
Porphyromonas gingivalis Conditioned Medium Induces Amyloidogenic Processing of the Amyloid-β Protein Precursor upon in vitro Infection of SH-SY5Y Cells
Abstract
Background:
Cleavage of the amyloid-β protein precursor (AβPP) mediated by host secretase enzymes, releases several fragments including amyloid-β (Aβ40 and Aβ42).
Objective:
To determine if Porphyromonas gingivalis conditioned medium cleaved AβPP to release Aβ40 and Aβ42.
Methods:
The SH-SY5Y cell line was challenged, in vitro, with P. gingivalis (Pg381) conditioned medium in the presence/absence of cytokines. The cells and their supernatants were assessed for AβPP cleavage fragments by immunoblotting and transmission electron microscopy.
Results:
Western blotting of the cell lysates with the anti-AβPP C-terminal antibody demonstrated variable molecular weight bands corresponding to full length and fragmented AβPP in lanes treated with the following factors: Tryptic soy broth (TSB), Pg381, IL-6, Pg381 + IL-1β, and Pg381 + TNF-α. The low molecular weight bands corresponding to the C99 dimerized fragment were observed in the Pg381 and interlukin-6 (IL-6) treated groups and were significantly more intense in the presence of Pg381 with either IL-6 or TNF-α. Bands corresponding to the dimerized C83 fragment were observed with cells treated with TNF-α alone and with Pg381 combined with IL-1β or IL-6 or TNF-α. The anti-Aβ antibody detected statistically significant Aβ40 and Aβ42, levels when these two Aβ species were pooled across test samples and compared to the untreated group. Electron microscopic examination of the supernatants demonstrated insoluble Aβ40 and Aβ42.
Conclusion:
These observations strongly imply that AβPP is an infection responsive protein cleaved via the amyloidogenic pathway on exposure to conditioned medium and in the presence of pro-inflammatory mediators.
INTRODUCTION
Periodontal disease is one of the most common diseases worldwide, affecting up to 50% of the global population [1]. This oral inflammatory disorder involves destruction of the soft and hard tissues which support teeth including gingivae, periodontal ligament, and alveolar bone. The chronic inflammation is a result of poor oral hygiene and the presence of bacterial infection from supra and subgingival microbiota, whereby Porphyromonas gingivalis has been identified as a keystone pathogen [2, 3]. P. gingivalis is a Gram-negative bacterium which has been extensively studied as one of the main etiological factors in periodontal disease due to its ability to produce several virulence factors and extracellular proteases such as lipopolysaccharides, fimbriae, and gingipains which can lead to the destruction of the tooth supporting structures and eventual tooth loss [4]. P. gingivalis has also been implicated in the instigation of inflammatory mediator (cytokines) release locally, which may lead to systemic spread via circulating blood [5, 6]. Hence, P. gingivalis attains greater significance not only in periodontitis but also as an initiator for pro-inflammatory cytokines (IL-1β, IL-6, and others) [7], which has been shown to contribute to the development of multiple systemic diseases including cardiovascular disease, hyperglycemia, and insulin resistance [8–14]. Periodontitis is a chronic condition, and its effective management requires frequent chairside periodontal therapy with patient’s adherence to oral hygiene and professional compliance.
Alzheimer’s disease (AD) is the most prevalent subtype of dementia comprising 60 to 80% of all dementia cases [15]. AD is a neurodegenerative disorder that is characterised by the clinical presentation and the two main neuropathological lesions: neurofibrillary tangles (NFTs) and amyloid-β (Aβ) plaques [16]. Advancing age and inheritance of the Apolipoprotein E, allele 4 (APOE ɛ4) susceptibility gene [17, 18] are widely recognised as risk factors contributing to the development of AD. An indirect effect of inheriting the APOE ɛ4 gene variant with respect to patients with periodontal disease is, considered to be, an increased susceptibility of the host to microbial infections [19–21]. This genotype has also been linked to increased inflammatory burden in terms of cytokines (systemic circulation and in the brain) in response to bacterial LPS [21–23], and further increase the risk for an individual to both periodontitis, and AD [24]. Previous studies have demonstrated that oral infections with P. gingivalis alone or introduction of its lipopolysaccharide (LPS), can lead to the development of key AD neuropathological hallmark lesions [25–30]. In addition, inflammatory markers and signalling pathways pertinent to impaired learning and spatial memory [25–28], were also observed in the periodontitis-AD infection mice models [29–31].
While the efforts to elucidate AβPP amyloidogenic pathway mechanisms that release Aβ under the influence of P. gingivalis infection in vivo are still in progress, the present study aimed to investigate, in vitro effects of P. gingivalis (FDC 381) conditioned medium on the amyloidogenic processing of AβPP in the wild-type SH-SY5Y cell line. To our knowledge, previous studies have utilized P. gingivalis infection for in-situ release of these virulence factors, which mainly contain proteinases such as gingipains, LPS, outer membrane vesicles, fimbriae, short chain fatty acids (butyric acid and propionic acids), and bacterial DNA [32]. Since gingipains is the major constituent of the conditioned medium, our hypothesis is that it is the gingipain enzymes from the crude conditioned medium that may be responsible for AβPP cleavage. This study used the non-differentiated SH-SY5Y cell line as an in vitro model of neurodegeneration in line with previous publications [33] for drug discovery and for AβPP processing [34]. These two studies motivated us to choose the SH-SY5Y cells [34], over modified in vitro cell lines like “SH-SY5Y.APP695” cells [34], which in our hands also demonstrated two phenotypes.
MATERIALS AND METHODS
P. gingivalis conditioned medium as a source of crude virulence factors
P. gingivalis (ATCC strain FDC 381) was cultured under anaerobic conditions to density of 5×109 per mL in Tryptone soya broth (VWR) supplemented with hemin (5μg/mL final concentration, Sigma-Aldrich UK), and menadione (1μg/mL final concentration) by a collaborator and co-author (SC) while based at the University of Florida, USA. The conditioned media containing crude virulence factors alongside the sterile growth medium Tryptone soya broth or TSB (as control) were received frozen on dry ice at the University of Central Lancashire and were kept at –80°C before use under specified cell culture conditions.
In vitro cell culture
The human neuroblastoma SH-SY5Y, CRL-2266™ cell line was obtained from the American Type Culture Collection (ATCC) (https://www.atcc.org/products/ SH-SY5Y CRL-2266™). The cell line was cultured in Dulbecco’s modified Eagle’s medium (DMEM, Lonza code BE12-604F) supplemented with 5% fetal bovine serum (FBS). Test cells at 80% confluence were challenged for 48 h with/without P. gingivalis medium containing virulence factors diluted 1 : 5 in DMEM only and with recombinant cytokine(s) IL-1β at 10 ng/ml (Life Technologies, Thermo Fisher Scientific, UK); TNF-α at 10 ng/ml and IL-6 at 5 ng/ml, (Gibco, Thermo Fisher Scientific, UK) alone and combined with P. gingivalis virulence factors. The sterile microbiological growth (TSB) medium, used as a control, was diluted 1 : 5 in DMEM (University of Florida, USA). This dilution factor was determined by a prior in-house cytotoxicity assay, which demonstrated P. gingivalis conditioned medium containing virulence factors diluted 1 : 5 was non-toxic to this non-differentiated neuroblastoma cell line in culture. The SH-SY5Y cells were grown in flasks, incubated at 37°C in a humidified atmosphere of 5% CO2, 95% air.
Cell lysate preparation
Following 48-h treatments, the supernatants from the SH-SY5Y cells were collected into sterile 15 mL Falcon tubes. They were centrifuged (Sigma3-16K, Sigma-Aldrich, UK) for 5 min at 3000 rpm to remove cell debris, and stored at –80°C. The remaining adhered cells in the flasks were washed twice with 5 mL phosphate buffered saline (PBS), detached, and suspended in PBS. Once detached, the cells were transferred into 15 mL sterile, Falcon tubes and centrifuged at 1000 rpm for 10 min. After draining off excess PBS, the cell pellet was lysed in 250μL volume of lysis buffer (RIPA buffer, pH 8.0: containing 50 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.5% Sodium deoxycholate, 0.5% (v/v) NP-40, 1% sodium dodecyl sulphate. 1/100 final of phenylmethanesulphonyl or PMSF and 5 mM dithiothreitol, 5% protease inhibitor cocktails 2 and 3 (Sigma-Aldrich, UK). The cells were vortex mixed and incubated on ice for 10 min with further vortex mixing in between. The cell homogenate now in Eppendorf tubes were centrifuged at 14,000 rpm for 20 min (Sigma 1–14 microfuge). The liquid phase was withdrawn, transferred into new pre-labelled 1.5 mL Eppendorf tubes, and used to determine total protein following a protein assay (see below). All cell lysates were stored at –80°C until needed for western blotting (AβPP cleavage products).
