
Perspectives on the Role of APOE4 as a Therapeutic Target for Alzheimer’s Disease
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease that is coupled with chronic cognitive dysfunction. AD cases are mostly late onset, and genetic risk factors like the Apolipoprotein E (APOE) play a key role in this process. APOE ɛ2, APOE ɛ3, and APOE ɛ4 are three key alleles in the human APOE gene. For late onset, APOE ɛ4 has the most potent risk factor while APOE ɛ2 plays a defensive role. Several studies suggests that APOE ɛ4 causes AD via different processes like neurofibrillary tangle formation by amyloid-β accumulation, exacerbated neuroinflammation, cerebrovascular disease, and synaptic loss. But the pathway is still unclear that which actions of APOE ɛ4 lead to AD development. Since APOE was found to contribute to many AD pathways, targeting APOE ɛ4 can lead to a hopeful plan of action in development of new drugs to target AD. In this review, we focus on recent studies and perspectives, focusing on APOE ɛ4 as a key molecule in therapeutic strategies.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease in which neuronal cell death occurs that leads to memory loss and impaired cognitive functions (Fig. 1). It is the most prevalent disease in all brain function disorders that is associated with cognitive decline [1]. AD is identified by extracellular amyloid-β (Aβ) aggregation, intracellular neurofibrillary tangles, with activated glia and neuronal death. The risk of progressing AD is increased by lifestyle and environmental factors, but genetic risk factors also play a key role in it. Several studies have shown that there are many loci connected with AD which are both susceptible and pathogenic genes. According to the age of onset, AD is defined by two forms, early onset (EOAD) and late onset (LOAD). EOAD occurs in people who are 65 years below and 1–5% cases of AD are early onset AD. LOAD occurs in people who are above 65 years and 95% of AD cases are late onset AD. There are three gene mutations that play a key role in EOAD and Aβ generation, such as amyloid precursor protein (APP), presenilin-1 (PSEN1), and presenilin-2 (PSEN2). On the other note, LOAD is connected with many genes, and it is more complicated. The allele APOE ɛ4 is considered as a preponderating determinant of LOAD via genetics [2].
Fig. 1
The symptoms of Alzheimer’s disease which are usually seen in a patient include: Amnesia (short term memory loss but later turns into long term memory loss); Aphasia (unable to speak); Apraxia (difficulty with skilled movement); Anosognosia (unaware of difficulties); and Dyscalculia (specific learning disabilities like math and number-based operations).
Single copy of APOE ɛ4 allele inheritance can cause higher risk for AD progression, and the people are at greater risk than those who have inherited two copies of APOE ɛ4 allele for AD progression. It has been found that APOE ɛ4 is also linked to the early onset of memory dysfunction and different symptoms in comparison with people who have AD but do not have the APOE ɛ4 gene. The reason behind the connection of APOE ɛ4 to AD progression is still not clear. Although amyloid plaques are greatly linked with the APOE ɛ4 genes in tissue of AD patient brains, accumulation of amyloid plaques in brain tissue leads to neuronal cell death. It has recently been reported that modifications in tau pathology and neurodegeneration are directly related to APOE isoforms [3].
Oxidative stress in AD patients is also linked with the APOE ɛ4 allele, and it has been found to reduce the antioxidant activity of enzymes in the hippocampus of AD brains. Some oxidative stress markers, including increased oxidized proteins, elevated levels of lipid peroxidation, glycosylated products, formation of aldehydes, alcohols, ketones, free carbonyls, and as well as modified RNA and mitochondrial and nuclear DNA due to oxidation, were found in the AD brain tissue and mitochondria isolated from initial phase of AD during postmortem. ApoE has shown to work as an antioxidant in B12 ApoE expressing cell lines against hydrogen peroxide mediated cytotoxicity. It has been found that ApoE-deficient mice have an increased level of plasma peroxide of low-density lipoproteins. The levels of lipid oxidation were found to be increased in the frontal cortex of APOE ɛ4 homozygous and heterozygous carriers of AD patients in comparison to homozygous APOE ɛ3 carriers. Catalase activity was upregulated in homozygous APOE ɛ4 carriers while superoxide dismutase and glutathione activity was not significantly upregulated in comparison to controls [4, 5].
APOE ɛ4 was identified to have role in AD development 25 years ago but to date there are no drugs approved for directly targeting it yet. APOE isoforms have become a propitious target to understand the progressive pathophysiology of AD patients and adding a novel therapeutic approach towards AD, due to its genetic preponderance. However, there is sufficient clinical research that shows APOE ɛ4 affects various pathways in the brain that lead to AD progression. In this review, we will accentuate APOE ɛ4 targeting studies as a therapeutic strategy to treat AD [6, 7].
AMYLOID-β THEORY OF AD
Amyloid-β is formed by the cleavage of the amyloid-β protein precursor (AβPP) by β-secretase and γ-secretase (see Fig. 2). AβPP is present on chromosome 21. Several alternative splicing isoforms have been observed in humans whose amino acid length ranges from 639–770 amino acids. AβPP has three domains: long extracellular N terminal region, short transmembrane segment, and short C terminal segment.
Fig. 2
In most of the cases, AβPP undergoes non-amyloidogenic processing through a consecutive cleavage by α- and γ-secretase. The α-secretase cleaves 639 amino acids longer AβPP into 83 amino acid fragments. γ-secretase further processes those fragments and generates a P3 fragment which has a role in signal transduction [2]. Alternatively, in case of amyloidogenic processing the AβPP is firstly cleaved by β-secretase (BACE 1) and then by γ-secretase. The β-secretase at first generates 99 amino acid long fragments which are cleaved by γ-secretase further. In 90% cases 40 amino acids long, amyloid-β is generated which is harmless, but in rarest condition, 42 amino longer amyloid-β is formed whose accumulation becomes a triggering cause of a neurodegenerative disease, i.e., Alzheimer’s disease [8].
![In most of the cases, AβPP undergoes non-amyloidogenic processing through a consecutive cleavage by α- and γ-secretase. The α-secretase cleaves 639 amino acids longer AβPP into 83 amino acid fragments. γ-secretase further processes those fragments and generates a P3 fragment which has a role in signal transduction [2]. Alternatively, in case of amyloidogenic processing the AβPP is firstly cleaved by β-secretase (BACE 1) and then by γ-secretase. The β-secretase at first generates 99 amino acid long fragments which are cleaved by γ-secretase further. In 90% cases 40 amino acids long, amyloid-β is generated which is harmless, but in rarest condition, 42 amino longer amyloid-β is formed whose accumulation becomes a triggering cause of a neurodegenerative disease, i.e., Alzheimer’s disease [8].](https://ip.ios.semcs.net:443/media/adr/2021/5-1/adr-5-1-adr210027/adr-5-adr210027-g002.jpg)
Amyloid-β 42 starts accumulating and forms clusters known as oligomers, which is the first stage of aggregation. Following this, chains of clusters form fibrils and the final stage is the formation of plaques which are composed of beta-sheet and other substances. These stages of aggregation disrupt cell-to-cell communication and activate immune cells. The activated immune cells stimulate inflammation in the brain, which ultimately leads to the brain cell destruction [9].
