
Temporal Lobe Epilepsy and Alzheimer’s Disease: From Preclinical to Clinical Evidence of a Strong Association
Abstract
Increasing evidence coming from both experimental and humans’ studies strongly suggest the existence of a link between epilepsy, in particular temporal lobe epilepsy (TLE), and Alzheimer’s disease (AD). Patients with mild cognitive impairment and AD are more prone to have seizures, and seizures seem to facilitate amyloid-β and tau deposits, thus promoting neurodegenerative processes. Consistent with this view, long-lasting drug-resistant TLE and AD have been shown to share several pathological and neuroimaging features. Even if studies addressing prevalence of interictal and subclinical epileptiform activity in these patients are not yet conclusive, their findings raise the possibility that epileptiform activity might negatively impact memory and hasten cognitive decline, either directly or by association with unrecognized silent seizures. In addition, data about detrimental effect of network hyperexcitability in temporal regions in the premorbid and early stages ofADopen up newtherapeutic opportunities for antiseizure medications and/or antiepileptic strategies that might complement or enhance existing therapies, and potentially modify disease progression. Here we provide a review of evidence linking epileptiform activity, network hyperexcitability, and AD, and their role promoting and accelerating neurodegenerative process. Finally, the effects of antiseizure medications on cognition and their optimal administration in patients with AD are summarized.
INTRODUCTION
It is well known that patients with Alzheimer’s disease (AD) have a greater risk of seizures than age-matched non-demented patients [1, 2]. Seizure prevalence in this group ranges from 1–22%[3, 4], and the incidence is well above the subjects of similar age without dementia. In the past, the general belief about relationship between seizures and AD was that they were a consequence of neurodegeneration characterizing AD, thus becoming a frequent complication of late stage of this condition [5, 6]. Recent evidence indicates that seizures and network hyperexcitability can occur also in the early stages of AD and contribute to cognitive decline [7–9]. In particular, younger age at AD onset was found to be an independent risk factor and significant predictor for seizures [10, 11].
Besides the overt seizures, estimates of subclinical epileptiform activity in AD vary depending on types of patients enrolled and different methods used (i.e., routine versus long-term electroencephalograms (EEG)). Recent findings suggest that subclinical epileptiform activity might be more common in AD than previously recognized, and raise the possibility that epileptiform activity and network hyperexcitability might accelerate cognitive decline, either directly or by association with unrecognized silent seizures [12]. Anyway, at the moment clinical data are not conclusive about this issue and there is a lack of clear guidelines indicating whether and how treat subclinical epileptiform abnormalities in AD patients without overt seizures. Indeed, antiseizure medications (ASMs) may have more negative than positive cognitive effects, and their use in AD as potential disease-modifying agents needs clear demonstration through further rigorous clinical trials.
In this review we will provide an update of the evidence and mechanisms subtending the relationship between epileptiform activity, network hyperexcitability, and AD, and their role promoting and accelerating neurodegenerative process. Finally, as the impact of epileptiform activity and/or ASMs on cognitive performances in patients with dementia is crucial [6], we will review the evidence related to cognitive effects of ASMs and their optimal administration in patients with AD.
EVIDENCE FROM ANIMAL MODELS OF ALZHEIMER’S DISEASE
Evidence from animal models helps us to better understand overlaps between AD and epilepsy.
Experimental studies in the past years demonstrated that amyloid-β (Aβ) and tau deposits might facilitate or even cause seizures [13]; at the same time there is evidence that seizures can increase brain deposits [14, 15] (Fig. 1).
Fig. 1
In the figure, the key points of the mutual relationship between epilepsy and Alzheimer’s disease (AD) are displayed. The experimental evidence in animal models has clearly evidenced the potential presence of a “vicious loop”: Aβ and tau deposits may be cause of seizures (box on the top) and seizures lead to increase in brain Aβ deposits and tau phosphorylation (box at the bottom). In the box on the right, the main findings supporting the association between temporal lobe epilepsy (TLE) and AD in humans are summarized. Aβ, amyloid-β; AβPP, amyloid-β protein precursor; NMDA, N-methyl-D-aspartate; CSF, cerebrospinal fluid; MCI, mild cognitive impairment; LEV, Levetiracetam.
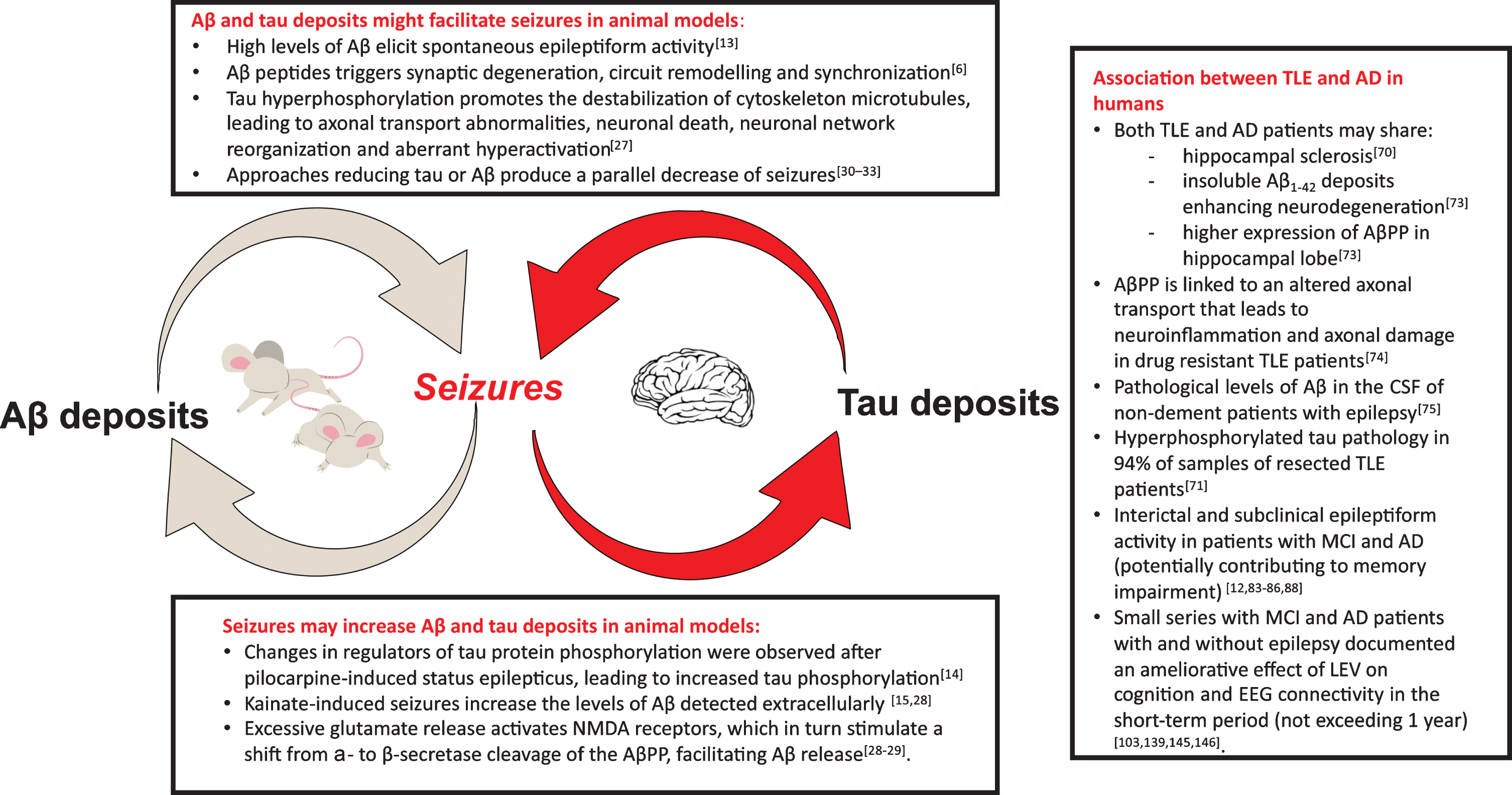
Evidence of a relationship between AD and epilepsy comes from two animal models primarily human amyloid precursor protein (hAPP) (J9, J20 and ARC48) [13] and APPSwPS1dE9 (APdE9) [16] transgenic mice. These strains of mice present sporadic unprovoked seizures along with increased Aβ deposits and cognitive impairment. Palop et al. (2007) [13] managed to prove for the first time that hAPP mice (J20 line), one of the most common models of AD, showed frequent subclinical epileptic activity. J20 mice do not have evidence of neural loss or clear neural damage, and this suggests that high levels of Aβ alone are sufficient to elicit spontaneous epileptiform activity in vivo in the absence of neurodegeneration [13]; moreover, sprouting of mossy fiber collaterals in the dentate gyrus was evident in hAPP-J20 mice but not in the nontransgenic controls. This was a shattering finding since it suggested that humans affected from AD could also be subject to subclinical seizures, and that these seizures could have a role in promoting the progression of AD [6]. Such findings were later also confirmed in the APdE9 mice model [16], in which it was shown an increase in cortico-thalamic excitability [17], that is, parenthetically, a typical feature of many patients with generalized epilepsy [18–20].
Many similarities emerged, comparing the histo-pathological features of hAPP mice and mice with temporal lobe epilepsy (TLE) induced by status epilepticus. Similar features among the two conditions include: hilar neuronal loss, reduced calbindin expression, mossy fiber sprouting, and GABAergic sprouting. The main difference is that TLE mice present a greater degree of mossy fiber sprouting, which is a hallmark of status epilepticus animal models [13, 21–24]. These similarities suggest that, ignoring brain deposits, the histopathological alterations in AD and in epilepsy appear to be much alike.
Moreover, one of the most relevant features involved in both AD and epilepsy is represented by tau pathology [25]. Tau protein belongs to the family of microtubule-associated proteins that ensures cytoskeleton stability in neuronal cells. In normal cell function, tau protein is processed via phosphorylation; however, with hyperphosphorylation, it loses its functional role and deposits in neuronal cells, forming intraneuronal neurofibrillary tangles [26]. Hyperphosphorylated tau may contribute to a series of events, implicated in epilepsy onset, such as neuronal network reorganization, unusual synchronization of networks, and aberrant hyperactivation. Even though the exact shared molecular mechanisms are unclear, hypotheses about it have been made. Altered activity of kinases and phosphatase implied in tau metabolism (mainly protein phosphatase 2A and Cyclin dependent kinase 5) is present in both AD and epilepsy animal models [27].