Protein assay
Coomassie Blue Protein Assay (Sigma-Aldrich, UK) was performed to determine total protein concentration as described fully elsewhere [13].
Western blot analysis of cell lysates
All lysates from the SH-SY5Y cell line (P. gingivalis virulence factors, cytokines, and combined P. gingivalis virulence factors with cytokines) were separated by SDS-PAGE on precast 12% mini-protean TGX stain-free linear gels (BioRad Laboratories, USA). Protein ladder, (PageRuler Plus, 26619, from Thermo Scientific) was loaded in the first well of each gel. All samples (10μg) of total protein in Laemmli reducing sample buffer containing 0.3% mercaptoethanol (Alfa Aesar) was electroblotted onto polyvinylidine difluoride (PVDF) membranes, as previously described by Poole et al. [13]. The membranes were incubated overnight in rabbit anti-AβPP C-terminal (cat no A8717) antibody diluted 1/4000 and rabbit anti-AβPP N-terminal (cat no A8967) diluted 1/1000, both from Sigma Aldrich UK, and mouse anti-Aβ (clone 6e10) from BioLegend was diluted 1/1000 in PBS/5% milk on a rotary device at 4°C. Chemiluminescent substrate (SuperSignal® West Pico, ThermoFisher Scientific, UK) was prepared and applied to the membranes according to the manufacturer’s instructions. Specific protein signal from the membranes was visualized using a ChemicDoc® (Bio-Rad, UK) and images captured with Image Lab® Software Version 3.0.1. India ink (Windsor & Newton) was used to stain the membrane to determine the amount of protein transferred onto the membrane(s) as a loading control. Briefly the PVDF membrane was washed free of the chemiluminescent substrate in PBS (3 changes for 1 min each while agitating the membrane in a plastic container). Neat India ink (100μL) in 50 mL of PBS was mixed and the membrane was transferred in it while shaking on a rotary platform. Within 3–5 min, the bands appeared on the membrane where the protein had electrophoresed. The membrane was gently washed free of the ink in distilled water (3 changes for 1 min each). The membrane was allowed to air dry at room temperature for at least 1 h and preferably overnight. Densitometry was carried out on the bands (in triplicate blots) using the Image J software, and the resulting data was normalized to the loading control.
Transmission electron microscopy detection of Aβ fibrils
For negative staining, all supernatants were dispensed as 50μL droplets onto Nescofilm (Bando Chemical Industries Ltd. Kobe, Japan) in a humidified chamber. Formvar/carbon coated nickel grids (400 mesh) were floated, film side down, on droplets of each supernatant for 20 min, transferred to 1% glutaraldehyde in PBS for 10 min, and washed by placing on droplets of PBS (3 x 1 min) and reverse osmosis-purified water for 6 x 1 min. Grids were then transferred to 2% uranium acetate for 10 min, lifted off with fine forceps, excess stain solution was drained with Whatman 50 filter paper and grids allowed to air dry before examination in a Philips CM12 TEM (FEI UK Ltd) at 80 kV. Images were captured with a Megaview III camera and AnalySIS software (Soft Imaging System GmbH, Germany). Aβ fibrils, prepared from a commercial peptide Aβ42 (Severn Biotech, UK) that was reconstituted to 1 mg/mL in Ringer’s buffer acted as a positive control.
Statistical analysis
The data was evaluated using the Statistical Package for the Social Sciences (SPSS). The data was non-parametric, and the Mann-Whitney U test was used to determine the differences between two independent groups which were SH-SY5Y untreated (TSB) versus Pg381; TNF-α versus Pg381 + TNF-α; IL-1β versus Pg381 + IL-1β; and IL-6) versus Pg381 + IL-6 treated groups. Aβ40 and Aβ42 levels were also evaluated statistically as per above groups and as relative Aβ40 and Aβ42 across all test conditions versus TSB control. A statistical probability (p value), less than or equal to≤0.05 was considered significant.
RESULTS
Western blot: Rabbit anti-AβPP C-terminal antibody
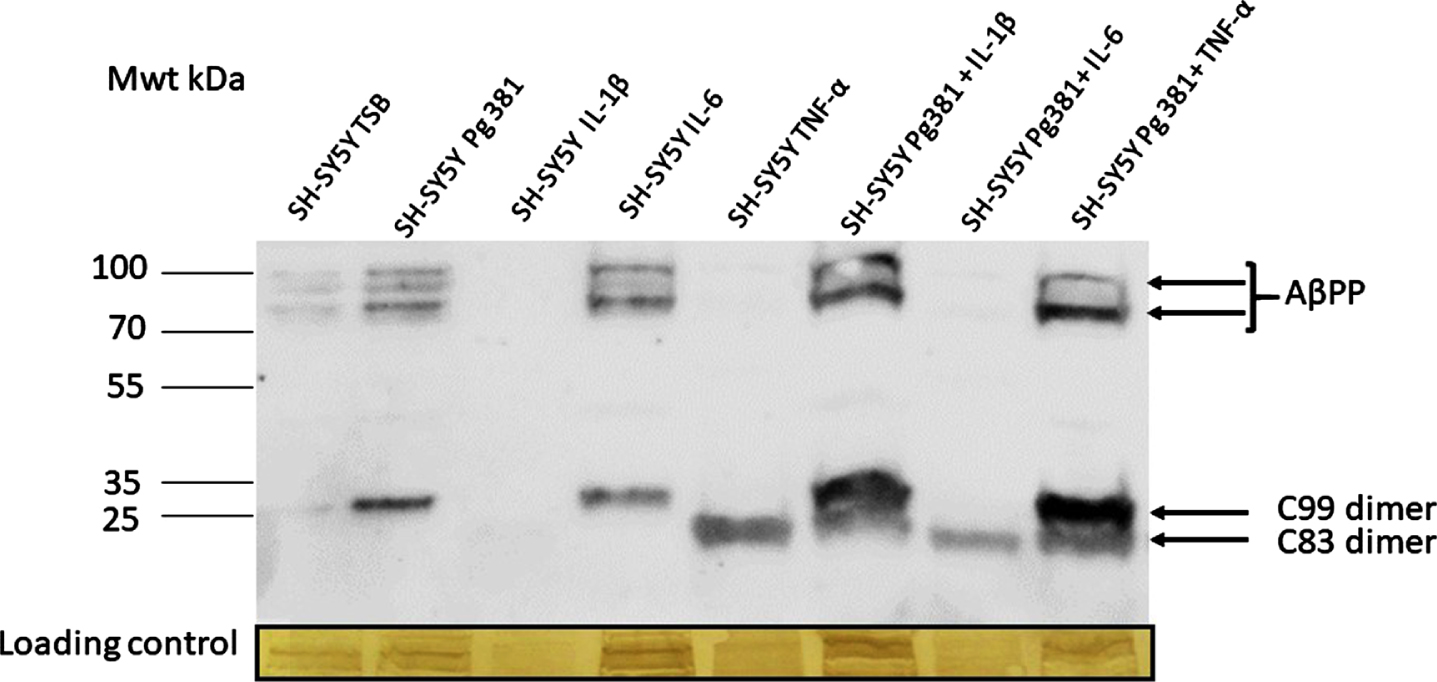
Following immunoblotting of the cell lysates, several high molecular weight bands in the region of 100-80 kDa corresponding to AβPP695, AβPP751, and AβPP770 (not quantified) were observed (Fig. 1A) in lanes control or TSB, Pg381, IL-6, Pg381 + IL-1β, and Pg381 + TNF-α. The low molecular bands corresponding to the AβPP C99 dimerized fragment were observed in the Pg381 treated group, IL-6 and significantly more intensely with Pg381 + IL-1β and Pg381 + TNF-α. The dimerized band corresponding to the AβPP C83 fragment was observed in groups with cells treated with TNF-α alone and with Pg381 + IL-1β, Pg381 + IL-6, and Pg381 + TNF-α.