TAU TANGLES PLAY A KEY ROLE IN NEURODEGENERATION
Tau is a microtubule associated protein which maintains cytoskeleton stability and plasticity. It also recruits signaling protein to regulate microtubule mediated axonal transport. Along with these, it has roles in neuronal development, cell signaling, neuroprotection, and apoptosis. To maintain the cytoskeleton plasticity, tau is present as a phosphorylated form and with age plasticity level reduces [10]. Abnormal or excessive phosphorylation (hyperphosphorylation) transforms the normal adult tau into neurofibrillary tangles (NFT). This consequence of tau hyperphosphorylation may arise due to several factors like interaction of ApoE4-tau, upregulation of kinases, or downregulation of phosphatases. Kinases and phosphatases are key enzymes. The phosphorylated tau binds to microtubules and stimulates their assembly, whereas the hyperphosphorylated ones lose their biological activity. In abnormally phosphorylated tau, tau neither binds to tubulin nor promotes microtubule assembly, instead it inhibits assembly and disrupts the microtubule organization required for maintaining the cytoskeleton plasticity [11]. In a hyperphosphorylated state, tau binds to other tau molecules and forms thread which eventually leads to tangle formation. These tangles block the neuron’s transport system and disrupt the synaptic communication between neurons. It was also found that tau hyperphosphorylation induced the over-production of reactive oxygen species which are responsible for cell damage and mitochondrial dysfunctions. Tauopathies dysregulated the balance between reactive oxygen species levels and antioxidant enzyme activity [12].
MUTATION IN PS1/PS2 IS RESPONSIBLE FOR INSOLUBLE AMYLOID AGGREGATION
Presenilin PS1 or PS2 are catalytic units of γ-secretase which is responsible for cleavage of many transmembrane proteins like AβPP. Following the cleavage of AβPP by β-secretase, processing by γ-secretase generates Aβ of varying lengths. Aβ40 accounts for 90% of population whereas minor Aβ42 is hydrophobic and acts to nucleate amyloid aggregation and plaque formation. Mutation in PS1 or PS2 causes overproduction of Aβ42 and acts as one of the responsible factors in causing pathogenic mechanisms in AD [13, 14].
APOLIPOPROTEINS ARE LESSER EXPLORED FROM AD PERSPECTIVE
Apolipoproteins are polar, and thus can travel in polar medium (an aqueous medium) despite being non-polar (lipophilic) in nature. Apolipoproteins interact with receptors and lipid transport proteins to participate in lipid processing and metabolism. They also serve as cofactors of enzymes participating in lipid metabolism.
There are various classes of apolipoproteins which are present on different types of lipoproteins and regulate their functions. Among them, ApoE has a role in the central nervous system. It plays a role in lipid homeostasis and transports cholesterol to neurons via ApoE receptors. ApoE is produced by microglia and astrocytes, whereas in the peripheral system, it is produced by liver and macrophages. Once ApoE is secreted from cells, the ATP binding cassette transporters (ABCA1 and ABCG1) import cholesterol and phospholipids to nascent ApoE to form lipidated apolipoprotein particles. ApoE acts as a ligand for low density lipoprotein receptors (LDLR) (e.g., ApoE receptor and LRP1) and very low-density lipoprotein receptors (VLDLR). ApoE binds with Aβ. LDLR and VLDLR enable ApoE-Aβ complex to pass through them by interacting with them and participating in the clearance of Aβ from the brain [3, 15].
ApoE has different isoforms which have been classified on the basis of difference in amino-acids at positions 112 and 158. E2 has cysteine at both the position, while E3 has cysteine at 112 and arginine at 158 position. It has a better binding affinity for LDLR to mediate lipolytic processing. E4 has arginine at both the positions due to which repulsion occurs between arginine 61 and arginine 112. This enables the formation of a salt bridge between arginine 61 and glutamate 251. Salt bridge disrupts normal functioning of apolipoproteins. E4 binds with VLDLR with better affinity which internalizes the ApoE-Aβ complex at a slower rate than LRP1. So, E4 was less efficient than E2 and E3 in clearing and promoting Aβ transport across the blood vessel walls [16, 17].
ABUNDANCE OF ApoE4 IS RESPONSIBLE FOR SLOW CLEARANCE OF INSOLUBLE AMYLOID AGGREGATES
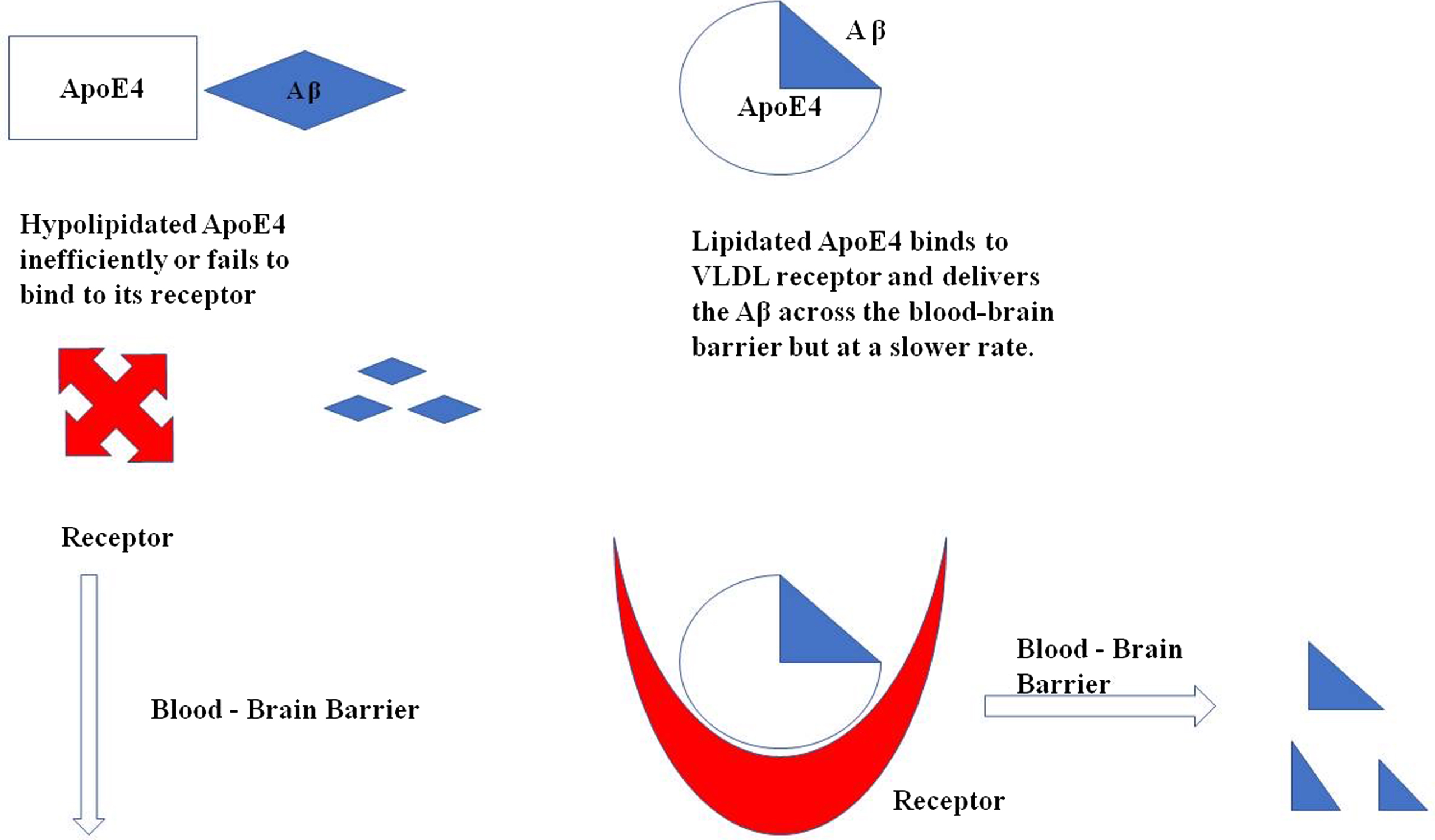
Apolipoproteins expressed by APOE ɛ4 are low in concentration and also hypolipidated as compared to other isoforms due to which it fails to bind to its receptors and even if it binds to the receptors, it is redirected to VLDLR which internalize the complex at slower rate. (Fig. 3)
Fig. 3
Illustration of ApoE4 binding with Aβ for its clearance across the blood-brain barrier. In a hypolipidated state ApoE4 fails to transport the Aβ across the cells as it does not bind to its receptor as efficiently as required, whereas the lipidated ApoE4 binds to its receptor VLDL and delivers the Aβ across the cells.
