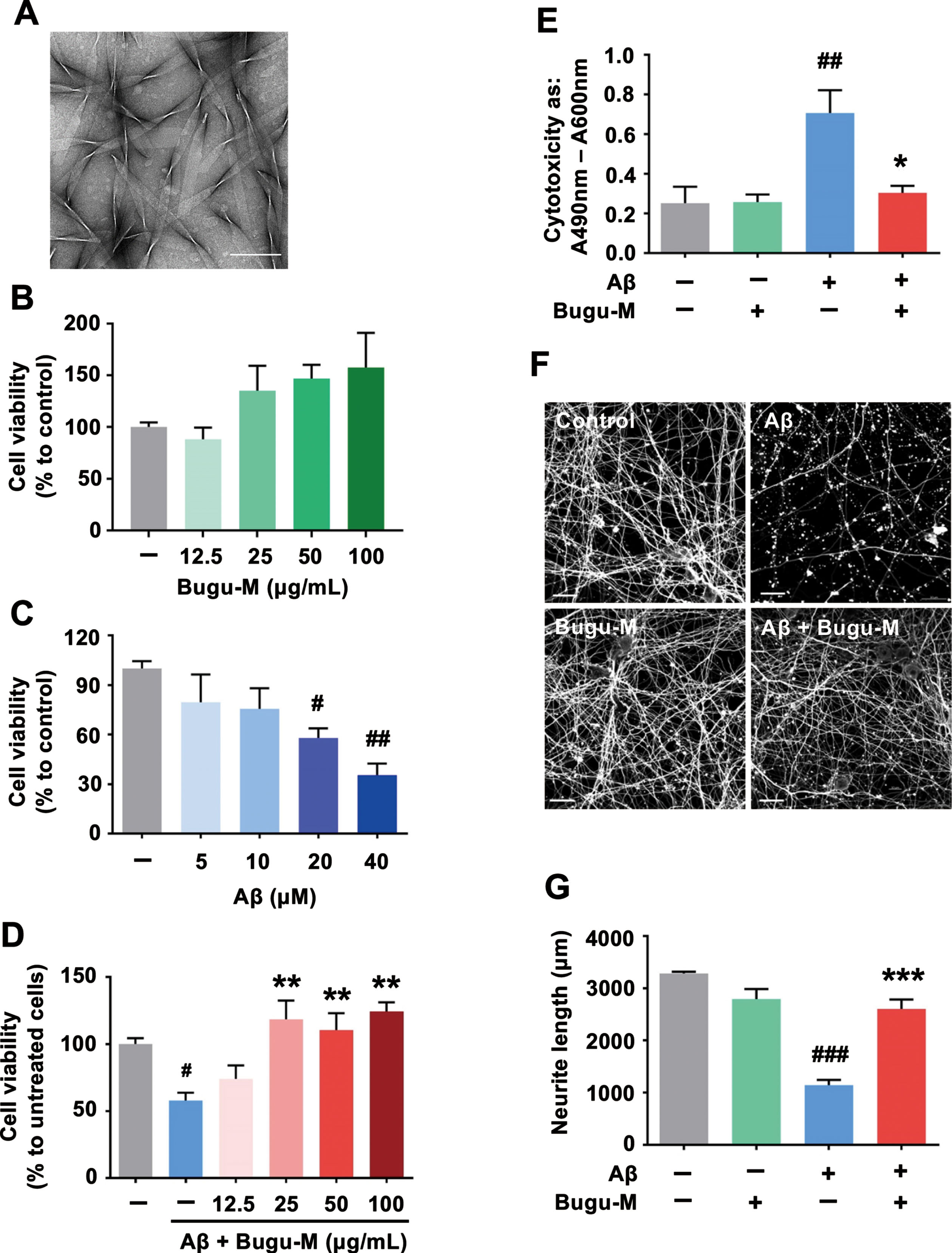
To further illustrate the neuroprotective function of Bugu-M, we determined the neurite outgrowth in the presence of Aβ25–35. β-III tubulin immunolabeling (Fig. 1F) showed pretreatment of neurons with 100 μg/mL Bugu-M significantly (p = 0.0003 versus Aβ; Fig. 1G) rescued the disrupted neuronal integrity by Aβ (p = 0.0001 versus control; Fig. 1F, G) by an average of ∼44% after 72 h compared to the Aβ alone group (Fig. 1G). These data support the protective effect of Bugu-M on reversing Aβ-elicited neurotoxicity.
Bugu-M alleviates Aβ-mediated apoptotic signaling in neurons
Activation of caspases and apoptosis by impaired mitochondrial function have been shown in brains from AD patients and AD mouse models [48, 49], where mitochondrial malfunction has been proposed as an early event in AD [50]. Thus, we next assessed the levels of caspase-3 that plays critical roles in apoptotic pathway. Western blot analysis displayed that 72 h of 20 μM Aβ25–35 treatment markedly increased caspase-3 activation fragment (Fig. 2A, lane 2; Cleaved caspase-3). However, Bugu-M (100 and 50 μg/mL) was able to reduce caspase-3 activation (Fig. 2A, lanes 5 and 6). Instead of initiator caspases, caspase-6 is often activated by caspase-3 [51], and thus we evaluated changes in α-tubulin [52] by immunoblotting. By doing so, we found that Aβ contributed to a significant increase in α-tubulin cleavage (Fig. 2A, lane 2; TubΔCsp6), whereas Bugu-M (100 and 50 μg/mL) administration prior to Aβ abrogated the elevation of cleaved fragments of α-tubulin by caspase-6 (Fig. 2A, lanes 5 and 6). Concomitantly, Bugu-M (50 and 100 μg/mL) resumed the HSP90 (significantly) and HSP40 (slightly) levels in the mitochondria of neurons exposed to Aβ, whereas HSP60 was reduced by Bugu-M (Fig. 2B, lanes 5 and 6).
Fig. 2
Bugu-M alleviates Aβ-triggered apoptotic signaling and axonal disruption in neurons. Rat cortical neurons were pretreated with Bugu-M (50 and 100 μg/mL) (A, B) or 100 μg/mL Bugu-M (C) for 1 h prior to Aβ25–35 incubation for 72 h. A, D) Whole lysates, (B) mitochondrial fractions and (C) membrane-enriched fractions analyzed by immunoblotting for apoptosis (cleaved caspase-3, caspase-6-cleaved α-tubulin (TubΔCsp6)), mitochondrial dysfunction (HSP60, HSP40, HSP90), axon (Tau, NfL), dendrite (MAP2), and TDP-43 pathology related proteins. GAPDH and β-actin were used as a loading control. Total TDP-43 shows C-terminal TDP-43 fragments (CTFs), while phospho-TDP-43 reveals high molecular weight smears (asterisk) and low levels of CTFs. Densitometric quantification of immunoreactive bands was determined using ImageJ, and the fold changes to control bands are analyzed in Supplementary Figure 3D.) Quantification of Tau by ELISA in cell culture supernatants. Data are represented as mean±SEM (n = 3). Statistical significance between groups were calculated by one-way ANOVA and Dunnett’s multiple comparisons test. ##p < 0.01 versus the untreated control group; **p < 0.01 versus the Aβ group.
Loss of transactive response DNA binding protein of 43 kDa (TDP-43) function due to pathological modifications and mislocalization is related to the severity of AD pathology, as evidenced by higher Braak neurofibrillary tangle stages and Thal amyloid phases [53]. In particular, caspase-3-cleaved TDP-43 accumulates in the AD brain and pathologically prone to promote disease progression [54]. TDP-43 is primarily located in the nucleus in the normal brain, whilst accumulation of pathlogical TDP-43 mainly occurs in the cytoplasm or neuronal processes [55, 56]. Based on our findings of caspase-3 inactivation by Bugu-M, we hypothesized that Bugu-M would block the aberrations of TDP-43. To address this, we conducted immunoblotting on neurons treated with Bugu-M and Aβ. The increase in full-length and fragmented TDP-43 and phospho-TDP-43 by 20 μM Aβ was declined by Bugu-M (100 μg/mL) after 72 h treatment (Fig. 2C, lane 4). In correspondence with this, TDP-43 distribution also appeared to be altered in Aβ-treated neurons (Supplementary Figure 1I-L), whereas Bugu-M prominently retained TDP-43 in the nucleus (Supplementary Figure 1M-P).
Bugu-M prevents Aβ-triggered axonal disruption in neurons
Defective axonal transport can trigger ‘dying back’ axonal degeneration and cell death [7–9]. As Aβ blocks axonal transport and amyloid plaques are related to organelle accumulation and cytoskeletal damage [57], we speculated that Bugu-M rescued neuronal death through preventing cytoskeletal damage. In line with the effects of Bugu-M on reserving β-III tubulin integrity in neurons treated with Aβ25–35 (Fig. 1F), 100 μg/mL Bugu-M also retained the expression levels of axonal cytoskeleton neurofilament light (NfL) (Fig. 2C, lane 4). In addition, Tau and MAP2, the microtubule-associated proteins, were elevated at 100 and 50 μg/mL Bugu-M-treated neurons upon exposure to 20 μM Aβ for 72 h (Fig. 2A, lanes 5 and 6). Consistent with this, a decrease of Tau (Supplementary Figure 2C) and MAP2 (Supplementary Figure 2G) immunoreactivity was observed in 20 μM Aβ-treated neurons. However, Bugu-M (100 μg/mL) administration was able to reverse the decrement of Tau (Supplementary Figure 2D) and MAP2 (Supplementary Figure 2H) by Aβ. Intriguingly, we found Bugu-M (100 μg/mL) significantly(p = 0.0087 versus Aβ) reduced extracellular Tau that was increased by 20 μM Aβ stimulation (p = 0.0011 versus control) in neurons (Fig. 2D). Altogether, these data demonstrate Bugu-M was capable to mitigate Aβ-induced axonal disruption in rat cortical neurons.