Fig. 1A
Representative western blot chemi doc image (A) from rabbit anti-AβPP C-terminal antibody; and related densitometry of bands from C99 (B) and C83 (C). A) Immunoblot of the cell lysate with anti-AβPP C-terminal antibody. Distinct bands around the 100 kDa molecular weight size corresponding to AβPP in lanes with the prefix SH-SY5Y and then TSB, Pg381, IL-6, Pg381 + IL-1β, and Pg381 + TNF-α. The low molecular bands correspond to the AβPP C99 fragment in the Pg381 treated group, IL-6 and significantly more intensely with Pg381 + IL-1β and Pg381 + TNF-α. The AβPP C83 band is in lanes TNF-α and with Pg381 + IL-1β, Pg381 + IL-6, and Pg381 + TNF-α.
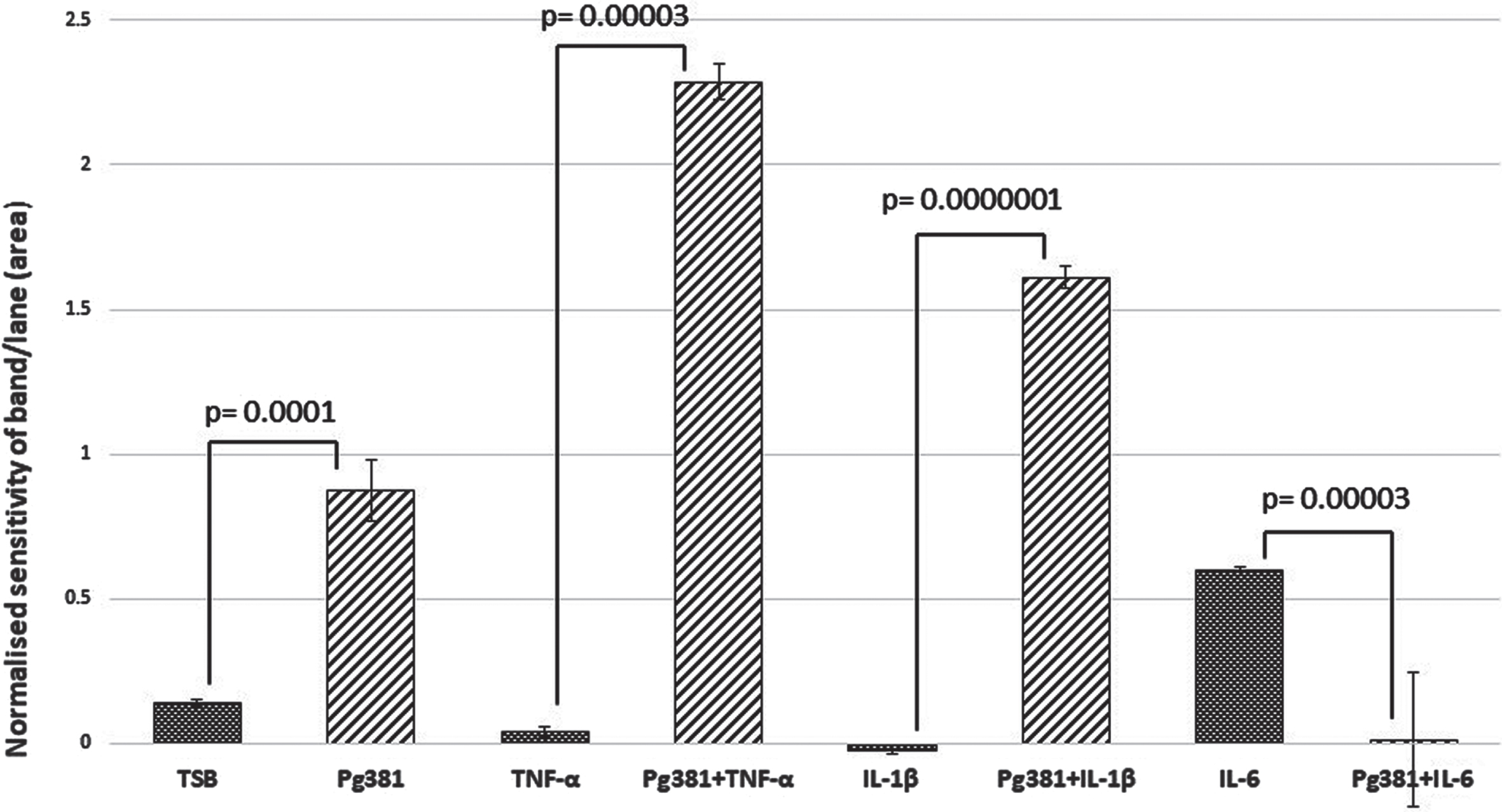
C99 dimer bands densitometry
Statistically the results for the C99 bands (Fig. 1B), were highly significant across (TSB) versus Pg381; TNF-α versus Pg381 + TNF-α; IL-1β versus Pg381 + IL-1β; and IL-6 versus Pg381 + IL-6.
Fig. 1B
Densitometric analysis of the cell lysate immunoblotted with anti-AβPP C-terminal antibody C99. All error bars represent standard error of mean. Figure 1B: Statistical analysis of the results for the C99 bands (B): p values were highly significant across (TSB) versus Pg381 (p = 0.0001); TNF-α versus Pg381 + TNF-α (p = 0.00003); IL-1β versus Pg381 + IL-1β (p = 0.0000001); and IL-6 versus Pg381 + IL-6 (p = 0.00003).
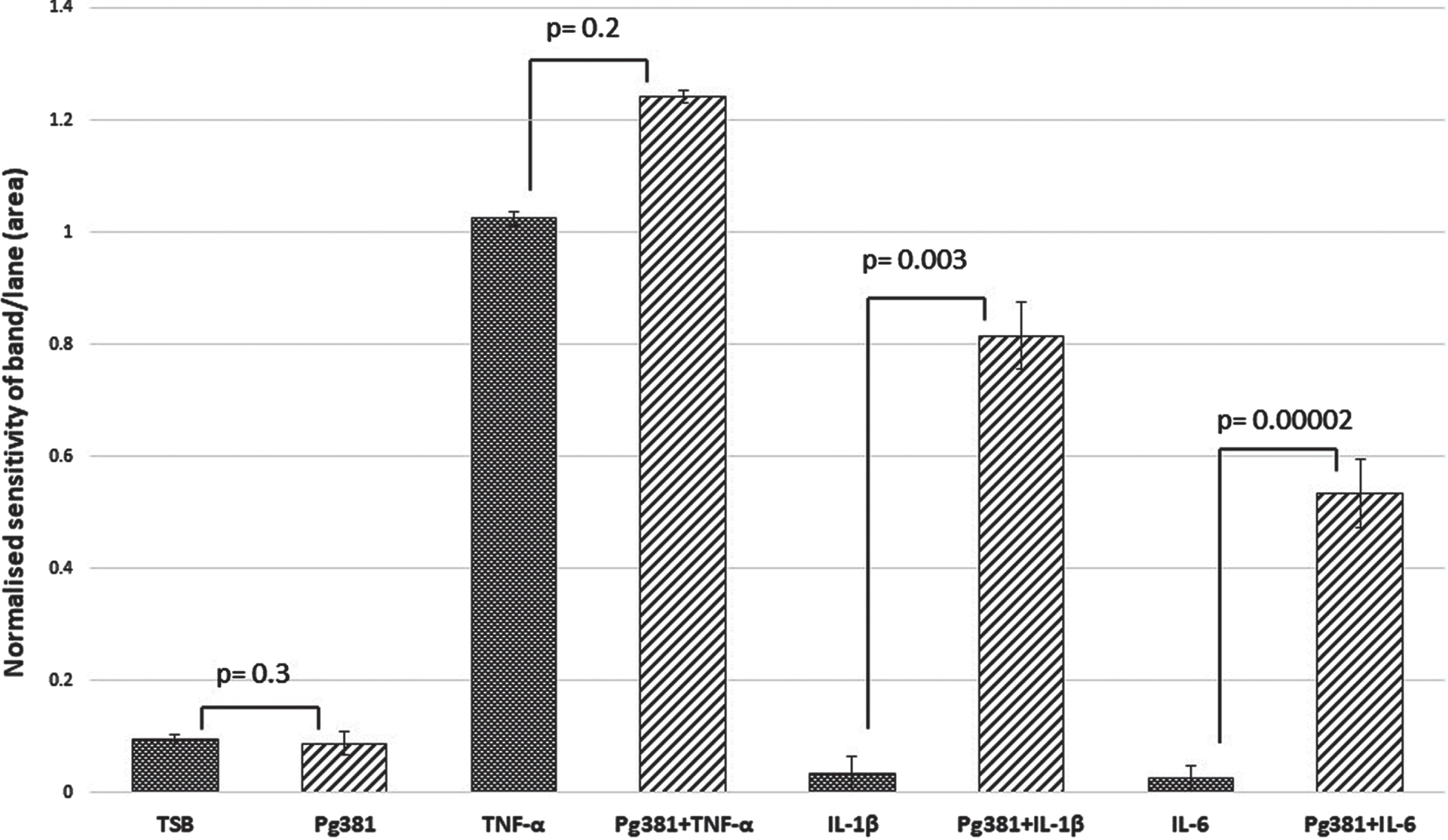
C83 dimer bands densitometry
Statistically the results for the AβPP C83 fragment (Fig. 1 C), were not significant across (TSB) versus Pg381 (p = 0.3); TNF-α versus Pg381 + TNF-α (p = 0.3); but were very significant for IL-1β versus Pg381 + IL-1β (p = 0.003); and highly significant for IL-6 versus Pg381 + IL-6 (p = 0.00002).