The link between AD and epilepsy is also confirmed in the opposite direction since seizures seem to facilitate the deposit of tau and Aβ. Changes in two regulators of tau protein phosphorylation (GSK3β and PP2A) were observed after pilocarpine-induced status epilepticus, leading to increased tau phosphorylation [14]. Experimental evidence showed that excessive synaptic activity such as seizure activity in animals increases the levels of Aβ detected extracellularly [15, 28, 29]. Further evidence of the correlation between AD and epilepsy in animal models is provided by the observation that several approaches aimed at reducing tau or Aβ produce a parallel decrease seizures [30–32] and that hAPP mice, knockout for the tau protein gene, suffers less seizures [33].
Altogether, these seminal studies grounded the hypothesis that Aβ and its byproducts might affect the balance between excitation/inhibition facilitating seizures. There is an interesting working hypothesis that proposes that Aβ and tau deposits can destabilize the synaptic cleft [9], influencing both ion channels [34, 35] and the development of neural plasticity [36, 37]. Based on transgenic mouse models, it has been proposed that seizures could be facilitated by the Aβ-related downregulation of the Nav1.1 sodium channel in a subset of GABAergic interneurons [38]. Other important seminal papers link Aβ to alteration in ion channel functioning [39–41]. Notably, Nav1.1 channel mutations are associated with two forms of genetic epilepsy: generalized epilepsy with febrile seizures plus (GEFS+) and Dravet syndrome [42]. Since the same channels result to be down regulated by Aβ, this co-occurrence strengthens the hypothesis of a potential link between deposits of neurofilament and epilepsy, mediated by Aβ interaction with ion channels known to be altered also in genetic forms of epilepsy.
Some limitations regarding the generalization of animal model findings to human brain pathology must be addressed. Most AD mice models come from genetic pedigrees, while in human population most cases of AD are sporadic [43]. Moreover, mice models do not replicate perfectly the clinical features of human AD [44, 45]. Anyway, despite these limitations, molecular models provided new insights and interesting proofs of concept regarding the connections between epilepsy and AD.
In conclusion, in animal models, Aβ and tau deposits may be cause of seizures [13, 25]. Their potential epileptogenicity is related to a destabilizing effect on the elements of the synaptic cleft. Likewise, seizures lead to increase in brain Aβ deposits and tau phosphorylation. This mutual relationship could potentially trigger a “vicious loop” fostering AD progression [44] (Fig. 1).
EVIDENCE FROM STUDIES IN HUMANS
Epidemiological studies in humans
Many epidemiological studies support a strong association between epilepsy and AD: patients with AD have an increased risk of developing epilepsy [11], and late onset or long-lasting epilepsy can be also complicated by cognitive impairment at a rate that is significantly higher than that of general population [46, 47]. The most significant association is probably represented by a causal link. Indeed, compared with healthy individuals of the same age, patients with sporadic AD present a 6- to 10-increased risk of developing clinical seizures during the course of their disease, with a lifetime prevalence of epilepsy in AD patients varying from 1.5 to 64%as demonstrated by both prospective and retrospective studies [1, 48–50]. In sporadic late onset AD patients with epilepsy, 70%of seizures are focal with impaired awareness [7, 8, 51]; in addition, generalized tonic-clonic seizures are also commonly observed in AD [52]. However, it is important to consider that the characterization of the clinical semiology of seizures is usually based on information provided by the patient or the caregiver, and it may challenging for caregivers to describe seizures’ manifestations and to distinguish them from other transitory manifestations commonly observed in AD patients (i.e., fluctuations in awareness, hallucinations, confusion, delusions) [48, 53]. Seizures occur more frequently in patients with AD than in those with non-AD dementias [54]. Myoclonus, which is generally considered to be due to cortical hyperexcitability, is also common in patients with AD, with a prevalence of 7–10%and a cumulative risk as high as 80%in the late stages of disease [55, 56]. Seizure prevalence seems to increase with AD duration, and it seems to occur more often in early-onset familial AD compared to sporadic one. Indeed, seizures have been described in 20–30%of patients with the presenilin 1 mutation (PSEN1) [57, 58], in 57–66%of patients with amyloid-β protein precursor (Aβ PP) duplication [58, 59], and in 15–30%of patients with presenilin 2 mutation (PSEN2) [58, 60]. In an 8-year prospective assessment [61], 41 (84%) of 49 patients with both AD and Down’s syndrome, carrying an additional copy of chromosome 21 that contains the Aβ PP gene, developed seizures. According to some studies, patients with a younger age of AD onset present a higher risk of seizures compared with age-matched population: seizure incidence increases 3-fold around the age of 70, and nearly 87-fold around the age of 50 [1, 48]. Moreover, most studies, but not all, have evidenced that seizures were more likely to occur in later stages of dementia [62–64]. The higher rates of seizures in more advanced dementia could be explained by different hypotheses. A possible explanation for these findings could be that severity of dementia and neurodegenerative process, rather than disease duration, may be associated with higher risk of seizures [11]. Of note, a prolonged preclinical phase of AD in which Aβ deposition reaches threshold of positivity many years before the onset of dementia has been previously demonstrated [65]. Alternatively, an intriguing hypothesis could be that seizures can cause a faster progression of cognitive decline. Observational studies [8, 11, 12] suggest that both mechanisms are possible, although the first scenario is usually considered more likely. There is still a relative lack of knowledge concerning the possible impact of seizures and seizures’ frequency in the progression of AD course and severity. Other risk factors for unprovoked seizures in patients with AD have been reported including African American ethnicity and focal epileptiform activity [11]. On the other hand, some small series [66] reported cognitive decline after epilepsy onset but are limited in sample size and did not account for the possible cognitive side effects of antiseizure drugs. Patients with epilepsy can experience cognitive impairment throughout the course of their illness [46]. The prevalence of dementia in patients with epilepsy has been described to range from 0.6 to 17.5%, with a higher rate for AD compared to vascular dementia [67]. In a large cohort of patients with AD, a history of epilepsy with onset in the adulthood before cognitive decline was found to be 17 times more frequent than in the general population [68]. In some cases, seizures may begin concurrently with onset of cognitive decline or even precede onset of AD [7, 8], possibly reflecting the epileptogenic potential of Aβ, which begins to accumulate more than 10 years before clinical signs of dementia [65]. Actually, it should be noted that the direction of the association between epilepsy and AD is tricky to detect since neurodegenerative disorders’ processes begin several years prior to the development of clinical symptoms.
Pathological association between epilepsy and Alzheimer’s disease
The pathological hallmark of AD is represented by the extracellular deposition of Aβ into insoluble plaques, known as “senile plaques”, and the accumulation of hyperphosphorylated tau protein in neurofibrillary tangles [69]. Growing evidence shows a possible link between cognitive impairment and certain forms of drug-resistant epilepsy. In particular, human TLE is strongly related to cognitive dysfunction and shares several pathological and neuroimaging features of AD, including hippocampal sclerosis [70]. Patients with drug-resistant TLE, who experienced higher frequency of seizures, are more likely to have cognitive comorbidity as well [71]. The association is even stronger as far as molecular pathways are concerned. Aβ peptides are derived from AβPP, which can be processed by either non-amyloidogenic α-secretase or amyloidogenic β-secretase pathway [72]. Insoluble Aβ1 - 42 deposits enhance neurodegeneration in hippocampal lobe in both AD and epilepsy [73]. A study conducted on a group of patients with drug-resistant TLE [73] who underwent anterior temporal lobe resection demonstrated a higher expression of AβPP in hippocampal lobe. Similar findings have been reported in brains of AD patients. Moreover, in these patients AβPP is processed mostly through amyloidogenic pathway, leading to a remarkable deposition of Aβ, which is associated with memory impairment. As far as AβPP expression is concerned, another study revealed a possible role of this protein in the pathogenesis of refractory epilepsy: AβPP is linked to an altered axonal transport that leads to neuroinflammation and axonal damage [74]. A study of Costa et al. [75] evaluated the cerebrospinal fluid (CSF) level of Aβ1 - 42 levels as a possible marker of cognitive status changes in patients with epilepsy of unknown origin. Pathological levels of Aβ were found in the CSF of a group of 40 non-dement patients with epilepsy of unknown origin. A significant percentage of these patients (17.5%) drives over a three-year period to AD. To summarize, Aβ pathology has a defined role in driving inflammation and neurodegeneration in both AD and epilepsy: addressing markers of amyloid in people with epilepsy could allow an early diagnosis of cognitive decline and AD. A study of Aboud et al. [76] exploring the possible associations between APOE genetics, neuroinflammation, and epilepsy found that APOE could directly influence acute adaptative response to stress factors, promote microglial and astrocytic activation and ultimately neuronal survival. APOE ɛ4/ɛ 4 is the allele combination less associated with neuronal resiliency, leading to early neuronal loss, altered inflammatory response in both AD and epilepsy [76].
In addition, other studies revealed the presence of tau pathology on cases of long-lasting chronic or drug-resistant epilepsy. In a postmortem study, chronic epilepsy was associated with increased tau neurofibrillary tangles at mid-Braak stages in patients aged 40–65 years [77]. Another clinicopathological study [71] on resected temporal lobe tissues of 33 patients with drug-resistant TLE demonstrated the presence of hyperphosphorylated tau pathology in 94%of samples; a positive correlation between post-operative cognitive decline and tau deposition was found in these patients. Moreover, an association between tau burden and history of secondary generalized seizures was identified, supporting the hypothesis of abnormal ictal activity contributing to an epilepsy-tau pathology in TLE. In this form of epilepsy, a high frequency of focal seizures gradually damages the hippocampal region of temporal lobe, leading to a significant memory loss. Synapse damage and synaptic protein loss are strongly related to the severity of dementia. Altered synaptic receptors endocytosis such as α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and N-methyl-D-aspartate (NMDA) has been reported to be crucial in the development of AD. Tau load could act as a promoting role in epilepsy development by enhancing excitotoxicity of NMDA receptors and increasing gamma-aminobutyric acid A (GABAA) receptors-mediated excitability [78].
