
Is There Any Evidence of Monocytes Involvement in Alzheimer’s Disease? A Pilot Study on Human Postmortem Brain
Abstract
Background:
The role of neuroinflammation has become more evident in the pathogenesis of neurodegenerative diseases. Increased expression of microglial markers is widely reported in Alzheimer’s disease (AD), but much less is known about the role of monocytes in AD pathogenesis. In AD animal models, bone marrow-derived monocytes appear to infiltrate the parenchyma and contribute to the phagocytosis of amyloid-β depositions, but this infiltration has not been established in systematic studies of the human brain postmortem.
Objective:
In addition to assessing the distribution of different subtypes of microglia by immunostaining for CD68, HLA-DR, CD163, and CD206, we focused on the involvement of C-chemokine receptor type2 (CCR2) positive monocytes during the AD course.
Methods:
We used formalin-fixed and paraffin-embedded tissue from four vulnerable brain regions (hippocampus, occipital lobe, brainstem, and cerebellum) from neuropathologically characterized AD cases at different Braak stages and age-matched controls.
Results:
Only singular migrated CCR2-positive cells were found in all brain regions and stages. The brainstem showed the highest number of positive cells overall, followed by the hippocampus. This mechanism of recruitment seems to work less efficiently in the human brain at an advanced age, and the ingress of monocytes obviously takes place in much reduced numbers or not at all.
Conclusion:
In contrast to studies on animal models, we observed only a quite low level of myeloid monocytes associated with AD pathology. Furthermore, we provide evidence associating early microglial reactions carried out in particular by pro-inflammatory cells with early effects on tangle- and plaque-positive vulnerable brain regions.
INTRODUCTION
Sporadic Alzheimer’s disease (AD) is the most common neurodegenerative disease in the elderly. Its incidence is increasing worldwide, especially in industrialized countries [1–3]. Tau and amyloid-β (Aβ) accumulation in specific brain regions is the major hallmark for the neuropathological diagnosis of AD [4–6]. Nevertheless, the pathogenesis and neuron loss during the progression of AD are still a matter of debate [7–10]. Chronologically and topographically selective vulnerability of distinct brain regions during the relentless progression of AD is the basis of disease staging [4, 11–15]. The cerebellar cortex seems to be less affected even in advanced stages [16], whereas, para- and hippocampal telencephalic regions display early and rapid progression of hyperphosphorylated tau accumulation [17]. Brainstem tau accumulation precedes that of the telencephalic cortex by decades [18–20], and noradrenergic locus coeruleus and serotoninergic raphe projections could trigger rapidly progressing telencephalic pathology [21, 22].
Many theories regarding the pathogenesis of AD-related cell death have been postulated (e.g., amyloid cascade, oxidative stress, prion-like transmission), but the mechanisms are not fully elucidated [17, 23–25]. In recent years, the role of neuroinflammation in the pathogenesis of neurogenerative disease has become more evident. Microglia, brain-specific immune cells, migrate from the yolk sac into the brain during embryogenesis and play important roles in synaptic plasticity and neuronal death. They also promote brain clearance and phagocytosis of Aβ in AD [26, 27]. The results of some studies have suggested that aberrant microglial activation might drive neuronal death and dysfunction in AD [28]. Others support the idea that rather than induction of microglial activation, the progressive, aging-related degeneration of microglia and loss of microglial neuroprotection contribute to the onset of sporadic AD [29].
In recent years, a scheme has been developed that divides microglia/macrophages into classically activated (M1) cells based on changes in responses to proinflammatory lipopolysaccharide and interferon gamma. These microglial cells could mediate inflammatory tissue damage. Alternative activation (M2) characterizes the microglia/macrophages responsible for removing cellular debris and promoting tissue repair [30], but these cells also commonly occur as tumor-promoting macrophages, as in malignant glioma [31]. This might be a simplified approach, but microglia is known to react differently to different stimuli and the nomenclature is still considered fundamental in the macrophage and microglia research field [32]. Moreover, a recent study showed that in contrast to M1 microglia, transplantation of M2-deviated microglia promotes recovery of motor function after spinal cord injury in mice [33].
A frequently used marker for activated microglia and peripheral macrophages is CD68, also known as macrosialin. Numerous studies in postmortem brain tissue have demonstrated brain region–dependent differences in CD68-positive cells in control brains and an increase during the course of AD [34–38].
The major histocompatibility complex class II protein HLA-DR identifies “activated” microglia and, especially, the M1 polarization status [39, 40]. In several studies, a significant accumulation of HLA-DR–positive microglial cells around amyloid plaques and neurofibrillary tangles in AD patients could be demonstrated [41].
The markers CD163 and CD206 are frequently used to detect the alternative M2 polarization status [42, 43]. Few findings relate to the role and distribution of CD163-expressing cells in the course of AD [35, 44]. CD206 represents a surface marker for perivascular anti-inflammatory, M2-polarized microglia, but the involvement of these cells in AD pathogenesis is unclear [45].
In AD animal models, bone marrow-derived monocytes appear to infiltrate the parenchyma and contribute to the phagocytosis of Aβ deposits [46], but detailed analyses in human postmortem AD brains are still lacking. The main receptor of these cells is the C-chemokine receptor type 2 (CCR2), which is expressed on the surface of monocytes and a small portion of natural killer and T cells, and mediates the migration of lymphocytes, peripheral blood dendritic cells, and monocytes [47].
The aim of this study was to identify the extent and distribution of the various microglia subpopulations in differentially vulnerable brain regions from human postmortem AD brains in comparison to control brains without clinical evidence of neurological or neurodegenerative disease. We expected the results to offer some data concerning the activation and the division of labor between two genetically distinct myeloid cells, the brain-specific immune cells (resident microglia) and the possible involvement of immune cell (monocyte) migration and their role in AD neuroinflammation.
MATERIAL AND METHODS
Sampling and processing
We used formalin-fixed and paraffin-embedded tissue from four brain regions (hippocampus, occipital lobe, brainstem, and cerebellum) from neuropathologically characterized AD with different Braak stages and age-matched controls collected from Brain Bank Center Wuerzburg, a member of the former BrainNet Europe Brain Bank Consortium Network. These brains were obtained with the consent of the next of kin and according to the guidelines of the National and Local Ethics Committees. The study was approved by the Local Ethics Committee of the University of Wuerzburg (internal application number 99/11) and was performed in keeping with the ethical standards described in the most recent version of the Declaration of Helsinki.
Three groups were created: controls (n = 9; Braak stage 0–I, no other neuropsychiatric disease), intermediate AD (n = 8; Braak stages III–IV), and late AD (n = 11; Braak stages V–VI). The postmortem intervals varied from 12 to 72 h, with no significant difference between groups. The ages at death ranged from 40 to 85 years in the control group, 62 to 86 years in the intermediate group, and 63 to 85 years in the late-AD group.
The male:female ratios were 6 : 3 among controls, 2 : 6 in the intermediate group, and 4 : 7 in the late-AD group. Demographic data of the cases are summarized in Table 1.
Table 1
Demographical data
| No. | Group | Age (y) | PMI (h) | Sex | Braak Stage | CERAD Score |
| 1 | Control | 80 | 23 | m | I | 0 |
| 2 | Control | 40 | 40 | f | 0 | 0 |
| 3 | Control | 62 | 48 | f | 0 | 0 |
| 4 | Control | 67 | 15 | m | 0 | A |
| 5 | Control | 64 | 9 | m | 0 | 0 |
| 6 | Control | 68 | 11 | m | I | 0 |
| 7 | Control | 70 | 2 | m | I | A |
| 8 | Control | 73 | 16 | f | I | 0 |
| 9 | Control | 69 | 48 | m | I | 0 |
| 10 | Interm.AD | 77 | 24 | f | III | B |
| 11 | Interm.AD | 78 | 38 | f | IV | B |
| 12 | Interm.AD | 75 | 20 | f | III | A |
| 13 | Interm.AD | 85 | 46 | f | III | B |
| 14 | Interm.AD | 64 | 24 | m | III | A |
| 15 | Interm.AD | 86 | 61 | f | III | B |
| 16 | Interm.AD | 77 | 28 | f | III | B |
| 17 | Interm.AD | 62 | 24 | m | III | A |
| 18 | Late AD | 74 | 26 | f | V | C |
| 19 | Late AD | 65 | 24 | f | V | B |
| 20 | Late AD | 80 | 24 | f | VI | C |
| 21 | Late AD | 83 | 22 | f | VI | C |
| 22 | Late AD | 70 | 39 | m | VI | C |
| 23 | Late AD | 62 | n.a. | m | V | B |
| 24 | Late AD | 86 | 21 | f | VI | C |
| 25 | Late AD | 76 | 7 | f | VI | C |
| 26 | Late AD | 74 | 50 | m | VI | C |
| 27 | Late AD | 71 | 12 | m | V | C |
| 28 | Late AD | 82 | 24 | f | VI | C |
AD, Alzheimer’s disease; f, female; m, male; n.a., not available; PMI; postmortem interval; Interm., Intermediate.