Fig. 1C
Densitometrical analysis of the cell lysate immunoblotted with anti-AβPP C-terminal antibody. Statistical analysis of the results for the AβPP C83 fragment (Fig. 1 C). p values were not significant across (TSB) versus Pg381 (p = 0.3); TNF-α versus Pg381 + TNF-α (p = 0.2); but are very significant for IL-1β versus Pg381 + IL-1β (p = 0.003); and highly significant for IL-6 versus Pg381 + IL-6 (p = 0.00002).
Western blot: Rabbit anti-AβPP N-terminal antibody
The cell lysates immunoblotted with the anti-AβPP N-terminal antibody only weakly detected the full AβPP bands at the 1/4000 dilution tested and other bands which were considered non-specific.
Western blot: Mouse anti-Aβ (clone 6e10) antibody
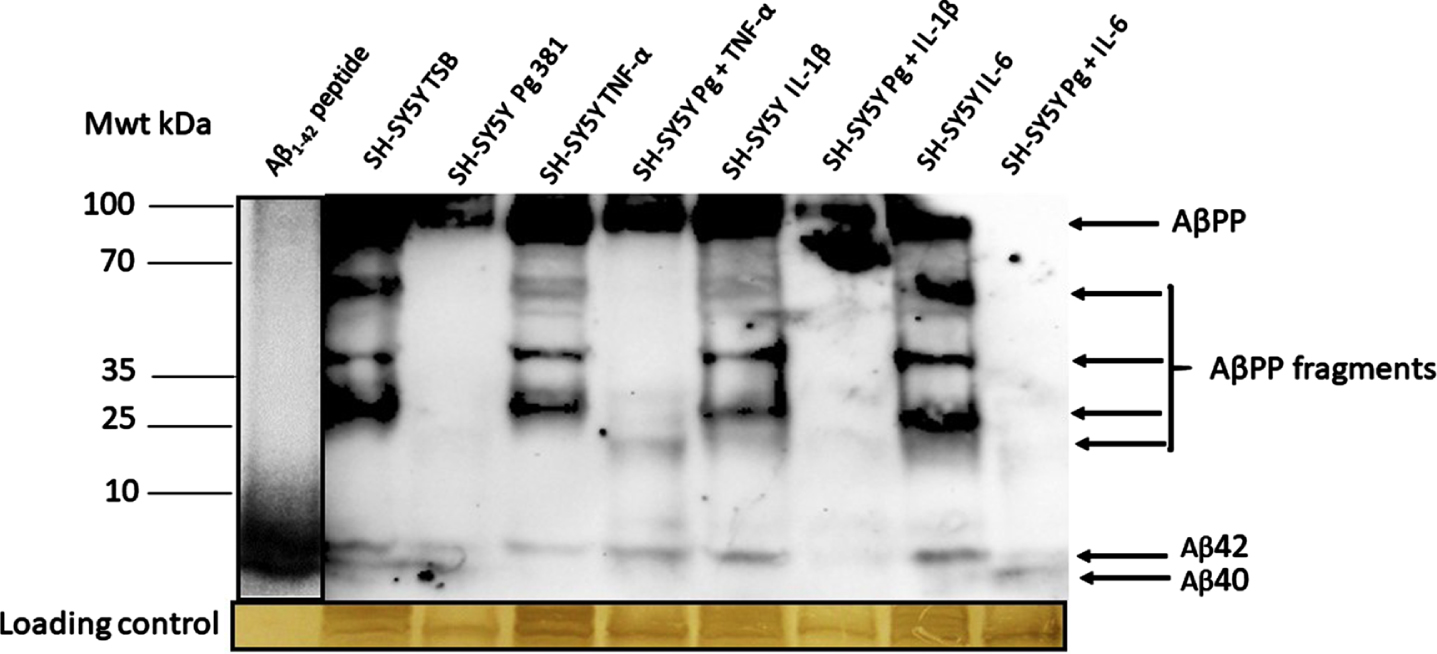
The anti-Aβ antibody apart from detecting the high molecular weight AβPP, also detected a range of AβPP cleavage fragments and above all detected the Aβ40 and Aβ42 bands as compared with the standard, reconstituted Aβ42 peptide (Fig. 2A). The Aβ40 and Aβ42 bands were statistically significant when Aβ40 and Aβ42 was pooled across test samples and compared to the untreated (TSB) for both Aβ species.
Fig. 2A
Western blot image from mouse anti-Aβ antibody; and related densitometric analysis of Aβ40 and Aβ42 bands (B, Aβ40; C, Aβ42) and when data was pooled for the relative abundance of Aβ. The anti-Aβ antibody apart from detecting the full AβPP and variable length cleavage fragments were also detected in addition to Aβ40 and Aβ42.