Bugu-M reverses Aβ-elicited synaptic disruption in neurons
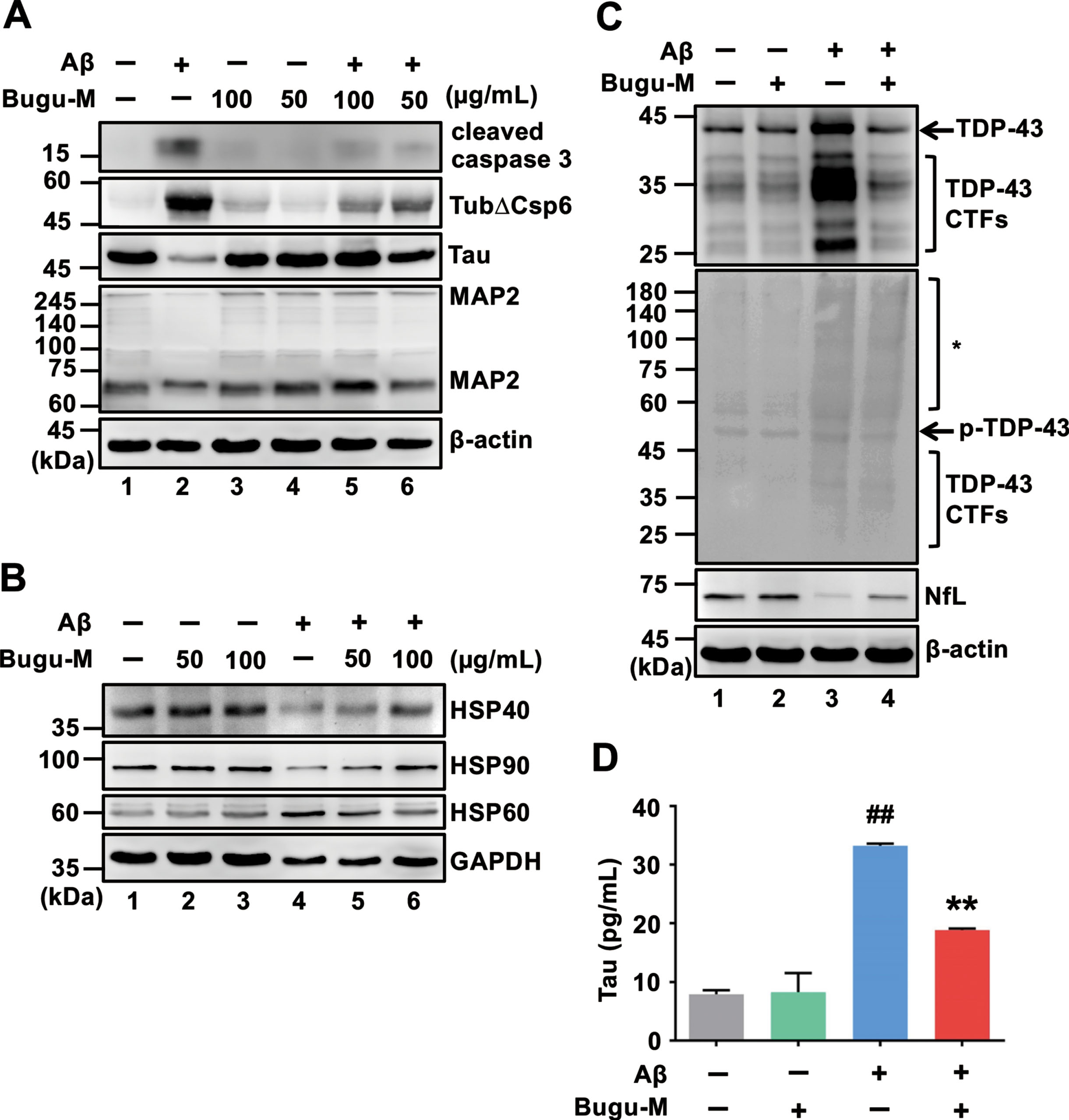
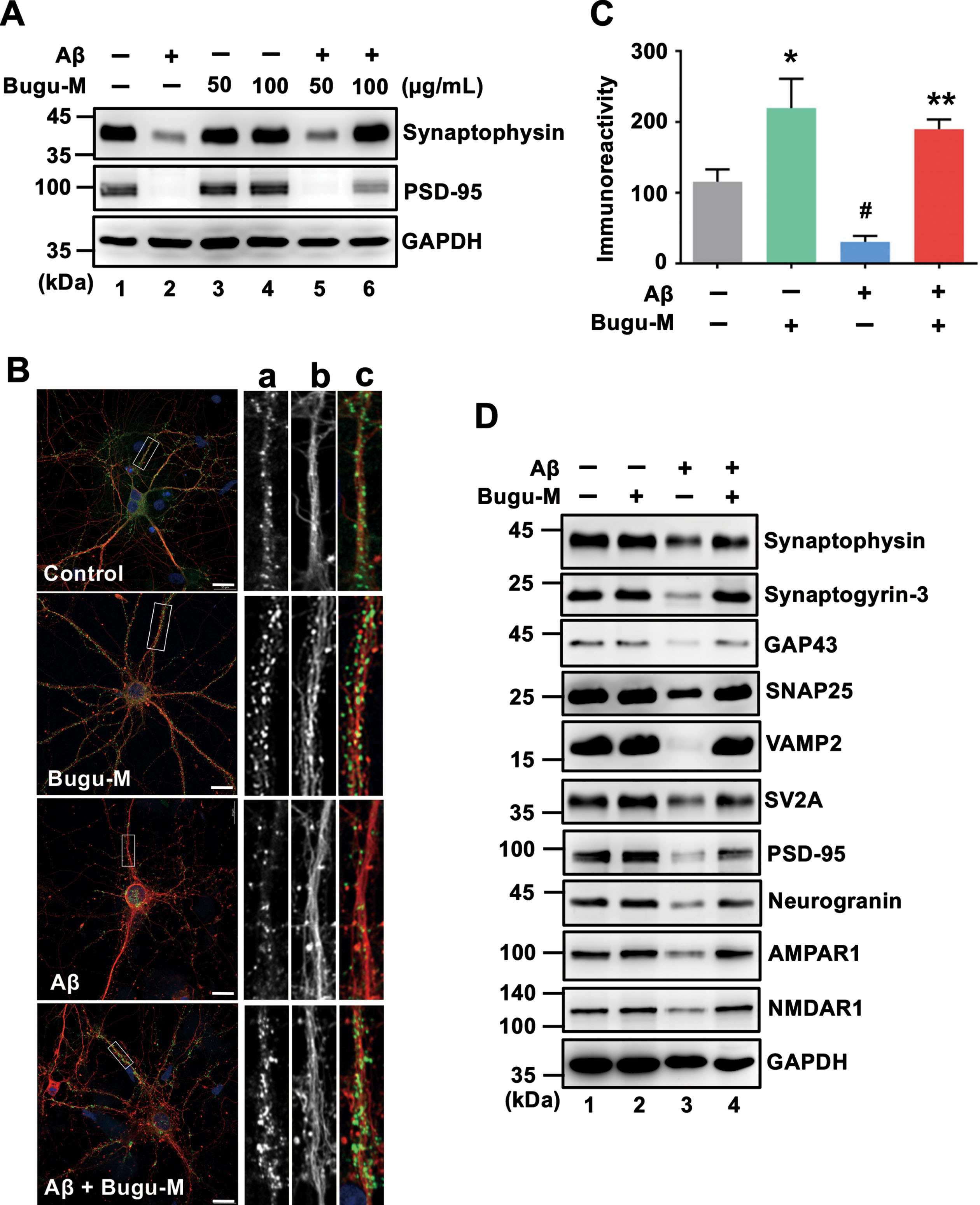
Presynaptic disturbances linking to axonal transport defects cause a great proportion of the critical early synapse loss that precedes cell death in AD [12, 13]. Since Bugu-M restores the levels of NfL, Tau and MAP2 that play a critical role on synaptic plasticity [58–60], we next investigated whether Bugu-M affects synaptic protein expression. We treated rat cortical neurons with Bugu-M (50 and 100 μg/mL) prior to incubation with 20 μM Aβ25–35. Administration of Aβ reduced synaptic proteins, synaptophysin and postsynaptic density protein 95 (PSD-95) (Fig. 3A, lane 2), which were elevated by Bugu-M at 100 μg/mL (Fig. 3A, lane 6). Concomitantly, PSD-95 immunolabeling also showed 100 μg/mL Bugu-M significantly enhanced staining intensity of PSD-95 that was diminished by Aβ (Fig. 3B, C).
To substantiate the aforementioned findings, we analyzed membrane-enriched fractions from Aβ-treated rat cortical neurons by immunoblotting. The levels of presynaptic (synaptophysin, synaptogyrin-3, GAP43, SNAP25, VAMP2, SV2A) and postsynaptic (PSD-95, neurogranin) proteins were increased in 100 μg/mL of Bugu-M-treated neurons (Fig. 3D, lane 4) relative to Aβ (20 μM) group (Fig. 3D, lane 3). These data corroborated that Bugu-M increased synaptic density post Aβ exposure.
Fig. 3
Bugu-M reverses Aβ-evoked synaptic disruption in neurons. A) Rat cortical neurons were pre-incubated with Bugu-M (50 and 100 μg/mL) for 1 h before Aβ25–35 stimulation for 72 h. Insoluble fractions were subject to immunoblotting for PSD-95 and synaptophysin. GAPDH was used as a loading control. B) Rat cortical neurons were pretreated with 100 μg/mL Bugu-M for 1 h and then incubated with Aβ25–35 for 72 h. Synaptic integrity was assessed by immunofluorescence co-labeling of PSD-95 (green, a) and β-III tubulin (red, b), and higher magnification images are shown in a-c (merge). Confocal images are single optical slices, and camera and microscope setting were equivalent for comparisons between groups. Shown are representative images. Scale bar, 10 μm. (C) PSD-95 immunoreactivity was quantified by ImageJ. Data are shown as mean±SEM (n = 3). Statistical significance between groups were analyzed by one-way ANOVA and Fisher‘s LSD test. #p < 0.05 versus the untreated control group; **p < 0.01 versus the Aβ group. D) Rat cortical neurons were pre-incubated with 100 μg/mL Bugu-M for 1 h followed by Aβ25–35 incubation for 72 h. The membrane-enriched fractions were subject to Westen blot analysis for presynaptic proteins (synaptophysin, synaptogyrin-3, GAP43, SNAP25, VAMP2, and SV2A), postsynaptic proteins (PSD-95 and neurogranin), and glutamate receptors (AMPAR1 and NMDAR1). GAPDH was used as a loading control. Densitometric quantification of immnoreactive bands was conducted using ImageJ, and the fold changes to control bands are analyzed in Supplementary Figure 4.
Bugu-M activates glutamate receptor signaling in neurons receiving Aβ
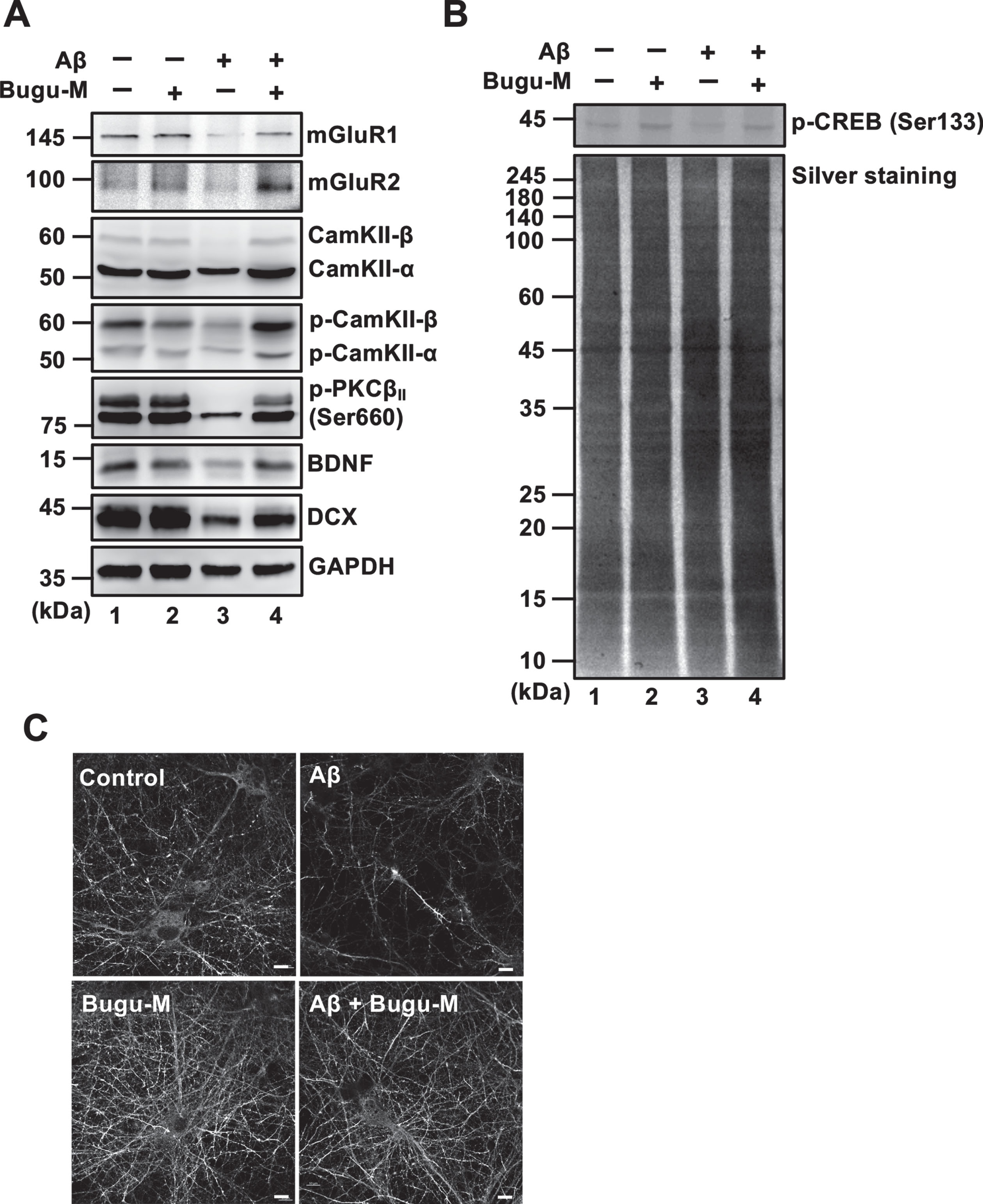
PSD-95 overexpression is reported to induce the maturation of glutamatergic presynaptic terminals [61], which could attribute to the elevation in synapse density detected by Bugu-M treatment. Synaptic expression and transmission of glutamate receptors are regulated by PSD-95 via direct physical interaction or coordination with auxiliary proteins [62] that is upregulated by Protein kinase C (PKC) activation [63]. Thus, we next investigated if Bugu-M increases cortical synapse density via a glutamate receptor-dependent mechanism. Aβ (20 μM) treatment for 72 h markedly reduced glutamate receptors including α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), N-methyl-D-aspartate receptor (NMDAR) (Fig. 3D, lane 3), mGluR1 and mGluR2 (Fig. 4A, lane 3); however, these effects were reversed by Bugu-M at 100 μg/mL (Fig. 3D and 4A, lane 4). In coincidence with this, phospho-calcium/calmodulin-dependent protein kinase II (CaMKII) and phospho-PKC were also elevated in Bugu-M treated cells compared to Aβ group (Fig. 4A, lane 4).
Given that the increase in NMDAR expression and postsynaptic CAMKII or PKC may activate cAMP response element-binding protein (CREB) phosphorylation [64], we addressed if Bugu-M affects CREB activation. As shown in Fig. 4B, Aβ (20 μM) diminished activated CREB levels after 72 h incubation, while 100 μg/mL Bugu-M increased phospho-CREB upon Aβ exposure (Fig. 4B, lane 4). These findings suggest that Bugu-M is capable to reserve the expression of glutamate receptors and their downstream signaling that is ablated by Aβ.
Fig. 4
Bugu-M activates glutamate receptor signaling in Aβ-treated neurons. Rat cortical neurons were pre-incubated with Bugu-M (100 μg/mL) for 1 h before Aβ25–35 incubation for 72 h. Whole lysates were analyzed by immunoblotting for glutamate receptors (mGluR1, mGluR2), CamKII, Phospho-CamKII, phospho-PKC, neurogenesis related factors (BDNF, DCX) (A), and phospho-CREB (B). GAPDH was used as a loading control. Quantification of immnoreactive bands was conducted using ImageJ, and the fold changes to control bands are analyzed in Supplementary Figure 5C.) Immunolabeling of DCX in rat cortical neurons. Scale bar, 10 μm. Confocal images are maximum projections, and camera and microscope setting were equivalent for comparisons between groups. Shown are representative images.