Consecutive 6-μm sections were mounted on microscope slides and stained immunohistochemically with commercially available monoclonal antibodies: CD68 (DakoCytomation, Glostrup, Denmark, 1 : 100), HLA-DR (DakoCytomation, Glostrup, Denmark, 1 : 80), CD163 (Novocastra, Leica Biosystems, UK, 1 : 100), CD206 (Bio-Rad, Marnes-la-Coquette, France, 1 : 500), and CCR2 (Abcam, Cambridge, UK, 1 : 800). Control slides were stained using solution without the primary antibodies to account for nonspecific staining. Ommission of primary antibodies in control experiments resulted in the expected absence of any cellular labelling. The microphotographs of the brain sections were recorded with an Olympus DP 27 digital camera mounted on an Olympus BX508 microscope using 20× magnification (both Olympus Corporation, Tokyo, Japan).
Ten images were selected at random from each slide to obtain a representative surface (10.34 mm2) for all sections. For quantitative evaluation of the sections, image processing software (Biomas, Erlangen, Germany) was used, as described previously [48]. Before electronic counting, parameters of cell morphology (size and staining intensity) for each antibody were defined. The software transferred the data automatically into an Excel macro table (Microsoft Corporation, Redmond, WA, USA). The number of cells discernible in 10 fields of view was set against the area investigated and calculated as the number of positive cells per square millimeter (density).
Statistical analysis
Statistical analysis was performed with SSPS software (IBM SPSS Statistics 26 Premium). To analyze the data, we used analysis of variance (one-way ANOVA) and post-hoc Bonferroni adjustment. p < 0.05 was considered statistically significant.
RESULTS
CD68
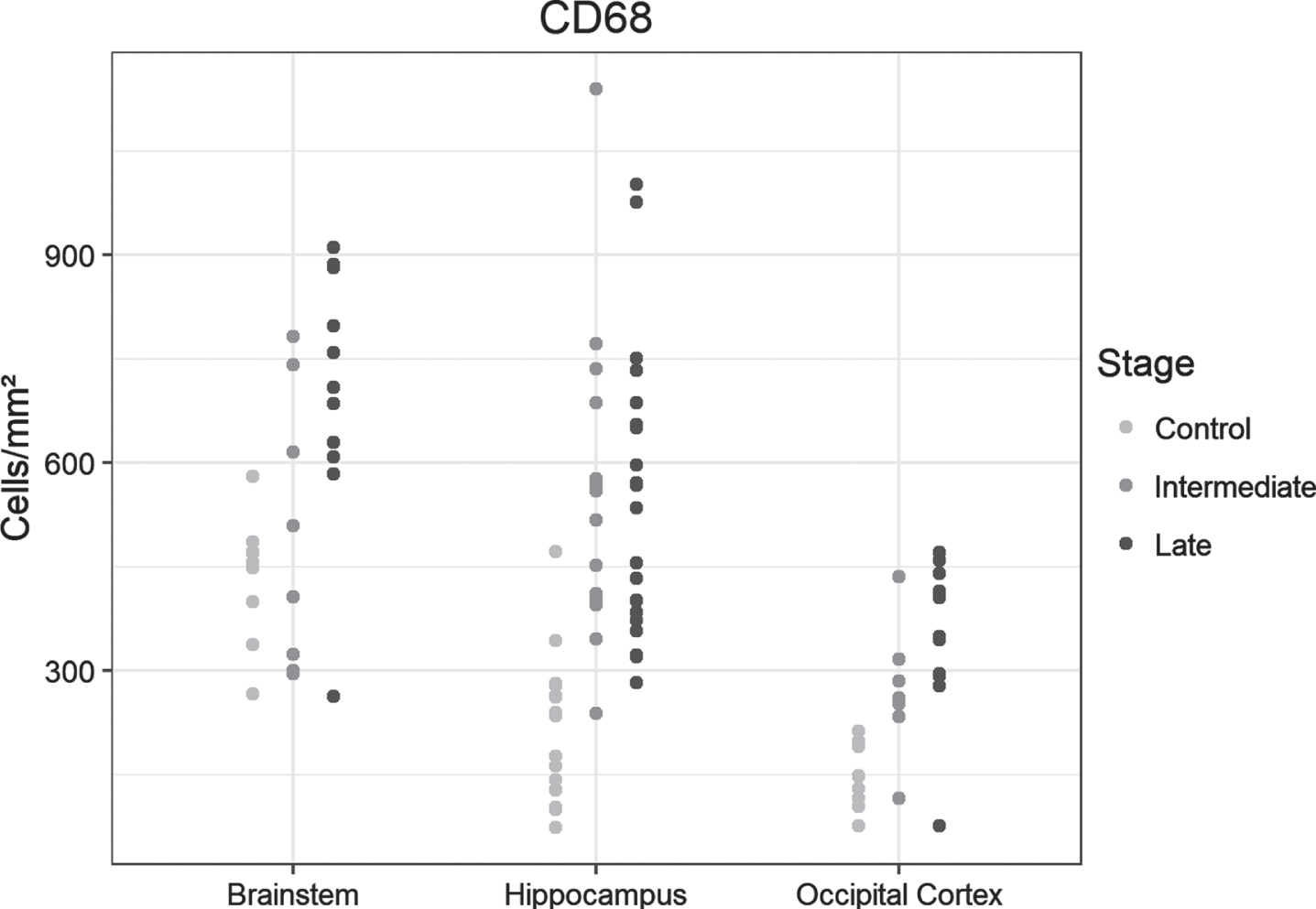
We observed a significantly greater cell density especially in the hippocampus of the intermediate-AD group versus controls (p < 0.0001), without augmentation in the late-AD group versus intermediate AD. The occipital lobe showed an overall lower density of positive cells compared to controls, but density increased in manner similar to that seen in the hippocampus, with a significant rise in the intermediate AD group (p = 0.029) and a significant gain in the late AD stage compared to controls (p < 0.0001) but not to intermediate AD. The density of positive cells was already surprisingly high in control brainstems, without a notable increase in the intermediate stages of AD. In the late AD stage, the brainstem showed a significantly higher density of positive cells compared to controls (p = 0.004) and to intermediate-AD cases respectively (p = 0.04) (Fig. 1A).
Fig. 1A
Dot plots presentation of CD68-positive cells in the analyzed regions of the control, intermediate and late AD groups.
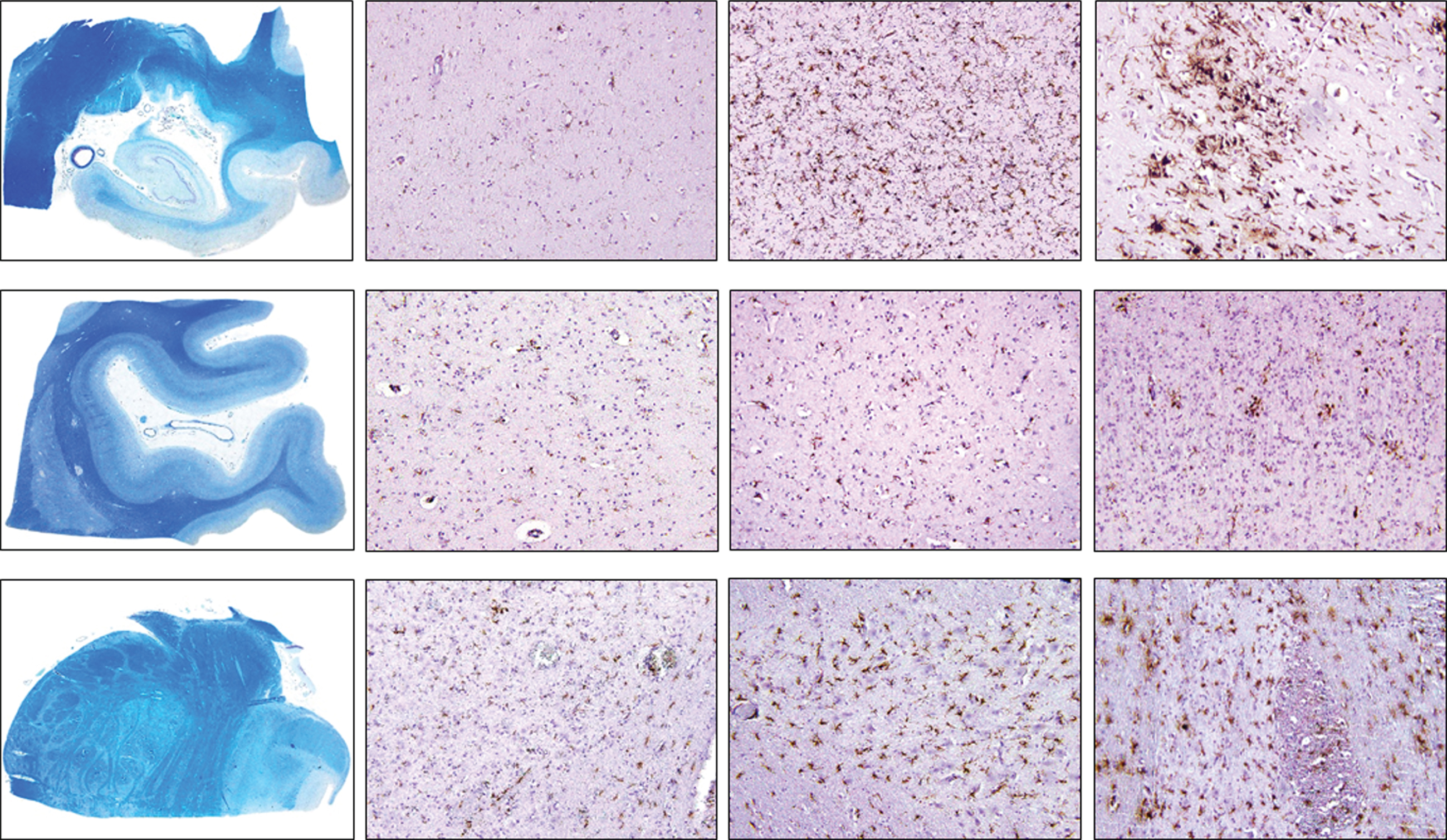
Examples of immunohistochemical examinations with CD68 are given in Fig. 1B.