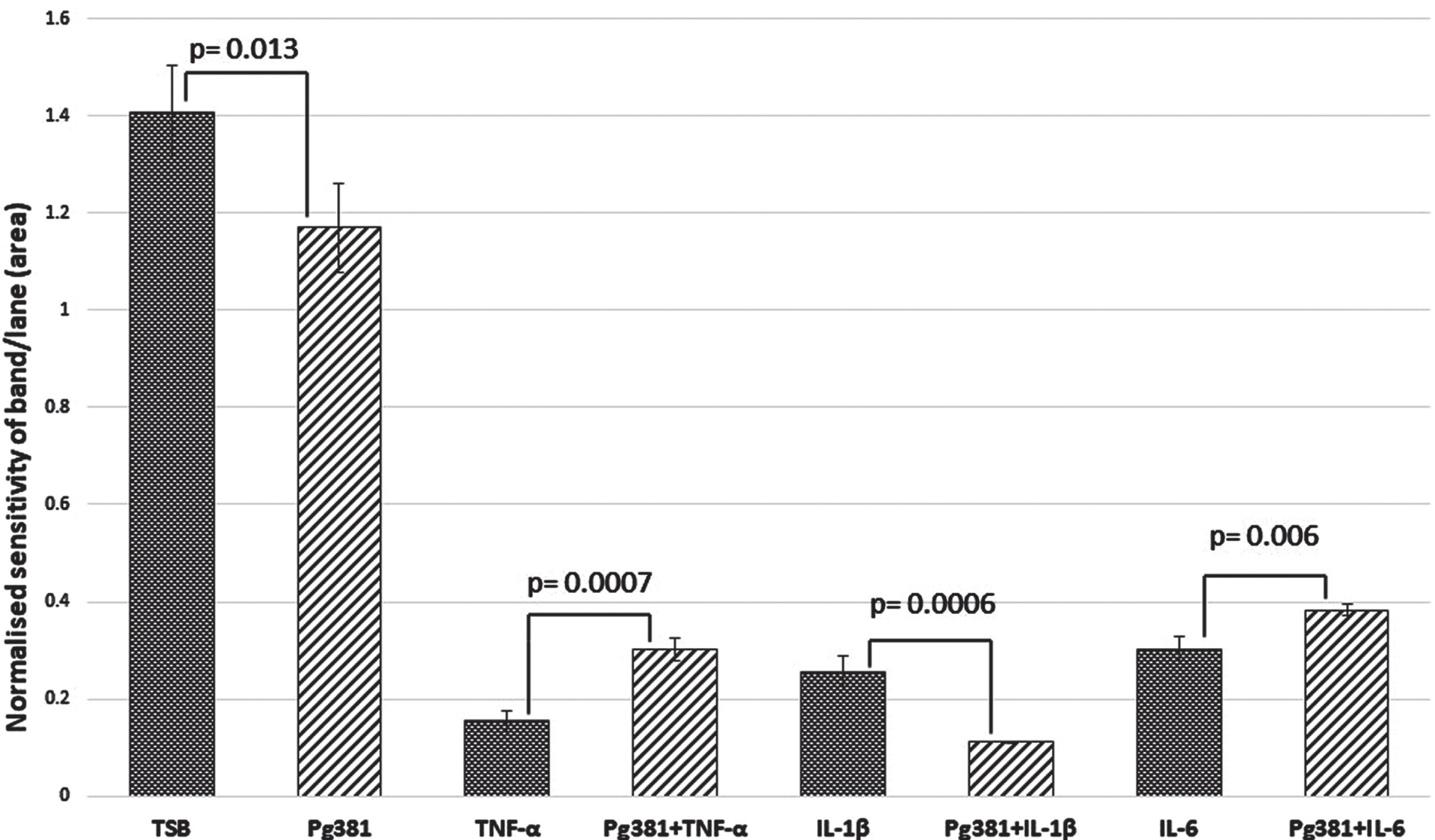
Aβ40 densitometry
Statistically the results for the Aβ40 band (Fig. 2B), were significant across (TSB) versus Pg381 (p = 0.013); TNF-α versus Pg381 + TNF-α (p = 0.0007). IL-1β was highly significant versus Pg381 + IL-1β (p = 0.0006); and IL-6 versus Pg381 + IL-6 (p = 0.006).
Fig. 2B
Aβ40 densitometry. Statistical analysis of the results for the Aβ40 band (B) p values were significant across TSB versus Pg381 (p = 0.013); TNF-α versus Pg381 + TNF-α (p = 0.0007). IL-1β was significant versus Pg381 + IL-1β (p = 0.0006); and IL-6 was significant versus Pg381 + IL-6 (p = 0.006).
Aβ42 densitometry
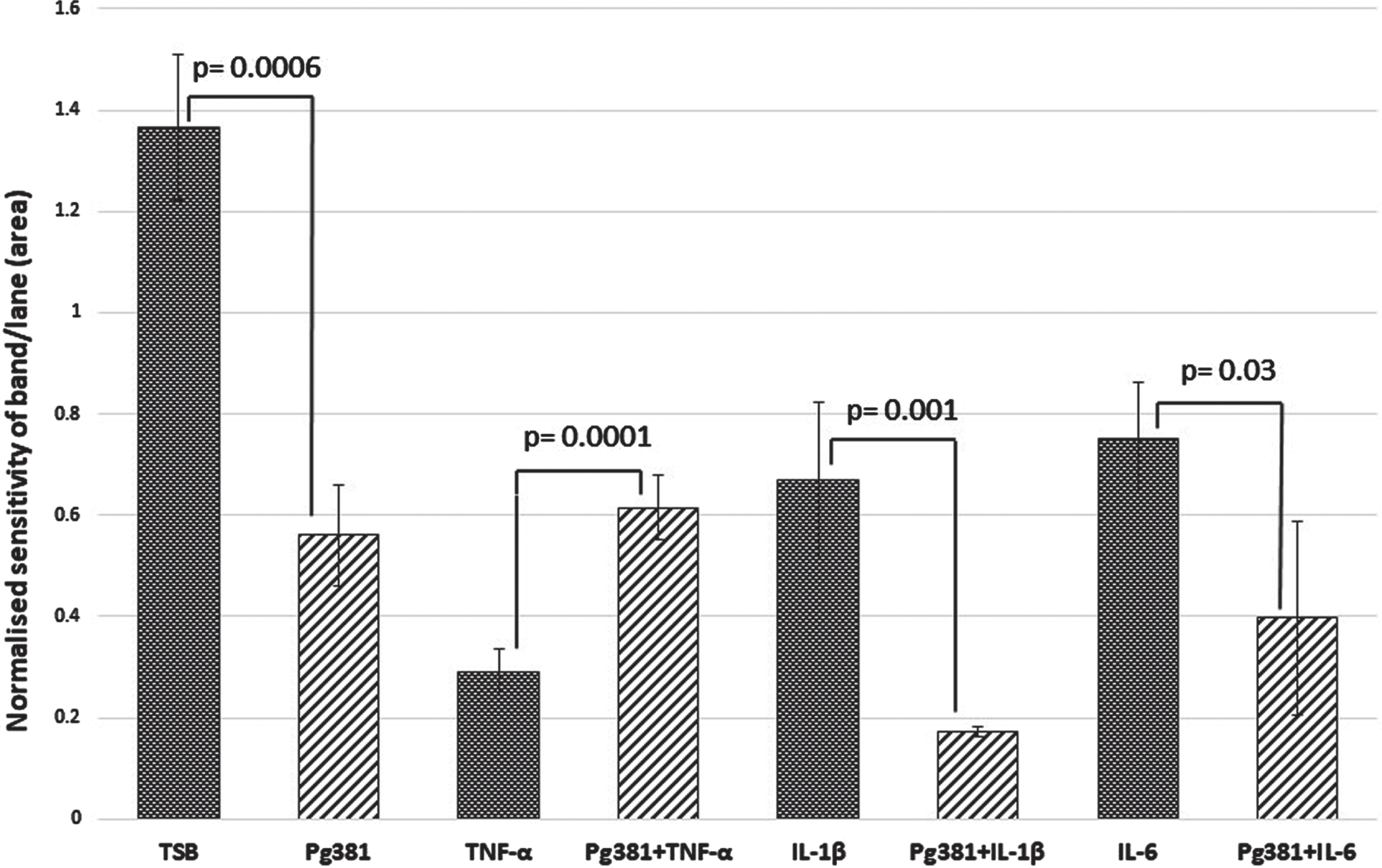
Statistically the results for the Aβ42 band (Fig. 2 C), were highly significant across control (TSB) over to Pg381 (p = 0.0006); TNF-α versus Pg381 + TNF-α (p = 0.0001). IL-1β was significant over Pg381 + IL-1β (p = 0.001); and IL-6 was significant over Pg381 + IL-6 (p = 0.03).
Fig. 2C
Aβ42 densitometry. Statistically the results for the Aβ42 band (C), were across control (TSB) significant over to Pg381 (p = 0.0006); TNF-α versus Pg381 + TNF-α (p = 0.0001). IL-1β was significant over Pg381 + IL-1β (p = 0.001); and IL-6 was significant over Pg381 + IL-6 (p = 0.03).