Bugu-M precludes Aβ-evoked decrement of neurogenic factors
CREB is essential not only for synaptic plasticity but also for neuronal survival [65]. Phosphorylation of CREB increases synaptic plasticity- and neuronal survival-related protein expression, including brain-derived neurotrophic factor (BDNF) and doublecortin (DCX) [65, 66]. Hence, we tested whether Bugu-M affects BDNF and DCX by Western blot analysis. Unsurprisingly, Aβ (20 μM) abolished the expression of BDNF and DCX (Fig. 4A, lane 3) in cortical neurons after 72 h incubation. Nevertheless, administration of Bugu-M (100 μg/mL) increased BDNF and DCX levels compared with its Aβ counterparts (Fig. 4A, lane 4). DCX immunolabeling also demonstrated Bugu-M at 100 μg/mL reserved staining intensity that was diminished by 20 μM Aβ treatment (Fig. 4C).
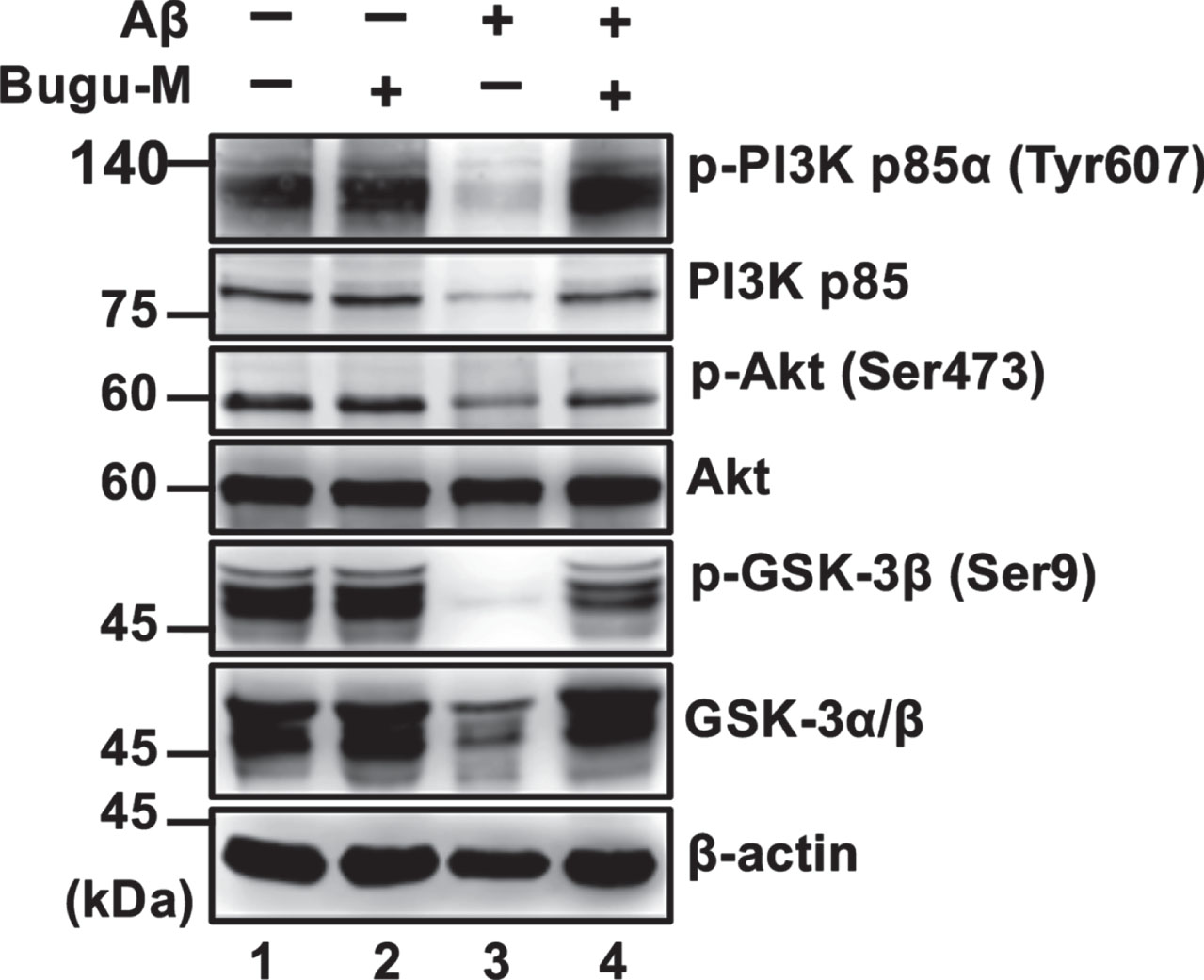
Bugu-M abrogates Aβ-induced suppression of PI3K/Akt/GSK-3β signaling
Besides CaMKII, Akt (commonly called protein kinase B, PKB) [67] can directly phosphorylate CREB. To scrutinize whether Bugu-M activates CREB through various cellular kinases, we evaluated Akt by treating neurons with Bugu-M and Aβ25–35. In accordance with remaining CREB activity by Bugu-M upon Aβ25–35 exposure in neurons, suppression of phospho-Akt by Aβ was also reversed in Bugu-M-treated neurons (Fig. 5, lane 4). Consistent with this, neurons receiving Bugu-M increased phospho-PI3K p85α and phospho-GSK-3β in the presence of Aβ compared to Aβ alone group (Fig. 5, lane 4). Our findings suggest that Bugu-M promotes CREB phosphorylation through PKC, PI3K/Akt/GSK-3β, and CaMKII signaling.
Fig. 5
Bugu-M abrogates Aβ-induced suppression of PI3K signaling in neurons. Rat cortical neurons were pre-incubated with Bugu-M (100 μg/mL) for 1 h followed by Aβ25–35 stimulation for 72 h. Membrane-enriched fractions were subject to Western blot analysis for phospho-PI3K, PI3K, Phospho-Akt, Akt, phospho-GSK-3β, and GSK-3. β-actin was used as a loading control. Quantification of immnoreactive bands was conducted by ImageJ, and the fold changes to control bands are analyzed in Supplementary Figure 6.
Bugu-M mitigates cognitive performance in 3×Tg-AD mice
In 3×Tg-AD mice, cognitive and synaptic deficits are known to develop from 3 months of age before Aβ deposition (6 months) and tauopathy (12 months) present [44]. Based on known abnormalities in presynaptic and postsynaptic signaling deficits linked to cognitive decline in early stages of AD [68], we extended the in vitro studies by asking if Bugu-M affects cognitive functions at early disease stages in the 3×Tg-AD mouse model, which displays both Aβ and tau pathology that mimics the neuropathology of the human form [44, 69]. Ideally, this is a period in which successful therapeutic intervention may still be achievable through preventing permanently neuronal loss stemmed from local and recoverable synapse loss.
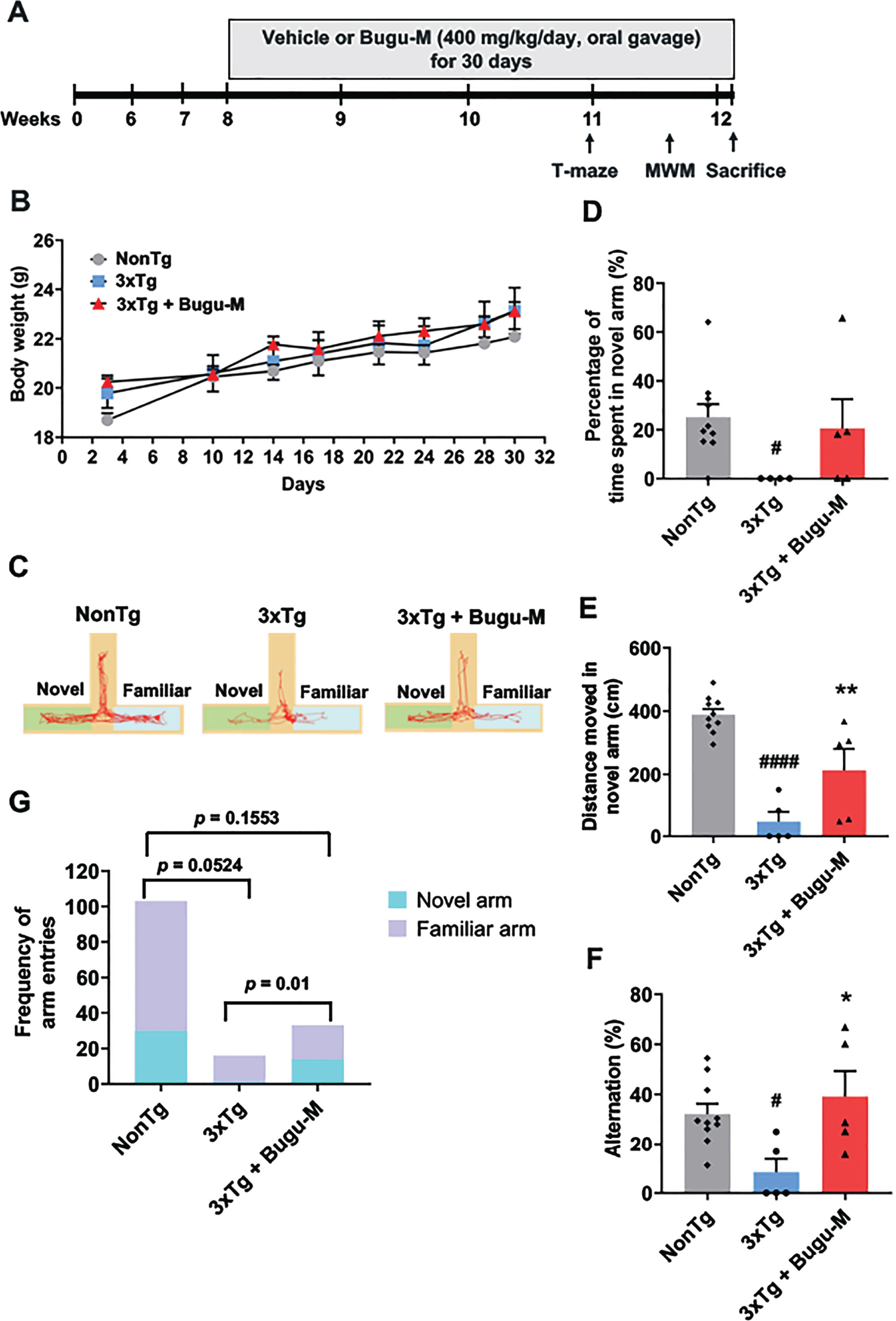
Vehicle (distilled water) and Bugu-M (400 mg/kg/day) were administered by oral gavage to 8-week-old NonTg and 3×Tg-AD mice for 30 days, followed by behavioral testing (Fig. 6A). Body weights (grams) were measured prior to behavioral testing in mice (Fig. 6B). No significant differences were observed between the body weights of 3×Tg-AD mice and NonTg mice at all time points, as well as the administration of vehicle or Bugu-M, indicating that Bugu-M did not exert toxic effects on the mice.
Fig. 6
Bugu-M improves T-maze performance in 3-month 3×Tg-AD mice. A) Illustration of experimental timeline. Acclimation was initiated 6 weeks of age. Bugu-M treatment was administered until the mice were 8 weeks of age. Bugu-M or water administration, and behavioral testing are shown along the experimental timeline of 30 days. B) Changes in body weight after Bugu-M treatment. Mice were treated with 400 mg/kg Bugu-M or distilled water by gavage for 30 days. Data are expressed as mean±SEM (n = 10 per group). No significance between groups were observed by two-way ANOVA. Spatial working memory was tested in T-maze by movement track (C), time spent in novel arm (D), distanced moved in novel arm (E), spontaneous alternation (F), and the frequency of entries into novel and familiar arms (G). Data are represented as mean±SEM (n = 3–10 per group). Statistic significance between groups were analyzed by one-way ANOVA and Fisher’s LSD test (D) or Dunnett’s multiple comparisons test (E, F), except the frequency of entries into novel and familiar arms was analyzed using Chi-square tests. #p < 0.05 and ####p < 0.0001 versus NonTg control; *p < 0.05 and **p < 0.01 versus 3×Tg-AD group.
To rule out a possible correction of motor deficits by Bugu-M as the main cause of its effects on cognitive performance, we conducted an open field test (OFT) to monitor the locomotor activity of the mice. Whilst vehicle-treated 3×Tg-AD mice showed less moving distance than their NonTg couterparts (p < 0.0001; Supplementary Figure 7A), we found no evidence that Bugu-M affected locomotor activity in 3×Tg-AD mice, as assessed by total distance travelled (p = 0.1568; Supplementary Figure 7A). However, compared to NonTg mice, both 3×Tg-AD groups had a significantly higher movement in the periphery and freezing response (p < 0.0001; Supplementary Figure 7B, C), but there was no significant difference upon Bugu-M treatment in 3×Tg-AD mice (Supplementary Figure 7B, C) for the former (p = 0.6291) and the latter (p = 0.9744) compared to the vehicle-treated 3×Tg-AD mice. Similarly, less ambulations in total distance travelled (p < 0.0001) and distance in open arms (p = 0.0012) were observed in 3×Tg-AD mice in the EPM (Supplementary Figure 7D, E), in which no alterations of total distance moved (p = 0.4573; Supplementary Figure 7D) and distance in open arms (p = 0.9583; Supplementary Figure 7E) were shown upon Bugu-M treatment in 3×Tg-AD mice. Also, the freezing response was not affected by Bugu-M relative to vehicle-treated 3×Tg-AD mice(p = 0.7949; Supplementary Figure 7F), which showed more immobility than NonTg mice(p < 0.0001; Supplementary Figure 7F). Taken together, these results suggest no apparent difference in locomotor activity and anxiety between vehicle- and Bugu-M-treated 3×Tg-AD mice.