Fig. 1B
Density of CD68-positive cells in control, intermediate and late AD, respectively (upper row, hippocampus; middle row, occipital cortex, lower row, brainstem) (magnification 100x).
HLA-DR
Similar to CD68, a significant increase in positive cell density was observed in the hippocampus and the occipital lobe between controls and the intermediate-AD group (p = 0.016 and p = 0.028, respectively), without significant augmentation in the late AD group versus intermediate AD. The brainstem showed a significantly higher cell density in intermediate-AD samples compared to controls (p = 0.043), without a notable rise in the late-AD stage compared to intermediate AD (Fig. 2A). Examples of immunohistochemical examinations with HLA-DR are given in Fig. 2B.
Fig. 2A
Dot plots presentation of HLA-DR-positive cells in the analyzed regions of control, intermediate and late AD groups.
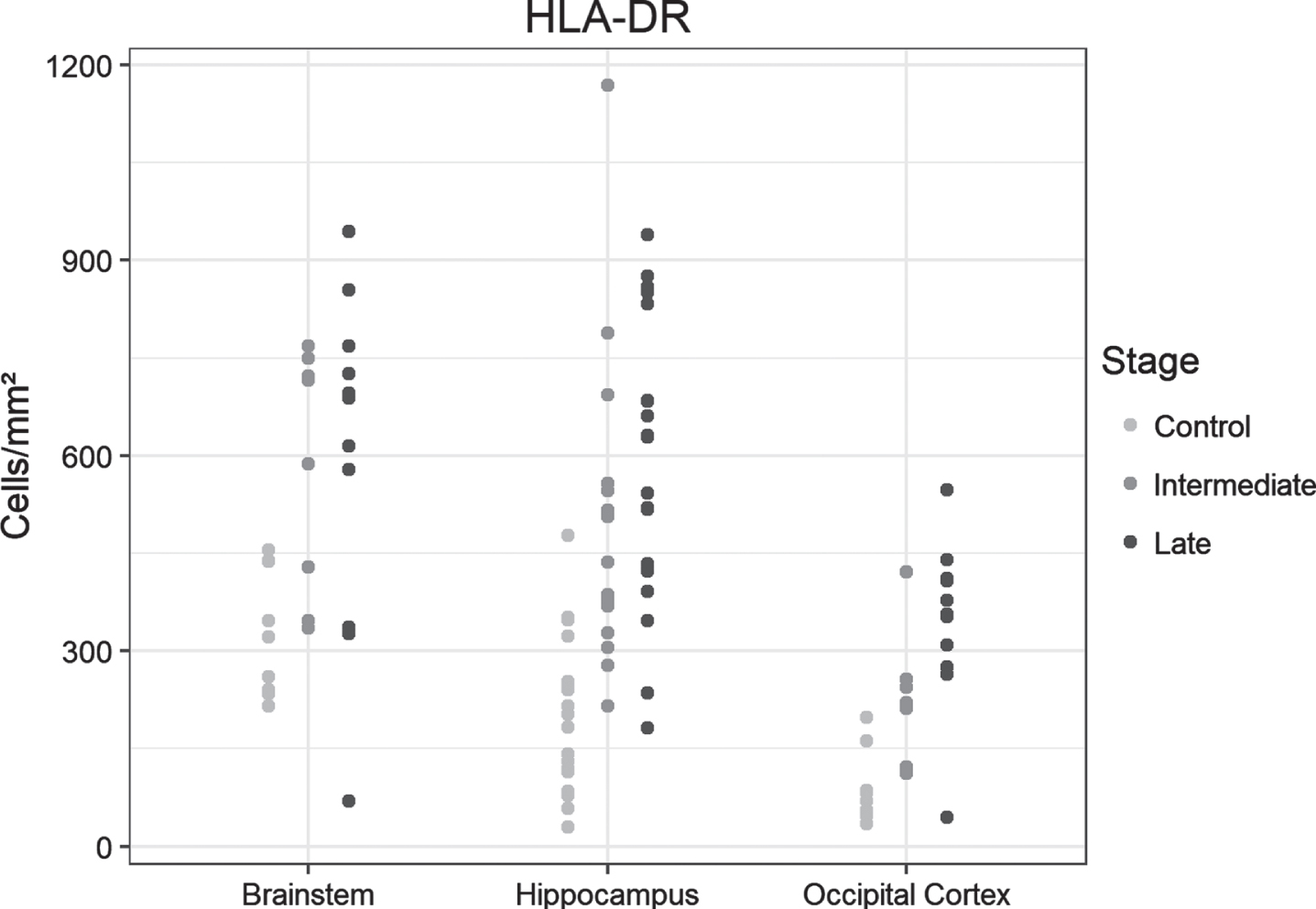
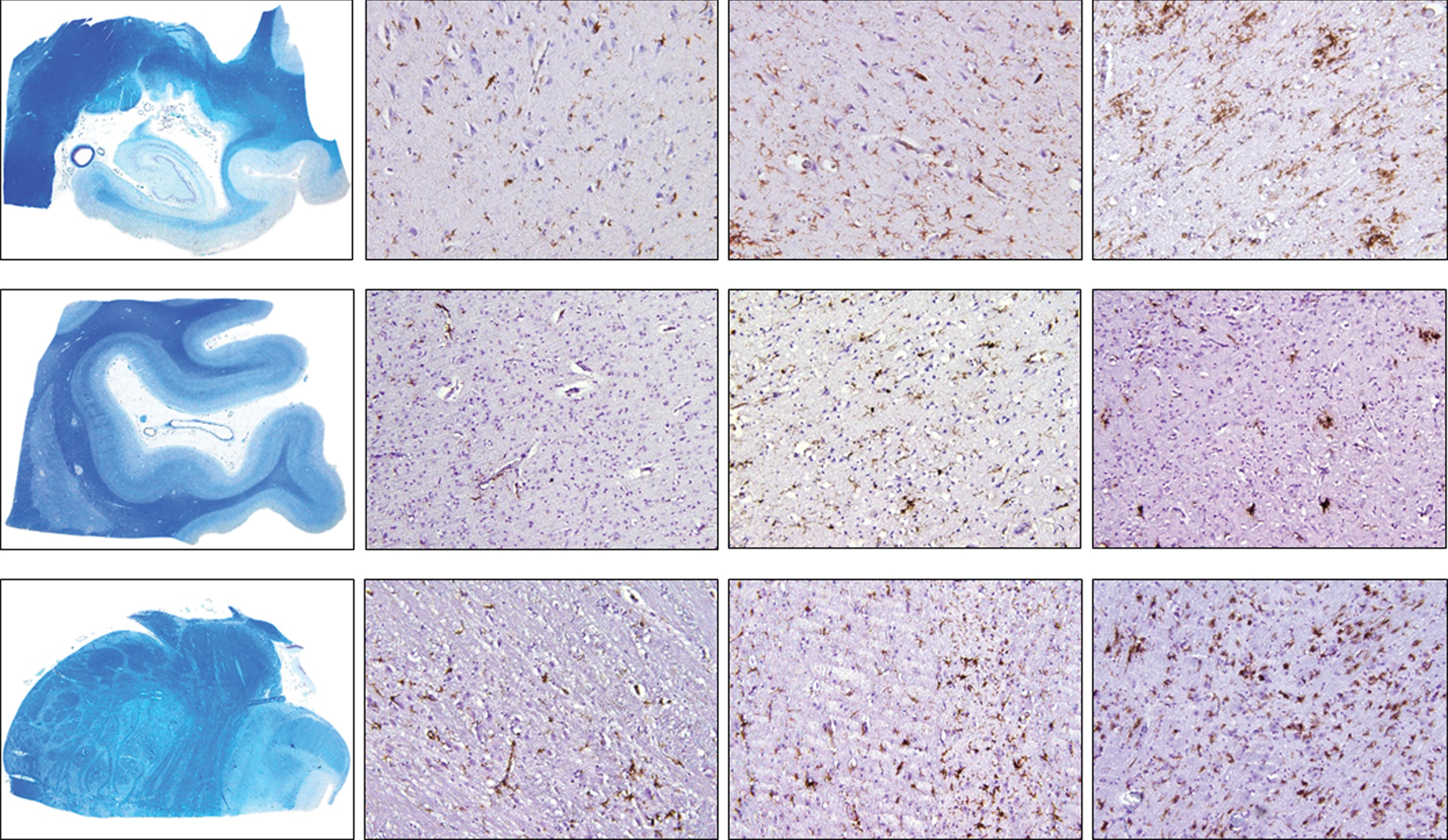
Fig. 2B
Density of HLA-DR-positive cells in control, intermediate and late AD, respectively (upper row, hippocampus; middle row, occipital cortex, lower row, brainstem) (magnification 100x).