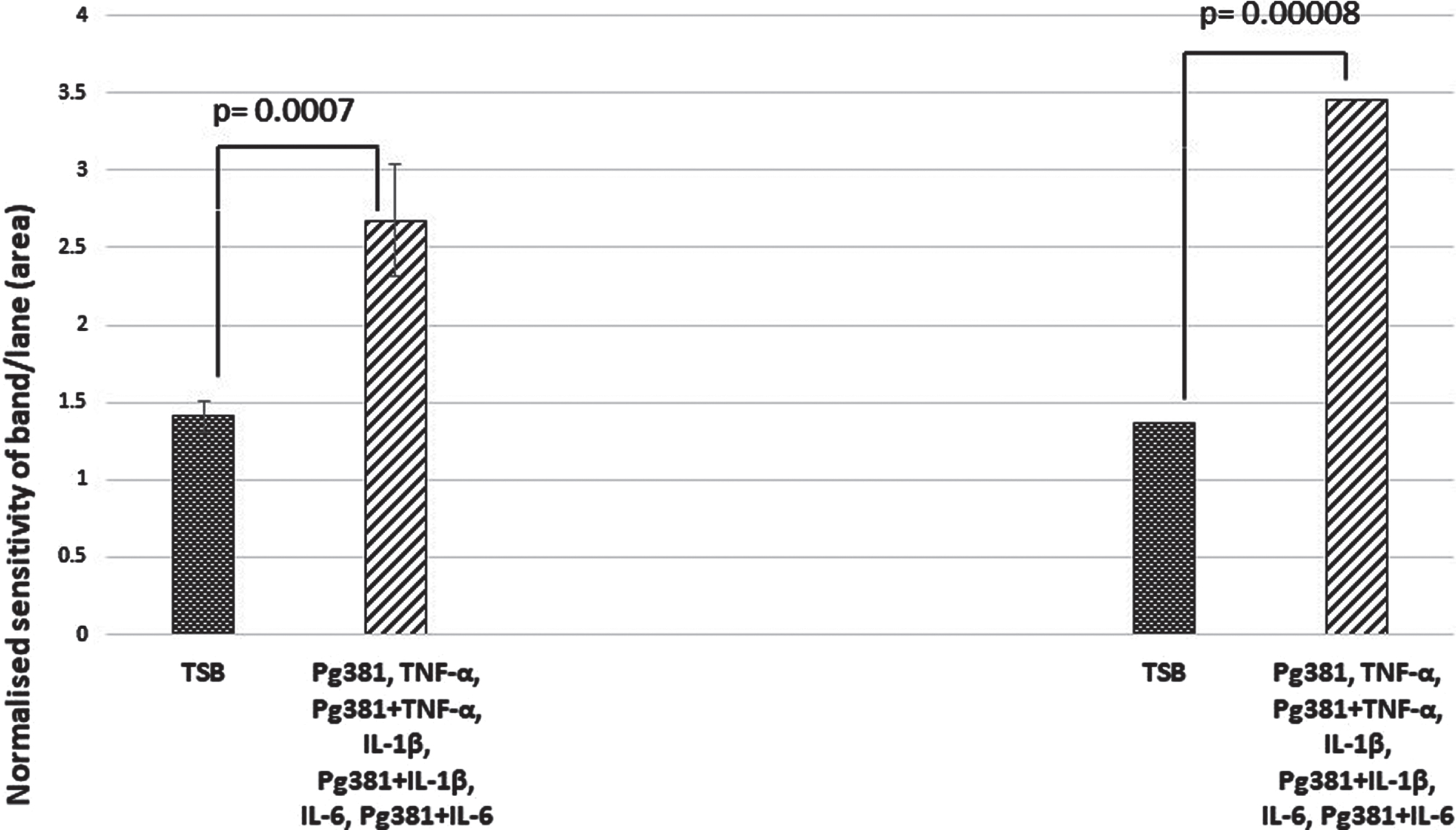
Aβ40 and Aβ42 relative abundance densitometry
When the relative abundance of both species Aβ40 and Aβ42 was statistically evaluated, the results for Aβ40 and Aβ42 across control (TSB) were highly significant over TSB versus all treatment groups for Aβ40 (p = 0.0007); and for Aβ42 TSB versus all treatment groups (p = 0.00008) (Fig. 2D).
Fig. 2D
Aβ40 and Aβ42 relative abundance densitometry. When relative abundance of both species Aβ40 and Aβ42 was statistically evaluated, the results for Aβ40 and Aβ42 (Fig. 2D), across control (TSB). TSB versus all treatment groups for Aβ40 (p = 0.0007); were highly significant, and for Aβ42 TSB versus all treatment groups were also significant (p = 0.00008). All error bars represent standard error of mean.
The ratio of the pooled Aβ40 and Aβ42 from the densitometry values from intracellular stores was around 1 : 2 for both species compared with the control (TSB).
Transmission electron microscopy for detection of Aβ fibril formation in supernatants
The test supernatants collected from the treatment of cells with P. gingivalis conditioned medium with or without cytokines demonstrated the presence of fibrils. The supernatant from SH-SY5Y treated with P. gingivalis 381 conditioned medium consistently demonstrated insoluble fibrils (Fig. 3A boxed area and B arrows) that were interpreted as being Aβ fibrils based on comparison with the Aβ42 peptide included as a positive control (Fig. 3 C).
Fig. 3
TEM detection of insoluble Aβ fibrils in Pg381 treated supernatant. Examination for insoluble Aβ in the supernatant from SH-SY5Y treated with P. gingivalis crude conditioned medium (A box and B arrows) demonstrated Aβ fibrils. The boxed area in panel C shows a positive control for Aβ fibrils from a commercial peptide Aβ42 under the TEM. MT = microtubule fragments. Micron bars represent 100 nm size.

DISCUSSION
The aim of the present study was to understand how the periodontal disease keystone pathogen P. gingivalis endo/exotoxins contribute to the amyloidogenic processing of AβPP and Aβ release because periodontitis is prevalent in individuals with poor oral hygiene and a high dental plaque index, which represents a risk factor for developing AD later in life [1–3, 35–40]. In the absence of an ideal immortalized neuronal cell line model and based on the knowledge that the non-differentiated SH-SY5Y cell line had been tested for AβPP processing via amyloidogenic pathway [34], we adopted this as a model cell line to elucidate the effect of bacterial factors on the AβPP processing to Aβ. In the present study, SH-SY5Y cells were tested without employing the use of chemical stimuli (rentenoic acid, tumor promoter 12-O-tetradecanoyl-phorbol-13-acetate, or brain-derived neurotrophic factor) as reported elsewhere [41] which enable a single phenotype shift toward neuronal cells because prior knowledge of any potential aberrant effect on cellular pathophysiology of the chemical stimuli on AβPP processing were poorly understood. Since the A βPP gene is expressed by all CNS cells due to its “housekeeping gene” function, the AβPP processing in this bi-phenotypic cell line can at least be attributed to a neuronal-like cell phenotype as the origins of the cell line after all are from a neuroblastoma. Immunoblotting with three different antibodies for detecting full-length AβPP of variable isotypes, demonstrated several bands of variable molecular weight sizes, some corresponding to the full-length AβPP and others representing its fragmented moieties including Aβ40 and Aβ42.
As inflammation is usually associated with an infection, we combined the virulence factors of P. gingivalis with the three most common proinflammatory cytokines in this in vitro study and demonstrated the risk was heightened for liberation of Aβ40 and Aβ42 in the presence of either the conditioned medium or TNF-α, IL-1β, and IL-6 and upon their combination. The putative C99 dimerized fragment was decided upon as the molecular weight of this band suggested it to a dimerized state. Next, we searched for the literature to support this view and in this context, literature on the crystal structure of AβPP supported the reasoning for this fragment existing in its dimerized form due to its N-terminal having growth factor-like domain [42, 43] and is prone to dimerization. Further cleavage of AβPP became apparent with the 4 and 3 kDa Aβ bands detected by western blotting as confirmed by the standard used (Aβ peptide) for these AβPP metabolites [44]. Statistically significant Aβ40 was released by the combination of Pg381 + TNF-α and Pg381 + IL-6. Statistically significant Aβ42 was released by the combination of Pg381 + TNF-α treatment only. These observations are of significance as understanding the amyloidogenic processing of AβPP via infection is important as Aβ moiety that forms the hallmark protein of AD plaques could potentially be a source of recurrent infections during life and AD.
Wu et al. [25] demonstrated the intracellular Aβ and this has led us to explore Aβ40 and Aβ42 levels using more sensitive assays on the SH-SY5Y cell lysates and supernatants. It has been suggested that the mechanism of Aβ cleavage in the wild-type AβPP (not mutated) is mediated by cathepsin B and potentially by gingipains (P. gingivalis protease) [45]. It is not entirely clear where the cleavage sites of AβPP mediated by gingipains lie in the AβPP protein, but the implication is around the β- and γ-secretase sites for Aβ40 and Aβ42 liberation. The present study is suggestive of enhanced AβPP C99 fragmentation by the P. gingivalis conditioned medium on SH-SY5Y cells. Therefore, the AβPP cleavage by gingipains at the β- and γ-secretase sites is plausible. The C83 band appears responsive to the inflammatory mediators, especially TNF-α, IL-1β, and IL-6 in combination with the P. gingivalis conditioned medium suggesting that the α secretase is also active under these experimental conditions. Alternatively, Aβ40 and Aβ42 release observed in the SH-SY5Y cell line could simply be a catabolic process, in which case it agrees with Haas et al. [44], who also demonstrated these two Aβ species in their cell lines (not SH-SY5Y) in vitro.