Fig. 7
Bugu-M improves MWM performance in 3-month 3×Tg-AD mice. MWM test for movement track (A), the assessment of escape latencies to arrive visible platform over days 1–3 (B) and with hidden platform on day 4 after removing the platform (C), and goal- versus non-goal-directed strategies (D) for 3×Tg-AD and NonTg mice. Data are presented as mean±SEM (n = 8–10 per group). For escape latencies, differences among groups were determined by one-way ANOVA and Fisher’s LSD test. #p < 0.05 and ##p < 0.01 versus NonTg control; *p < 0.05 versus 3×Tg-AD group. The frequency of use of various search strategies (operationally defined as per [42]) was calculated across trials and analyzed using logistic regression. Statistical results are provided below each panel.
![Bugu-M improves MWM performance in 3-month 3×Tg-AD mice. MWM test for movement track (A), the assessment of escape latencies to arrive visible platform over days 1–3 (B) and with hidden platform on day 4 after removing the platform (C), and goal- versus non-goal-directed strategies (D) for 3×Tg-AD and NonTg mice. Data are presented as mean±SEM (n = 8–10 per group). For escape latencies, differences among groups were determined by one-way ANOVA and Fisher’s LSD test. #p < 0.05 and ##p < 0.01 versus NonTg control; *p < 0.05 versus 3×Tg-AD group. The frequency of use of various search strategies (operationally defined as per [42]) was calculated across trials and analyzed using logistic regression. Statistical results are provided below each panel.](https://ip.ios.semcs.net:443/media/adr/2023/7-1/adr-7-1-adr220056/adr-7-adr220056-g007.jpg)
T-maze alternation task is widely used for working memory in mice, which relies on the natural exploratory behavior of rodents and is considered the most rudimentary task to assess spatial working memory [45]. The representative activity of the mice was shown in Fig. 6C. Examination of the percentage of time spent in the novel arm revealed less duration of 3×Tg-AD compared to NonTg mice (p = 0.0363; Fig. 6D). However, Bugu-M showed a trend to increase the time of the mice into the novel arm relative to 3×Tg-AD group (p = 0.1185; Fig. 6D). Similarily, 3×Tg mice showed less distance travelled in novel arm (p < 0.0001 vs NonTg; Fig. 6E), which was reversed by Bugu-M treatment (p = 0.0183; Fig. 6E). In addition, the 3×Tg-AD mice showed significant reductions in the percentage of correct decisions compared to NonTg mice (p = 0.0235; Fig. 6F), whereas Bugu-M treatment prevented the decline significantly relative to 3×Tg-AD group(p = 0.0117; Fig. 6F). Finally, 3×Tg-AD mice showed less novel arm preference compared to NonTg mice (p = 0.0524; Fig. 6G), which was altered by Bugu-M (p = 0.0100; Fig. 6G) in a trend smiliar to NonTg mice (p = 0.1553; Fig. 6G).
To assess the effect of Bugu-M on spatial learning and memory, we performed MWM in 3×Tg-AD mice. The representative track of the mice was shown in Fig. 7A. No obvious difference was shown in escape latency between groups on day 1 and day 2. However, on day 3, the escape latency of 3×Tg-AD mice was significantly higher than NonTg mice (p = 0.002; Fig. 7B), indicating that 3×Tg-AD mice exhibited cognitive and spatial memory loss. On the contrary, Bugu-M treatment significantly reduced the escape latency compared to 3×Tg-AD mice receiving water (p = 0.0365; Fig. 7B). On the 4th day, the platform was removed to perform a probe trial. The 3×Tg-AD mice spent more time to the first target cross than NonTg mice (p = 0.0090; Fig. 7C), which was reduced by Bugu-M treatment (p = 0.0401; Fig. 7C). Intriguingly, when animals were analyzed by the strategies of navigational paths, random search was observed in the majority of the NonTg and 3×Tg-AD mice in the first trial (T1; Fig. 7D). In the successive trials, the number of 3×Tg-AD mice showed less goal-directed search than NonTg mice (p < 0.001; Fig. 7D), which was significantly reversed by Bugu-M treatment that exhibited more goal-directed strategy than 3×Tg-AD mice (p = 0.030; Fig. 7D).
DISCUSSION
The tremendous burden from AD in an aging society is growing in the coming decades [70]. This recognition magnifies the critical need for prevention strategies to stop or slow this disease in either preclinical or early AD (mild cognitive impairment to mild dementia). In this study, we assessed the effects of Bugu-M, a multi-herbal extract containing antler-shaped Ganoderma lucidum, Ziziphus jujuba Mill., Dimocarpus longan, and Nelumbo nucifera Gaertn. on axonal damage and synaptic dysfunction. We demonstrated that Bugu-M was capable to reverse deficits in synapse density in primary cortical neurons receiving Aβ and in cognition in 3×Tg-AD mice. These effects were along with restoration of key synaptic proteins, reversal of axonal damage, inactivation of apoptotic pathway, and enhancement of neurite outgrowth and neurogenesis by Bugu-M in neurons exposed to Aβ. As Bugu-M provides a robust preclinical evidence that targeting multiple pathways is a potential therapeutic approach in AD, and thus the efficacy of Bugu-M is of high translational significance.
Aβ toxicity triggers synaptic and neuronal degeneration in patients with AD and transgenic mouse models, which is one of the mechanisms involving in the pathogenesis of AD [8–13]. Hence, we illustrated the efficacy of Bugu-M on Aβ exposure in rat cortical neurons, in anticipation that it could provide insights into Bugu-M mechanisms or assist with the identification of new therapeutics. The fact that Bugu-M rescues cell death, increases neurite outgrowth and inhibits apoptosis by Aβ25–35 (Fig. 1D-G, 2A) corroborates earlier studies showing that Aβ evokes cytoskeletal disruption, mitochondrial dysfunction, synaptic loss, and apoptosis [4] and strongly supports the protective effects of Bugu-M on Aβ-triggered neurotoxicity. Of note, Bugu-M diminished the TDP-43 and α-tubulin fragments cleaved by caspases (Fig. 2A, C). TDP-43 aberrations are dominant in human AD brains, including phosphorylated, ubiquitinated and truncated forms of TDP-43 [71], which are associated with greater memory loss and worse brain atrophy [72]. Intriguingly, full length TDP-43 alongside caspases-3, -4, and -7 truncated variants reduce Aβ fibrillization due to their cross-seeding [73, 74].
Similar to lentiviral expression of Aβ1-42 in the rat motor cortex, Aβ25–35 triggered TDP-43 pathogenic alteration in rat cortical neurons, including upregulation, cleavage, and phosphorylation [75]. Also, nuclear localization of TDP-43 was concomitantly depleted in Aβ25-35-treated neurons as that observed in rat cortex injected with lentiviral-Aβ1-42. Truncation, phosphorylation, and ubiquitination alters TDP-43’s conformation, size, and charge, contributing to the decrease of nuclear shuttling with diffuse cytoplasmic and nuclear distribution [71, 76]. Both cytoplasmic and oligomeric aggregates of TDP-43 are neurotoxic in cultured neurons and in mouse [77, 78] through reducing the DNA binding capacity of TDP-43, thereby leading to either toxic gain of function or loss of physiological function [73]. Our previous [41] and present findings show Aβ25–35 induced caspase-3 and caspase-6 activation and mitochondrial dysfunction in rat hippocampal and cortical neurons, which might give rise to TDP-43 fragmentation and exacerbate TDP-43 toxicity that in turn, amplifies oxidative stress [79]. These results suggest that caspase activation and mitochondrial dysfunction may contribute to TDP-43 proteinopathies by Aβ25–35. In line with the effects of Bugu-M on blocking the aberrations of TDP-43, it is presumed that Bugu-M may be a potential therapy for AD as a disease-modifying agent.
Impaired mitochondrial function presents in early-stage AD, resulting in activation of caspases and apoptosis in AD patients and transgenic mouse models [48, 50]. In particular, abnormalities of Hsp60, Hsp70/HSP40, and Hsp90 are found in AD [80]. Whereas Bugu-M increased HSP40 and HSP90 levels, HSP60 was depleted by Bugu-M upon Aβ exposure in cortical neurons (Fig. 2B). This was not surprising as the complex of Hsp90 with Hsp70/Hsp40 can inhibit Aβ formation [81], while HSP60 participates in the translocation of amyloid-β protein precursor (AβPP) and Aβ to the mitochondria and induces mitochondrial dysfunction in both 3×Tg-AD mouse model and human AD subjects [82]. Overall, our studies identify an alleviating effect of Bugu-M on mitochondrial HSPs that were altered by Aβ, and thus extrapolated Bugu-M was able to restore mitochondrial function.
Accumulation of Aβ plaques in the brain poteintially damages axons, leading to cytoskeletal alterations that are essential in the pathology of neurofibrillary tangle and neurodegeneration [57, 83]. In fact, defective axonal transport by disruption of microtubular cytoskeleton in diseased axons has been demonstrated in AD brains [84], which might increase Aβ accumulation [85]. A changed proportion in the stabilization and destabilization of microtubules contributes to neuronal cytoskeletal alterations, and thus altering the dynamics of axons and synapses. In line with these findings, the immunoreactivity of β-III tubulin upon Aβ stimulation was low in rat cortical neurons (Fig. 1F), suggesting that in this amyloidogenic model, AD pathology may also attribute to unstable and depolymerized microtubules. Of importance, Bugu-M treatment caused an increase of β-III tubulin intensity and thus in the neuronal integrity (Fig. 1F, G), as revealed previously in tauopathy [86].
Tau is a microtubule-associated protein and plays a role in axonal transport [87]. Partial decrease or loss of tau expression may cause axonal degeneration, short-term memory loss, hyperactivities, and synaptic plasticity deficits [88, 89]. Of interest, extracellular tau has been identified as a possible cause of memory loss and synaptic dysfunction [90], and its release could occur prior to neuronal death or from dying neurons [91]. Concurrently, degeneration affected brain areas show progressive neuron loss, while in human brains without tangles, tau species can seed misfolding [13]. In line with this, our findings showed Aβ decreased tau expression levels (Fig. 2A) and increased extracellular Tau (Fig. 2D), which was ablated by Bugu-M treatment (Fig. 2A, D), suggesting Bugu-M may possess the ability to prevent tau abnormalities and seeding.
MAP2 is expressed in cell bodies and dendrites and is considered a microtubule stabilizer [92] as it increases microtubule rigidity [93]. Similar to tau protein, loss of MAP2 has been widely linked to AD in human samples and in animal models [94, 95]. Within the current study as well, Aβ triggered a decrease in MAP2 expression level. This ablation of MAP2 protein levels was reversed by Bugu-M treatment at 100 μg/mL (Fig. 2A). Along these lines, NfL, a crucial axonal cytoskeletal protein involved in axonal and dendritic branching and growth, and axonal integrity, has been measured for axonal degeneration in AD [96]. As cytoskeletal proteins, neurofilaments are primarily expressed in neuronal axons, with NfL the smallest of the neurofilament proteins. Importantly, CSF and plasma NfL levels are correlated to each other and are elevated in AD [97]. Our findings are similar to those reported by Tohda and colleagues wherein the expression levels of neurofilament heavy and synaptophysin were reduced in the cerebral cortex and the hippocampus of Aβ25–35-treated mice [98]; however, Bugu-M restored the NfL levels in neurons exposed to Aβ (Fig. 2C). Axons near amyloid deposits encouter changes that further promote Aβ accumulation [99]. Given that Bugu-M exerts axoprotective effects as observed for β-III tubulin, Tau, MAP2, and NfL and published observations that immunotherapy resulting in Aβ clearance can ameliorate Aβ-related axonal degeneration in AD transgenic models [100, 101], we surmise that Bugu-M may reduce Aβ neurotoxicity through protection against axonal disruption.
Aβ-related dystrophic axons contribute to synaptic damage [102], and synaptic loss is correlated with cognitive impairment in AD. Therefore, synaptic proteins are postulated as prognostic biomarkers for AD progression as well as assessment for the efficacy of disease-modifying therapies [103]. In fact, SV2A, a vesicle membrane protein expressed at synapses, is currently employed as a diagnostic marker in AD patients [104]. Increased Aβ not only affects neuronal communication in both presynaptic and postsynaptic mechanisms but also impair overall brain activity [105]. Thus, motivated by these lines of evidence, we sought to untangle the effects of Bugu-M on presynaptic and postsynaptic proteins in Aβ-treated neurons, in which Aβ had impact on synaptic loss and axonal integrity. We used presynaptic markers (e.g., synaptophysin, synaptogyrin-3, GAP43, SNAP25, VAMP2, and SV2A) and postsynaptic markers (e.g., neurogranin and PSD-95) because of their roles in synaptic communication, regulating synaptic strength and plasticity, and promoting synapse maturation [106–109]. We found that increases in presynaptic and postsynaptic proteins occurred in Bugu-M-treated neurons upon exposure to 20 μM Aβ (Fig. 3A, D). Along with the elevated number of PSD-95-positive elements, Bugu-M treatment significantly increased synapse-like puncta immunopositive for the postsynaptic scaffolding protein PSD-95 (Fig. 3B, C). Previous report has demonstrated that the synaptic content of PSD-95 is positively correlated with spine size and synaptic efficacy [110, 111]. Along with the increase of PSD-95, neurogranin was also elevated by Bugu-M in neurons receiving Aβ (Fig. 3D). Neurogranin deletion causes deficits in spatial memory and synaptic plasticity in mice [112]. On the contrary, high levels of neurogranin in CSF during prodromal AD is predictive of more rapid progression toward AD [113, 114]. Altogether, these findings indicate that Bugu-M could prevent synaptic disruption and might be able to restore synaptic function.