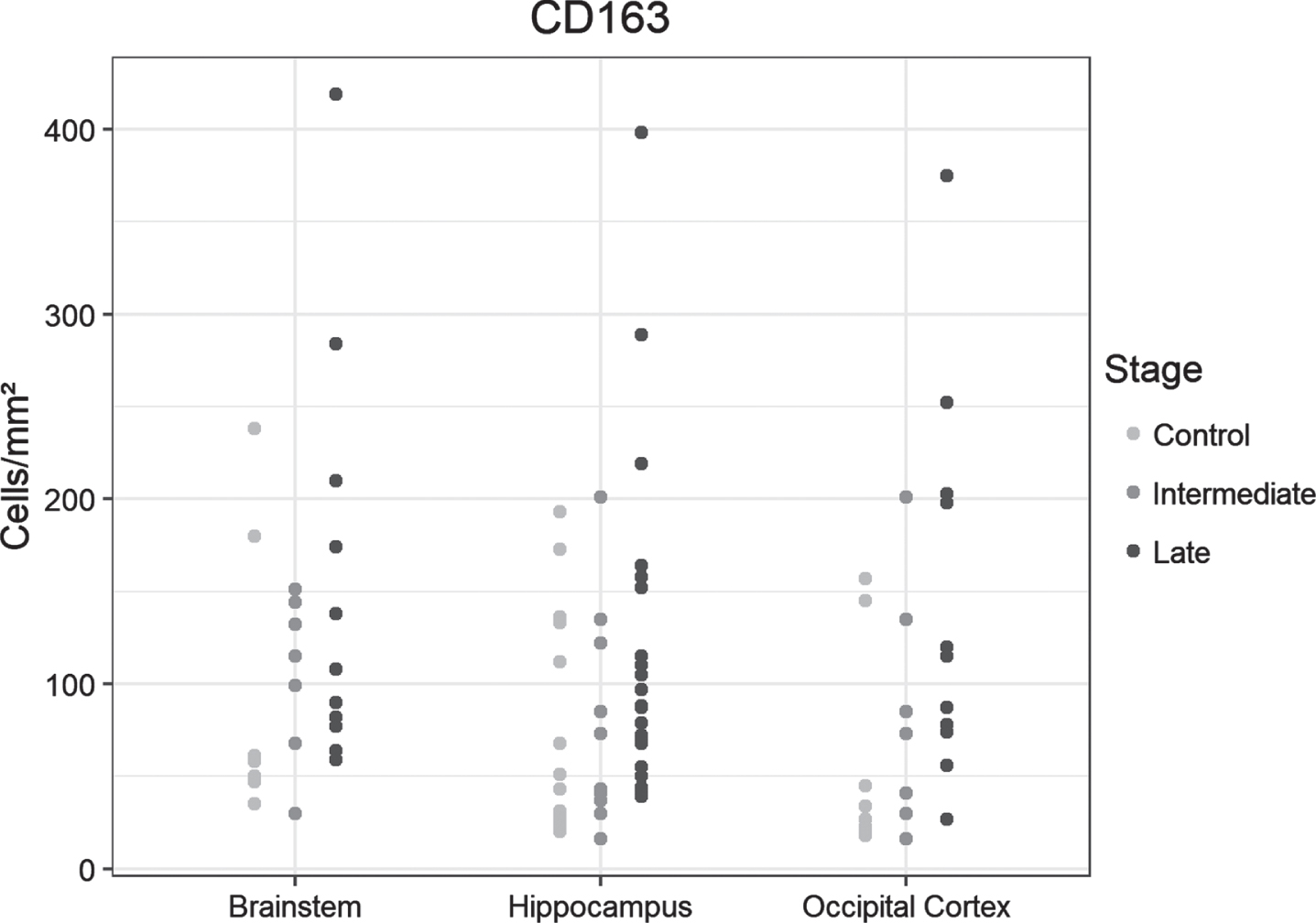
CD163
The number of positive cells increased stepwise in the hippocampus, occipital lobe, and brainstem samples from controls to the intermediate- and late-AD stages, but the groups did not differ significantly (Fig. 3).
Fig. 3
Dot plots presentation of CD163-positive cells in the analyzed regions of the control, intermediate and late AD groups.
CD 206
The overall density of positive cells was considerably lower in all regions of interest, and stained cells were predominantly encountered in perivascular regions. Cell density increased incrementally from control cases to late-AD stages, with significant differences between the control and late-AD samples from hippocampus, occipital and brainstem, but also between intermediate and late AD in hippocampus and occipital (Fig. 4A). Examples of immunohistochemical examinations with CD206 are given in Fig. 4B.
Fig .4A