Further studies to clarify the role of AβPP cleavage by gingipains are needed because of the plethora of publications, which provide evidence towards the oral pathogen P. gingivalis being implicated in the initiation and/or progression AD. The present study builds on the previous in vitro and in vivo studies [25, 29, 44, 46] and suggests the Aβ peptide release as an antimicrobial peptide innate immune protein [46]. This links in with the brains limited ability to clear microbial pathogens because of its reliance on the innate immune inflammatory responses, which by becoming chronic (due to genetic defects) result in enhancing neurodegeneration associated with AD [47, 48].
Evidence suggests that low levels of endogenous Aβ seen in the healthy brain is essential to enhance hippocampal long-term potentiation and synaptic plasticity, ultimately boosting memory formation [49]. This helps to explain why the antibody-based therapies developed for AD patients to mop up Aβ have not resulted in improved memory [50] and alternative therapies inclusive of bacterial aspects should now be investigated [51].
In the present study, we also show Aβ fiber formation with a similar fiber appearance to those reported by Kumar et al. [46]. The present study suggests that the presence of virulence factors or the live P. gingivalis bacterium itself can contribute to AβPP processing and enhance the release of Aβ40 in combination with IL-6 and Aβ42 release with TNF-α via the amyloidogenic pathway in vitro. It is, therefore, plausible to suggest that P. gingivalis virulence factors acting as antigens of the immune system can trigger the release of Aβ that eventually deposits as the extracellular plaques seen in AD autopsy brains where P. gingivalis and its virulence factors and pro-inflammatory mediators have been detected [13, 14, 52, 53].
Limitations of the study
We have used the wild-type SH-SY5Y in this study. This cell line is a neuroblastoma derived, non-differentiated immortalized cell line representing a mixed phenotype with predominantly cancer cells. Therefore, the translational value of the results presented here may vary and not fully simulate the results of other fully differentiated cell types.
Conclusions
The SH-SY5Y cell line when challenged alone with P. gingivalis virulence factors and/or combined with TNF-α and IL-1β enhances the AβPP cleavage. The release of C99 fragment appears to be influenced by P. gingivalis virulence factors, especially gingipains acting on the β- and γ-secretase sites, but further work needs to be carried out to definitively confirm this observation. The present study shows that both inflammation and infection can contribute to excessive release of Aβ40 and Aβ42.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
SK and SKS in 2017 and again with RW and SKS in 2018 (UK) received PreViser awards from the Oral and Dental Research Trust. SC (USA) acknowledges funding from NIH/ NIDCR: R03DE02896.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Nazir MA ((2017) ) Prevalence of periodontal disease, its association with systemic diseases and prevention. Int J Health Sci (Qassim) 11: , 72–80. |
[2] | Hajishengallis G , Darveau RP , Curtis MA ((2012) ) The keystone-pathogen hypothesis. Nat Rev Microbiol 10: , 717–725. |
[3] | Chapple ILC , Mealey BL , Van Dyke TE , Bartold PM , Dommisch H , Eickholz P , Geisinger ML , Genco RJ , Glogauer M , Goldstein M , Griffin TJ , Holmstrup P , Johnson GK , Kapila Y , Lang NP , Meyle J , Murakami S , Plemons J , Romito GA , Shapira L , Tatakis DN , Teughels W , Trombelli L , Walter C , Wimmer G , Xenoudi P , Yoshie H ((2018) ) Periodontal health and gingival diseases and conditions on an intact and a reduced periodontium: Consensus report of workgroup 1 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Clin Periodontol 20: , S68–S77. |
[4] | Papapanou PN ((1996) ) Periodontal diseases: Epidemiology. Ann Periodontol 1: , 1–36. |
[5] | Vernal R , León R , Silva A , van Winkelhoff AJ , García-SanzJ , Sanz M ((2009) ) Differential cytokine expression by human dendriticcells in response to different porphyromonas gingivalis capsularserotypes. J Clin Periodontol 36: , 823–829. |
[6] | Díaz-Zúñiga J , More J , Melgar-Rodríguez S , Jiménez-Unión M , Villalobos-Orchard F , Muñoz-Manríquez C , Monasterio G , Valdés JL , Vernal R , Paula-Lima A ((2020) ) Alzheimer’s disease-like pathology triggered byPorphyromonas gingivalis in wild type rats is serotype dependent. Front Immunol 11: , 588036. |
[7] | Sansores-España LD , Melgar-Rodríguez S , Olivares-Sagredo K , Cafferata EA , Martínez-Aguilar VM , Vernal R , Paula-Lima AC , Díaz-Zúñiga J ((2021) ) Oral-gut-brain axis inexperimental models of periodontitis: Associating gut dysbiosis withneurodegenerative diseases. Front Aging 2: , 781582. |
[8] | Chiu H-C , Fu MM-J , Yang T-S , Fu E , Chiang C-Y , Tu H-P , Chin Y-T , Lin F-G , Shih K-C ((2016) ) Effect of high glucose, lipopolysaccharide and advanced glycation end-products on production of interleukin-6/-8 by gingival fibroblasts. J Periodontal Res 52: , 268–276. |
[9] | Makiura N , Ojima M , Kou Y , Furuta N , Okahashi N , Shizukuishi S , Amano A ((2008) ) Relationship of Porphyromonas gingivalis with glycemic level in patients with type 2 diabetes following periodontal treatment. Oral Microbiol Immunol 23: , 348–351. |
[10] | Bale BF , Doneen AL , Vigerust DJ ((2017) ) High-risk periodontal pathogens contribute to the pathogenesis of atherosclerosis. Postgrad Med J 93: , 215–220. |
[11] | Demmer RT , Norby FL , Lakshminarayan K , Walker KA , Pankow JS , Aaron R , Folsom AR , Mosley T , Beck J , Lutsey PL ((2020) ) Periodontal disease and incident dementia: The Atherosclerosis Risk in Communities Study (ARIC). Neurology 95: , e1660–e1671. |
[12] | Stein PS , Desrosiers M , Donegan SJ , Yepes JF , Kryscio RJ ((2007) ) Tooth loss, dementia and neuropathy in the Nun study. J Am Dent Assoc 138: , 1314–1322. |
[13] | Poole S , Singhrao SK , Kesavalu L , Curtis MA , Crean St J ((2013) ) Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis 36: , 665–677. |
[14] | Dominy SS , Lynch C , Ermini F , Benedyk M , Marczyk A , Konradi A , Nguyen M , Haditsch U , Raha D , Griffin C , Holsinger LJ , Arastu-Kapur S , Kaba S , Lee A , Ryder MI , Potempa B , Mydel P , Hellvard A , Adamowicz K , Hasturk H , Walker GD , Reynolds EC , Faull RLM , Curtis MA , Dragunow M , Potempa J ((2019) ) in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5: , eaau3333. |
[15] | Duong S , Patel ST , Chang F ((2017) ) Dementia. What pharmacists need to know. Can Pharm J (Ott) 150: , 118–129. |
[16] | Alzheimer A ((1907) ) Ueber eine eigenartige Erkrankung der Hirnrinde. Allgemeine Z Psychiatr Psychisch-gerichtliche Med 64: , 146–148. |
[17] | Jin YP , Østbye T , Feightner JW , Hachinski ((2008) ) Joint effect of stroke and APOE E4 on dementia risk: the Canadian Study of Health and Aging. Neurology 70: , 9–16. |
[18] | Li L , Cavuoto M , Biddiscombe K , Pike KE ((2020) ) Diabetes mellitus increases risk of incident dementia in APOEɛ4 carriers: A meta-analysis. J Alzheimers Dis 74: , 1295–1308. |
[19] | de Bont N , Netea MG , Demacker PN ((1999) ) Apolipoprotein E knock-out mice are highly susceptible to endotoxemia and infection. J Lipid Res 40: , 680–685. |