Furthermore, we observed increased levels of mGluR1, mGluR2, NMDAR1, and AMPAR1 (Fig. 3D, 4A) as well as phosphorylation of CamKII, PKCβII, CREB, PI3K, Akt, and GSK-3β by Bugu-M treatment (Fig. 4A, 4B, 5). Previous reports have shown that elevated Aβ attenuates glutamatergic synaptic transmission strength and plasticity [115] by the number of surface NMDARs and AMPARs [116, 117] that are regulated by PSD-95 via direct physical interaction or coordination with auxiliary proteins [62]. Activated NMDARs induces CaMKII phosphorylation and promotes its translocation to the post-synapse [118], leding to the activation of CREB associated with the formation of long-term memory [119, 120]. Besides, the activation of PI3K/Akt pathway by NMDARs induces long-term potentiation [121] and alterations in Akt phosphorylation are correlated with the staging and severity of AD [122]. Notably, binding of BDNF to TrkB can also activate Akt and subsequently phosphorylates CREB [123]. BDNF is decreased significantly in the hippocampus in AD, which affects hippocampal long-term potentiation and thus memory [124, 125]. In accordance with our findings, Aβ diminished neurogenesis-associated BDNF and DCX whose production is dependent on CREB activation [126]. Nevertheless, Bugu-M was capable to remain the expression levels of BDNF and DCX (Fig. 4A, C). Overall, these findings might open possibilities for Bugu-M as a new therapeutic agent for AD based on its effects on restoration of axonal and synaptic integrity.
Deficits in hippocampal-dependent navigation and spatial memory are frequently observed in patients with AD as the key cognitive symptoms [127, 128]. To illustrate the effect of Bugu-M on cognitive impairment, we used 3×Tg-AD mice with cognitive and synaptic deficits developed between 3 and 5 months of age before the presence of Aβ plaques and tau tangles [129, 130]. Consistent with this, we observed cognitive dysfunction in 3×Tg-AD mice at 3 months of age, which was reduced by 30-day dosing of Bugu-M at 400 mg/kg. Indeed, Bugu-M improved spontaneous alteration in T-maze and preference to novel arm (Fig. 6F, G), as well as escape latency and goal-directed search strategies (Fig. 7B-D) in MWM performance in 3×Tg-AD mice, indicating Bugu-M is beneficial to early-stage AD. It is important to note that cognitive improvement by Bugu-M is not attributed to locomotor ability as we found no significant difference in overall distance moved between Bugu-M- and vehicle-treated 3×Tg-AD mice in the OFT and EPM (Supplementary Figure 7A, D). Also, it is not because of anxiety as revealed by either unaltered time in the periphery of the open field (Supplementary Figure 7B) or distance traveled in the open arms in EPM (Supplementary Figure 7E) by Bugu-M treatment compared to 3×Tg-AD group. However, compared to NonTg mice, 3×Tg-AD mice showed greater freezing and limited exploratory behavior in the open field and EPM, implying a basal difference in performance (Supplementary Figure 7C, F). These observations are consistent with previous findings [69, 131, 132] indicating 3×Tg mice may have a lowered fear response threshold, leading to a higher level of anxiety or and further a predisposition due to AD pathology. The characteristics of an increased freezing behavior and time spent in the peripheries of the open field might be due to the increase in corticosterone responses to stressful tasks [133] or elevated brain inflammatory responses [134, 135].
Taking these measures into account, we found 3×Tg-AD mice exhibited short-term spatial memory deficits at a young age, at which Bugu-M demonstrated its capability to resume the spatial learning and memory. Therefore, our results suggest that 30-day Bugu-M treatment may be beneficial to prevent cognitive decline in AD, which is commonly first recognized as memory impairment. Despite these findings, the current studies have limitations. One limitation is the absence of the degree of AD neuropathological development in 3×Tg-AD mice. Nonetheless, as the triple transgenic mouse model (APPSwe, PS1M146V, and tauP301L) is a well-characterized model that mimics synaptic changes prior to plaque and tangle pathologies [44], we assumed 3-month 3×Tg-AD mice would display impairment in both synaptic plasticity and spatial working memory [44, 129, 134], as well as altered expression of genes involving neuronal death, apoptosis, mitochondrial functioning, synaptic modulation, and cognitive dysfunction [134, 136]. However, the exact mechanisms underlying Bugu-M effects on AD progression in 3×Tg-AD mice remains to be elucidated. Another limitation is the low number of 3×Tg-AD mice to meet the inclusion criteria of total arm entries equal or more than 3 in the T-maze test, preventing us to draw a conclusion on the potential effect of Bugu-M on 3×Tg-AD mice; for example, the time spent in the novel arm. Nevertheless, rodents’ spontaneous alternation behavior in T-maze demonstrates their memory for the previous spatial location, which is different from motivated learning-based spatial tasks [45]. Hence, the increase in the preference to novel arm and spontaneous alternation in Bugu-M-treated 3×Tg-AD displays the beneficial effects of Bugu-M on spatial working memory. Furthermore, we only used Aβ25–35 peptide to treat rat neurons. Albeit the most frequent Aβ isoforms in human AD brains are Aβ1–40 [137] and Aβ1-42 [138], the functional domain comprising amino acids 25–35 of Aβ, the actual biologically active region of Aβ, is essential for its neurotoxicity [139–145]. Physiologically, the Aβ25–35 fragment has also been found in elderly people [146], suggesting it might be involved in the development of AD. Albeit Aβ25–35 is the shortest fragment to form large β-sheet aggregates, it reserves the toxicity of the full-length Aβ [144], without requirement of aging to form aggregate and gain toxicity like its full-length counterpart [144, 147–149]. Also, it has been shown that in vitro Aβ25–35 is aggregation-prone, and its aggregation states are concentration-dependent [150]. In our experiments, we prepared Aβ25–35 stock at a concentration of 1 mM by dissolving it directly in sterile distilled water and incubated for 7 days. Under these conditions, Aβ25–35 readily formed fibrils that are more than 1 μm long and 8–12 nm in diameter (Fig. 1A). After being diluted to desired concentrations to treat primary rat neurons, however, we do not know whether Aβ25–35 formed monomers, oligomers, protofibrils, or stayed as fibrils, nor do we know the correlation between the specific aggregation states and their toxicity toward neurons. These are limitations of the present study. Although amyloid fibrils were initially recognized to be the primary toxic species, a steadily accumulating body of evidence has indicated that soluble oligomers or prefibrils can be toxic even more [151]. Additional studies will be required to determine the molecular and structural basis of Aβ25–35 aggregation and toxicity, which may correlate with the neurodegenerative process of AD.
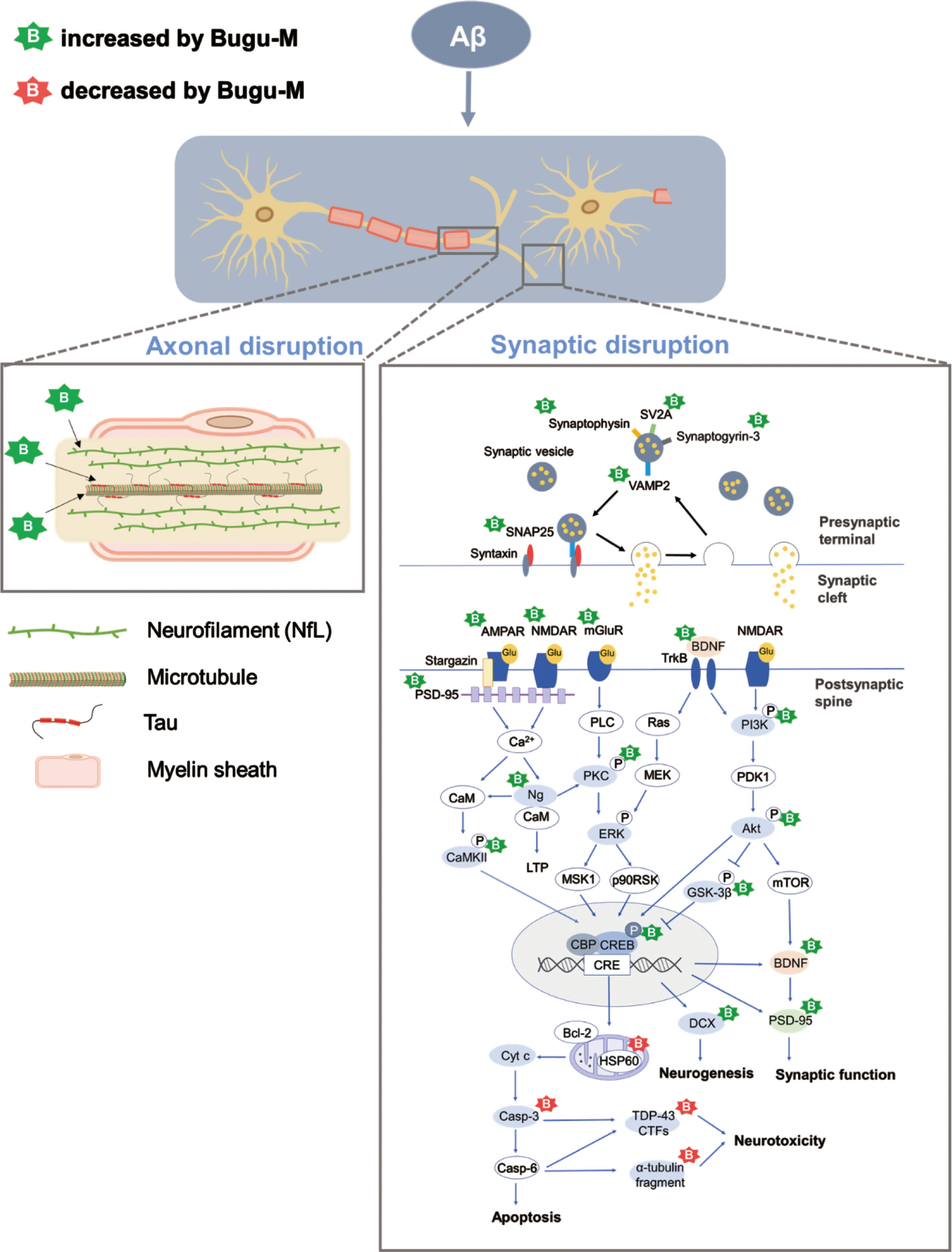
In conclusion, we found that Bugu-M preserves axonal and synaptic integrity that are ablated by Aβ treatment in rat cortical neurons (Fig. 8). Mechanistically, the neuroprotective effects of Bugu-M involve reduction of cell death, suppression of apoptotic signaling, decrease of mitochondrial dysfunction, and promotion of neurogenesis. Also, Bugu-M reverses cognitive deficits in female 3×Tg-AD mice at 3 months of age. These findings highlight the potential of Bugu-M for preventing the development of AD. Further clinical study to investigate the efficacy of Bugu-M in the early stage of AD is warranted.
Fig. 8
Proposed mechanisms of Bugu-M acting on Aβ25–35 neurotoxicity in rat cortical neurons. Aβ results in a multitude of biochemical alterations that closely mirror the pathophysiological and functional changes that characterize the progression of AD. Decrease of NfL and tau at the presynaptic nerve terminal leads to cytoskeletal disruption, concomitant with axonal damage and thus limiting the rate of axonal transport and damages presynaptic vesicle cycling. Of note, Aβ gives rise to synaptic protein depletion along with decrease in glutamate receptor (NMDAR, AMPAR, and mGluR) expression, both of which result in synaptic dysfunction and neurotoxicity. These are manifested by downstream dysregulation of CAMKII, PKC, PI3K/Akt, and CREB-associated pathways related to neurogenesis, synaptic function, and apoptosis. Bugu-M, a multi-component agent, sustained cytoskeletal and axonal integrity and its associated protein levels when given at 100 μg/mL for 72 h. In parallel, Bugu-M rescues Aβ damage by reserving synaptic proteins, remaining glutamate receptor levels, activating CREB, promoting neurogenesis, decreasing apoptosis, and modulating mitochondrial function.
ACKNOWLEDGMENTS
The authors would like to acknowledge Dr. Horng-Huei Liou and Cheng-Chiang Chen for scientific discussions. The authors are grateful to Dr. Sheng-Mao Chang for help with statistical analysis.
FUNDING
This work was supported by grants from the Ministry of Economic Affairs (Grant No. 109-EC-17-A-21-0337, 110-EC-17-A-21-0337, and 111-EC-17-A-21-0337) to Biomedical Technology and Device Research Laboratories. NSL and MDP have received research support from the Ministry of Science and Technology (Grant No. 110-2320-B-007-002, 109-2320-B-007-002, and 108-2918-1-007-013) and National Tsing Hua University (Grant No. 109A2008V6 and 108A0117V6).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-220056.
REFERENCES
[1] | Aisen PS , Jimenez-Maggiora GA , Rafii MS , Walter S , Raman R ((2022) ) Early-stage Alzheimer disease: Getting trial-ready. Nat Rev Neurol 4: , 1–11. |
[2] | Silva MVF , Loures CMG , Alves LCV , de Souza LC , Borges KBG , Carvalho MDG ((2019) ) Alzheimer’s disease: Risk factors and potentially protective measures. J Biomed Sci 26: , 33. |
[3] | Cummings J , Lee G , Nahed P , Kambar MEZN , Zhong K , Fonseca J , Taghva K ((2022) ) Alzheimer’s disease drug development pipeline: 2022. Alzheimers Dement (N Y) 8: , e12295. |
[4] | Fan L , Mao C , Hu X , Zhang S , Yang Z , Hu Z , Sun H , Fan Y , Dong Y , Yang J , Shi C , Xu Y ((2020) ) New insights into the pathogenesis of Alzheimer’s disease. Front Neurol 10: , 1312. |
[5] | Black MM , Slaughter T , Moshiach S , Obrocka M , Fischer I ((1996) ) Tau is enriched on dynamic microtubules in the distal region of growing axons. J Neurosci 16: , 3601–3619. |
[6] | Kempf M , Clement A , Faissner A , Lee G , Brandt R ((1996) ) Tau binds to the distal axon early in development of polarity in a microtubule- and microfilament-dependent manner. J Neurosci 16: , 5583–5592. |
[7] | Wang JT , Medress ZA , Barres BA ((2012) ) Axon degeneration: Molecular mechanisms of a self-destruction pathway. J Cell Biol 196: , 7–18. |
[8] | Sadleir KR , Kandalepas PC , Buggia-Prevot V , Nicholson DA , Thinakaran G , Vassar R ((2016) ) Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol 132: , 235–256. |
[9] | Kandalepas PC , Sadleir KR , Eimer WA , Zhao J , Nicholson DA , Vassar R ((2013) ) The Alzheimer’s beta-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol 126: , 329–352. |
[10] | Domínguez-Álvaro M , Montero-Crespo M , Blazquez-Llorca L , Insausti R , DeFelipe J , Alonso-Nanclares L ((2018) ) Three-dimensionalanalysis of synapses in the transentorhinal cortex of Alzheimer’sdisease patients. Acta Neuropathol Commun 6: , 20. |
[11] | Price KA , Varghese M , Sowa A , Yuk F , Brautigam H , Ehrlich ME , Dickstein DL ((2014) ) Altered synaptic structure in the hippocampus in a mouse model of Alzheimer’s disease with soluble amyloid-β oligomers and no plaque pathology. Mol Neurodegener 9: , 41. |
[12] | Lleó A , Núñez-Llaves R , Alcolea D , Chiva C , Balateu-Paños D , Colom-Cadena M , Gomez-Giro G , Muñoz L , Querol-Vilaseca M , Pegueroles J , Rami L , Lladó A , Molinuevo JL , Tainta M , Clarimón J , Spires-Jones T , Blesa R , Fortea J , Martínez-Lage P , Sánchez-Valle R , Sabidó E , BayésÁ , Belbin O ((2019) ) Changes in synaptic proteins precede neurodegeneration markers in preclinical Alzheimer’s diseasecerebrospinal fluid. Mol Cell Proteomics 18: , 546–560. |
[13] | DeVos SL , Corjuc BT , Oakley DH , Nobuhara CK , Bannon RN , Chase A , Commins C , Gonzalez JA , Dooley PM , Frosch MP , Hyman BT ((2018) ) Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Front Neurosci 12: , 267. |
[14] | Kennedy DO , Wightman EL ((2011) ) Herbal extracts and phytochemicals: Plant secondary metabolites and the enhancement of human brain function. Adv Nutr 2: , 32–50. |
[15] | Scott LJ , Goa KL ((2000) ) Galantamine: A review of its use in Alzheimer’s disease. Drugs 60: , 1095–1122. |
[16] | Kumar V ((2006) ) Potential medicinal plants for CNS disorders: An overview. Phytother Res 20: , 1023–1035. |
[17] | Chen X , Drew J , Berney W , Lei W ((2021) ) Neuroprotective natural products for Alzheimer’s disease. Cells 10: , 1309. |
[18] | Jiao YN , Zhang JS , Qiao WJ , Tian SY , Wang YB , Wang CY , Zhang YH , Zhang Q , Li W , Min DY , Wang ZY ((2022) ) Kai-Xin-San inhibits tau pathology and neuronal apoptosis in aged SAMP8 mice. Mol Neurobiol 59: , 3294–3309. |
[19] | Pradeep S , Jain AS , Dharmashekara C , Prasad SK , Kollur SP , Syed A , Shivamallu C ((2020) ) Alzheimer’s disease and herbal combination therapy: A comprehensive review. Alzheimers Dis Rep 4: , 417–429. |
[20] | Huang S , Mao J , Ding K , Zhou Y , Zeng X , Yang W , Wang P , Zhao C , Yao J , Xia P , Pei G ((2017) ) Polysaccharides from Ganoderma lucidum promote cognitive function and neural progenitor proliferation in mouse model of Alzheimer’s disease. Stem Cell Reports 8: , 84–94. |
[21] | Yu N , Huang Y , Jiang Y , Zou L , Liu X , Liu S , Chen F , Luo J , Zhu Y ((2020) ) Ganoderma lucidum triterpenoids (GLTs) reduce neuronal apoptosis via inhibition of ROCK signal pathway in APP/PS1 transgenic Alzheimer’s disease mice. Oxid Med Cell Longev 2020: , 9894037. |
[22] | Lai G , Guo Y , Chen D , Tang X , Shuai O , Yong T , Wang D , Xiao C , Zhou G , Xie Y , Yang BB , Wu Q ((2019) ) Alcohol extracts from Ganoderma lucidum delay the progress of Alzheimer’s disease by regulating DNA methylation in rodents. Front Pharmacol 10: , 272. |
[23] | Choi YJ , Yang HS , Jo JH , Lee SC , Park TY , Choi BS , Seo KS , Huh CK ((2015) ) Anti-amnesic effect of fermented Ganoderma lucidum water extracts by lactic acid bacteria on scopolamine-induced memory impairment in rats. Prev Nutr Food Sci 20: , 126–132. |
[24] | Zhang Y , Wang X , Yang X , Yang X , Xue J , Yang Y ((2021) ) Ganoderic acid A to alleviate neuroinflammation of Alzheimer’s disease in mice by regulating the imbalance of the Th17/Tregs axis. J Agric Food Chem 69: , 14204–14214. |
[25] | Zhang Y , Li H , Song L , Xue J , Wang X , Song S , Wang S ((2021) ) Polysaccharide from ameliorates cognitive impairment by regulating the inflammation of the brain-liver axis in rats. Food Funct 12: , 6900–6914. |
[26] | Zhao HL , Cui SY , Qin Y , Liu YT , Cui XY , Hu X , Kurban N , Li MY , Li ZH , Xu J , Zhang YH ((2021) ) Prophylactic effects of sporoderm-removed Ganoderma lucidum spores in a rat model of streptozotocin-induced sporadic Alzheimer’s disease. J Ethnopharmacol 269: , 113725. |
[27] | Zhou Y , Qu ZQ , Zeng YS , Lin YK , Li Y , Chung P , Wong R , Hägg U ((2012) ) Neuroprotective effect of preadministration with Ganoderma lucidum spore on rat hippocampus. Exp Toxicol Pathol 64: , 673–680. |
[28] | Sharma P , Tulsawani R ((2020) ) Ganoderma lucidum aqueous extract prevents hypobaric hypoxia induced memory deficit by modulating neurotransmission, neuroplasticity and maintaining redox homeostasis. Sci Rep 10: , 8944. |
[29] | Zhang Y , Song S , Li H , Wang X , Song L , Xue J ((2022) ) Polysaccharide from Ganoderma lucidum alleviates cognitive impairment in a mouse model of chronic cerebral hypoperfusion by regulating CD4+CD25+Foxp3+ regulatory T cells. Food Funct 13: , 1941–1952. |
[30] | Kim MJ , Jung JE , Lee S , Cho EJ , Kim HY ((2021) ) Effects of the fermented Zizyphus jujube in the amyloid β25-35-induced Alzheimer’s disease mouse model. Nutr Res Pract 15: , 173–186. |
[31] | Djeuzong E , Kandeda AK , Djiogue S , Stéphanie L , Nguedia D , Ngueguim F , Djientcheu JP , Kouamouo J , Dimo T ((2021) ) Antiamnesic andneuroprotective effects of an aqueous extract of Mill. (Rhamnaceae) on scopolamine-induced cognitiveimpairments in rats.}. Evid Based Complement Alternat Med 2021: , 5577163. |
[32] | Kandeda AK , Nguedia D , Ayissi ER , Kouamouo J , Dimo T ((2021) ) Ziziphus jujuba (Rhamnaceae) alleviates working 1291 memory impairment and restores neurochemical alter-ations in the prefrontal cortex of D-galactose-treated rats.. Evid Based Complement Alternat Med 2021: , 6610864. |
[33] | Chen CJ , Liu X , Chiou JS , Hang LW , Li TM , Tsai FJ , Ko CH , Lin TH , Liao CC , Huang SM , Liang WM , Lin YJ ((2021) ) Effects of Chinese herbal medicines on dementia risk in patients with sleep disorders in Taiwan. J Ethnopharmacol 264: , 113267. |
[34] | Li H , Lei T , Zhang J , Yan Y , Wang N , Song C , Li C , Sun M , Li J , Guo Y , Yang J , Kang T ((2021) ) Longan (Dimocarpus longan Lour.) Aril ameliorates cognitive impairment in AD mice induced by combination of D-gal/AlCl 3 and an irregular diet via RAS/MEK/ERK signaling pathway. J Ethnopharmacol 267: , 113612. |
[35] | Lee DS , Choi J , Kim SH , Kim S ((2014) ) Ameliorating effects of HX106N, a water-soluble botanical formulation, on Aβ25-35-induced memory impairment and oxidative stress in mice. Biol Pharm Bull 37: , 954–960. |
[36] | Yin Shuaizeng , Ran Q , Yang J , Zhao Y , Li C ((2020) ) Nootropic effect of neferine on aluminium chloride-indueced Alzheimer’s disease in experimental models. J Biochem Mol Toxicol 34: , e22429. |
[37] | Jung HA , Jin SE , Choi RJ , Kim DH , Kim YS , Ryu JH , Kim DW , Son YK , Park JJ , Choi JS ((2010) ) Anti-amnesic activity of neferine with antioxidant and anti-inflammatory capacities, as well as inhibition of ChEs and BACE1. Life Sci 87: , 420–430. |
[38] | Ahn YJ , Park SJ , Woo H , Lee HE , Kim HJ , Kwon G , Gao Q , Jang DS , Ryu JH ((2014) ) Effects of allantoin on cognitive function and hippocampal neurogenesis. Food Chem Toxicol 64: , 210–216. |
[39] | Kim ES , Weon JB , Yun BR , Lee J , Eom MR , Oh KH , Ma CJ ((2014) ) Cognitive enhancing and neuroprotective effect of the embryo of the Nelumbo nucifera seed. Evid Based Complement Alternat Med 2014: , 869831. |
[40] | Wu XL , Deng MZ , Gao ZJ , Dang YY , Li YC , Li CW ((2020) ) Neferine alleviates memory and cognitive dysfunction in diabetic mice through modulation of the NLRP3 inflammasome pathway and alleviation of endoplasmic-reticulum stress. Int Immunopharmacol 84: , 106559. |
[41] | Chuang KA , Li MH , Lin NH , Chang CH , Lu IH , Pan IH , Takahashi T , Perng MD , Wen SF ((2017) ) Rhinacanthin C alleviates amyloid-β fibrils’ toxicity on neurons and attenuates neuroinflammatoion triggered by LPS, amyloid-ß, and interferon-γ in glial cells. Oxid Med Cell Longev 2017: , 5414297. |
[42] | Pan IH , Li KC , Kuo ZK , Lu CH , Hsieh YW , Wen SF (2021) Composition for modulating intestinal permeability and/or treating and/or preventing leaky gut related diseases, and method for modulating intestinal permeability and/or treating and/or preventing leaky gut related diseases. US20210290710A1. |
[43] | Lin NH , Yang AW , Chang CH , Perng MD ((2021) ) Elevated GFAP isoform expression promotes protein aggregation and compromises astrocyte function. FASEB J 35: , e21614. |
[44] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 39: , 409–421. |
[45] | Wenk GL ((2001) ) Assessment of spatial memory using the T maze. Curr Protoc Neurosci 4: , 8.5B.1–8.5A.7. |
[46] | Morris R ((1984) ) Developments of a water-maze procedure for studying a spatial learning in the rat. J Neurosci Methods 11: , 47–60. |
[47] | Baeta-Corral R , Giménez-Llort L ((2015) ) Persistent hyperactivityand distinctive strategy features in the Morris water maze in3×Tg-AD mice at advanced stages of disease. BehavNeurosci 129: , 129–137. |
[48] | Rizzuto R , De Stefani D , Raffaello A , Mammucari C ((2012) ) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13: , 566–578. |
[49] | Bernardi P ((1999) ) Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol Rev 79: , 1127–1155. |
[50] | Yao J , Irwin RW , Zhao L , Nilsen J , Hamilton RT , Brinton RD ((2009) ) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106: , 14670–14675. |
[51] | Slee EA , Harte MT , Kluck RM , Wolf BB , Casiano CA , Newmeyer DD , Wang HG , Reed JC , Nicholson DW , Alnemri ES , Green DR , Martin SJ ((1999) ) Ordering the cytochrome c–initiated caspase cascade: Hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9–dependent manner. J Cell Biol 144: , 281–292. |
[52] | Klaiman G , Petzke TL , Hammond J , LeBlanc AC ((2008) ) Targets of caspase-6 activity in human neurons and Alzheimer disease. Mol Cell Proteomics 7: , 1541–1555. |
[53] | Katsumata Y , Fardo DW , Kukull WA , Nelson PT ((2018) ) Dichotomous scoring of TDP-43 proteinopathy from spe-cific brain regions in 27 academic research centers: Associations with Alzheimer⁉s disease and cerebrovas-cular disease pathologies.. Acta Neuropathol Commun 6: , 142. |
[54] | Rohn TT ((2008) ) Caspase-cleaved TAR DNA-binding protein-43 is a major pathological finding in Alzheimer’s disease. Brain Res 1228: , 189–198. |
[55] | Arai T , Hasegawa M , Akiyama H , Ikeda K , Nonaka T , Mori H , Mann D , Tsuchiya K , Yoshida M , Hashizume Y , Oda T ((2006) ) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351: , 602–611. |
[56] | Neumann M , Sampathu DM , Kwong LK , Truax AC , Micsenyi MC , Chou TT , Bruce J , Schuck T , Grossman M , Clark CM , McCluskey LF , Miller BL , Masliah E , Mackenzie IR , Feldman H , Feiden W , Kretzschmar HA , Trojanowski JQ , Lee VM ((2006) ) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: , 130–133. |
[57] | Phinney AL , Deller T , Stalder M , Calhoun ME , Frotscher M , Sommer B , Staufenbiel M , Jucker M ((1999) ) Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci 19: , 8552–8559. |
[58] | Sainio MT , Rasila T , Molchanova SM , Järvilehto J , Torregrosa-Muñumer R , Harjuhaahto S , Pennonen J , Huber N , Herukka SK , Haapasalo A , Zetterberg H , Taira T , Palmio J , Ylikallio E , Tyynismaa H ((2022) ) Neurofilament light regulates axon caliber, synaptic activity, and organelle trafficking in cultured human motor neurons. Front Cell Dev Biol 9: , 820105. |
[59] | Caceres A , Binder LI , Payne MR , Bender P , Rebhun L , Steward O ((1984) ) Differential subcellular localization of tubulin and themicrotubule-associated protein MAP2 in brain tissue as revealed byimmunocytochemistry with monoclonal hybridoma antibodies. JNeurosci 4: , 394–410. |
[60] | Elie A , Prezel E , Guérin C , Denarier E , Ramirez-Rios S , Serre L , Andrieux A , Fourest-Lieuvin A , Blanchoin L , Arnal I ((2015) ) Tauco-organizes dynamic microtubule and actin networks. Sci Rep 5: , 9964. |
[61] | El-Husseini AE , Schnell E , Chetkovich DM , Nicoll RA , Bredt DS ((2000) ) PSD-95 involvement in maturation of excitatory synapses. Science 290: , 1364–1368. |
[62] | Chen X , Levy JM , Hou A , Winters C , Azzam R , Sousa AA , Leapman RD , Nicoll RA , Reese TS ((2015) ) PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc Natl Acad Sci U S A 112: , E6983–E6992. |
[63] | Lazarevic V , Yang Y , Ivanova D , Fejtova A , Svenningsson P ((2018) ) Riluzole attenuates the efficacy of glutamatergic transmission by interfering with the size of the readily releasable neurotransmitter pool. Neuropharmacology 143: , 38–48. |
[64] | Roberson ED , English JD , Adams JP , Selcher JC , Kondratick C , Sweatt JD ((1999) ) The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci 19: , 4337–4348. |
[65] | Mayr B , Montminy M ((2001) ) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2: , 599–609. |
[66] | Shmueli O , Gdalyahu A , Sorokina K , Nevo E , Avivi A , Reiner O ((2001) ) DCX in PC12 cells: CREB-mediated transcription and neurite outgrowth. Hum Mol Genet 10: , 1061–1070. |
[67] | Du K , Montminy M ((1998) ) CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem 273: , 32377–32379. |
[68] | Chakroborty S , Hill ES , Christian DT , Helfrich R , Riley S , Schneider C , Kapecki N , Mustaly-Kalimi S , Seiler FA , Peterson DA , West AR , Vertel BM , Frost WN , Stutzmann GE ((2019) ) Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer’s disease. Mol Neurodegener 14: , 7. |
[69] | Sterniczuk R , Antle MC , Laferla FM , Dyck RH ((2010) ) Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: Part 2. Behavioral and cognitive changes. Brain Res 1348: , 149–155. |
[70] | Ashford JW , Schmitt FA , Bergeron MF , Bayley PJ , Clifford JO , Xu Q , Liu X , Zhou X , Kumar V , Buschke H , Dean M , Finkel SI , Hyer L , Perry G ((2022) ) Now is the time to improve cognitive screening andassessment for clinical and research advancement. J AlzheimersDis 87: , 305–315. |
[71] | Meneses A , Koga S , O’Leary J , Dickson DW , Bu G , Zhao N ((2021) ) TDP-43 pathology in Alzheimer’s disease. Mol Neurodegener 16: , 84. |
[72] | Josephs KA , Whitwell JL , Weigand SD , Murray ME , Tosakulwong N , Liesinger AM , Petrucelli L , Senjem ML , Knopman DS , Boeve BF , Ivnik RJ , Smith GE , Jack CR Jr , Parisi JE , Petersen RC , Dickson DW ((2014) ) TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127: , 811–824. |
[73] | Fang YS , Tsai KJ , Chang YJ , Kao P , Woods R , Kuo PH , Wu CC , Liao JY , Chou SC , Lin V , Jin LW , Yuan HS , Cheng IH , Tu PH , Chen YR ((2014) ) Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat Commun 5: , 4824. |
[74] | Li Q , Yokoshi M , Okada H , Kawahara Y ((2015) ) The cleavage pattern of TDP-43 determines its rate of clearance and cytotoxicity. Nat Commun 6: , 6183. |
[75] | Herman AM , Khandelwal PJ , Stanczyk BB , Rebeck GW , Moussa CE ((2011) ) β-amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res 1386: , 191–199. |
[76] | Zhang YJ , Xu YF , Cook C , Gendron Zhang YJ TF , Roettges P , Link CD , Lin WL , Tong J , Castanedes-Casey M , Ash P , Gass J , Rangachari V , Buratti E , Baralle F , Golde TE , Dickson DW , Petrucelli L ((2009) ) Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A 106: , 7607–7612. |
[77] | Johnson BS , Snead D , Lee JJ , McCaffery JM , Shorter J , Gitler AD ((2009) ) TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 284: , 20329–20339. |
[78] | Shih YH , Tu LH , Chang TY , Ganesan K , Chang WW , Chang PS , Fang YS , Lin YT , Jin LW , Chen YR ((2020) ) TDP-43 interacts with amyloid-beta, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease. Nat Commun 11: , 5950. |
[79] | Wang W , Wang L , Lu J , Siedlak SL , Fujioka H , Liang J , Jiang S , Ma X , Jiang Z , da Rocha EL , Sheng M , Choi H , Lerou PH , Li H , Wang X ((2016) ) The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med 22: , 869–878. |
[80] | Marino Gammazza A , Bavisotto CC , Barone R , de Macario EC , Macario AJ ((2016) ) Alzheimer’s disease and molecular chaperones: Current knowledge and the future of chaperonotherapy. Curr Pharm Des 22: , 4040–4049. |
[81] | Luo GR , Le WD ((2010) ) Collective roles of molecular chaperones in protein degradation pathways associated with neurodegenerative diseases. Curr Pharm Biotechnol 11: , 180–187. |
[82] | Walls KC , Coskun P , Gallegos-Perez JL , Zadourian N , Freude K , Rasool S , Blurton-Jones M , Green KN , LaFerla FM ((2012) ) Swedish Alzheimer mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein (APP) and beta-amyloid. J Biol Chem 287: , 30317–30327. |
[83] | Muresan V , Muresan Z ((2009) ) Is abnormal axonal transport a cause, a contributing factor or a consequence of the neuronal pathology in Alzheimer’s disease? . Future Neurol 4: , 761–773. |
[84] | Fiala JC ((2007) ) Mechanisms of amyloid plaque pathogenesis. Acta Neuropathol 114: , 551–571. |
[85] | Zhang B , Carroll J , Trojanowski JQ , Yao Y , Iba M , Potuzak JS , Hogan AM , Xie SX , Ballatore C , Smith AB , 3rd Lee VM , Brunden KR ((2012) ) The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci 32: , 3601–3611. |
[86] | Gao Y , Tan L , Yu JT , Tan L ((2018) ) Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr Alzheimer Res 15: , 283–300. |
[87] | Biundo F , Del Prete D , Zhang H , Arancio O , D’Adamio L ((2018) ) A role for tau in learning, memory and synaptic plasticity. Sci Rep 8: , 3184. |
[88] | Dawson HN , Cantillana V , Jansen M , Wang H , Vitek MP , Wilcock DM , Lynch JR , Laskowitz DT ((2010) ) Loss of tau elicits axonal degeneration in a mouse model of Alzheimer’s disease. Neuroscience 169: , 516–531. |
[89] | Puzzo D , Piacentini R , Fá M , Gulisano W , Li Puma DD , Staniszewski A , Zhang H , Tropea MR , Cocco S , Palmeri A , Fraser P , D’Adamio L , Grassi C , Arancio O ((2017) ) LTP and memory impairmentcaused by extracellular Aβ and tau oligomers isAPP-dependent. Elife 6: , e26991. |
[90] | Simón D , García-García E , Royo F , Falcón-Pérez JM , Avila J ((2012) ) Proteostasis of tau. Tau overexpression resultsin its secretion via membrane vesicles. FEBS Lett 586: , 47–54. |
[91] | Yamada K , Cirrito JR , Stewart FR , Jiang H , Finn MB , Holmes BB , Binder LI , Mandelkow EM , Diamond MI , Lee VM , Holtzman DM ((2011) ) Invivo microdialysis reveals age-dependent decrease of braininterstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci 31: , 13110–13117. |
[92] | Goodson HV , Jonasson EM ((2018) ) Microtubules and microtubule-associated proteins. Cold Spring Harb Perspect Biol 10: , a022608. |
[93] | Felgner H , Frank R , Biernat J , Mandelkow EM , Mandelkow E , Ludin B , Matus A , Schliwa M ((1997) ) Domains of neuronal microtubule-associated proteins and flexural rigidity of microtubules. J Cell Biol 138: , 1067–1075. |
[94] | Manczak M , Kandimalla R , Yin X , Reddy PH ((2018) ) Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet 27: , 1332–1342. |
[95] | Reddy PH , Yin X , Manczak M , Kumar S , Pradeepkiran JA , Vijayan M , Reddy AP ((2018) ) Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum Mol Genet 27: , 2502–2516. |
[96] | Gaetani L , Blennow K , Calabresi P , Di Filippo M , Parnetti L , Zetterberg H ((2019) ) Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry 90: , 870–881. |
[97] | Mattsson N , Andreasson U , Zetterberg H , Blennow K ((2017) ) Alzheimer’s Disease Neuroimaging I. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol 74: , 557–566. |
[98] | Tohda C , Matsumoto N , Zou K , Meselhy MR , Komatsu K ((2004) ) Abeta(25-35)-induced memory impairment, axonal atrophy, and synaptic loss are ameliorated by M1, A metabolite of protopanaxadiol-type saponins. Neuropsychopharmacology 29: , 860–868. |
[99] | Tsai J , Grutzendler J , Duff K , Gan WB ((2004) ) Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci 7: , 1181–1183. |
[100] | Brendza RP , Bacskai BJ , Cirrito JR , Simmons KA , Skoch JM , Klunk WE , Mathis CA , Bales KR , Paul SM , Hyman BT , Holtzman DM ((2005) ) Anti-Aβ antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest 115: , 428–433. |
[101] | Lombardo JA , Stern EA , McLellan ME , Kajdasz ST , Hickey GA , Bacskai BJ , Hyman BT ((2003) ) Amyloid-β antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci 23: , 10879–10883. |
[102] | Sanchez-Varo R , Trujillo-Estrada L , Sanchez-Mejias E , Torres M , Baglietto-Vargas D , Moreno-Gonzalez I , De Castro V , Jimenez S , Ruano D , Vizuete M , Davila JC , Garcia-Verdugo JM , Jimenez AJ , Vitorica J , Gutierrez A ((2012) ) Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol 123: , 53–70. |
[103] | Pei YA , Davies J , Zhang M , Zhang HT ((2020) ) The role of synaptic dysfunction in Alzheime’s disease. J Alzheimers Dis 76: , 49–62. |
[104] | Chen MK , Mecca AP , Naganawa M , Finnema SJ , Toyonaga T , Lin SF , Najafzadeh S , Ropchan J , Lu Y , McDonald JW , Michalak HR , Nabulsi NB , Arnsten AFT , Huang Y , Carson RE , van Dyck CH ((2018) ) Assessing synaptic density in Alzheimer disease with synaptic vesicle glycoprotein 2A positron emission tomographic imaging. JAMA Neurol 75: , 1215–1224. |
[105] | Gulisano W , Melone M , Ripoli C , Tropea MR , Li Puma DD , Giunta S , Cocco S , Marcotulli D , Origlia N , Palmeri A , Arancio O , Conti F , Grassi C , Puzzo D ((2019) ) Neuromodulatory action of picomolar extracellular Aβ42 oligomers on presynaptic and postsynaptic mechanisms underlying synaptic function and memory. J Neurosci 39: , 5986–6000. |
[106] | Brinkmalm A , Brinkmalm G , Honer WG , Frölich L , Hausner L , Minthon L , Hansson O , Wallin A , Zetterberg H , Blennow K , Öhrfelt A ((2014) ) SNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer’s disease. Mol Neurodegener 9: , 53. |
[107] | Allegra Mascaro AL , Cesare P , Sacconi L , Grasselli G , Mandolesi G , Maco B , Knott GW , Huang L , De Paola V , Strata P , Pavone FS ((2013) ) Invivo single branch axotomy induces GAP-43-dependent sprouting andsynaptic remodeling in cerebellar cortex. Proc Natl Acad Sci US A 110: , 10824–10829. |
[108] | Zetterberg H , Blennow K ((2015) ) Neurogranin levels in cerebrospinal fluid: A new addition to the Alzheimer disease diagnostic toolbox. JAMA Neurol 72: , 1237–1238. |
[109] | Kim MJ , Futai K , Jo J , Hayashi Y , Cho K , Sheng M ((2007) ) Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron 56: , 488–502. |
[110] | Matsuzaki M , Ellis-Davies GCR , Nemoto T , Miyashita Y , Iino M , Kasai H ((2001) ) Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4: , 1086–1092. |
[111] | Cane M , Maco B , Knott G , Holtmaat A ((2014) ) The relationship betweenPSD-95 clustering and spine stability in vivo.. J Neurosci 34: , 2075–2086. |
[112] | Huang K-P , Huang FL , Jäger T , Li J , Reymann KG , Balschun D ((2004) ) Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. J Neurosci 24: , 10660–10669. |
[113] | Kvartsberg H , Duits FH , Ingelsson M , Andreasen N , Öhrfelt A , Andersson K , Brinkmalm G , Lannfelt L , Minthon L , Hansson O , Andreasson U , Teunissen CE , Scheltens P , Van der Flier WM , Zetterberg H , Portelius E , Blennow K ((2014) ) Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement 11: , 1180–1190. |
[114] | Portelius E , Zetterberg H , Skillbäck T , Törnqvist U , Andreasson U , Trojanowski JQ , Weiner MW , Shaw LM , Mattsson N , Blennow K Alzheimer’s Disease Neuroimaging Initiative ((2015) ) Cerebrospinal fluid neu-rogranin: Relation to cognition and neurodegeneration in Alzheimer’s disease. Brain 138: , 3373–3385. |
[115] | Hsia AY , Masliah E , McConlogue L , Yu GQ , Tatsuno G , Hu K , Kholodenko D , Malenka RC , Nicoll RA , Mucke L ((1999) ) Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A 96: , 3228–3233. |
[116] | Chapman PF , White GL , Jones MW , Cooper-Blacketer D , Marshall VJ , Irizarry M , Younkin L , Good MA , Bliss TV , Hyman BT , Younkin SG , Hsiao KK ((1999) ) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2: , 271–276. |
[117] | Walsh DM , Klyubin I , Fadeeva JV , Cullen WK , Anwyl R , Wolfe MS , Rowan MJ , Selkoe DJ ((2002) ) Naturally secreted oligomers of amyloidβ protein potently inhibit hippocampal long-termpotentiation in vivo. Nature 416: , 535–539. |
[118] | Stan TL , Sousa VC , Zhang X , Ono M , Svenningsson P ((2015) ) Lurasidone and fluoxetine reduce novelty-induced hypophagia and NMDA receptor subunit and PSD-95 expression in mouse brain. Eur Neuropsychopharmacol 25: , 1714–1722. |
[119] | Muller M , Cardenas C , Mei L , Cheung KH , Foskett JK ((2011) ) Constitutive cAMP response element binding protein (CREB) activation by Alzheimer’s disease presenilin-driven inositol trisphosphate receptor (InsP3R) Ca2+ signaling. Proc Natl Acad Sci U S A 108: , 13293–13298. |
[120] | Pugazhenthi S , Wang M , Pham S , Sze CI , Eckman CB ((2011) ) Downregulation of CREB expession in Alzheimer’s brain and in Abeta-treated rat hippocampal neurons. Mol Neurodegener 6: , 60. |
[121] | Abbott JJ , Howlett DR , Francis PT , Williams RJ ((2008) ) Abeta(1-42) modulation of Akt phosphorylation via alpha7 nAChR and NMDA receptors. Neurobiol Aging 29: , 992–1001. |
[122] | Pei JJ , Khatoon S , An WL , Nordlinder M , Tanaka T , Braak H , Tsujio I , Takeda M , Alafuzoff I , Winblad B , Cowburn RF , Grundke-Iqbal I , Iqbal K ((2003) ) Role of protein kinase B in Alzheimer’s neurofibrillary pathology. Acta Neuropathol 105: , 381–392. |
[123] | Yin C , Deng Y , Liu Y , Gao J , Yan L , Gong Q ((2018) ) Icariside II ameliorates cognitive impairments induced by chronic cerebral hypoperfusion by inhibiting the amyloidogenic pathway: Involvement of BDNF/TrkB/CREB signaling and up-regulation of PPARα and PPARγ in rats. Front Pharmacol 9: , 1211. |
[124] | Bambah-Mukku D , Travaglia A , Chen DY , Pollonini G , Alberini CM ((2014) ) A positive autoregulatory BDNF feedback loop via C/EBPβ mediates hippocampal memory consolidation. J Neurosci 34: , 12547–12559. |
[125] | Zhang Z , Liu X , Schroeder JP , Chan CB , Song M , Yu SP , Weinshenker D , Ye K ((2014) ) 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 39: , 638–650. |
[126] | Jagasia R , Steib K , Englberger E , Herold S , Faus-Kessler T , Saxe M , Gage FH , Song H , Lie DC ((2009) ) GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J Neurosci 29: , 7966–7977. |
[127] | Jheng SS , Pai MC ((2009) ) Cognitive map in patients with mild Alzheimer’s disease: A computer-generated arena study. Behav Brain Res 200: , 42–47. |
[128] | Kalová E , Vlcek K , Jarolímová E , Bures J ((2005) ) Allothetic orientation and sequential ordering of places is impairedin early stages of Alzheimer’s disease: Corresponding results inreal space tests and computer tests. Behav Brain Res 159: , 175–186. |
[129] | Clark JK , Furgerson M , Crystal JD , Fechheimer M , Furukawa R , Wagner JJ ((2015) ) Alterations in synaptic plasticity coincide with deficits in spatial working memory in presymptomatic 3xTg-AD mice. Neurobiol Learn Mem 125: , 152–162. |
[130] | Stevens LM , Brown RE ((2015) ) Reference and working memory deficits in the 3xTg-AD mouse between 2 and 15-months of age: A cross-sectional study. Behav Brain Res 278: , 496–505. |
[131] | Gimenez-Llort L , Blazquez G , Canete T , Johansson B , Oddo S , Tobena A , Laferla FM , Fernandez-Teruel A ((2007) ) Modeling behavioral and neuronal symptoms of Alzheimer’s disease in mice: A role for intraneuronal amyloid. Neurosci Biobehav Rev 31: , 125–147. |
[132] | Hebda-Bauer EK , Simmons TA , Sugg A , Ural E , Stewart JA , Beals JL , Wei Q , Watson SJ , Akil H ((2013) ) 3xTg-AD mice exhibit an activatedcentral stress axis during early-stage pathology. J AlzheimersDis 33: , 407–422. |
[133] | Clinton LK , Billings LM , Green KN , Caccamo A , Ngo J , Oddo S , McGaugh JL , LaFerla FM ((2007) ) Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis 28: , 76–82. |
[134] | Janelsins MC , Mastrangelo MA , Oddo S , LaFerla FM , Federoff HJ , Bowers WJ ((2005) ) Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein1 expression specifically within the entorhinal cortex of triple trans-genic Alzheimer’s disease mice.. J Neuroinflammation 2: , 23. |
[135] | Kitazawa M , Cheng D , Tsukamoto MR , Koike MA , Wes PD , Vasilevko V , Cribbs DH , LaFerla FM ((2011) ) Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol 187: , 6539–6549. |
[136] | Gatta V , D’Aurora M , Granzotto A , Stuppia L , Sensi SL ((2014) ) Early and sustained altered expression of aging-related genes in young 3xTg-AD mice. Cell Death Dis 5: , e1054. |
[137] | Mori H , Takio K , Ogawara M , Selkoe DJ ((1992) ) Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J Biol Chem 267: , 17082–17086. |
[138] | Näslund J , Schierhorn A , Hellman U , Lannfelt L , Roses AD , Tjernberg LO , Silberring J , Gandy SE , Winblad B , Greengard P , Nordstedt C , Terenius L ((1994) ) Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci U S A 91: , 8378–8382. |
[139] | Stepanichev MY , Moiseeva YV , Lazareva NA , Gulyaeva NV ((2005) ) Studies of the effects of fragment (25-35) of beta-amyloid peptide on the behavior of rats in a radial maze. Neurosci Behav Physiol 35: , 511–518. |
[140] | Limón ID , Díaz A , Mendieta L , Chamorro G , Espinosa B , Zenteno E , Guevara J ((2009) ) Amyloid-beta(25-35) impairs memory andincreases NO in the temporal cortex of rats.. Neurosci Res 63: , 129–137. |
[141] | Gao L , Zhou F , Wang KX , Zhou YZ , Du GH , Qin XM ((2020) ) Baicalein protects PC12 cells from Abeta(25)(-)(35)-induced cytotoxicity via inhibition of apoptosis and metabolic disorders. Life Sci 248: , 117471. |
[142] | Zhang L , Guo Y , Wang H , Zhao L , Ma Z , Li T , Liu J , Sun M , Jian Y , Yao L , Du Y , Zhang G ((2019) ) Edaravone reduces Abeta-induced oxidative damage in SH-SY5Y cells by activating the Nrf2/ARE signaling pathway. Life Sci 221: , 259–266. |
[143] | Lin CI , Chang YC , Kao NJ , Lee WJ , Cross TW , Lin SH ((2020) ) 1,25(OH)(2)D(3) alleviates Abeta(25-35)-induced Tau hyperphosphorylation, excessive reactive oxygen species, and apoptosis through interplay with glial cell line-derived neurotrophic factor signaling in SH-SY5Y cells. Int J Mol Sci 21: , 4215. |
[144] | Pike CJ , Walencewicz-Wasserman AJ , Kosmoski J , Cribbs DH , Glabe CG , Cotman CW ((1995) ) Structure-activity analyses of beta-amyloid peptides: Contributions of the beta 25-35 region to aggregation and neurotoxicity. J Neurochem 64: , 253–265. |
[145] | D’Errico G , Vitiello G , Ortona O , Tedeschi A , Ramunno A , D’Ursi AM ((2008) ) Interaction between Alzheimer’s Abeta(25-35) peptide and phospholipid bilayers: The role of cholesterol. Biochim Biophys Acta 1778: , 2710–2716. |
[146] | Kubo T , Nishimura S , Kumagae Y , Kaneko I ((2002) ) In vivo conversionof racemized beta-amyloid ([D-Ser 26]A beta 1-40) to truncated andtoxic fragments ([D-Ser 26]A beta 25-35/40) and fragment presence inthe brains of Alzheimer’s patients. J Neurosci Res 70: , 474–483. |
[147] | Lau L , Barnham KJ , Curtain CC , Masters CL , Separovic F ((2003) ) Magnetic resonance studies of β-amyloid peptides. Aust J Chem 56: , 349–356. |
[148] | Varadajaran S , Kanski J , Aksenova M , Landerback C , Butterfield DA ((2001) ) Different mechanisms of oxidative stress and neurotoxicity for Alzheimer’s A beta(1-42) and A beta(25-35). J Am Chem Soc 123: , 5625–5631. |
[149] | Ali FE , Separovic F , Barrow CJ , Cherny RA , Fraser F , Bush AI , Masters CL , Barnham KJ ((2005) ) Methionine regulates copper/hydrogenperoxide oxidation products of Abeta. J Pept Sci 11: , 353–360. |
[150] | Millucci L , Ghezzi L , Bernardini G , Santucci A ((2010) ) Conformations and biological activities of amyloid beta peptide 25-35. Curr Protein Pept Sci 11: , 54–67. |
[151] | Holscher C , Gengler S , Gault VA , Harriott P , Mallot HA ((2007) ) Soluble beta-amyloid[25-35] reversibly impairs hippocampal synapticplasticity and spatial learning. Eur J Pharmacol 561: , 85–90. |