Dot plots presentation of CD206-positive cells in the analyzed regions of the control, intermediate and late AD groups.
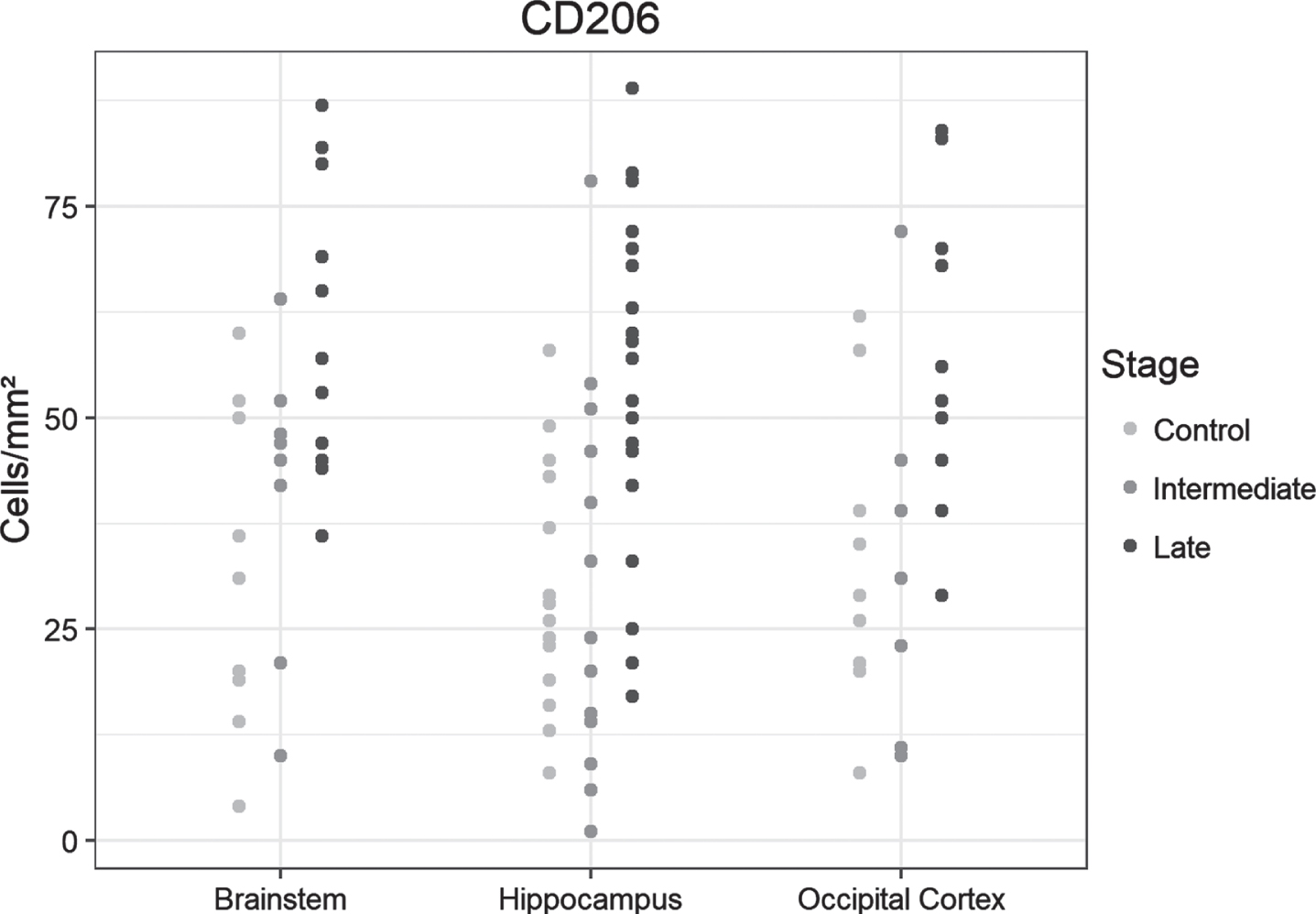
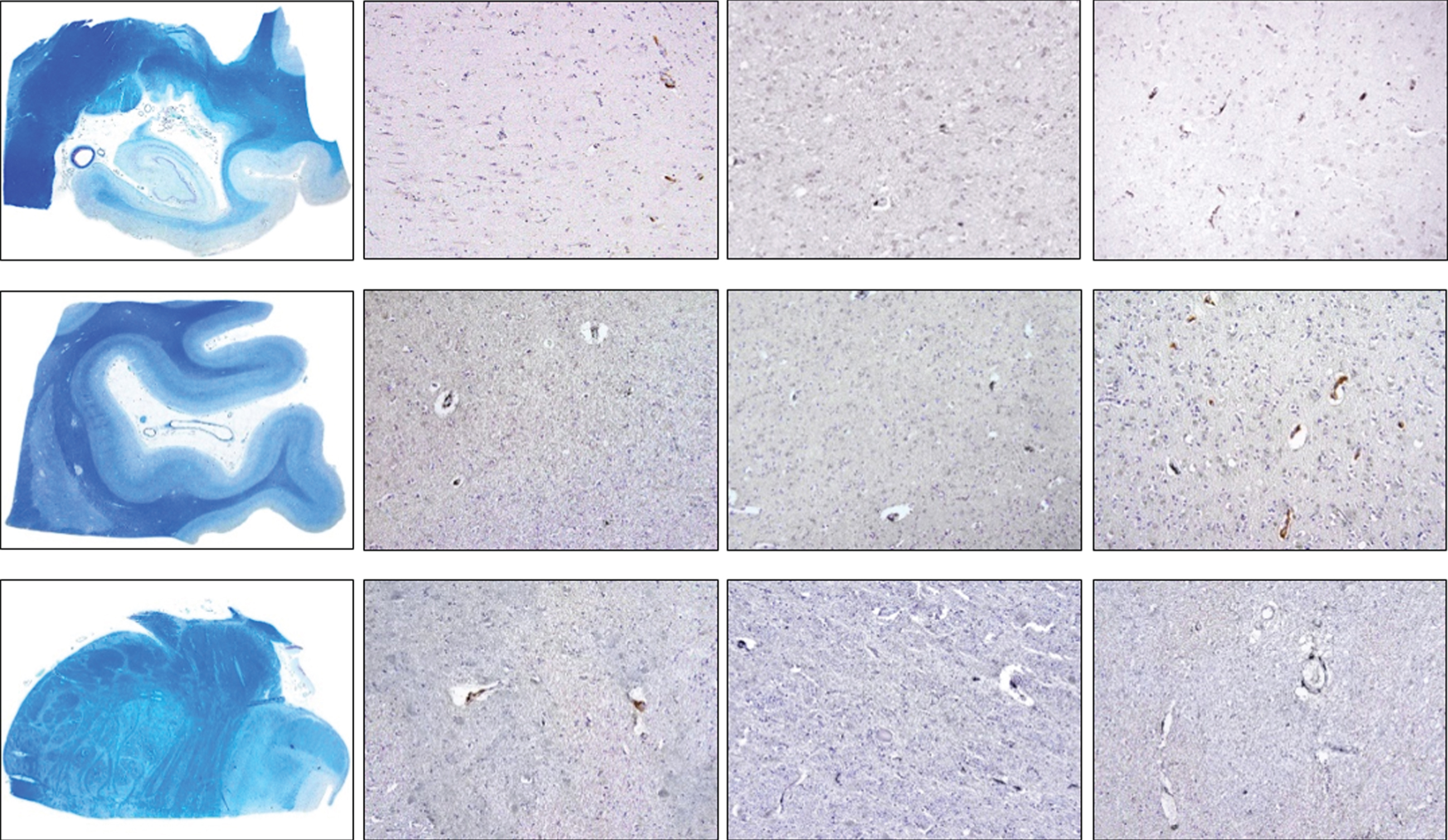
Fig. 4B
Density of CD206-positive cells in control, intermediate and late AD, respectively (upper row, hippocampus; middle row, occipital cortex, lower row, brainstem) (magnification 100x).
CCR2
Few cells were positive for this antibody. We found no significant differences in CCR2-positive cell densities between samples from the controls and the two AD groups (Fig. 5A). Examples of immunohistochemical examinations with CCR2 are given in Fig. 5B.
Fig. 5A
Dot plots presentation of CCR2-positive cells in the analyzed regions of the control, intermediate and late AD groups.
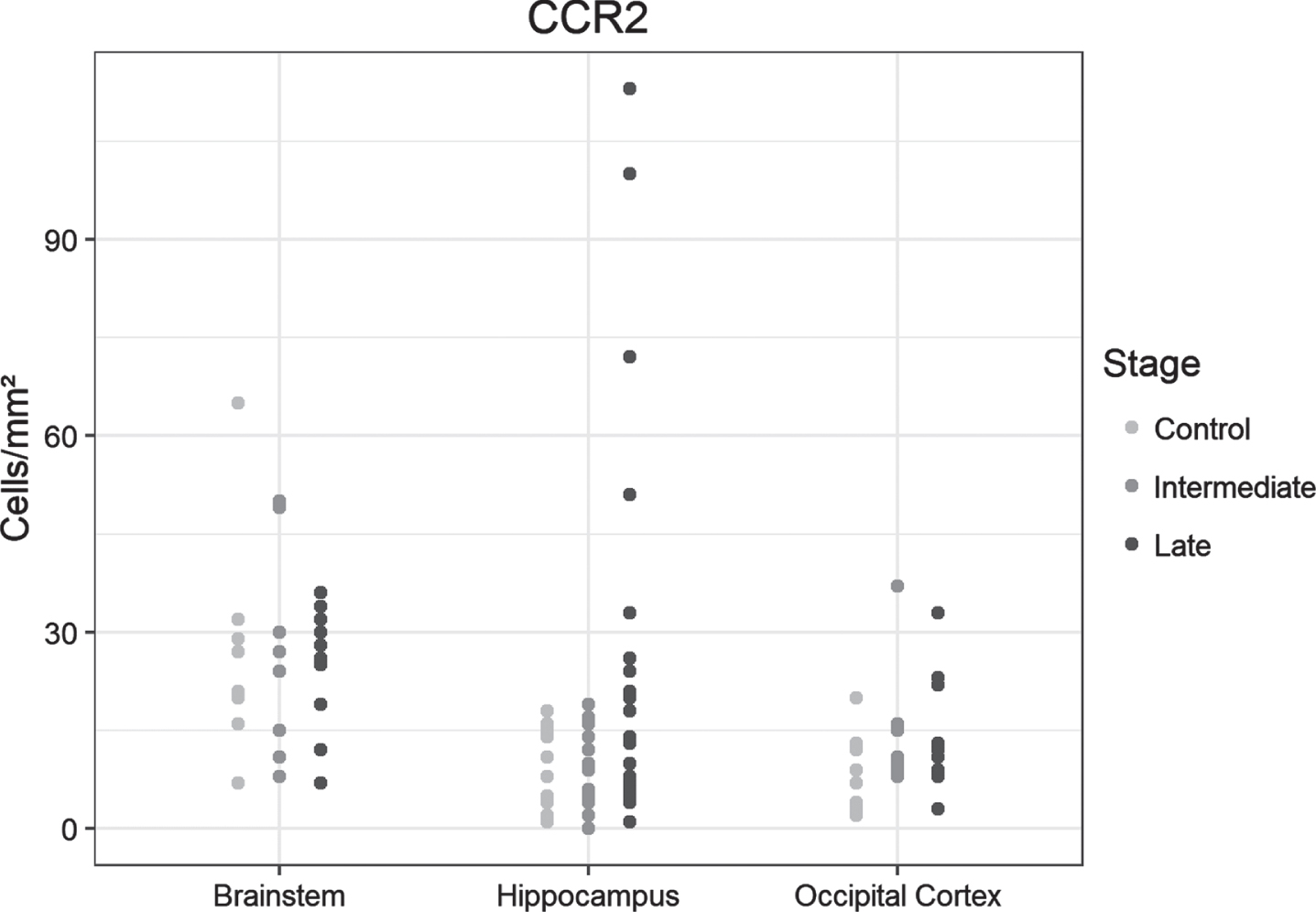
Fig. 5B
Density of CCR2-positive cells in control, intermediate and late AD, respectively (upper row, hippocampus; middle row, occipital cortex, lower row, brainstem) (magnification 200x).