ApoE4 AFFECTS AND CAUSES THE CONSEQUENCES IN ALZHEIMER’S DISEASE IN NUMEROUS WAYS
ApoE4 is less efficient in clearing the accumulation of Aβ from the brain due to its salt bridge structure and also plays a key role in the over-production of Aβ by affecting the activity of γ-secretase. With respect to clearance, ApoE4 competes with Transcription factor EB to bind with CLEAR (Co-ordinated Lysosomal Expression & Regulation) DNA motif to interfere in the expression of three genes (Sequestosome1, LC3B, and LAMP2) required for the process of autophagy [18]. When Aβ accumulation occurs inside the brain cells, different kinds of proteases are appointed to clear these kinds of aggregation. Neprilysin, a metallo-protease responsible for clearing the Aβ, is usually impaired due to the presence of allele APOE ɛ4 [12]. NMDAR (N-methyl D aspartate receptor) which is a glutamate receptor and ion channel protein in nerve cells gets activated once glutamate and glycine bind to it. After activation, it allows positive charged ions to flow across the cell membrane. The NMDA receptor is responsible for maintaining synaptic plasticity and regulating the formation of synapse underlying the memory. It also leads to the formation of neural networks during developments. ApoE4 blocks this receptor and disrupt the functions carried out by NMDAR [19, 20]. Along with this, ApoE4 blocks certain other receptors like insulin due to which glucose metabolism lowers down. ApoE4 shows the downregulation of brain peroxisome proliferator activated receptor γ (PPARγ) and PPARγ co-activator 1 (PGC1α) signaling, which is involved in maintaining the glucose uptake and metabolism [21]. It also blocks growth factor. ApoE4 is quite prone to proteolytic cleavage due to its unique salt bridge structure. The proteolysis of ApoE4 generates neurotoxic fragments which causes inflammation in the brain [22]. It is also one of the prominent reasons behind the hyperphosphorylation of tau, since it manages to escape the secretory pathways and reaches to cytoplasm where it interacts with zinc and induces the excessive phosphorylation of tau [19]. It acts as an inhibitor of the expression of oxidative respiratory complexes I, IV, and V. This causes mitochondrial dysfunction [14, 19], since Aβ accumulation is high and clearance is low in the carriers of APOE ɛ4, and as a result, they are at greater risk of having cerebral amyloid angiopathy. Cerebral amyloid angiopathy is a condition in which amyloid deposits across the arteries and leads to their shrinkage [23]. ApoE4 is also responsible for other diseases like diabetes, atherosclerosis, and other heart-related diseases as it disturbs lipid homeostasis in cells. See Figs. 4 and 5.
Fig. 4
ApoE4 plays a key role in AD pathophysiology [24]. As mentioned above, ApoE4 is inefficient as compared to other isoforms in clearing Aβ. As ApoE4 fails to clear the accumulation and triggering the over-production of Aβ by increasing the activity of γ-secretase, it eventually leads to the formation of plaques. C) ApoE4 increases the neuroinflammation by elevating the production of cytokines through the microglial activation. D) ApoE4 also plays a significant role in inducing the hyperphosphorylation of tau through ERK activation.
![ApoE4 plays a key role in AD pathophysiology [24]. As mentioned above, ApoE4 is inefficient as compared to other isoforms in clearing Aβ. As ApoE4 fails to clear the accumulation and triggering the over-production of Aβ by increasing the activity of γ-secretase, it eventually leads to the formation of plaques. C) ApoE4 increases the neuroinflammation by elevating the production of cytokines through the microglial activation. D) ApoE4 also plays a significant role in inducing the hyperphosphorylation of tau through ERK activation.](https://ip.ios.semcs.net:443/media/adr/2021/5-1/adr-5-1-adr210027/adr-5-adr210027-g004.jpg)
Fig. 5
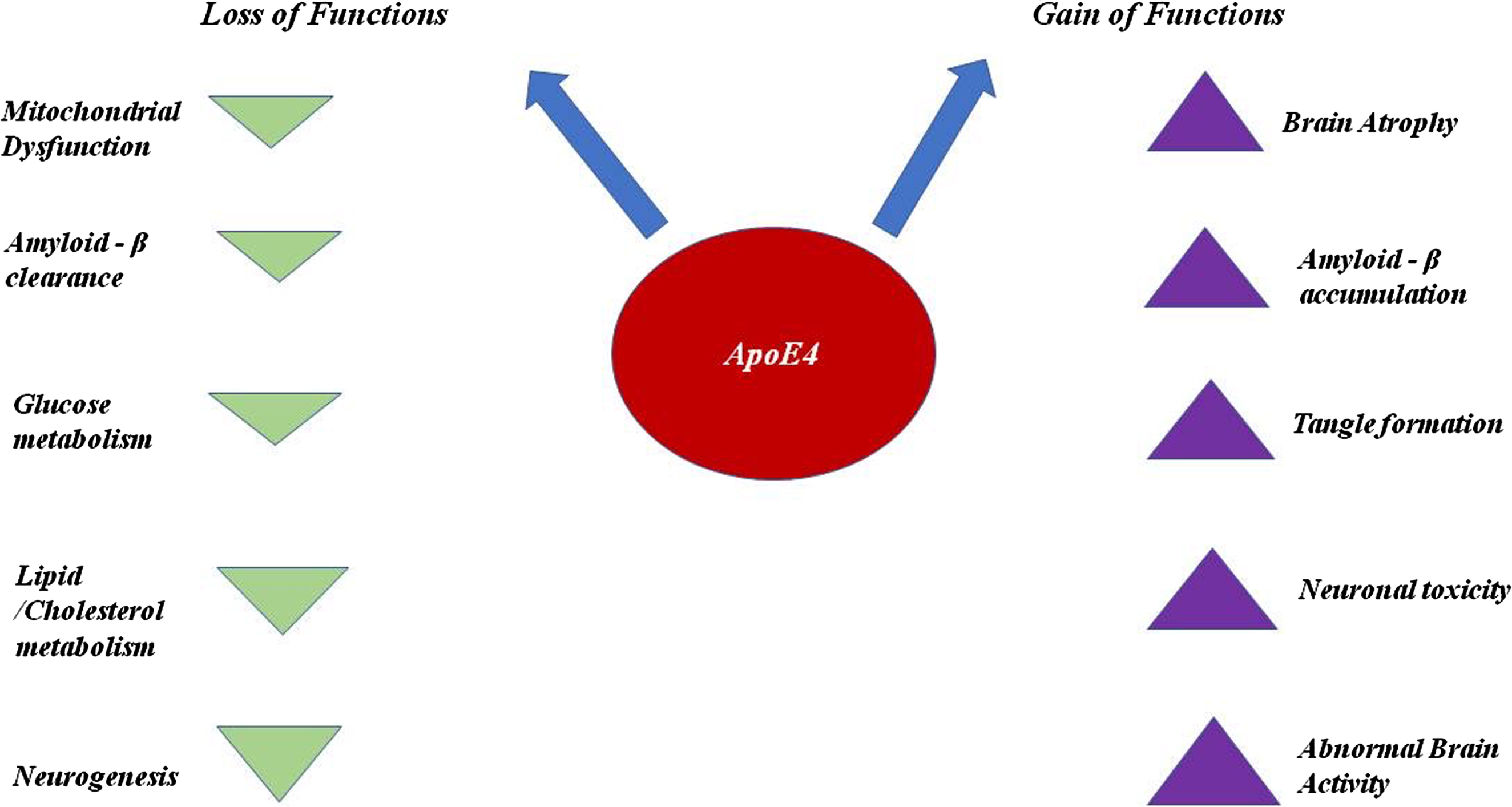
ApoE4 plays a significant role in increasing the risk of causing AD, as ApoE4 interferes in numerous pathways and downregulates as well as upregulates the phenomena which eventually contributes to the pathological consequences in AD.
POSSIBLE THERAPEUTICS DEVELOPMENT APPROACHES BY TARGETING APOE4
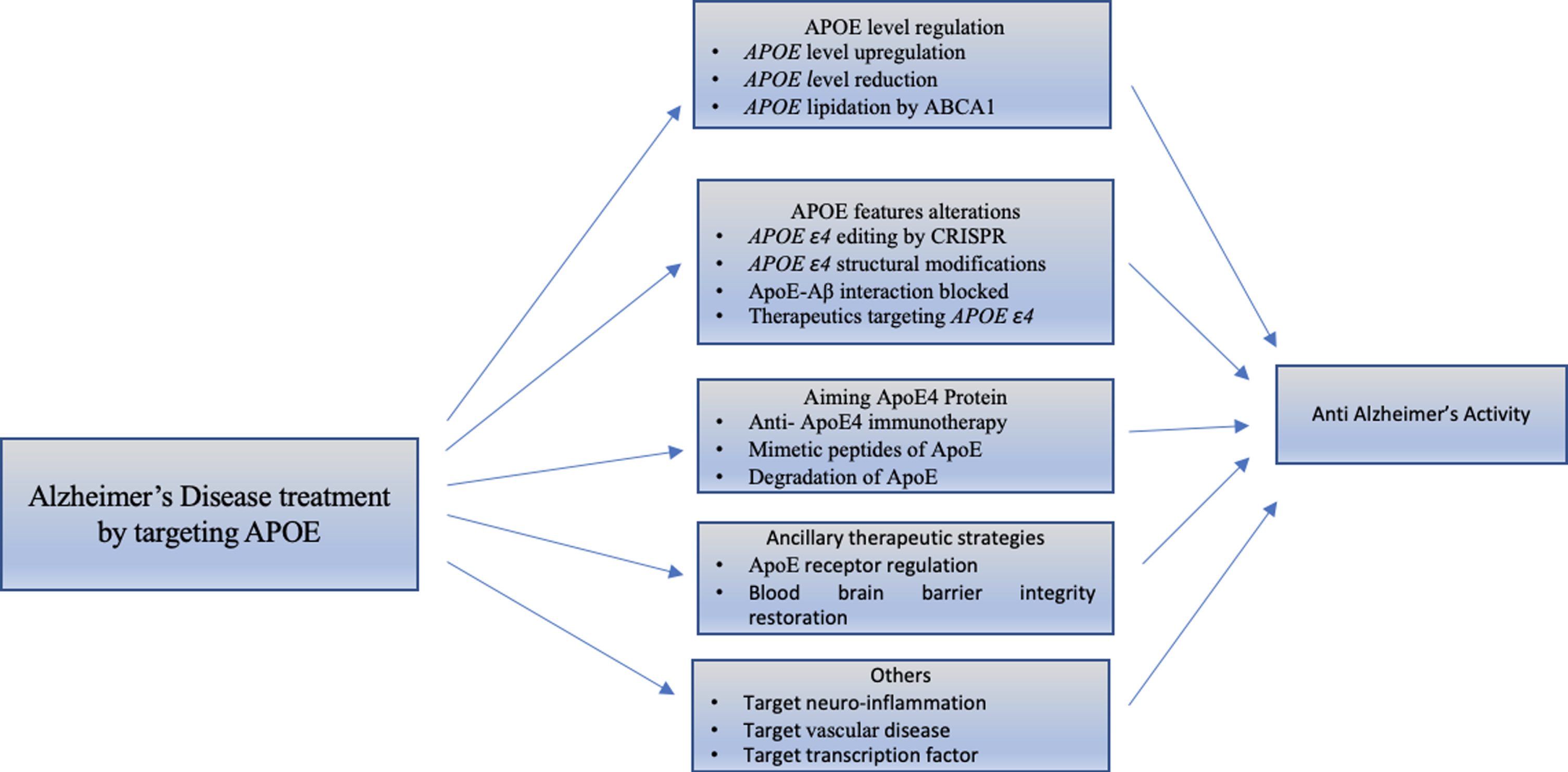
Many studies have suggested that ApoE4 can alter different biological pathways that lead to progression of AD, although the mechanism that links the ApoE to neurodegeneration is still unclear. However, the views to find novel targets for the treatment of AD suggest altering the APOE allele and APOE structure [25]. Different AD treatment strategies targeting APOE4 are shown in Fig. 6.
Fig. 6
Classification of strategies that target APOE for treatment of Alzheimer’s disease. There are different studies that demonstrate the molecular mechanism showing effects of APOE4. We emphasized the advanced therapeutic strategies for different APOE4 characteristics, discussed below.
APOE level regulation
APOE level upregulation
Many reports suggest that the quantity of ApoE in cerebrospinal fluid and plasma of AD patients decreased as compared to healthy individuals. Retinoid X receptor (RXRs), nuclear receptors and Liver X Receptors (LXRs) positively control the APOE transcription [26–28]. ApoE levels in the brain were increased by taking RXR agonist with bexarotene orally, which also decreased Aβ accumulation. Bexarotene alone was found to reduce cognitive dysfunction, and it also helps with restoration of age dependent synaptic plasticity in mice models [29–36]. Although it has negative effects, such as hepatic failure in mice and also failed to clear amyloid plaques from the brain in human models.
ApoE level reduction
Recent studies suggest that ApoE haploinsufficiency instead of APOE genotypes weakens the Aβ accumulation in targeting APOE replacement mice [37–39]. It has also been shown that anti-ApoE antibodies were able to increase spatial learning ability by decreasing ApoE level and Aβ accumulation in animal models [40]. ApoE has bidirectional properties like it increases the AD pathogenesis. So, ApoE removal can help in reducing the toxicity. ApoE isoforms and ApoE fragments deposition can be harmful, so removal of these fragments by immune-depletion like anti monoclonal antibody can be a new therapeutic approach [40–43].
ApoE lipidation by ABCA1
Biologically active ApoE is lipidated and ABCA1 helps in loading of ApoE protein in the brain [44]. Removal of ABCA1 leads to less lipidated ApoE which further leads to amyloidogenic pathogenesis. Moreover, ABCA1 overexpression can reduce the Aβ accumulation in mice models. RXR-LXR agonists can help in transcription of APOE and ABCA1, that leads to Aβ clearance and helps in different other functions like synapse regeneration, cholesterol/lipid transport, and modulation of immune cells [45–47]. So, it can be a new therapeutic strategy without increasing the ApoE level.
APOE features alterations
APOE ɛ4 editing by CRISPR (clustered regularly interspaced short palindromic repeats)
The APOE ɛ4 concentration termination and transformation of APOE ɛ4 to APOE ɛ2 and APOE ɛ3 have potential to play an important role in treatment of AD [48, 49]. The DNA coding of APOE ɛ4 and APOE ɛ3 varies by one nucleotide (112th position of APOE4 is Arg and its cysteine in APOE ɛ3, and chromosome 19 produces APOE ɛ4 allele. CRISPR technology can be employed to modify the APOE ɛ4 to APOE ɛ3 [50, 51]. Here, CRISPR editing and portable off target gene modification at its initial stage provide promising approaches for AD treatment.
ApoE4 structural modifications
The amino acid residues 112 and 158 at N terminal are markers of ApoE polymorphism, while Glu255 and Arg 61 interaction is the structural characteristic of ApoE4 and ApoE3 respectively [52–54]. An anomalous structure of ApoE4 can be produced by this domain-domain interaction, which leads to neurotoxicity [55, 56]. ApoE4 structure-correctors (PH002 and GIND25) interacts with Arg61 which disrupt this domain-domain (Arg61 at N-terminal and Glu255 at C-terminal) interaction leads to ApoE4 to ApoE3 [53]. Therefore, the pathological confirmation change can provide an alternative therapeutic approach.
ApoE -Aβ interaction inhibition
ApoE and Aβ interaction cause Aβ accumulation in the AD brain. An artificial peptide Aβ12 - 28P, analogous to the ApoE binding site on Aβ, reduced the interaction between ApoE and Aβ, as well as Aβ aggregation in animal models [57–61]. Aβ20–29 peptides were also found to reduce the ApoE and Aβ interaction in vitro, apart from helping in decreasing fibrillogenesis and cytotoxicity of Aβ [62]. Immunotherapy of ApoE -like antibodies against human ApoE3 and ApoE4 was found to be useful against non-lipidated ApoE, and helps in reducing the Aβ accumulation in mice models [63]. So, inhibiting ApoE and Aβ interaction might be a promising strategy to treat AD.
Therapeutics targeting ApoE2
ApoE2 occurrence in AD brains is two times lower than normal individuals. ApoE2 has been found to decrease cognitive dysfunction and improve neuroprotective activity [64–67]. Viral vectors expressing ApoE2 injection counteract ApoE4 pathological effects in mice models. So, it is a novel approach to upregulate ApoE2 expression in AD patients [68–70].
Aiming ApoE4 protein
Immunotherapy for anti-APOE4
Antibodies against Aβ and tau are introduced into the periphery where it can neutralize the toxic effect of ApoE4 because here ApoE level is 10 times more than brain [71]. It has been found that anti-ApoE antibodies used in the periphery reduce the amyloid deposition even before the amyloid plaque formation. Anti-ApoE monoclonal antibodies have potential to become a promising strategy for anti- ApoE4 immunotherapy [72, 73].
Mimetic peptides of APOE
These tiny peptides have ApoE domains which includes the amphipathic helix domain. It helps in reducing the neurodegeneration after brain insult and reduces the Aβ and tau pathogenetic effects in mice models [74–76]. Some studies have revealed that these peptides have anti- inflammatory effect and are found to play defensive role in ApoE3 and ApoE4 mice after brain insult [77–79].
Degradation of APOE4
As described earlier, ApoE3 and ApoE4 domain-domain interaction makes it more vulnerable to proteases and contributes to enhanced neurotoxicity through fragments of ApoE4 [80]. ApoE4 fragment generation leads to stressful neurotoxic actions of ApoE4. So, the inhibitor against these ApoE4 fragments could lead to a neutralizing strategy against ApoE4 [81, 82].
Ancillary therapeutic strategies
ApoE receptor regulation
Endocytosis of ApoE is mediated by LDLR and LRP1 (low density lipoprotein receptor related protein 1). These receptors are responsible for the uptake of Aβ either by making ApoE-Aβ complex or by Aβ interaction inhibition to these receptors by competitive inhibition [83–87]. Hence, it is clear that LDLR and LRP1 helps in clearing the Aβ plaques [87–89].
Blood-brain barrier integrity restoration
Blood-brain barrier breaks down in AD by activated astrocytes (that depends on ApoE4 expression on astrocytes) which activates the cyclophilin A- (CyA-) pro-inflammatory molecule, and nuclear factor κB-matrix metalloproteinase 9 (MMP-9) and this further disrupts the tight junctions [90, 91]. Cyclosporin-A was found to be effective in the restoration of blood-brain barrier (BBB) in ApoE4 mice. CyA- and MP-9 was found to be increased in age dependent AD cerebrospinal fluid. A prospective approach including Cyclosporin-A can provide promising strategy to treat AD [92, 93].
Others approaches
Targeting neuro-inflammation
Activated microglia is mostly responsible for the inflammation and the microglia activation varies for different gene expressions. This microglia activation can be controlled [94, 95]. The neuro-inflammation plays protective role at initial stage but causes increased pathogenesis at later stage of AD. So, the therapeutic strategies targeting this inflammation may be helpful in relieving the patient from AD symptoms [96, 97].
Targeting vascular disease
It has been reported that AD risk increases due to vascular atherosclerosis, diabetes and hypertension as ApoE4 is related to vascular atherosclerosis that lead to dementia and disrupted BBB [98–100]. So, the therapeutic strategies that aim to find molecules related to ApoE4 and vascular disease may be helpful in AD treatment also [100–102].
Target transcription factors
ApoE has recently been discovered to translocate to the nucleus and bind with DNA at different locations, some of which are present on the promoter, implying that it functions as a transcription factor that suppresses promoters of genes implicated to AD [103–106]. Though the complete mechanism of its escape from the endoplasmic reticulum and translocation into nucleus is still unknown but it provides a hope for transcriptional regulation-based approach for AD treatment.
CONCLUSION
Recent advancements in technology have helped in understanding AD in a better manner. Studies suggest that multiple factors may be involved in neurodegenerative diseases like AD, and it may be difficult to target a single factor for therapeutic purposes. Diagnosis and therapeutic strategies require clear understanding of the factors involved in disease development and progression at molecular level. Recently APOE ɛ4 has emerged as a promising therapeutic target for treatment of AD as it plays key role in triggering the disease [107]. Studies suggest that AD progression can be avoided by targeting APOE ɛ4 gene and gene products as well as its functions with the aid of the above mentioned approaches [108]. Immunomodulation with ApoE4 monoclonal antibodies, mimesis through peptides of ApoE and ApoE4 degradation may be helpful as a promising approach for delaying the onset of disease.
ACKNOWLEDGMENTS
All authors contributed to the literature survey and manuscript writing.
FUNDING
AM is highly thankful to SERB New Delhi for a research grant (CRG/2018/00912).
CONFLICT OF INTEREST
The authors declare no conflict of interests.
REFERENCES
[1] | Weller J , Budson A ((2018) ) Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 7: , F1000 Faculty Rev-1161. |
[2] | Serrano-Pozo A , Frosch MP , Masliah E , Hyman BT ((2011) ) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1: , a006189. |
[3] | Uddin MS , Kabir MT , Al Mamun A , Abdel-Daim MM , Barreto GE , Ashraf GM ((2019) ) APOE and Alzheimer’s disease: evidence mounts that targeting APOE4 may combat Alzheimer’s pathogenesis. Mol Neurobiol 56: , 2450–2465. |
[4] | Uddin MS , Kabir MT , Jeandet P , Mathew B , Ashraf GM , Perveen A , Bin-Jumah MN , Mousa SA , Abdel-Daim MM ((2020) ) Novel anti-Alzheimer’s therapeutic molecules targeting amyloid precursor protein processing. Oxid Med Cell Longev 2020: , 7039138. |
[5] | Rahman MA , Rahman MR , Zaman T , Uddin MS , Islam R , Abdel-Daim MM , Rhim H ((2020) ) Emerging potential of naturally occurring autophagy modulators against neurodegeneration. Curr Pharm Des 26: , 772–779. |
[6] | Sultana R , Mecocci P , Mangialasche F , Cecchetti R , Baglioni M , Butterfield DA ((2011) ) Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer’s disease: insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. J Alzheimers Dis 24: , 77–84. |
[7] | Miyata M , Smith JD ((1996) ) Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptides. Nat Genet 14: , 55–61. |
[8] | Chen GF , Xu TH , Yan Y , Zhou YR , Jiang Y , Melcher K , Xu HE ((2017) ) Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 38: , 1205–1235. |
[9] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297: , 353–356. |
[10] | Noble W , Hanger DP , Miller CCJ , Lovestone S ((2013) ) The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol 4: , 83. |
[11] | Kolarova M , García-Sierra F , Bartos A , Ricny J , Ripova D ((2012) ) Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis 2012: , 731526. |
[12] | Alavi Naini SM , Soussi-Yanicostas N ((2015) ) Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid Med Cell Longev 2015: , 151979. |
[13] | Wang X , Wang W , Li L , Perry G , Lee H gon , Zhu X ((2014) ) Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta 1842: , 1240–1247. |
[14] | Deane R , Sagare A , Hamm K , Parisi M , Lane S , Finn MB , Holtzman DM , Zlokovic BV ((2008) ) ApoE isoform–specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest 118: , 4002–4013. |
[15] | Goyal M , Grover S , Dhanjal JK , Goyal S , Tyagi C , Chacko S , Grover A ((2013) ) Novel natural structure corrector of apoe4 for checking Alzheimer’s disease: Benefits from high throughput screening and molecular dynamics simulations. Biomed Res Int 2013: , 620793. |
[16] | Luo J , Maréchal JD , Wärmländer S , Gräslund A , Perálvarez-Marín A ((2010) ) In silico analysis of the apolipoprotein E and the amyloid β peptide interaction: misfolding induced by frustration of the salt bridge network. PLoS Comput Biol 6: , e1000663. |
[17] | Parcon PA , Balasubramaniam M , Ayyadevara S , Jones RA , Liu L , Shmookler Reis RJ , Barger SW , Mrak RE , Griffin WST ((2018) ) Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement 14: , 230–242. |
[18] | Safieh M , Korczyn AD , Michaelson DM ((2019) ) ApoE4: an emerging therapeutic target for Alzheimer’s disease. BMC Med 17: , 64. |
[19] | Chen Y , Durakoglugil MS , Xian X , Herz J ((2010) ) ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A 107: , 12011–12016. |
[20] | Yamazaki Y , Zhao N , Caulfield TR , Liu CC , Bu G ((2019) ) Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol 15: , 501. |
[21] | Bano S , Rasheed MA , Jamil F , Ibrahim M , Kanwal S ((2019) ) In silico identification of novel apolipoprotein E4 inhibitor for Alzheimer’s disease therapy. Curr Comput Aided Drug Des 15: , 97–103. |
[22] | Liu CC , Kanekiyo T , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9: , 106–118. |
[23] | Dhiman K , Blennow K , Zetterberg H , Martins RN , Gupta VB ((2019) ) Cerebrospinal fluid biomarkers for understanding multiple aspects of Alzheimer’s disease pathogenesis. Cell Mol Life Sci 76: , 1833–1863. |
[24] | Boehm-Cagan A , Bar R , Liraz O , Bielicki JK , Johansson JO , Michaelson DM ((2016) ) ABCA1 agonist reverses the ApoE4-driven cognitive and brain pathologies. J Alzheimers Dis 54: , 1219–1233. |
[25] | Boehm-Cagan A , Michaelson DM ((2014) ) Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J Neurosci 34: , 7293–7301. |
[26] | Veeraraghavalu K , Zhang C , Miller S , Hefendehl JK , Rajapaksha TW , Ulrich J , Jucker M , Holtzman DM , Tanzi RE , Vassar R , Sisodia SS ((2013) ) Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models.”. Science 340: , 924. |
[27] | Cramer PE , Cirrito JR , Wesson DW , Lee CYD , Karlo JC , Zinn AE , Casali BT , Restivo JL , Goebel WD , James MJ , Brunden KR , Wilson DA , Landreth GE ((2012) ) ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335: , 1503–1506. |
[28] | Tachibana M , Shinohara M , Yamazaki Y , Liu CC , Rogers J , Bu G , Kanekiyo T ((2016) ) Rescuing effects of RXR agonist bexarotene on aging-related synapse loss depend on neuronal LRP1. Exp Neurol 277: , 1–9. |
[29] | Laclair KD , Manaye KF , Lee DL , Allard JS , Savonenko AV , Troncoso JC , Wong PC ((2013) ) Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice. Mol Neurodegener 8: , 18. |
[30] | Tai LM , Koster KP , Luo J , Lee SH , Wang YT , Collins NC , Aissa M Ben , Thatcher GRJ , LaDu MJ ((2014) ) Amyloid-β pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J Biol Chem 289: , 30538–30555. |
[31] | Tesseur I , Lo AC , Roberfroid A , Dietvorst S , Broeck B Van , Borgers M , Gijsen H , Moechars D , Mercken M , Kemp J , Hooge RD’ , De Strooper B ((2012) ) Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models.”. Science 340: , 924-e. |
[32] | Burns MP , Vardanian L , Pajoohesh-Ganji A , Wang L , Cooper M , Harris DC , Duff K , Rebeck GW ((2006) ) The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J Neurochem 98: , 792–800. |
[33] | Koldamova RP , Lefterov IM , Staufenbiel M , Wolfe D , Huang S , Glorioso JC , Walter M , Roth MG , Lazo JS ((2005) ) The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J Biol Chem 280: , 4079–4088. |
[34] | Casali BT , Corona AW , Mariani MM , Karlo JC , Ghosal K , Landreth GE ((2015) ) Omega-3 fatty acids augment the actions of nuclear receptor agonists in a mouse model of Alzheimer’s disease. J Neurosci 35: , 9173–9181. |
[35] | Riddell DR , Zhou H , Comery TA , Kouranova E , Lo CF , Warwick HK , Ring RH , Kirksey Y , Aschmies S , Xu J , Kubek K , Hirst WD , Gonzales C , Chen Y , Murphy E , Leonard S , Vasylyev D , Oganesian A , Martone RL , Pangalos MN , Reinhart PH , Jacobsen JS ((2007) ) The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Neurosci 34: , 621–628. |
[36] | Vanmierlo T , Rutten K , Dederen J , Bloks VW , van Vark-van der Zee LC , Kuipers F , Kiliaan A , Blokland A , Sijbrands EJG , Steinbusch H , Prickaerts J , Lütjohann D , Mulder M ((2011) ) Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging 32: , 1262–1272. |
[37] | Suidan GL , Ramaswamy G ((2019) ) Targeting Apolipoprotein E for Alzheimer’s disease: an industry perspective. Int J Mol Sci 20: , 2161. |
[38] | Bien-Ly N , Gillespie AK , Walker D , Yoon SY , Huang Y ((2012) ) Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci 32: , 4803–4811. |
[39] | Kim J , Jiang H , Park S , Eltorai AEM , Stewart FR , Yoon H , Basak JM , Finn MB , Holtzman DM ((2011) ) Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J Neurosci 31: , 18007–18012. |
[40] | Kim J , Eltorai AEM , Jiang H , Liao F , Verghese PB , Kim J , Stewart FR , Basak JM , Holtzman DM ((2012) ) Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J Exp Med 209: , 2149–2156. |
[41] | Mak ACY , Pullinger CR , Tang LF , Wong JS , Deo RC , Schwarz JM , Gugliucci A , Movsesyan I , Ishida BY , Chu C , Poon A , Kim P , Stock EO , Schaefer EJ , Asztalos BF , Castellano JM , Wyss-Coray T , Duncan JL , Miller BL , Kane JP , Kwok PY , Malloy MJ ((2014) ) Effects of the absence of apolipoprotein E on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol 71: , 1228. |
[42] | Bu G ((2009) ) Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10: , 333–344. |
[43] | Al Mamun A , Sahab Uddin M , Fahim Bin Bashar M , Zaman S , Begum Y , Bulbul IJ , Siddiqul Islam M , Shahid Sarwar M , Mathew B , Shah Amran M , Md Ashraf G , Bin-Jumah MN , Mousa SA , Abdel-Daim MM ((2020) ) Molecular insight into the therapeutic promise of targeting APOE4 for Alzheimer’s disease. Oxid Med Cell Longev 2020: , 5086250. |
[44] | Hauser PS , Narayanaswami V , Ryan RO ((2011) ) Apolipoprotein E: from lipid transport to neurobiology. Prog Lipid Res 50: , 62–74. |
[45] | Wahrle SE , Jiang H , Parsadanian M , Legleiter J , Han X , Fryer JD , Kowalewski T , Holtzman DM ((2004) ) ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem 279: , 40987–40993. |
[46] | Wahrle SE , Jiang H , Parsadanian M , Hartman RE , Bales KR , Paul SM , Holtzman DM ((2005) ) Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem 280: , 43236–43242. |
[47] | Wahrle SE , Jiang H , Parsadanian M , Kim J , Li A , Knoten A , Jain S , Hirsch-Reinshagen V , Wellington CL , Bales KR , Paul SM , Holtzman DM ((2008) ) Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest 118: , 671. |
[48] | Doudna JA , Charpentier E ((2014) ) The new frontier of genome engineering with CRISPR-Cas9. Science 346: , 1258096. |
[49] | Komor AC , Kim YB , Packer MS , Zuris JA , Liu DR ((2016) ) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533: , 420–424. |
[50] | Rabinowitz R , Kadair A , Ben-Zur T , Michaelson D , Offen D ((2019) ) ApoE4 allele specific knockout using a synthetic Cas9 variant as a potential gene therapy approach for Alzheimer’s disease. Cytotherapy 21: , e7. |
[51] | Zhong N , Scearce-Levie K , Ramaswamy G , Weisgraber KH ((2008) ) Apolipoprotein E4 domain interaction: synaptic and cognitive deficits in mice. Alzheimers Dement 4: , 179–192. |
[52] | Chen HK , Ji ZS , Dodson SE , Miranda RD , Rosenblum CI , Reynolds IJ , Freedman SB , Weisgraber KH , Huang Y , Mahley RW ((2011) ) Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J Biol Chem 286: , 5215–5221. |
[53] | Brodbeck J , McGuire J , Liu Z , Meyer-Franke A , Balestra ME , Jeong DE , Pleiss M , McComas C , Hess F , Witter D , Peterson S , Childers M , Goulet M , Liverton N , Hargreaves R , Freedman S , Weisgraber KH , Mahley RW , Huang Y ((2011) ) Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J Biol Chem 286: , 17217–17226. |
[54] | Cacabelos R , Torrellas C ((2015) ) Epigenetics of aging and Alzheimer’s disease: implications for pharmacogenomics and drug response. Int J Mol Sci 16: , 30483–30543. |
[55] | Smith JA , Zagel AL , Sun Y V , Dolinoy DC , Bielak LF , Peyser PA , Turner ST , Mosley TH , Kardia SL ((2014) ) Epigenomic indicators of age in African Americans. Hered Genet 3: , 137. |
[56] | Ma Y , Smith CE , Lai CQ , Irvin MR , Parnell LD , Lee YC , Pham L , Aslibekyan S , Claas SA , Tsai MY , Borecki IB , Kabagambe EK , Berciano S , Ordovás JM , Absher DM , Arnett DK ((2015) ) Genetic variants modify the effect of age on APOE methylation in the Genetics of Lipid Lowering Drugs and Diet Network study. Aging Cell 14: , 49–59. |
[57] | Verghese PB , Castellano JM , Garai K , Wang Y , Jiang H , Shah A , Bu G , Frieden C , Holtzman DM ((2013) ) ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci U S A 110: , E1807–E1816. |
[58] | Namba Y , Tomonaga M , Kawasaki H , Otomo E , Ikeda K ((1991) ) Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res 541: , 163–166. |
[59] | Kuszczyk MA , Sanchez S , Pankiewicz J , Kim J , Duszczyk M , Guridi M , Asuni AA , Sullivan PM , Holtzman DM , Sadowski MJ ((2013) ) Blocking the interaction between apolipoprotein E and Aβ reduces intraneuronal accumulation of Aβ and inhibits synaptic degeneration. Am J Pathol 182: , 1750–1768. |
[60] | Sadowski MJ , Pankiewicz J , Scholtzova H , Mehta PD , Prelli F , Quartermain D , Wisniewski T ((2006) ) Blocking the apolipoprotein E/amyloid-β interaction as a potential therapeutic approach for Alzheimer’s disease. Proc Natl Acad Sci U S A 103: , 18787–18792. |
[61] | Liu S , Breitbart A , Sun Y , Mehta PD , Boutajangout A , Scholtzova H , Wisniewski T ((2014) ) Blocking the apolipoprotein E/amyloid β interaction in triple transgenic mice ameliorates Alzheimer’s disease related amyloid β and tau pathology. J Neurochem 128: , 577–591. |
[62] | Hao J , Zhang W , Zhang P , Liu R , Liu L , Lei G , Su C , Miao J , Li Z ((2010) ) Aβ20–29 peptide blocking apoE/Aβ interaction reduces full-length Aβ42/40 fibril formation and cytotoxicity in vitro. Neuropeptides 44: , 305–313. |
[63] | Liao F , Li A , Xiong M , Bien-Ly N , Jiang H , Zhang Y , Finn MB , Hoyle R , Keyser J , Lefton KB , Robinson GO , Serrano JR , Silverman AP , Guo JL , Getz J , Henne K , Leyns CEG , Gallardo G , Ulrich JD , Sullivan PM , Lerner EP , Hudry E , Sweeney ZK , Dennis MS , Hyman BT , Watts RJ , Holtzman DM ((2018) ) Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest 128: , 2144–2155. |
[64] | Hyman BT , Gomez-Isla T , Briggs M , Chung H , Nichols S , Kohout F , Wallace R ((1996) ) Apolipoprotein E and cognitive change in an elderly population. Ann Neurol 40: , 55–66. |
[65] | Staehelin HB , Perrig-Chiello P , Mitrache C , Miserez AR , Perrig WJ ((1999) ) Apolipoprotein E genotypes and cognitive functions in healthy elderly persons. Acta Neurol Scand 100: , 53–60. |
[66] | Wilson RS , Bienias JL , Berry-Kravis E , Evans DA , Bennett DA ((2002) ) The apolipoprotein E ɛ2 allele and decline in episodic memory. J Neurol Neurosurg Psychiatry 73: , 672–677. |
[67] | Conejero-Goldberg C , Gomar JJ , Bobes-Bascaran T , Hyde TM , Kleinman JE , Herman MM , Chen S , Davies P , Goldberg TE ((2014) ) APOE2 enhances neuroprotection against Alzheimer’s disease through multiple molecular mechanisms. Mol Psychiatry 19: , 1243–1250. |
[68] | Dodart JC , Marr RA , Koistinaho M , Gregersen BM , Malkani S , Verma IM , Paul SM ((2005) ) Gene delivery of human apolipoprotein E alters brain Aβ burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 102: , 1211–1216. |
[69] | Suri S , Heise V , Trachtenberg AJ , Mackay CE ((2013) ) The forgotten APOE allele: a review of the evidence and suggested mechanisms for the protective effect of APOE ɛ2. Neurosci Biobehav Rev 37: , 2878–2886. |
[70] | Hu J , Liu CC , Chen XF , Zhang YW , Xu H , Bu G ((2015) ) Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Aβ metabolism in apoE4-targeted replacement mice. Mol Neurodegener 10: , 6. |
[71] | Bittar A , Sengupta U , Kayed R ((2018) ) Prospects for strain-specific immunotherapy in Alzheimer’s disease and tauopathies. NPJ Vaccines 3: , 9. |
[72] | Mahley RW ((2016) ) Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler Thromb Vasc Biol 36: , 1305–1315. |
[73] | Luz I , Liraz O , M. Michaelson D ((2016) ) An anti-apoE4 specific monoclonal antibody counteracts the pathological effects of apoE4 in vivo. Curr Alzheimer Res 13: , 918–929. |
[74] | Ghosal K , Stathopoulos A , Thomas D , Phenis D , Vitek MP , Pimplikar SW ((2013) ) The apolipoprotein-E-mimetic COG112 protects amyloid precursor protein intracellular domain-overexpressing animals from Alzheimer’s disease-like pathological features. Neurodegener Dis 12: , 51–58. |
[75] | Vitek MP , Christensen DJ , Wilcock D , Davis J , Van Nostrand WE , Li FQ , Colton CA ((2012) ) APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer’s disease transgenics. Neurodegener Dis 10: , 122–126. |
[76] | Wang W , Zhu X ((2018) ) HDL mimetic peptides affect apolipoprotein E metabolism: equal supplement or functional enhancer? J Neurochem 147: , 580–583. |
[77] | Laskowitz DT , Song P , Wang H , MacE B , Sullivan PM , Vitek MP , Dawson HN ((2010) ) Traumatic brain injury exacerbates neurodegenerative pathology: improvement with an apolipoprotein E-based therapeutic. J Neurotrauma 27: , 1983–1995. |
[78] | Yao X , Vitek MP , Remaley AT , Levine SJ ((2012) ) Apolipoprotein mimetic peptides: A new approach for the treatment of asthma. Front Pharmacol 3: , 37. |
[79] | Wang H , Anderson LG , Lascola CD , James ML , Venkatraman TN , Bennett ER , Acheson SK , Vitek MP , Laskowitz DT ((2013) ) ApolipoproteinE mimetic peptides improve outcome after focal ischemia. Exp Neurol 241: , 67–74. |
[80] | Brecht WJ , Harris FM , Chang S , Tesseur I , Yu GQ , Xu Q , Fish JD , Wyss-Coray T , Buttini M , Mucke L , Mahley RW , Huang Y ((2004) ) Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci 24: , 2527–2534. |
[81] | Huang Y , Liu XQ , Wyss-Coray T , Brecht WJ , Sanan DA , Mahley RW ((2001) ) Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A 98: , 8838–8843. |
[82] | Harris FM , Brecht WJ , Xu Q , Tesseur I , Kekonius L , Wyss-Coray T , Fish JD , Masliah E , Hopkins PC , Scearce-Levie K , Weisgraber KH , Mucke L , Mahley RW , Huang Y ((2003) ) Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci U S A 100: , 10966–10971. |
[83] | Holtzman DM , Herz J , Bu G ((2012) ) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006312. |
[84] | Kanekiyo T , Zhang J , Liu Q , Liu CC , Zhang L , Bu G ((2011) ) Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci 31: , 1644–1651. |
[85] | Liu Q , Trotter J , Zhang J , Peters MM , Cheng H , Bao J , Han X , Weeber EJ , Bu G ((2010) ) Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci 30: , 17068–17078. |
[86] | Kim J , Castellano JM , Jiang H , Basak JM , Parsadanian M , Pham V , Mason SM , Paul SM , Holtzman DM ((2009) ) Overexpression of low density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron 64: , 632. |
[87] | Kanekiyo T , Cirrito JR , Liu CC , Shinohara M , Li J , Schuler DR , Shinohara M , Holtzman DM , Bu G ((2013) ) Neuronal clearance of amyloid-β by endocytic receptor LRP1. J Neurosci 33: , 19276–19283. |
[88] | Shinohara M , Sato N , Kurinami H , Takeuchi D , Takeda S , Shimamura M , Yamashita T , Uchiyama Y , Rakugi H , Morishita R ((2010) ) Reduction of brain β-amyloid (Aβ) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Aβ clearance. J Biol Chem 285: , 22091. |
[89] | Qosa H , Abuznait AH , Hill RA , Kaddoumi A ((2012) ) Enhanced brain amyloid-β clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. J Alzheimers Dis 31: , 151–165. |
[90] | Bell RD , Winkler EA , Singh I , Sagare AP , Deane R , Wu Z , Holtzman DM , Betsholtz C , Armulik A , Sallstrom J , Berk BC , Zlokovic BV ((2012) ) Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485: , 512–516. |
[91] | Alata W , Ye Y , St-Amour I , Vandal M , Calon F ((2015) ) Human apolipoprotein E ɛ4 expression impairs cerebral-vascularization and blood–brain barrier function in mice. J Cereb Blood Flow Metab 35: , 86. |
[92] | Bien-Ly N , Boswell CA , Jeet S , Beach TG , Hoyte K , Luk W , Shihadeh V , Ulufatu S , Foreman O , Lu Y , DeVoss J , van der Brug M , Watts RJ ((2015) ) Lack of widespread BBB disruption in Alzheimer’s disease models: focus on therapeutic antibodies. Neuron 88: , 289–297. |
[93] | Keren-Shaul H , Spinrad A , Weiner A , Matcovitch-Natan O , Dvir-Szternfeld R , Ulland TK , David E , Baruch K , Lara-Astaiso D , Toth B , Itzkovitz S , Colonna M , Schwartz M , Amit I ((2017) ) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169: , 1276–1290.e17. |
[94] | Brown GC , St George-Hyslop PH ((2017) ) Deciphering microglial diversity in Alzheimer’s disease. Science 356: , 1123–1124. |
[95] | Wattananit S , Tornero D , Graubardt N , Memanishvili T , Monni E , Tatarishvili J , Miskinyte G , Ge R , Ahlenius H , Lindvall O , Schwartz M , Kokaia Z ((2016) ) Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci 36: , 4182–4195. |
[96] | Uddin MS , Kabir MT , Mamun A Al , Barreto GE , Rashid M , Perveen A , Ashraf GM ((2020) ) Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. Int Immunopharmacol 84: , 106479. |
[97] | Bell RD ((2012) ) The imbalance of vascular molecules in Alzheimer’s disease. J Alzheimers Dis 32: , 699–709. |
[98] | Altman R , Rutledge JC ((2010) ) The vascular contribution to Alzheimer’s disease. Clin Sci 119: , 407–421. |
[99] | Rohn TT ((2014) ) Is apolipoprotein E4 an important risk factor for vascular dementia? Int J Clin Exp Pathol 7: , 3504. |
[100] | Mahley RW , Weisgraber KH , Huang Y ((2009) ) Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res 50 Suppl: , S183–S188. |
[101] | Davey DA ((2014) ) Alzheimer’s disease and vascular dementia: one potentially preventable and modifiable disease? Part II: Management, prevention and future perspective. Neurodegener Dis Manag 4: , 261–270. |
[102] | Theendakara V , Peters-Libeu CA , Spilman P , Poksay KS , Bredesen DE , Rao RV ((2016) ) Direct transcriptional effects of apolipoprotein E. J Neurosci 36: , 685–700. |
[103] | Anne UB , Urfer R ((2017) ) Identification of a nuclear respiratory factor 1 recognition motif in the apolipoprotein E variant APOE4 linked to Alzheimer’s disease. Sci Rep 7: , 40668. |
[104] | Rohn TT ((2017) ) Nuclear localization of apolipoprotein E4: a new trick for an old protein. Int J Neurol Neurother 4: , 067. |
[105] | Theendakara V , Peters-Libeu CA , Bredesen DE , Rao R V. ((2018) ) Transcriptional effects of ApoE4: relevance to Alzheimer’s disease. Mol Neurobiol 55: , 5243–5254. |
[106] | Huang HJ , Chen HY , Lee CC , Chen CYC ((2014) ) Computational design of apolipoprotein e4 inhibitors for Alzheimer’s disease therapy from traditional Chinese medicine. Biomed Res Int 2014: , 452625. |
[107] | Tun MKM , Herzon SB ((2012) ) The pharmacology and therapeutic potential of (–)-huperzine A. J Exp Pharmacol 4: , 113. |
[108] | Bossu P , Salani F , Ciaramella A , Sacchinelli E , Mosca A , Banaj N , Assogna F , Orfei MD , Caltagirone C , Gianni W , Spalletta G ((2018) ) Anti-inflammatory effects of homotaurine in patients with amnestic mild cognitive impairment. Front Aging Neurosci 10: , 285. |