[20] | Moretti EW , Morris RW , Podgoreanu M , Schwinn DA , Newman MF , Bennett E , Moulin VG , Mba UU , Laskowitz DT ((2005) ) APOE polymorphism is associated with risk of severe sepsis in surgical patients. Perioperative Genetics and Safety Outcomes Study (PEGASUS) Investigative Team. Crit Care Med 33: , 2521–2526. |
[21] | Watts A , Crimmins EM , Gatz M ((2008) ) Inflammation as a potential mediator for the association between periodontal disease and Alzheimer’s disease. Neuropsychiatr Dis Treat 4: , 865–876. |
[22] | Tsoi LM , Wong KY , Liu YM , Ho YY ((2007) ) Apoprotein E isoform-dependent expression and secretion of pro-inflammatory cytokines TNF-α and IL-6 in macrophages. Arch Biochem Biophys 460: , 33–40. |
[23] | Hubacek JA , Peasey A , Pikhart H , Stavek P , Kubinova R , Marmot M , Bobak M ((2010) ) APOE polymorphism and its effect on plasma C-reactive protein levels in a large general population sample. Hum Immunol 71: , 304–308. |
[24] | Singh A , Rouxel P , Watt RG , Tsakos G ((2013) ) Social inequalities in clustering of oral health related behaviors in a national sample of British adults. Prev Med 57: , 102–106. |
[25] | Wu Z , Ni J , Liu Y , Teeling JL , Takayama F , Collcutt A , Ibbett P , Nakanishi H ((2017) ) Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun 65: , 350–361. |
[26] | Ishida N , Ishihara Y , Ishida K , Tada H , Funaki-Kato Y , Hagiwara M , Ferdous T , Abdullah M , Mitani A , Michikawa M , Matsushita K ((2017) ) Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech Dis 6: , 15. |
[27] | Ding Y , Ren J , Yu H , Yu W , Zhou Y ((2018) ) , a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun Ageing 215: , 6. |
[28] | Zhang J , Yu C , Zhang X , Chen H , Dong J , Lu W , Song Z , Zhou W ((2018) ) Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J Neuroinflammation 15: , 37. |
[29] | Ilievski V , Zuchowska PK , Green SJ , Toth PT , Ragozzino ME , Le K , Aljewari HW , O’Brien-Simpson NM , Reynolds EC , Watanabe K ((2018) ) Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One 13: , e0204941. |
[30] | Poole S , Singhrao SK , Chukkapalli S , Rivera M , Velsko I , Kesavalu L , Crean StJ ((2015) ) Active infection of and infection-induced complement activation in ApoE-/-mice brains. J Alzheimers Dis 43: , 67–80. |
[31] | Rokad F , Moseley R , Hardy SR , Chukkapalli S , Crean S , Kesavalu L , Singhrao SK ((2017) ) Cerebral oxidative stress and microvasculature defects in TNF-α expressing transgenic and-infected ApoE–/–mice. J Alzheimers Dis 60: , 359–369. |
[32] | Holt SC , Kesavalu L , Walker S , Genco CA ((1999) ) Virulence factors of Porphyromonas gingivalis. Periodontol 2000 20: , 168–238. |
[33] | Cetin S , Knez D , Gobec S , Kos J , Pislar A ((2022) ) Cell models forAlzheimer’s and Parkinson’s disease: At the interface of biology anddrug discovery. Biomed Pharmacother 149: , 112924. |
[34] | Belyaev ND , Kellett KAB , Beckett C , Markova NZ , Revett TJ , Nalivaeva NN , Hooper NM , Turner AJ ((2010) ) The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a ${$beta$}$-secretase-dependent pathway. J Biol Chem 285: , 41443–41454. |
[35] | Harding A , Robinson S , Crean S , Singhrao SK ((2017) ) Can better management of periodontal disease delay the onset and progression of Alzheimer’s disease? . J Alzheimers Dis 58: , 337–348. |
[36] | Harding A , Gonder U , Robinson SJ , Crean S , Singhrao SK , (2017b) Exploring the association between Alzheimer’s disease, oral health, microbial endocrinology and nutrition. Front Aging Neurosci 9: , 398. |
[37] | Armitage GC , Dickinson WR , Jenderseck RS , Levine SM , Chambers DW ((1982) ) Relationship between the percentage of subgingival spirochetes and the severity of periodontal disease. J Periodontol 53: , 550–556. |
[38] | Kirst ME , Li EC , Alfant B , Chi YY , Walker C , Magnusson I , Wang GP ((2015) ) Dysbiosis and alterations in predicted functions of the subgingival microbiome in chronic periodontitis. Appl Environ Microbiol 81: , 783–793. |
[39] | Diaz PI , Hoare A , Hong BY ((2016) ) Subgingival microbiome shifts and community dynamics in periodontal diseases. J Calif Dent Assoc 44: , 421–435. |
[40] | Curtis MA , Diaz PI , Van Dyke TE ((2020) ) The role of the microbiota in periodontal disease. Periodontol 2000 83: , 14–25. |
[41] | Dwane S , Durack E , Kiely PA ((2013) ) Optimising parameters for the differentiation of SH-SY5Y cells to study cell adhesion and cell migration. BMC Res Notes 6: , 366. |
[42] | Rossjohn J , Cappai R , Feil SC , Henry A , McKinstry WJ , Galatis D , Hesse L , Multhaup G , Beyreuther K , Masters CL , Parker MW ((1999) ) Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol 6: , 327–331. |
[43] | Gralle M , Ferreira ST ((2007) ) Structure and functions of the human amyloid precursor prote, The whole is more than the sum of itsarts, Prog Neurobiol 82: , 11–32. |
[44] | Haass C , Schlossmacher MG , Hung AY , Vigo-Pelfrey C , Mellon A , Ostaszewski BL , Lieberburg I , Koo EH , Schenk D , Teplow DB ((1992) ) Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 359: , 322–325. |
[45] | Olsen I , Taubman MA , Singhrao SK ((2016) ) suppresses adaptive immunity in periodontitis, atherosclerosis and Alzheimer’s disease. . J Oral Microbiol 8: , 33029. |
[46] | Kumar DK , Choi SH , Washicosky KJ , Eimer WA , Tucker S , Ghofrani J , Lefkowitz A , McColl G , Goldstein LE , Tanzi RE , Moir RD ((2016) ) Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 8: , 340ra72. |
[47] | Singhrao SK , Olsen I ((2018) ) Are outer membrane vesicles, microbullets for sporadic Alzheimer’s disease manifestation? Rep 2: , 219–228. |
[48] | Olsen I , Singhrao SK ((2019) ) Is there a link between genetic defects in the complement cascade and in Alzheimer’s disease? J Oral Microbiol 12: , 167648. |
[49] | Palmeri A , Ricciarelli R , Gulisano W , Rivera D , Rebosio C , Calcagno E , Tropea MR , Conti S , Das U , Roy S , Pronzato MA , Arancio O , Fedele E , Puzzo D ((2017) ) Amyloid-β peptide is needed for cGMP-induced long-term potentiation and memory. J Neurosci 37: , 6926–6937. |
[50] | Doody RS , Thomas RG , Farlow M , Iwatsubo T , Vellas B , Joffe S , Kieburtz K , Raman R , Sun X , Aisen PS , Siemers E , Liu-Seifert H , Mohs R , Alzheimer’s Disease Cooperative Study Steering Committee; Solanezumab Study Group ((2014) ) Phase 3 trial of solanezumab for mild to moderate Alzheimer’s disease. N Engl J Med 370: , 311–321. |
[51] | Panza F , Lozupone M , Logroscino G , Imbimbo BP ((2019) ) A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol 15: , 73–88. |
[52] | Akiyama H , Barger S , Barnum S , Bradt B , Bauer J , Cole GM , Akiyama H , Barger S , Barnum S , Cooper NR , Eikelenboom P , Emmerling M , Fiebich BL , Finch CE , Frautschy S , Griffin WS , Hampel H , Hull M , Landreth G , Lue L , Mrak R , Mackenzie IR , McGeer PL , O’Banion MK , Pachter J , Pasinetti G , Plata-Salaman C , Rogers J , Rydel R , Shen Y , Streit W , Strohmeyer R , Tooyoma I , Van Muiswinkel FL , Veerhuis R , Walker D , Webster S , Wegrzyniak B , Wenk G , Wyss-Coray T ((2000) ) Inflammation and Alzheimer’s disease. Neurobiol Aging 21: , 383–421. |
[53] | Heneka MT , Carson MJ , El Khoury J , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |