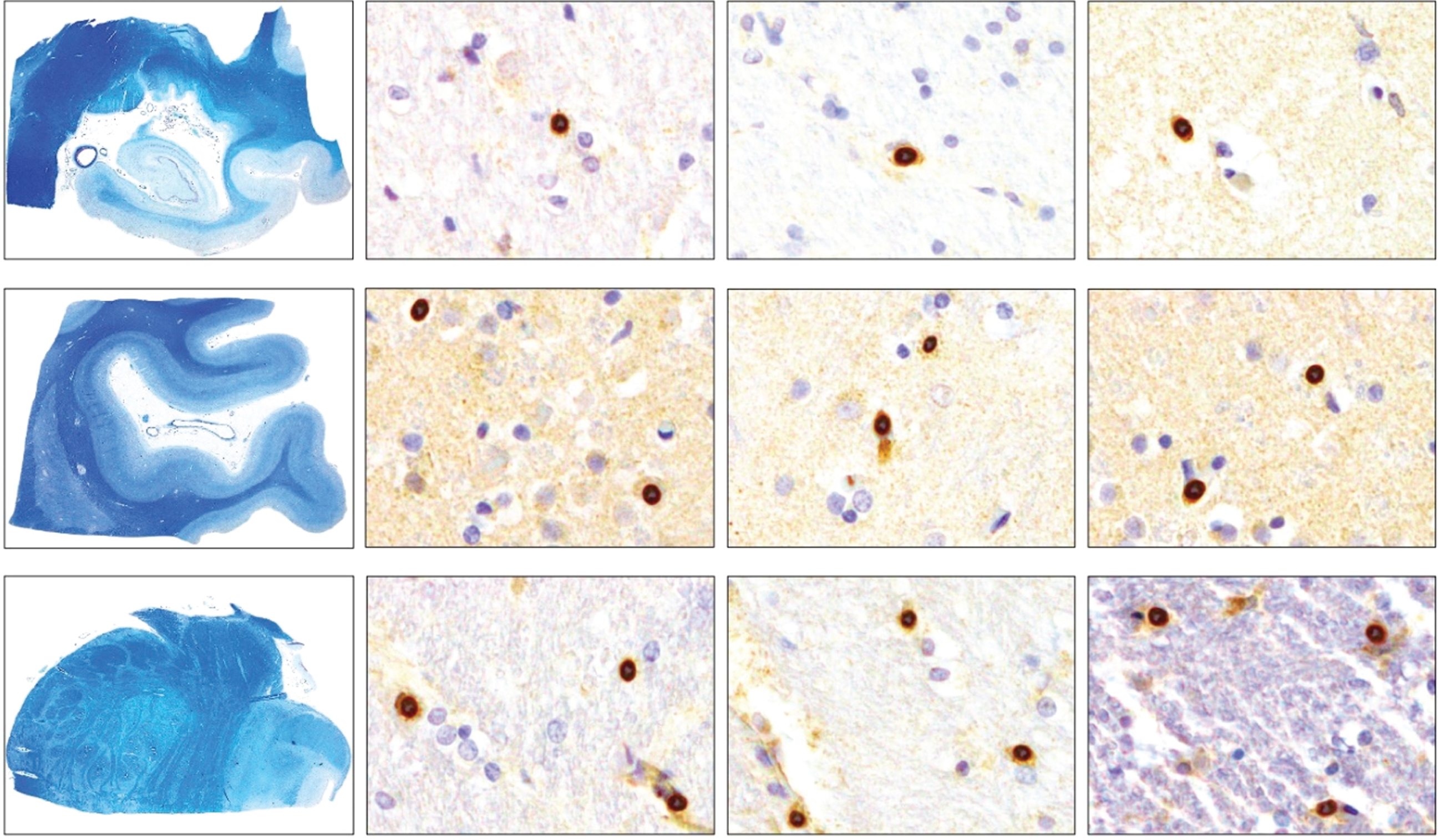
Within the cerebellum, positive cells represented a low and almost constant number in all analyzed groups and with all markers used (data not shown).
For the relative proportions of HLA-DR–positive microglia (pro-inflammatory type) and CD163- and CD206-positive microglia (anti-inflammatory type), we used two-way ANOVA to evaluate differences among groups. We found that the proportions in intermediate and late AD were higher in the hippocampus compared to controls (p < 0.001), but the intermediate- and late-AD groups did not differ with each other. Stepwise increases from controls to the intermediate- and late-AD groups were not significant in the occipital lobe, and neither was an increase in brainstem in intermediate-AD compared to control samples or a decrease in the late-AD group versus controls.
DISCUSSION
The aim of this study was to analyze the extent and distribution of different subtypes of microglia in differently vulnerable brain regions during the AD course and to examine the participation of monocytes in plaque clearance. With the common macrophage marker CD68, we gained an overview of the overall distribution of tmicroglia in the selected brain regions at different disease stages, intermediate and late.
We found a significant increase in CD68-positive cell density with disease progression in all regions examined except the cerebellum. Similar results have been eported in studies on human postmortem tissue [49], but others have reported only an agglomeration of such cells in the vicinity of plaques [44]. In particular, the early affected hippocampus displayed a significant increase in the intermediate-AD samples compared with unaffected controls, without any further increase seen in late-AD samples. In previous studies, a significant increase was seen by other authors only in late AD [50]. One explanation could be the different study design including cases with Braak stage II in the control group by these authors.
In both, controls and AD samples, we identified the most CD68-positive cells in the brainstem regions around the pontine locus coeruleus. Mittelbronn et al. who examined microglial distribution in healthy brains, also found the highest number of CD68-positive cells in brainstem, especially in the pons [51]. The role of brainstem in AD is controversial. Some authors have described tau deposits in the locus coeruleus in young patients, assuming an involvement in very early AD stages [52]. In our samples, we could not detect neurofibrillary tangles in this region in the intermediate-stage group and found them only sporadically in the late-stage samples. The analysis of thick sections versus 5-μm thick sections may explain this divergence from recently reported findings.
The characteristic neuromelanin-rich cells of the locus coeruleus are arranged in an elongated slender cell column that extends up to 20 mm in its rostro-caudal course. With extensive serial sectioning, a focal and disease-specific progression of tau pathology and neurodegeneration has been described [53, 54]. In the current work, staining for HLA-DR, which marks activated and predominantly inflammatory M1 microglia, also was significantly higher in the brainstem compared to the other regions. These data may indicate the involvement of neuroinflammation in early AD. In comparison, we found no significant increase in HLA-DR activity in late-stage AD brainstem samples. With the exception of the hippocampus, the inflammatory microglial reaction thus appears to flatten. Our density measurements are in line with the the data of Carpenter et al. [38]. The number of microglia per area may increase by a factor of 2 to 3 in the course of AD. Density increase is associated with marked hypertrophy and clustering in CD68- and HLA-DR-positive microglia.
The primary sensory cortex of the occipital lobe is affected by AD-typical changes only in later stages (V–VI). Compared with the hippocampus and brain stem data, the results in the occipital lobe showed only an incremental increase from controls to intermediate and late AD, with glial densities comparable to those in hippocampus and brainstem. The primary visual cortex has received little attention in studies on postmortem AD tissue. Our data are in accord with the results of Rezaie et al., who reported moderate to intense CD68 and HLA-DR reactivity, especially around senile plaques, in area 17 of the occipital lobe of patients with AD [55]. The highest density of HLA-DR–positive cells was observed in late-stage samples, with a significant increase compared with controls detectable already in the intermediate-AD samples. This result agrees in part with the neuropathological Braak staging, in which the occipital area is affected only in the late stages. On this basis, microglial involvement would be expected in later stages, with increased rates of tau-positive neuron death and selective region-specific vulnerability of tau-positive neurons [56]. Our results suggest that in regions such as the occipital lobe and brainstem, an inflammatory reaction or microglial activation occurs before the AD-typical neuropathology becomes visible, with an unclear influence on the course of the disease (neuroprotective versus harmful).
The M2 polarization state of the microglia seems to play a rather subordinate role in the pathogenesis of AD, at least quantitatively. Overall, we detected a lower density of CD163- and CD206-positive cells, with an incremental and non-significant increase from control through intermediate- to late-stage AD samples. A slight increase in CD163-positive microglial cells during the disease course has previously been reported in the hippocampus and occipital lobe [43]. In an AD mouse model, however, only a slight increase through the disease course was reported in perivascular CD206-positive microglial cells [57, 58]. These studies also confirm a limited involvement of the CD206-positive microglia in the pathogenesis of neurodegenerative diseases, with the M2 polarization known to be more characteristic of tumor-associated macrophages, which obviously support tumor growth [59].
In contrast to acute events such as trauma, inflammation, ischemia, and hemorrhage, in which numerous monocytes migrate into the damaged brain parenchyma [60, 61], the role of these cells appears to be at least numerically insignificant in human AD. In our study, we found only very few cells in all brain regions and stages studied. The brainstem contained the highest number of positive cells overall, followed by the hippocampus. Studies using an AD mouse model showed that CCR2-positive monocytes migrate into the brain parenchyma to participate in the breakdown of amyloid plaques [62]. Other studies have shown that CCR2 antagonists, used in chronic inflammatory diseases, can accelerate AD progression [46]. It is possible that the mechanism of monocytes involvement works less efficiently in an aged human brain. Consequently, the “recruitment” of monocytes takes place in only a quite limited way or not at all. In addition, monocytes from patients with AD have a significantly lower level of phagocytic activity than monocytes from healthy individuals [63]. Indeed, transplantation of bone marrow stem cells from healthy people into patients with AD has recently been considered as a therapeutic option [62]. Similar observations have been reported in Parkinson’s disease, with relatively numerous CCR2-positive monocytes found to migrate to the brain parenchyma in animal models but not in postmortem human brains [64]. However, animal models for neurodegenerative diseases do not fully reproduce the chronicity of degeneration observed in humans.
In our comparison of the hippocampal distribution ratios of pro-inflammatory M1 microglia (HLA-DR) and anti-inflammatory M2 microglia (CD163 and CD206), we found a quantitatively significant difference between control versus intermediate- and late-AD samples. This pattern implies that the pro-inflammatory microglial cells predominate over anti-inflammatory microglia, which also showed a slight increase from controls to intermediate- and late-stage disease samples. A similar tendency, but not significant, was found in the occipital lobe. In the brainstem, we found a significant increase in intermediate-stage samples but a decrease in the late-AD group. Increasing evidence attributes an initial protective function of microglia with ensuing senescence, followed by deficits in the immunological response that accelerates disease progression [65, 66].
This study is limited by its small sample size, biased postmortem intervals and sex-specific bias, that may have influenced staining quality and quantitative assessment. Nevertheless, the samples represent the tissue quality of routine autopsy material. Future studies with an appropriate number of controls and AD cases in all stages should detail the role of monocytes with respect to regional selective vulnerability, focus on the regional and chronological impact of tau on neuronal survival/death, and disentangle the impact of genetic and environmental factors on the pathobiology of AD subtypes [67].
CONCLUSIONS
The present study provides additional insights into the role and distribution of microglia and migrated monocytes in the course of AD in postmortem brains. In contrast to studies on animal models, we observed only a quite low level of myeloid monocytes associated with AD pathology. Our data suggest that the immune response in the human brain is supported mainly by local microglial cells. In addition, we provide evidence associating early microglial reactions carried out in particular by pro-inflammatory cells with early effects on tangle- and plaque-positive vulnerable brain regions. The reaction increases during the disease course in some regions but flattens or even decreases in others.
ACKNOWLEDGMENTS
We thank Michaela Hartmann and Petra Herud for expert technical assistance, and Erwin Schmitt for help with figure design.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Dumurgier J , Tzourio C ((2020) ) Epidemiology of neurological diseases in older adults. Rev Neurol 176: , 642–648. |
[2] | Nichols E , Szoeke CEI , Vollset SE , Abbasi N , Abd-Allah F , Abdela J , Aichour MTE , Akinyemi RO , Alahdab F , Asgedom SW , et al. ((2019) ) Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18: , 88–106. |
[3] | Hebert LE , Weuve J , Scherr PA , Evans DA ((2013) ) Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80: , 1778–1783. |
[4] | Montine TJ , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Trojanowski JQ , Vinters HV , Hyman BT ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123: , 1–11. |
[5] | Besser LM , Kukull WA , Teylan MA , Bigio EH , Cairns NJ , Kofler JK , Montine TJ , Schneider JA , Nelson PT ((2018) ) The Revised National Alzheimer’s Coordinating Center’s Neuropathology Form-available data and new analyses. J Neuropathol Exp Neurol 77: , 717–726. |
[6] | Hyman BT , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Carrillo MC , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Thies B , Trojanowski JQ , Vinters HV , Montine TJ ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8: , 1–13. |
[7] | Husain M ((2017) ) Alzheimer’s disease: time to focus on the brain, not just molecules. Brain 140: , 251–253. |
[8] | Castellani RJ , Lee HG , Zhu XW , Nunomura A , Perry G , Smith MA ((2006) ) Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol 111: , 503–509. |
[9] | Mudher A , Lovestone S ((2002) ) Alzheimer’s disease - do tauists and baptists finally shake hands? Trends Neurosci 25: , 22–26. |
[10] | Duyckaerts C ((2011) ) Tau pathology in children and young adults: can you still be unconditionally baptist? Acta Neuropathol 121: , 145–147. |
[11] | Thal DR , Rüb U , Orantes M , Braak H ((2002) ) Phases of Ab-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[12] | Stratmann K , Heinsen H , Korf HW , Del Turco D , Ghebremedhin E , Seidel K , Bouzrou M , Grinberg LT , Bohl J , Wharton SB , den Dunnen W , Rub U ((2016) ) Precortical phase of Alzheimer’s disease (AD)-related tau cytoskeletal pathology. Brain Pathol 26: , 371–386. |
[13] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[14] | Alafuzoff I , Arzberger T , Al-Sarraj S , Bodi I , Bogdanovic N , Braak H , Bugiani O , Del-Tredici K , Ferrer I , Gelpi E , Giaccone G , Graeber MB , Ince P , Kamphorst W , King A , Korkolopoulou P , Kovacs GG , Larionov S , Meyronet D , Monoranu C , Parchi P , Patsouris E , Roggendorf W , Seilhean D , Tagliavini F , Stadelmann C , Streichenberger N , Thal DR , Wharton SB , Kretzschmar H ((2008) ) Staging of neurofibrillary pathology in Alzheimer’s disease: a study of the BrainNet Europe Consortium. Brain Pathol 18: , 484–496. |
[15] | Braak H , Del Tredici K ((2015) ) Neuroanatomy and pathology of sporadic Alzheimer’s disease. Adv Anat Embryol Cell Biol 215: , 1–162. |
[16] | Mavroudis IA , Fotiou DF , Adipepe LF , Manani MG , Njau SD , Psaroulis D , Costa VG , Baloyannis SJ ((2010) ) Morphological changes of the human purkinje cells and deposition of neuritic plaques and neurofibrillary tangles on the cerebellar cortex of Alzheimer’s disease. Am J Alzheimers Dis Other Demen 25: , 585–591. |
[17] | Braak H , Del Tredici K ((2011) ) Alzheimer’s pathogenesis: is there neuron-to-neuron propagation? Acta Neuropathol 121: , 589–595. |
[18] | Grinberg LT , Rub U , Ferretti RE , Nitrini R , Farfel JM , Polichiso L , Gierga K , Jacob-Filho W , Heinsen H , Brazilian Brain Bank Study G ((2009) ) The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol Appl Neurobiol 35: , 406–416. |
[19] | Simic G , Stanic G , Mladinov M , Jovanov-Milosevic N , Kostovic I , Hof PR ((2009) ) Does Alzheimer’s disease begin in the brainstem? Neuropathol Appl Neurobiol 35: , 532–554. |
[20] | Braak H , Thal DR , Ghebremedhin E , Del Tredici K ((2011) ) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70: , 960–969. |
[21] | Kaufman SK , Del Tredici K , Thomas TL , Braak H , Diamond MI ((2018) ) Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol 136: , 57–67. |
[22] | Heinsen H , Grinberg LT ((2018) ) On the origin of tau seeding activity in Alzheimer’s disease. Acta Neuropathol 136: , 815–817. |
[23] | Berridge MJ ((2010) ) Calcium hypothesis of Alzheimer’s disease. Pflugers Arch 459: , 441–449. |
[24] | Shigenaga MK , Hagen TM , Ames BN ((1994) ) Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A 91: , 10771–10778. |
[25] | Mudher A , Colin M , Dujardin S , Medina M , Dewachter I , Alavi Naini SM , Mandelkow EM , Mandelkow E , Buee L , Goedert M , Brion JP ((2017) ) What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun 5: , 99. |
[26] | Aguzzi A , Barres BA , Bennett ML ((2013) ) Microglia: scapegoat, saboteur, or something else? Science 339: , 156–161. |
[27] | Katsumoto A , Lu H , Miranda AS , Ransohoff RM ((2014) ) Ontogeny and functions of central nervous system macrophages. J Immunol 193: , 2615–2621. |
[28] | Griffin WS ((2006) ) Inflammation and neurodegenerative diseases. Am J Clin Nutr 83: , 470S–474S. |
[29] | Streit WJ , Braak H , Xue QS , Bechmann I ((2009) ) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 118: , 475–485. |
[30] | Walker DG , Lue LF ((2015) ) Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 7: , 56. |
[31] | Wei J , Gabrusiewicz K , Heimberger A ((2013) ) The controversial role of microglia in malignant gliomas. Clin Dev Immunol 2013: , 285246. |
[32] | Franco R , Lillo A , Rivas-Santisteban R , Reyes-Resina I , Navarro G ((2021) ) Microglial adenosine receptors: from preconditioning to modulating the M1/M2 balance in activated cells. Cells 10: , 1124. |
[33] | Kobashi S , Terashima T , Katagi M , Nakae Y , Okano J , Suzuki Y , Urushitani M , Kojima H ((2020) ) Transplantation of M2-deviated microglia promotes recovery of motor function after spinal cord injury in mice. Mol Ther 28: , 254–265. |
[34] | Holness CL , Simmons DL ((1993) ) Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 81: , 1607–1613. |
[35] | Hopperton KE , Mohammad D , Trepanier MO , Giuliano V , Bazinet RP ((2018) ) Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: a systematic review. Mol Psychiatry 23: , 177–198. |
[36] | Hoozemans JJ , van Haastert ES , Veerhuis R , Arendt T , Scheper W , Eikelenboom P , Rozemuller AJ ((2005) ) Maximal COX-2 and ppRb expression in neurons occurs during early Braak stages prior to the maximal activation of astrocytes and microglia in Alzheimer’s disease. J Neuroinflammation 2: , 27. |
[37] | Kellner A , Matschke J , Bernreuther C , Moch H , Ferrer I , Glatzel M ((2009) ) Autoantibodies against beta-amyloid are common in Alzheimer’s disease and help control plaque burden. Ann Neurol 65: , 24–31. |
[38] | Carpenter AF , Carpenter PW , Markesbery WR ((1993) ) Morphometric analysis of microglia in Alzheimer’s disease. J Neuropathol Exp Neurol 52: , 601–608. |
[39] | McGeer PL , Itagaki S , Tago H , McGeer EG ((1988) ) Occurrence of HLA-DR reactive microglia in Alzheimer’s disease. Ann N Y Acad Sci 540: , 319–323. |
[40] | Perry VH , Nicoll JA , Holmes C ((2010) ) Microglia in neurodegenerative disease. Nat Rev Neurol 6: , 193–201. |
[41] | Akiyama H , Barger S , Barnum S , Bradt B , Bauer J , Cole GM , Cooper NR , Eikelenboom P , Emmerling M , Fiebich BL , Finch CE , Frautschy S , Griffin WS , Hampel H , Hull M , Landreth G , Lue L , Mrak R , Mackenzie IR , McGeer PL , O’Banion MK , Pachter J , Pasinetti G , Plata-Salaman C , Rogers J , Rydel R , Shen Y , Streit W , Strohmeyer R , Tooyoma I , Van Muiswinkel FL , Veerhuis R , Walker D , Webster S , Wegrzyniak B , Wenk G , Wyss-Coray T ((2000) ) Inflammation and Alzheimer’s disease. Neurobiol Aging 21: , 383–421. |
[42] | Lau SK , Chu PG , Weiss LM ((2004) ) CD163: a specific marker of macrophages in paraffin-embedded tissue samples. Am J Clin Pathol 122: , 794–801. |
[43] | Pey P , Pearce RK , Kalaitzakis ME , Griffin WS , Gentleman SM ((2014) ) Phenotypic profile of alternative activation marker CD163 is different in Alzheimer’s and Parkinson’s disease. Acta Neuropathol Commun 2: , 21. |
[44] | Dal Bianco A , Bradl M , Frischer J , Kutzelnigg A , Jellinger K , Lassmann H ((2008) ) Multiple sclerosis and Alzheimer’s disease. Ann Neurol 63: , 174–183. |
[45] | Kerrigan AM , Brown GD ((2009) ) C-type lectins and phagocytosis. Immunobiology 214: , 562–575. |
[46] | El Khoury J , Toft M , Hickman SE , Means TK , Terada K , Geula C , Luster AD ((2007) ) Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med 13: , 432–438. |
[47] | Mack M , Cihak J , Simonis C , Luckow B , Proudfoot AE , Plachy J , Bruhl H , Frink M , Anders HJ , Vielhauer V , Pfirstinger J , Stangassinger M , Schlondorff D ((2001) ) Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol 166: , 4697–4704. |
[48] | Pretscher D , Distel L , Grabenbauer G , Wittlinger M , Buettner M , Niedobitek G ((2009) ) Distribution of immune cells in head and neck cancer: CD8+ T-cells and CD20+ B-cells in metastatic lymph nodes are associated with favourable outcome in patients with oro-and hypopharyngeal carcinoma. BMC Cancer 9: , 292. |
[49] | Bachstetter AD , Van Eldik LJ , Schmitt FA , Neltner JH , Ighodaro ET , Webster SJ , Patel E , Abner EL , Kryscio RJ , Nelson PT ((2015) ) Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun 3: , 32. |
[50] | Sanchez-Mejias E , Navarro V , Jimenez S , Sanchez-Mico M , Sanchez-Varo R , Nunez-Diaz C , Trujillo-Estrada L , Davila JC , Vizuete M , Gutierrez A , Vitorica J ((2016) ) Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol 132: , 897–916. |
[51] | Mittelbronn M , Dietz K , Schluesener HJ , Meyermann R ((2001) ) Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol 101: , 249–255. |
[52] | Braak H , Del Tredici K ((2011) ) The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol 121: , 171–181. |
[53] | Arendt T , Brückner MK , Morawski M , Jäger C , Gertz HJ ((2015) ) Early neurone loss in Alzheimer’s disease: cortical or subcortical? Acta Neuropathol Commun 3: , 10. |
[54] | Theofilas P , Ehrenberg AJ , Dunlop S , Di Lorenzo Alho AT , Nguy A , Leite RE , Rodriguez RD , Mejia MB , Suemoto CK , Ferretti-Rebustini RE , Polichiso L , Nascimento CF , Seeley WW , Nitrini R , Pasqualucci CA , Jacob Filho W , Rueb U , Neuhaus J , Heinsen H , Grinberg LT ((2017) ) Locus coeruleus volume and cell population changes during Alzheimer’s disease progression: a stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimers Dement 13: , 236–246. |
[55] | Rezaie P , Pontikis CC , Hudson L , Cairns NJ , Lantos PL ((2005) ) Expression of cellular prion protein in the frontal and occipital lobe in Alzheimer’s disease, diffuse Lewy body disease, and in normal brain: an immunohistochemical study. J Histochem Cytochem 53: , 929–940. |
[56] | Eser RA , Ehrenberg AJ , Petersen C , Dunlop S , Mejia MB , Suemoto CK , Walsh CM , Rajana H , Oh J , Theofilas P , Seeley WW , Miller BL , Neylan TC , Heinsen H , Grinberg LT ((2018) ) Selective vulnerability of brainstem nuclei in distinct tauopathies: A postmortem study. J Neuropathol Exp Neurol 77: , 149–161. |
[57] | Mammana S , Fagone P , Cavalli E , Basile MS , Petralia MC , Nicoletti F , Bramanti P , Mazzon E ((2018) ) The role of macrophages in neuroinflammatory and neurodegenerative pathways of Alzheimer’s disease, amyotrophic lateral sclerosis, and multiple sclerosis: pathogenetic cellular effectors and potential therapeutic targets. Int J Mol Sci 19: , 831. |
[58] | Scholtzova H , Chianchiano P , Pan J , Sun Y , Goni F , Mehta PD , Wisniewski T ((2014) ) Amyloid beta and Tau Alzheimer’s disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol Commun 2: , 101. |
[59] | Roesch S , Rapp C , Dettling S , Herold-Mende C ((2018) ) When immune cells turn bad-tumor-associated microglia/macrophages in glioma. Int J Mol Sci 19: , 436. |
[60] | Deng YY , Lu J , Ling EA , Kaur C ((2009) ) Monocyte chemoattractant protein-1 (MCP-1) produced via NF-kappaB signaling pathway mediates migration of amoeboid microglia in the periventricular white matter in hypoxic neonatal rats. Glia 57: , 604–621. |
[61] | Dimitrijevic OB , Stamatovic SM , Keep RF , Andjelkovic AV ((2007) ) Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38: , 1345–1353. |
[62] | Theriault P , ElAli A , Rivest S ((2015) ) The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res Ther 7: , 41. |
[63] | Avagyan H , Goldenson B , Tse E , Masoumi A , Porter V , Wiedau-Pazos M , Sayre J , Ong R , Mahanian M , Koo P , Bae S , Micic M , Liu PT , Rosenthal MJ , Fiala M ((2009) ) Immune blood biomarkers of Alzheimer disease patients. J Neuroimmunol 210: , 67–72. |
[64] | Hirsch EC , Standaert DG ((2021) ) Ten unsolved questions about neuroinflammation in Parkinson’s disease. Mov Disord 36: , 16–24. |
[65] | Ulland TK , Song WM , Huang SC , Ulrich JD , Sergushichev A , Beatty WL , Loboda AA , Zhou Y , Cairns NJ , Kambal A , Loginicheva E , Gilfillan S , Cella M , Virgin HW , Unanue ER , Wang Y , Artyomov MN , Holtzman DM , Colonna M ((2017) ) TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170: , 649–663 e613. |
[66] | Sarlus H , Heneka MT ((2017) ) Microglia in Alzheimer’s disease. J Clin Invest 127: , 3240–3249. |
[67] | Jellinger KA ((2021) ) Pathobiological subtypes of Alzheimer disease. Dement Geriatr Cogn Disord 49: , 321–333. |