Neurophysiological evidence of cortical hyperexcitability in epilepsy and Alzheimer’s disease
The excitability of cerebral cortex has been ext-ensively investigated using transcranial magnetic stimulation (TMS), a noninvasive technique of stimulation of the human brain, both in AD and in epileptic patients. Using TMS related techniques, it is possible to perform a functional evaluation of glutamatergic, GABAergic, and cholinergic circuits of the intact human brain [79]. Several abnormalities have been described in AD (see [80] for a review) and in epilepsy [81] with some overlaps in these two conditions, in particular an enhanced cortical excitability has been extensively reported in both diseases.
Subclinical and interictal epileptiform activity in AD patients
When approaching the review of literature about AD and epileptiform activity, there are several limitations hindering comparison among studies: lack of definition of interictal epileptiform abnormalities (IEAs), different electroencephalogram (EEG) techniques used, methods of evaluation of IEAs by different researchers, etc. Criteria for IEAs definition were only recently validated [82]. In particular, the IEAs are defined as paroxysmal sharp waveforms (di- or tri-phasic waves with sharp or spiky morphology) on EEG, lasting 20 to 200 ms and followed by an associated slow after-wave, that disrupt background activity. Moreover, estimates of epileptiform activity in AD are limited by the use of non-invasive scalp recordings, a limitation that does not occur in animal models, in which subdural or depth electrodes are commonly used.
Epileptiform activity is named interictal when detected in patients with seizures, and subclinical when detected in patients without known seizures.
Interictal epileptiform activity
Consistent with studies addressing epilepsy in older adults, routine EEG detects interictal epileptiform activity in about a third of patients with mild cognitive impairment (MCI) or dementia and comorbid seizures. The sensitivity of the EEG increases with extended recordings that capture sleep states and with serial EEGs [83]. The retrospective evaluation by Vossel and colleagues (2013) [8] revealed epileptiform activity in 62%of patients with amnesic MCI or AD who were evaluated for seizures and in 6%(7 of 113) of those without known seizures, by means of the analysis of 20-min EEGs. In AD patients with epilepsy, epileptic foci were mainly focal and unilateral with a temporal lobe predominance. Notably, authors reported an electro-clinical topographical matching between epileptiform activity and impaired cognitive domains. Sarkis et al. (2016) [84] retrospectively evaluating patients with dementia and unprovoked seizures reported a 36%prevalence of epileptiform activity. They did not find any difference of occurrence of epileptiform activity between early and late-onset AD patients. However, in both studies neither an epileptiform activity definition nor an EEG interpretation by expert epileptologists has been provided.
Subclinical epileptiform activity
The occurrence of subclinical epileptiform activity in AD may vary and the usefulness of EEG monitoring in AD patients without suspected seizures has not yet been defined. Liedorp and colleagues (2010) [85] retrospectively examined 1.674 consecutive patients of a memory clinic and found prevalence of subclinical epileptiform activity in only 42 (3%) patients, without any association with specific dementia diagnosis. However, authors examined only wakefulness routine EEGs in different types of cognitive impaired patients (AD, MCI, other dementia). Later, Vossel and colleagues (2016) [12] prospectively evaluated subclinical epileptiform activity in 33 AD patients and 19 controls without cognitive impairment through overnight long-term video-EEG and a 1-h resting magnetoencephalography exam with simultaneous EEG. Experienced epileptologists and clinical neurophysiologists reviewed EEG recordings and authors provided an accurate definition of epileptiform activity even if not completely fulfilling the recent criteria [82]. They detected subclinical epileptiform activity in 42.4%of AD patients and 10.5%of controls and reported its prevalence in temporal lobes and during NREM sleep. At the time of monitoring, AD patients with epileptiform activity did not differ clinically from those without such activity, but they showed faster declines in global cognition as evaluated by Mini-Mental State Examination (MMSE) and in executive function. Opposite findings were reported by Brunetti and colleagues [86] who recently explored nocturnal EEG-polysomnography in a prospective cohort of non-epileptic AD, MCI patients, and healthy controls; they found low frequency of epileptiform activity, without any difference among groups (about 6%). The different prevalence of epileptiform activity in these two studies could be partially explained by the different techniques the authors used: consecutive 24-h EEG in Vossel et al. [12] versus full night EEG in the work of Brunetti et al. [86]. In addition, in the Brunetti study the epileptic discharges were automatically detected by a dedicated software and then visually reviewed by clinical neurophysiologists. Finally, differences in the clinical features of patients’ samples may further explain these conflicting results: while in the work of Brunetti et al., the enrolled patients probably suffered from the typical form of AD, Vossel et al. recruited for their study both typical and atypical AD with younger age and more atypical symptoms, potentially subtended by different neuropathological features [87] that may affect detectable spiking activity on EEG.
Horvath and colleagues (2018) [83] prospectively and retrospectively evaluated 42 AD patients using 24-h ambulatory EEG and identified subclinical epileptic activity (detected by two expert epileptologists) in 28%of subjects and seizures confirmed by EEG in 24%. Recently, Lam and coworkers (2020) [88] in a cross-sectional study compared the incidence of epileptiform activity in 24-h ambulatory scalp EEGs of 43 cognitively normal elderly healthy controls, 41 participants with early-stage AD with no history or risk factors for epilepsy, and 15 participants with early-stage AD with late-onset epilepsy related to AD. A panel of 9 concordant epileptologists confirmed the occurrence of epileptiform abnormalities, respectively in 4.7%, 22%, and 53%of subjects.
In conclusion, current literature about the prevalence and significance of interictal and subclinical epileptiform activity in AD patients is limited and not conclusive, due the low number and heterogeneity of available studies. In particular, at the moment it is not absolutely known whether the epileptiform abnormalities are the consequence of the neurodegenerative process or can contribute to it. Anyway, these findings raise the possibility that epileptiform activity might negatively impact memory and accelerate cognitive decline, either directly or by association with unrecognized silent seizures. Recently, clinically silent hippocampal seizures and epileptiform spikes during sleep were identified in two AD patients without a history or EEG evidence of seizures, using intracranial foramen ovale electrodes [89]. A careful clinical and EEG assessment for epileptiform activity and silent seizures in MCI and AD patients should be performed, especially if they present fluctuations in cognition and/or myoclonus.
COGNITION AND EPILEPTIFORM ACTIVITY
Effects of ictal and interictal epileptiform activity on cognition
Even if the role of epileptiform activity detected in some cases of patients with AD is not clearly understood, the detrimental effects of this activity on cognition should be taken into account. Both ictal (subclinical) and interictal epileptiform activity could interfere with cognition. If the ictal and post-ictal effects of seizures on cognition are clearly understandable, the IEAs are typically considered to be asymptomatic. Anyway, in recent years robust evidence has been gathered that IEAs are also related to brief lapses in cognition [90–92]. Several studies have found that the occurrence of IEAs recorded with intracranial electrodes correlates with impaired behavioral performance in working memory [92, 93] and delayed free recall tasks [94]. Moreover, it was found that IEAs outside a left hemispheric seizure onset zone impacted memory encoding, recall, and retrieval, while those inside the seizure onset zone did not [92]. Very recently, utilizing single-neuron recordings in patients with medically refractory epilepsy, some authors [95] demonstrated that hippocampal IEAs transiently changed firing of hippocampal neurons and disrupted selectively the retrieval, but not encoding, of declarative memories; the extent of the modulation of the individual firing of hippocampal neurons by IEAs predicted the extent of reduction of subjective retrieval confidence. These findings have been observed in patients with refractory seizures by use of depth electrodes. However, such invasive testing is rarely performed in patients with AD, and the exact extent and acute cognitive effects of hippocampal epileptiform discharges in the AD population are unknown.
In addition to these ‘acute’ effects on cognition due to the epileptiform activity, the impact of comp-ensatory (inhibitory) changes in brain networks and cortical excitability should be considered. Alt-hough such changes can limit overall network hyperexcitability, they can also have deleterious consequences for neural plasticity, particularly in the hippocampus [6]. Experimental evidence from animal studies demonstrated that soluble forms of Aβ are cytotoxic, induce the appearance of aberrant excitatory neuronal network activity in vivo, and trigger complex molecular and cellular patterns of compensatory inhibitory and excitatory mechanisms in hippocampal circuitry; on the other hand at least two positive feedback mechanisms intersecting with seizure-related pathophysiology have been identified that synergistically enhance the further release of Aβ by seizures, thus providing a molecular basis an unchecked vicious cycle at the neuronal network level capable of accelerating AD pathology progression [6] (Fig. 1). In mouse models [96–101] of AD, interventions aiming at suppressing the epileptiform activity, including genetic ablation of tau and cellular prion protein [97], and treatment with some ASMs [96], reduce synaptic and cognitive deficits. In these mouse models, the acute suppression of epileptiform activity with ASMs does not improve cognitive function, whereas prolonged treatment over 3 weeks or longer improves both synaptic function and cognition [96, 98, 99, 101]. Thus, even if we are well aware that these findings coming from animal models’ studies are not directly transferable to the human clinical setting of AD patients with seizures, they raised the hypothesis that deleterious effects of epileptiform activity on cognition in AD may occur more frequently through chronic remodeling of neuronal circuits than through abrupt disruptions of network functions [6, 9].
Network hyperactivation in patients with Alzheimer’s disease
Also in humans, network hyperexcitability in temporal regions has been related to memory dysfunction and AD pathogenesis. In patients with MCI due to AD, task-based functional magnetic resonance imaging (fMRI) studies have shown increased hippocampal activation during memory encoding [102], probably linked to compensatory recruitment of neural reserves. Nevertheless, the findings of the study indicate that compensatory hyperactivation of the hippocampus and multiple other cortical and subcortical brain regions during encoding is ineffective, might contribute to AD pathogenesis and predict future cognitive decline [102]. Interestingly, treatment with Levetiracetam (LEV) (125 mg twice a day) in patients with amnestic MCI, suppressing hippocampal dentate gyrus and CA3 hyperactivation, normalized fMRI changes and improved their performance on a hippocampal-based pattern-separation task [103]. Additionally, functional studies with PET [104] evidenced that network hyperactivation may spread to regions outside the hippocampus in early stages of AD, including the default mode network (precuneus and posterior cingulate cortex), indicated by failure to deactivate this network during memory encoding. Finally, resting-state MEG has shown network hypersynchronization across multiple frequency bands in frontoparietal and interhemispheric networks in patients with amnestic MCI [105].
ANTISEIZURE MEDICATIONS AND ALZHEIMER’S DISEASE
Antiseizure medications that worsen cognitive performance and dementia
ASMs are increasingly used in the elderly not only for treatment of epilepsy but also for management of psychiatric diseases, neuropathic pain, and behavioral disorders observed in dementia with a special regard for AD [106, 107]. It has been an object of debate if use of ASMs in the geriatric population, notoriously more susceptible to drugs’ adverse effects in reason of age-related altered pharmacodynamics, polytherapy, and comorbidities, could represent a predisposing factor for dementia itself. Currently only two studies have explored this correlation. The first one is a case-control study of Carter and colleagues [47] who analyzed a cohort of more than 5,000 elderly subjects with a five-year follow-up period and concluded that people reporting ASMs use at baseline had an overall odds ratio (OR) of 2.11 for developing dementia compared to controls. Taipale et al. [108] taking into consideration the same topic found a less strong correlation (with an OR of 1.28 and 1.15 for developing dementia and AD, respectively) in a German and Finnish population analyzed with a lag time of two years between ASM use and dementia diagnosis with a higher risk for drugs associated to known cognitive adverse effects (CAEs). Anyway, these studies have been criticized by scientific community for their methodological approach [109], thus still leaving unsolved the question whether ASMs could directly cause dementia.
ASMs are notoriously associated with both negative and positive cognitive effects. Therapeutic compliance depends mostly on tolerability, and CAEs are one of the main reasons for drug discontinuation [110]. Studies that explored the relationship between ASMs and cognition are difficult to summarize because they are usually heterogeneous in patient selection and in neuropsychiatric tasks used to assess cognitive functions. It is also noteworthy that not only ASMs, but several other factors such as age of patients, type of epilepsy, pre-existing brain damage, seizure frequency, and co-morbid neuropsychiatric disorders, could play an important role.
ASMs exert their antiepileptic effect with a suppression of neuronal excitability or with an enhancement of neuronal inhibition; thus, it has been proposed that their mechanism of action may explain mostly of their cognitive profile. Actually, a specific relationship between the action of individual drugs and specific cognitive deficit has not yet been clarified [111]. Among drugs with a prominent mechanism of neuronal inhibition, benzodiazepines (BZD) remain the most commonly prescribed in the elderly in many developed countries for their anxiolytic effect [112]. Both short-term [113] and long-term BDZ treatment has been associated with increased sedation and impairment in several cognitive domains such as visuospatial ability, speed of processing, and verbal learning [114]. Furthermore, chronic BDZ use in an older population has been associated with increased risk of developing AD [115]. Two potential mechanisms have been proposed to explain the correlation between long-term use of BDZ and dementia [116]: first, BZD could potentiate GABA-secreting activity of astrocytes located in the area of amyloid plaques in pre-symptomatic patients by precipitating them in the symptomatic phase of AD; second, these drugs, by lowering brain activation level, could drastically reduce cognitive reserve which is a well-known protective factor for the onset of dementia.
The majority of first generation ASMs (developed before the 1990s) are sedative and may induce psychomotor slowing by increasing reaction time [117] (Table 1). These effects may have significant consequences in the geriatric population already predisposed to falls and domestic accidents. Among these drugs, phenobarbital (PHB) seems to be associated with worst tolerability profile especially for intelligence and cognitive scores impairment [118].
Table 1
Antiseizure medications (ASMs) and their effect on cognition
| Drug | Dose© (mg per day) | Mechanism of Action | Tolerability | Cognitive side effects | Potential effect on cognition and mood |
| Phenobarbital (PHB) | 50–100 | Enhances GABAA receptor function by increasing chloride channel open time | Poor | Yes | Cognitive side-effects |
| Carbamazepine (CBZ) | 400–1200 (Probably 400–600) | Blocks Na channels (enhances fast inactivation) | Poor | Yes | Cognitive side-effects can be more marked in older people Mood stabilizer |
| Phenytoin (PHT) | 200–300 | Blocks Na channels (enhances fast inactivation) | Poor | Yes | Can be associated with adverse effects on cognition Can sometimes have adverse effects on mood |
| Valproic Acid (VPA) | 250–1000 | Blocks T-type Ca channels Enhances GABA action | Fair | Yes | Can affect cognition; also hyperammonemic encephalopathy Mood stabilizer |
| Levetiracetam (LEV) | 250–2000 | Modulate synaptic vesicle protein SV2A | Good | No | May improve cognition Can have adverse effects on mood (e.g., irritability, anxiety, low mood) |
| Gabapentin (GBP) | 300–1500 | Ligand of α2δ subunit of T-type Ca channels | Good | No | Usually cognitively neutral Can be anxiolytic and benefit mood |
| Pregabalin (PGB) | 100–300 | Ligand of α2δ subunit of T-type Ca channels | Good | No | Usually cognitively neutral Can be anxiolytic and benefit mood |
| Lamotrigine (LTG) | 50–500 (probably ≤200) | Blocks Na and T-type Ca channels | Good | No | Usually cognitively neutral Mood stabilizer |
| Topiramate (TPM) | 100–400 (probably ≤150) | Blocks Na channels (enhances fast inactivation) AMPA/kainate receptor antagonist GABAA receptor allosteric modulator Inhibitor of brain carbonic anhydrase | Poor | Yes | Can have adverse effects on cognition; word-finding difficulty in particular Can have adverse effects on mood |
| Zonisamide (ZNS) | 100–500 (probably ≤300) | Blocks Na channels Inhibitor of T-Type Ca channels Inhibitor of brain carbonic anhydrase | Poor | Yes | Can have adverse effects on cognition; word-finding difficulty in particular Can have adverse effects on mood |
| Oxcarbazepine (OXC) | 600–1200 (probably ≤600) | Block Na (enhances fast inactivation) and N-type Ca channels Increases potassium conductance | Fair | No | Can be associated with hyponatremia which could result in confusion, apathy, and lethargy Mood stabilizer |
| Eslicarbazepine (ESL) | 800–1200 (probably ≤800) | Blocks Na channels (enhances slow inactivation) Blocks T-type Ca channels | Fair | No | Can be associated with hyponatremia which could result in confusion, apathy, and lethargy Not thought to cause substantial adverse effect on mood |
| Lacosamide (LCM) | 100–400 (probably ≤300) | Blocks Na channels (enhances slow inactivation) | Good | No | Usually cognitively neutral Generally thought to have a benign psychological profile, but can occasionally have adverse effects on mood |
| Perampanel* (PER) | 4–8 (probably ≤4) | AMPA receptor antagonist | Fair | No | Likely to be cognitively neutral, but data are scarce. Might have adverse effects on mood with risk of unusual thoughts especially at higher doses |
| Brivaracetam* (BRV) | 50–200 (probably ≤100) | Modulate synaptic vesicle protein SV2A with higher affinity than LEV | Good | No | Likely to be cognitively neutral, but data are scarce. Might have adverse effects on mood |
GABA, gamma-aminobutyric acid; Na, sodium; Ca, calcium; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid. Information collected from several sources [5, 107, 108, 146, 155]. *These ASMs may be prescribed only as add-on therapy; © based on trials with AD patients and expert opinion.
Evidence for using phenytoin (PHT) in patients with AD is mostly limited to observational studies reporting variability in its efficacy in seizure control and neurological side-effects, including ataxia, delirium, sedation, and accelerated cognitive decline [119]. Consistently, deleterious effects of PHT on seizures and cognition in hAPP–J20 mice models have been recently demonstrated [120], probably related to the depletion of Nav1.1 in parvalbumin-positive interneurons in the parietal cortex, a change also observed in brain samples from patients with AD. Moreover, PTH has a poor therapeutic index and a high risk of pharmacokinetic interactions with other drugs.
Carbamazepine (CBZ) is largely used in the elderly for management of epilepsy, bipolar disorder, trigeminal neuralgia, and neuropathic pain. Several studies reported CBZ-related impairment in attention, verbal fluency, memory, and arithmetic performance [121]. CBZ has also been found to be useful in the management of behavioral disorders but its use in clinical practice is limited because of tolerability, CAE, and drug-drug interactions [122, 123]. Valproic acid (VPA) is commonly used to treat epilepsy and many psychiatric conditions in the elderly such as manic episodes, bipolar disorder, and impulse control, although there is a warning on its use in this population for treating behavioral symptoms in dementia in reason of an excessive sedative effect [124]. Studies in animals observed that VPA could also affect memory performance by reducing cell proliferation within the hippocampus and by lowering levels of brain-derived neurotrophic factor [125]. Moreover, in a multicenter, randomized, double-blind, placebo-controlled trial [126] assessing the efficacy of VPA (10–12 mg/kg per day), given over 2 years, to treat agitation in 313 patients with moderate AD without epilepsy, VPA did not reduce incidence of agitation or psychosis, and was associated with higher rates of somnolence, gait disturbance, tremor, diarrhea, and muscle weakness; patients who were taking VPA also showed greater brain volume loss and faster decline in MMSE at 1 year than patients in the placebo group.
With the exception of Topiramate (TPM) and Zonisamide, new generation drugs [127] have a better tolerability profile in terms of global cognitive functioning with particular regard for LEV, Lamotrigine (LTG), Oxcarbazepine (OXC), Eslicarbazepine, Gabapentin, Lacosamide [128], and probably also for Brivaracetam (BRV) [129] and Perampanel (PER) [130] (Table 1). TPM represents an exception because of its detrimental effect on verbal fluency and other frontal lobe-associated neuropsychological functions like cognitive speed and visual short-term memory [131]. Negative effects of TPM seems to be irrespective of titration schedule [132] and more evident in patients with left temporal EEG epileptic focus [133] with a consequently high rate of drug discontinuation [134]. TPM could cause impairment in working memory and verbal fluency tasks by reducing specific activation of some cerebral regions (left superior temporal gyrus and left inferior frontal region) as evidenced in fMRI studies of patients with temporal lobe epilepsy taking TPM compared to controls [135].
Polytherapy is also a well-recognized factor that contributes to increase ASM-related CAEs probably through a potentiation in adverse effects of two or more drugs or for contextual presence of comorbidities and drug-resistant epilepsy in patients where polytherapy is necessary [111]. Furthermore, the number of ASMs in a treatment regimen seems more strongly associated with changes in cognition than the type of ASM [136].
Antiseizure medications that may improve cognition
As mentioned above, some ASMs with a special regard for new generation molecules could improve cognitive performance (Table 1). Seizure control and a better management of psychiatric comorbidities could explain this evidence, but some molecules have demonstrated to directly improve cognition in different ways [121]. In several studies, LEV seems to improve a range of cognitive abilities such as visual short-term memory, working memory, psychomotor speed, concentration, and fluid intelligence [121]. Some authors attributed this effect to LEV-related reduction in seizure frequency [137], while others argued that this is irrespective of seizure control [138]. In the elderly with MCI or overt dementia and focal epilepsy, LEV seems to improve global cognitive performance [139]. Several hypotheses have been raised to explain this effect in addition to seizure control. First, an improvement of some electroencephalographic patterns observed in patients with dementia [140] could be related to an amelioration of cognitive performances. In one study when a single dose of LEV (at a dosage of 7.5 mg/kg) was administered to mild AD patients, a normalizing effect on the EEG with a decrease in coherence in slower frequency bands and an increase in coherence in the faster bands was observed suggesting a role of this drug in restoring altered patterns of cortico-cortical connections [141, 142]. Second, the neuroprotective potential of LEV, and in general of ASMs (except for those with well-known negative cognitive effects as BDZ, VPA, PHB, and PHT), could play a crucial role as the excitotoxic mechanisms are involved in the pathogenesis of many neurodegenerative diseases including AD [143]. In this regard, fMRI studies have shown that this cortical hyperexcitability in MCI patients is concentrated in specific region (dentate gyrus/CA3) of hippocampus notoriously involved in memory tasking [144]. LEV has been demonstrated to reduce this cortical hyperactivation [145], and its long-term use seems also to contrast hippocampal remodeling and synaptic dysfunction observed in hAPP transgenic mice models [96]. Finally, as mentioned a low-dose LEV (125 mg twice a day) improved hippocampal-based memory performance in patients with MCI [103]. However, for clarity, it is mandatory to specify that studies [103, 139, 145, 146] evaluating cognitive impact of LEV in MCI and AD patients with and without epilepsy documented an ameliorative effect only in the short-term period (not exceeding 1 year) and in small patient samples. Anyway, these promising results encouraged further research. For this reason, the effect of LEV in AD patients contrasting network hyperexcitability and possibly also cognitive decline in a longer time period has been currently tested in several Phase II ongoing randomized controlled trials (e.g., LEV-AD, NCT02002819; NCT03875638; ILiAD, NCT03489044; LAPSE, NCT04004702) [147]. Third, Stockburger and colleagues [148] observed that the synaptic vesicle protein 2A (SV2a), the molecular target of LEV, is expressed in mitochondria so it could represent a target of action that contributes to further explain its positive effect in cognitive performances in AD patients. Indeed, mitochondrial dysfunction has been demonstrated to play a fundamental role in the pathogenesis of AD through reduction in cellular energy supply and increase of oxidative stress [149].
Although less prominent than hyperactivation of NMDA receptors, AMPA and kainate receptors also have a role in glutamate mediated excitotoxicity suggesting that ASMs which exert an AMPA-kainate antagonism could exert a role in neuroprotection. Interestingly, it has been suggested that the motor cortex hyperexcitability demonstrated by TMS studies in AD might be interpreted as the consequence of an imbalance between non-NMDA and NMDA neurotransmission in favor of the non-NMDA transmission [150], thus drugs with an AMPA antagonism might be beneficial. TPM proved to be protective against selective hypoxic–ischemic white matter injury in rodent models in consideration of its action as AMPA–kainate receptor antagonist [151]. Among newer generation drugs, PER also expresses its action as selective noncompetitive AMPA receptor antagonist, and it has shown to improve post-stroke cognitive function by contrasting neuronal death of hippocampal CA1 neurons in animal models [152]. A similar mechanism of neuroprotection mediated by reduction of glutamate and aspartate concentrations in the CSF with a consequent decrease in neuronal excitability has been proposed also for LTG [153]. Consistently, a small, double-blind, crossover trial of LTG (150 or 300 mg per day) for patients with AD without epilepsy showed that treatment with 300 mg of LTG was associated with improved performance on recognition and naming tasks as well as depressed mood on the ADAS behavior subscale after 8 weeks of treatment [154].
In a three-group parallel case-control study confronting the action of LEV (500–2000 mg per day), LTG (25–100 mg per day), and PHB (50–100 mg per day) in 95 patients with AD and epilepsy and 68 age-matched control patients with AD but without epilepsy, who did not receive ASMs, all ASMs had equivalent effects on seizure reduction, but LEV and LTG treatment resulted in better cognitive outcomes than PHB, that provoked more adverse events, in particular somnolence in 30%of patients [146]. LEV use resulted in improved attention, short-term memory, and oral fluency thus sustaining its role in attenuating AD cognitive deficits [146]. However, it has been commonly associated with psychiatric side effects such as anxiety, confusion, and irritability [5, 155], thus it should be used with caution in AD patients with behavioral and psychological symptoms (Table 1). Instead psychiatric adverse effects are not commonly associated with LTG which has excellent effectiveness and tolerability rate and improves mood in AD-related epilepsy [155]. LTG and Gabapentin seem to have a better tolerability profile than CBZ in AD patients with epilepsy with similar seizure control [156].
Among the newest ASMs, ELS has no documented effectiveness in AD patients but cognitive side effects were relatively rare, except at higher doses [155] (Table 1). Other ASMs, such as OXC and Lacosamide, are being used successfully as monotherapy to treat seizures in older adults, but limited data are available for their use in patients with AD. Patients on OXC or CBZ and Eslicarbazepine should have sodium levels monitored regularly, as these drugs can cause hyponatremia, particularly with advanced age. In a similar way, there are no head-to-head comparison trials of PER and Brivaracetam in elderly population, nor evidence about their use in people with AD, but from observational studies it seems that they have similar efficacy in elderly as that in adult patients [122].
In conclusions, ASMs are the first choice treatment for management of epilepsy in people with AD [157] and, even if a single antiseizure drug at low dose is generally adequate for the management of epilepsy at any stage of dementia, tolerability should be a primary target in such vulnerable patients [5, 155]. In these terms it seems reasonable to avoid cognitively harmful, sedative, or unpredictable ASMs in favor of use of cognitively neutral or stimulating molecules, as it has been shown for the majority of new generation ASMs, in particular LEV and LTG.
CONCLUSIONS
There is robust and increasing experimental evidence coming from both animals and humans’ studies clearly demonstrating a deep link between epilepsy and AD: patients with MCI and AD are more prone to have seizures and seizures seem to facilitate the deposit of tau and Aβ, thus promoting and accelerating neurodegenerative processes. Consistent with this view, TLE and AD have been proven to share several pathological and neuroimaging features. Even if studies addressing prevalence of interictal and subclinical epileptiform activity in these patients are not yet conclusive, their findings raise the possibility that epileptiform activity might negatively impact memory and hasten cognitive decline, either directly or by association with unrecognized silent seizures. In addition, data about detrimental effect of network hyperexcitability in temporal regions in the premorbid and early stages of AD open new therapeutic opportunities for ASMs and/or antiepileptic strategies that might complement or enhance existing therapies, and potentially modify disease progression. To this aim, there is a compelling need for large longitudinal epidemiological studies in MCI and early phase AD patients addressing with an accurate and long-term neurophysiological monitoring (possibly including sleep) the effect of epileptiform activity on cognition and disease progression. More clinical trials are required to clearly establish the benefits and risks of treating network hyperexcitability in patients with AD. Actually, there is a lack of clear guidelines indicating whether and how to treat subclinical epileptiform abnormalities in AD without overt seizures. Indeed, ASMs may have more negative than positive cognitive effects: if the first generation ASMs could usually impair attention, vigilance, and psychomotor speed, the new generation ones are better tolerated and proved to improve cognitive functions and mood. In this regard, LEV and LTG seem to be the best therapeutic options, but further information should be gathered for the last generation drugs. Finally, neuromodulatory approaches (such as deep brain stimulation, TMS, transcranial direct current stimulation, and vagus nerve stimulation) well known to decrease cortical hyperexcitability and epileptiform abnormalities [158, 159], should also be explored to evaluate also their contribution to improve cognition and potentially treat AD patients.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Scarmeas N , Honig LS , Choi H , Cantero J , Brandt J , Blacker D , Albert M , Amatniek JC , Marder K , Bell K , Hauser WA ((2009) ) Seizures in Alzheimer disease: Who, when, and how common? Arch Neurol 66: , 992–997. |
[2] | Friedman D , Honig LS , Scarmeas N ((2012) ) Seizures and epilepsy in Alzheimer’s disease. CNS Neurosci Ther 18: , 285–294. |
[3] | Sjogren T , Sjogren H , Lindgren AG ((1952) ) Morbus Alzheimer and morbus Pick; A genetic, clinical and patho-anatomical study. Acta Psychiatr Neurol Scand Suppl 82: , 1–152. |
[4] | Hesdorffer DC , Hauser WA , Annegers JF , Kokmen E , Rocca WA ((1996) ) Dementia and adult-onset unprovoked seizures. Neurology 46: , 727–730. |
[5] | Vossel KA , Tartaglia MC , Nygaard HB , Zeman AZ , Miller BL ((2017) ) Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurol 16: , 311–322. |
[6] | Noebels J ((2011) ) A perfect storm: Converging paths of epilepsy and Alzheimer’s dementia intersect in the hippocampal formation. Epilepsia 52: , 39–46. |
[7] | Cretin B , Sellal F , Philippi N , Bousiges O , Di Bitonto L , Martin-Hunyadi C , Blanc F ((2016) ) Epileptic prodromal Alzheimer’s disease, a retrospective study of 13 new cases: Expanding the spectrum of Alzheimer’s disease to an epileptic variant? J Alzheimers Dis 52: , 1125–1133. |
[8] | Vossel KA , Beagle AJ , Rabinovici GD , Shu H , Lee SE , Naasan G , Hegde M , Cornes SB , Henry ML , Nelson AB , Seeley WW , Geschwind MD , Gorno-Tempini ML , Shih T , Kirsch HE , Garcia PA , Miller BL , Mucke L ((2013) ) Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol 70: , 1158–1166. |
[9] | Palop JJ , Mucke L ((2010) ) Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat Neurosci 13: , 812–818. |
[10] | Irizarry MC , Jin S , He F , Emond JA , Raman R , Thomas RG , Sano M , Quinn JF , Tariot PN , Galasko DR , Ishihara LS , Weil JG , Aisen PS ((2012) ) Incidence of new-onset seizures in mild to moderate Alzheimer disease. Arch Neurol 69: , 368–372. |
[11] | Amatniek JC , Hauser WA , DelCastillo-Castaneda C , Jacobs DM , Marder K , Bell K , Albert M , Brandt J , Stern Y ((2006) ) Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia 47: , 867–872. |
[12] | Vossel KA , Ranasinghe KG , Beagle AJ , Mizuiri D , Honma SM , Dowling AF , Darwish SM , Van Berlo V , Barnes DE , Mantle M , Karydas AM , Coppola G , Roberson ED , Miller BL , Garcia PA , Kirsch HE , Mucke L , Nagarajan SS ((2016) ) Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann Neurol 80: , 858–870. |
[13] | Palop JJ , Chin J , Roberson ED , Wang J , Thwin MT , Bien-Ly N , Yoo J , Ho KO , Yu GQ , Kreitzer A , Finkbeiner S , Noebels JL , Mucke L ((2007) ) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55: , 697–711. |
[14] | Cloyd RA , Abisambra J , Smith B ((2019) ) Common mechanisms underlying epilepsy and tauopathy. J Clin Transl Sci 3: , 6. |
[15] | Cirrito JR , Yamada KA , Finn MB , Sloviter RS , Bales KR , May PC , Schoepp DD , Paul SM , Mennerick S , Holtzman DM ((2005) ) Synaptic activity regulates interstitial fluid amyloid–ॆ levelsin vivo. Neuron 48: , 913–922. |
[16] | Minkeviciene R , Rheims S , Dobszay MB , Zilberter M , Hartikainen J , Fülöp L , Penke B , Zilberter Y , Harkany T , Pitkänen A , Tanila H ((2009) ) Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 29: , 3453–3462. |
[17] | Gurevicius K , Lipponen A , Tanila H ((2013) ) Increased cortical and thalamic excitability in freely moving APPswe/PS1dE9 mice modeling epileptic activity associated with Alzheimer’s disease. Cereb Cortex 23: , 1148–1158. |
[18] | Assenza G , Lanzone J , Dubbioso R , Coppola A , Boscarino M , Ricci L , Insola A , Bilo L , Tombini M , Di Lazzaro V ((2020) ) Thalamic and cortical hyperexcitability in juvenile myoclonic epilepsy. Clin Neurophysiol 131: , 2041–2046. |
[19] | Kostopoulos GK ((2001) ) Involvement of the thalamocortical system in epileptic loss of consciousness. Epilepsia 42: (Suppl 3), 13–19. |
[20] | Blumenfeld H ((2003) ) From molecules to networks: Cortical/subcortical interactions in the pathophysiology of idiopathic generalized epilepsy. Epilepsia 44: , 7–15. |
[21] | Sperk G , Bellmann R , Gruber B , Greber S , Marksteiner J , Röder C , Rupp E ((1996) ) Neuropeptide Y expression in animal models of temporal lobe epilepsy. Epilepsy Res Suppl 12: , 197–203. |
[22] | Nägerl UV , Mody I , Jeub M , Lie AA , Elger CE , Beck H ((2000) ) Surviving granule cells of the sclerotic human hippocampus have reduced Ca2+influx because of a loss of calbindin-D(28k) in temporal lobe epilepsy. J Neurosci 20: , 1831–1836. |
[23] | Palop JJ , Jones B , Kekonius L , Chin J , Yu G-Q , Raber J , Masliah E , Mucke L ((2003) ) Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci U S A 100: , 9572–9577. |
[24] | Palop JJ , Chin J , Bien-Ly N , Massaro C , Yeung BZ , Yu GQ , Mucke L ((2005) ) Vulnerability of dentate granule cells to disruption of Arc expression in human amyloid precursor protein transgenic mice. J Neurosci 25: , 9686–9693. |
[25] | Garg N , Joshi R , Medhi B ((2018) ) Cracking novel shared targets between epilepsy and Alzheimer’s disease: Need of the hour. Rev Neurosci 29: , 425–442. |
[26] | Amos LA , Schlieper D ((2005) ) Microtubules and maps. Adv Protein Chem 71: , 257–298. |
[27] | Sánchez MP , García-Cabrero AM , Sánchez-Elexpuru G , Burgos DF , Serratosa JM ((2018) ) Tau-induced pathology in epilepsy and dementia: Notions from patients and animal models. Int J Mol Sci 19: , 1092. |
[28] | Jang SS , Royston SE , Lee G , Wang S , Chung HJ ((2016) ) Seizure-induced regulations of amyloid-β, STEP 61, and STEP 61 substrates involved in hippocampal synaptic plasticity. Neural Plast 2016: , 2123748. |
[29] | Hoey SE , Williams RJ , Perkinton MS ((2009) ) Synaptic NMDA receptor activation stimulates α-secretase amyloid precursor protein processing and inhibits amyloid-β Production. J Neurosci 29: , 4442–4460. |
[30] | Cheng IH , Scearce-Levie K , Legleiter J , Palop JJ , Gerstein H , Bien-Ly N , Puoliväli J , Lesné S , Ashe KH , Muchowski PJ , Mucke L ((2007) ) Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem 282: , 23818–23828. |
[31] | DeVos SL , Goncharoff DK , Chen G , Kebodeaux CS , Yamada K , Stewart FR , Schuler DR , Maloney SE , Wozniak DF , Rigo F , Bennett CF , Cirrito JR , Holtzman DM , Miller TM ((2013) ) Antisense reduction of tau in adult mice protects against seizures. J Neurosci 33: , 12887–12897. |
[32] | Holth JK , Bomben VC , Graham Reed J , Inoue T , Younkin L , Younkin SG , Pautler RG , Botas J , Noebels JL ((2013) ) Tau loss attenuates neuronal network hyperexcitability in mouse and drosophila genetic models of epilepsy. J Neurosci 33: , 1651–1659. |
[33] | Liu G , Thangavel R , Rysted J , Kim Y , Francis MB , Adams E , Lin Z , Taugher RJ , Wemmie JA , Usachev YM , Lee G ((2019) ) Loss of tau and Fyn reduces compensatory effects of MAP2 for tau and reveals a Fyn-independent effect of tau on calcium. J Neurosci Res 97: , 1393–1413. |
[34] | Kamenetz F , Tomita T , Hsieh H , Seabrook G , Borchelt D , Iwatsubo T , Sisodia S , Malinow R ((2003) ) APP processing and synaptic function. Neuron 37: , 925–937. |
[35] | Mucke L , Selkoe DJ ((2012) ) Neurotoxicity of amyloid β-protein: Synatic and network dysfunction. Cold Spring Harb Perspect Med 2: , a006338. |
[36] | Walsh DM , Klyubin I , Fadeeva JV , Cullen WK , Anwyl R , Wolfe MS , Rowan MJ , Selkoe DJ ((2002) ) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416: , 535–539. |
[37] | Venkitaramani DV , Chin J , Netzer WJ , Gouras GK , Lesne S , Malinow R , Lombroso PJ ((2007) ) β-amyloid modulation of synaptic transmission and plasticity. J Neurosci 27: , 11832–11837. |
[38] | Corbett BF , Leiser SC , Ling HP , Nagy R , Breysse N , Zhang X , Hazra A , Brown JT , Randall AD , Wood A , Chin J , Reinhart PH ((2013) ) Sodium channel cleavage is associated with aberrant neuronal activity and cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci 33: , 7020–7026. |
[39] | Querfurth HW , LaFerla FM ((2010) ) Alzheimer’s disease. N Engl J Med 362: , 329–344. |
[40] | Busche MA , Eichhoff G , Adelsberger H , Abramowski D , Wiederhold KH , Haass C , Staufenbiel M , Konnerth A , Garaschuk O ((2008) ) Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321: , 1686–1689. |
[41] | Costa C , Parnetti L , D’Amelio M , Tozzi A , Tantucci M , Romigi A , Siliquini S , Cavallucci V , Di Filippo M , Mazzocchetti P , Liguori C , Nobili A , Eusebi P , Mercuri NB , Calabresi P ((2016) ) Epilepsy, amyloid-β, and D1 dopamine receptors: A possible pathogenetic link? Neurobiol Aging 48: , 161–171. |
[42] | Catterall WA , Kalume F , Oakley JC ((2010) ) NaV1.1 channels and epilepsy. J Physiol 588: , 1849–1859. |
[43] | Reitz C , Brayne C , Mayeux R ((2011) ) Epidemiology of Alzheimer disease. Nat Rev Neurol 7: , 137–152. |
[44] | Scharfman HE ((2012) ) Alzheimer’s disease and epilepsy: Insight from animal models. Future Neurol 7: , 177–192. |
[45] | Kandratavicius L , Alves Balista P , Lopes-Aguiar C , Ruggiero RN , Umeoka EH , Garcia-Cairasco N , Bueno-Junior LS , Leite JP ((2014) ) Animal models of epilepsy: Use and limitations. Neuropsychiatr Dis Treat 10: , 1693–1705. |
[46] | Beghi E , Beghi M ((2020) ) Epilepsy, antiepileptic drugs and dementia. Curr Opin Neurol 33: , 191–197. |
[47] | Carter MD , Weaver DF , Joudrey HR , Carter AO , Rockwood K ((2007) ) Epilepsy and antiepileptic drug use in elderly people as risk factors for dementia. J Neurol Sci 252: , 169–172. |
[48] | Pandis D , Scarmeas N ((2012) ) Seizures in alzheimer disease: Clinical and epidemiological data. Epilepsy Curr 12: , 184–187. |
[49] | McAreavey MJ , Ballinger BR , Fenton GW ((1992) ) Epileptic seizures in elderly patients with dementia. Epilepsia 33: , 657–660. |
[50] | Hauser WA , Kurland LT ((1975) ) The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967. Epilepsia 16: , 1–66. |
[51] | Rao SC , Dove G , Cascino GD , Petersen RC ((2009) ) Recurrent seizures in patients with dementia: Frequency, seizure types, and treatment outcome. Epilepsy Behav 14: , 118–120. |
[52] | Mendez MF , Catanzaro P , Doss RC , Arguello R , Frey WH ((1994) ) Seizures in Alzheimer’s disease: Clinicopathologic study. J Geriatr Psychiatry Neurol 7: , 230–233. |
[53] | Tombini M , Koch G , Placidi F , Sancesario G , Marciani MG , Bernardi G ((2005) ) Temporal lobe epileptic activity mimicking dementia: A case report. Eur J Neurol 12: , 805–6. |
[54] | Sherzai D , Losey T , Vega S , Sherzai A ((2014) ) Seizu-res and dementia in the elderly: Nationwide inpatient sample 1999-2008. Epilepsy Behav 36: , 53–56. |
[55] | Hauser WA , Morris ML , Heston LL , Anderson VE ((1986) ) Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 36: , 1226–1230. |
[56] | Chen JY , Stern Y , Sano M , Mayeux R ((1991) ) Cumulative risks of developing extrapyramidal signs, psychosis, or myoclonus in the course of Alzheimer’s disease. Arch Neurol 48: , 1141–1143. |
[57] | Larner AJ ((2011) ) Presenilin-1 mutation Alzheimer’s disease: A genetic epilepsy syndrome? Epilepsy Behav 21: , 20–22. |
[58] | Shea YF , Chu LW , Chan AOK , Ha J , Li Y , Song YQ ((2016) ) A systematic review of familial Alzheimer’s disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J Formos Med Assoc 115: , 67–75. |
[59] | Cabrejo L , Guyant-Maréchal L , Laquerrière A , Vercelletto M , De La Fournière F , Thomas-Antérion C , Verny C , Letournel F , Pasquier F , Vital A , Checler F , Frebourg T , Campion D , Hannequin D ((2006) ) Phenotype associated with APP duplication in five families. Brain 129: , 2966–2976. |
[60] | Jayadev S , Leverenz JB , Steinbart E , Stahl J , Klunk W , Yu CE , Bird TD ((2010) ) Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 133: , 1143–1154. |
[61] | Lai F , Williams RS ((1989) ) A prospective study of Alzheimer disease in Down syndrome. Arch Neurol 46: , 849–53. |
[62] | Risse SC , Lampe TH , Bird TD , Nochlin D , Sumi SM , Keenan T , Cubberley L , Peskind E , Raskind MA ((1990) ) Myoclonus, seizures, and paratonia in Alzheimer disease. Alzheimer Dis Assoc Disord 4: , 217–225. |
[63] | Romanelli MF , Ashkin K , Morris JC , Coben LA ((1990) ) Advanced Alzheimer’s disease is a risk factor for late-onset seizures. Arch Neurol 47: , 847–850. |
[64] | Förstl H , Burns A , Levy R , Cairns N , Luthert P , Lantos P ((1992) ) Neurologic signs in Alzheimer’s disease: Results of a prospective clinical and neuropathologic study. Arch Neurol 49: , 1038–1042. |
[65] | Villemagne VL , Burnham S , Bourgeat P , Brown B , Ellis KA , Salvado O , Szoeke C , Macaulay SL , Martins R , Maruff P , Ames D , Rowe CC , Masters CL ((2013) ) Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol 12: , 357–367. |
[66] | Volicer L , Smith S , Volicer BJ ((1995) ) Effect of seizures on progression of dementia of the alzheimer type. Dement Geriatr Cogn Disord 6: , 258–263. |
[67] | Subota A , Pham T , Jetté N , Sauro K , Lorenzetti D , Holroyd-Leduc J ((2017) ) The association between dementia and epilepsy: A systematic review and meta-analysis. Epilepsia 58: , 962–972. |
[68] | Difrancesco JC , Tremolizzo L , Polonia V , Giussani G , Bianchi E , Franchi C , Nobili A , Appollonio I , Beghi E , Ferrarese C ((2017) ) Adult-onset epilepsy in presymptomatic Alzheimer’s disease: A retrospective study. J Alzheimers Dis 60: , 1267–1274. |
[69] | Zheng P , Shultz SR , Hovens CM , Velakoulis D , Jones NC , O’Brien TJ ((2014) ) Hyperphosphorylated tau is implicated in acquired epilepsy and neuropsychiatric comorbidities. Mol Neurobiol 49: , 1532–1539. |
[70] | Davidson YS , Raby S , Foulds PG , Robinson A , Thompson JC , Sikkink S , Yusuf I , Amin H , Duplessis D , Troakes C , Al-Sarraj S , Sloan C , Esiri MM , Prasher VP , Allsop D , Neary D , Pickering-Brown SM , Snowden JS , Mann DMA ((2011) ) TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s Syndrome: Association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol 122: , 703–713. |
[71] | Tai XY , Koepp M , Duncan JS , Fox N , Thompson P , Baxendale S , Liu JYW , Reeves C , Michalak Z , Thom M ((2016) ) Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: A study of temporal lobe resections. Brain 139: , 2441–2455. |
[72] | Andrew RJ , Kellett KAB , Thinakaran G , Hooper NM ((2016) ) A Greek tragedy: The growing complexity of Alzheimer amyloid precursor protein proteolysis. J Biol Chem 291: , 19235–19244. |
[73] | Gourmaud S , Shou H , Irwin DJ , Sansalone K , Jacobs LM , Lucas TH , Marsh ED , Davis KA , Jensen FE , Talos DM ((2020) ) Alzheimer-like amyloid and tau alterations associated with cognitive deficit in temporal lobe epilepsy. Brain 143: , 191–209. |
[74] | Sima X , Xu J , Li J , Zhong W , You C ((2014) ) Expression of β-amyloid precursor protein in refractory epilepsy. Mol Med Rep 9: , 1242–1248. |
[75] | Costa C , Romoli M , Liguori C , Farotti L , Eusebi P , Bedetti C , Siliquini S , Cesarini EN , Romigi A , Mercuri NB , Parnetti L , Calabresi P ((2019) ) Alzheimer’s disease and late-onset epilepsy of unknown orig: Two faces of beta amyloid pathology. Neurobiol Aging 73: , 61–67 . |
[76] | Aboud O , Mrak RE , Boop FA , Griffin WST ((2014) ) Epilepsy: Neuroinflammation, neurodegeneration, and APOE genotype. Acta Neuropathol Commun 1: , 41. |
[77] | Thom M , Liu JYW , Thompson P , Phadke R , Narkiewicz M , Martinian L , Marsdon D , Koepp M , Caboclo L , Catarino CB , Sisodiya SM ((2011) ) Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: A post-mortem study. Brain 134: , 2969–2981. |
[78] | Limon A , Reyes-Ruiz JM , Miledi R ((2012) ) Loss of functional GABA A receptors in the Alzheimer diseased brain. Proc Natl Acad Sci U S A 109: , 10071–10076. |
[79] | Di Lazzaro V , Rothwell J , Capogna M ((2018) ) Noninvasive stimulation of the human brain: Activation of multile cortical circuits. Neuroscientist 24: , 246–260. |
[80] | Cantone M , Di Pino G , Capone F , Piombo M , Chiarello D , Cheeran B , Pennisi G , Di Lazzaro V ((2014) ) The contribution of transcranial magnetic stimulation in the diagnosis and in the management of dementia. Clin Neurophysiol 125: , 1509–1532. |
[81] | de Goede AA , ter Braack EM , van Putten MJAM ((2016) ) Single and paired pulse transcranial magnetic stimulation in drug naïve epilepsy. Clin Neurophysiol 127: , 3140–3155. |
[82] | Kural MA , Duez L , Sejer Hansen V , Larsson PG , Rampp S , Schulz R , Tankisi H , Wennberg R , Bibby BM , Scherg M , Beniczky S ((2020) ) Criteria for defining interictal epileptiform discharges in EEG: A clinical validation study.. Neurology 94: , e2139–e2147. |
[83] | Horváth A , Szcs A , Hidasi Z , Csukly G , Barcs G , Kamondi A ((2018) ) Prevalence, semiology, and risk factors of epilepsy in Alzheimer’s disease: An ambulatory EEG study. J Alzheimers Dis 63: , 1045–1054. |
[84] | Sarkis RA , Dickerson BC , Cole AJ , Chemali ZN ((2016) ) Clinical and neurophysiologic characteristics of unprovoked seizures in patients diagnosed with dementia. J Neuropsychiatry Clin Neurosci 28: , 56–61. |
[85] | Liedorp M , Stam CJ , Van Der Flier WM , Pijnenburg YAL , Scheltens P ((2010) ) Prevalence and clinical significance of epileptiform EEG discharges in a large memory clinic cohort. Dement Geriatr Cogn Disord 29: , 432–437. |
[86] | Brunetti V , D’Atri A , Della Marca G , Vollono C , Marra C , Vita MG , Scarpelli S , De Gennaro L , Rossini PM ((2020) ) Subclinical epileptiform activity during sleep in Alzheimer’s disease and mild cognitive impairment. Clin Neurophysiol 131: , 1011–1018. |
[87] | Murray ME , Graff-Radford NR , Ross OA , Petersen RC , Duara R , Dickson DW ((2011) ) Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol 10: , 785–796. |
[88] | Lam AD , Sarkis RA , Pellerin KR , Jing J , Dworetzky BA , Hoch DB , Jacobs CS , Lee JW , Weisholtz DS , Zepeda R , Westover MB , Cole AJ , Cash SS ((2020) ) Association of epileptiform abnormalities and seizures in Alzheimer disease. Neurology 95: , e2270–e2270. |
[89] | Lam AD , Deck G , Goldman A , Eskandar EN , Noebels J , Cole AJ ((2017) ) Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat Med 23: , 678–680. |
[90] | Aldenkamp AP , Arends J ((2004) ) Effects of epileptiform EEG discharges on cognitive function: Is the concept of “transient cognitive impairment” still valid? Epilepsy Behav 5: , S25–S34. |
[91] | Aldenkamp AP , Arends J , Verspeek S , Berting M ((2004) ) The cognitive impact of epileptiform EEG-discharges; relationship with type of cognitive task. Child Neuropsychol 10: , 297–305. |
[92] | Ung H , Cazares C , Nanivadekar A , Kini L , Wagenaar J , Becker D , Krieger A , Litt LT , Davis Ka ((2017) ) Interictal epileptiform activity outside the seizure onset zone impacts cognition. Brain 140: , 2157–2168. |
[93] | Krauss GL , Summerfield M , Brandt J , Breiter S , Ruchkin D ((1997) ) Mesial temporal spikes interfere with working memory. Neurology 49: , 975–980. |
[94] | Kleen JK , Scott RC , Holmes GL , Roberts DW , Rundle MM , Testorf M , Lenck-Santini PP , Jobst BC ((2013) ) Hippocampal interictal epileptiform activity disrupts cognition in humans. Neurology 81: , 18–24. |
[95] | Reed CM , Mosher CP , Chandravadia N , Chung JM , Mamelak AN , Rutishauser U ((2020) ) Extent of single-neuron activity modulation by hippocampal interictal discharges predicts declarative memory disruption in humans. J Neurosci 40: , 682–693. |
[96] | Sanchez PE , Zhu L , Verret L , Vossel KA , Orr AG , Cirrito JR , Devidze N , Ho K , Yu GQ , Palop JJ , Mucke L ((2012) ) Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci U S A 109: , E2895–E2903. |
[97] | Roberson ED , Halabisky B , Yoo JW , Yao J , Chin J , Yan F , Wu T , Hamto P , Devidze N , Yu GQ , Palop JJ , Noebels JL , Mucke L ((2011) ) Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci 31: , 700–711. |
[98] | Nygaard HB , Kaufman AC , Sekine-Konno T , Huh LL , Going H , Feldman SJ , Kostylev MA , Strittmatter SM ((2015) ) Brivaracetam, but not ethosuximide, reverses memory impairments in an Alzheimer’s disease mouse model. Alzheimers Res Ther 7: , 25. |
[99] | Zhang MY , Zheng CY , Zou MM , Zhu JW , Zhang Y , Wang J , Liu CF , Li QF , Xiao ZC , Li S , Ma QH , Xu RX ((2014) ) Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol Aging 35: , 2713–2725. |
[100] | Um JW , Nygaard HB , Heiss JK , Kostylev MA , Stagi M , Vortmeyer A , Wisniewski T , Gunther EC , Strittmatter SM ((2012) ) Alzheimer amyloid-Î 2 oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15: , 1227–1235. |
[101] | Shi JQ , Wang BR , Tian YY , Xu J , Gao L , Zhao SL , Jiang T , Xie HG , Zhang YD ((2013) ) Antiepileptics topiramate and levetiracetam alleviate behavioral deficits and reduce neuropathology in APPswe/PS1dE9 transgenic mice. CNS Neurosci Ther 19: , 871–881. |
[102] | Huijbers W , Mormino EC , Schultz AP , Wigman S , Ward AM , Larvie M , Amariglio RE , Marshall GA , Rentz DM , Johnson KA , Sperling RA ((2015) ) Amyloid-β deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain 138: , 1023–1035. |
[103] | Bakker A , Albert MS , Krauss G , Speck CL , Gallagher M ((2015) ) Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. Neuroimage Clin 7: , 688–698. |
[104] | Sperling RA , LaViolette PS , O’Keefe K , O’Brien J , Rentz DM , Pihlajamaki M , Marshall G , Hyman BT , Selkoe DJ , Hedden T , Buckner RL , Becker JA , Johnson KA ((2009) ) Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63: , 178–188. |
[105] | Maestú F , Peña JM , Garcés P , González S , Bajo R , Bagic A , Cuesta P , Funke M , Mäkelä JP , Menasalvas E , Nakamura A , Parkkonen L , López ME , Del Pozo F , Sudre G , Zamrini E , Pekkonen E , Henson RN , Becker JT ((2015) ) A multicenter study of the early detection of synaptic dysfunction in mild cognitive impairment using magnetoencephalography-derived functional connectivity. Neuroimage Clin 9: , 103–109. |
[106] | Konovalov S , Muralee S , Tampi RR ((2008) ) Anticonvulsants for the treatment of behavioral and psychological symptoms of dementia: A literature review. Int Psychogeriatrics 20: , 293–308. |
[107] | Sen A , Jette N , Husain M , Sander JW ((2020) ) Epilepsy in older people. Lancet 10225: , 735–748. |
[108] | Taipale H , Gomm W , Broich K , Maier W , Tolppanen AM , Tanskanen A , Tiihonen J , Hartikainen S , Haenisch B ((2018) ) Use of antiepileptic drugs and dementia risk—an analysis of Finnish Health Register and German health insurance data. J Am Geriatr Soc 66: , 1123–1129. |
[109] | Sarkis RA , Meador KJ ((2018) ) Comment on: Use of antiepileptic drugs and dementia risk. J Am Geriatr Soc 66: , 1852–1853. |
[110] | Bootsma HP , Ricker L , Hekster YA , Hulsman J , Lambrechts D , Majoie M , Schellekens A , de Krom M , Aldenkamp AP ((2009) ) The impact of side effects on long-term retention in three new antiepileptic drugs. Seizure 18: , 327–331. |
[111] | Mula M , Trimble MR ((2009) ) Antiepileptic drug-induced cognitive adverse effects: Potential mechanisms and contributing factors. CNS Drugs 23: , 121–137. |
[112] | Paulose-Ram R , Safran MA , Jonas BS , Gu Q , Orwig D ((2007) ) Trends in psychotropic medication use among U.S. adults. Pharmacoepidemiol Drug Saf 16: , 560–570. |
[113] | Buffett-Jerrott S , Stewart S ((2005) ) Cognitive and seda-tive effects of benzodiazepine use. Curr Pharm Des 8: , 45–58. |
[114] | Stewart SA ((2005) ) The effects of benzodiazepines on cognition. J Clin Psychiatry 66: , 9–13. |
[115] | De Gage SB , Moride Y , Ducruet T , Kurth T , Verdoux H , Tournier M , Pariente A , Bégaud B ((2014) ) Benzodiazepine use and risk of Alzheimer’s disease: Case-control study. BMJ 349: , g5205. |
[116] | Pariente A , De Gage SB , Moore N , Bégaud B ((2016) ) The benzodiazepine-dementia disorders link: Current state of knowledge. CNS Drugs 30: , 1–7. |
[117] | Lagae L ((2006) ) Cognitive side effects of anti-epileptic drugs. The relevance in childhood epilepsy. Seizure 15: , 235–241. |
[118] | AP A ((2001) ) Effects of antiepileptic drugs on cognition. Epilepsia 42: , 46–49. |
[119] | Tsiouris JA , Patti PJ , Tipu O , Raguthu S ((2002) ) Adverse effects of phenytoin given for late-onset seizures in adults with Down syndrome. Neurology 59: , 779–780. |
[120] | Verret L , Mann EO , Hang GB , Barth AMI , Cobos I , Ho K , Devidze N , Masliah E , Kreitzer AC , Mody I , Mucke L , Palop JJ ((2012) ) Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in alzheimer model. Cell 149: , 708–721. |
[121] | Eddy CM , Rickards HE , Cavanna AE ((2011) ) The cognitive impact of antiepileptic drugs. Ther Adv Neurol Disord 4: , 385–407. |
[122] | Kaur U , Chauhan I , Gambhir IS , Chakrabarti SS ((2019) ) Antiepileptic drug therapy in the elderly: A clinical pharmacological review. Acta Neurol Belg 119: , 163–173. |
[123] | Bessey LJ , Walaszek A ((2019) ) Management of behavioral and psychological symptoms of dementia. Curr Psychiatry Rep 21: , 66. |
[124] | Gallagher D , Herrmann N ((2014) ) Antiepileptic drugs for the treatment of agitation and aggression in dementia: Do they have a place in therapy? Drugs 74: , 1747–1755. |
[125] | Umka J , Mustafa S , ElBeltagy M , Thorpe A , Latif L , Bennett G , Wigmore PM ((2010) ) Valproic acid reduces spatial working memory and cell proliferation in the hippocampus. Neuroscience 166: , 15–22. |
[126] | Fleisher AS , Truran D , Mai JT , Langbaum JBS , Aisen PS , Cummings JL , Jack CR , Weiner MW , Thomas RG , Schneider LS , Tariot PN ((2011) ) Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology 77: , 1263–1271. |
[127] | Mula M ((2012) ) Recent and future antiepileptic drugs and their impact on cognition: What can we expect? Expert Rev Neurother 12: , 667–671. |
[128] | Li KY , Huang LC , Chang YP , Yang YH ((2020) ) The effects of lacosamide on cognitive function and psychiatric profiles in patients with epilepsy. Epilepsy Behav 113: , 107580. |
[129] | Witt JA , Elger CE , Helmstaedter C ((2018) ) Short-term and longer-term effects of brivaracetam on cognition and behavior in a naturalistic clinical setting—Preliminary data. Seizure 62: , 49–54. |
[130] | Meador KJ , Yang H , Piña-Garza JE , Laurenza A , Kumar D , Wesnes KA ((2016) ) Cognitive effects of adjunctive perampanel for partial-onset seizures: A randomized trial. Epilepsia 57: , 243–251. |
[131] | Gomer B , Wagner K , Frings L , Saar J , Carius A , Härle M , Steinhoff BJ , Schulze-Bonhage A ((2007) ) The influence of antiepileptic drugs on cognition: A comparison of levetiracetam with topiramate. Epilepsy Behav 10: , 486–494. |
[132] | Thompson PJ , Baxendale SA , Duncan JS , Sander JWAS ((2000) ) Effects of topiramate on cognitive function. J Neurol Neurosurg Psychiatry 69: , 636–641. |
[133] | Mula M , Trimble MR , Thompson P , Sander JW ((2003) ) Topiramate and word-finding difficulties in patients with epilepsy. Neurology 60: , 1104–1107. |
[134] | Tatum IV WO , French JA , Faught E , Morris GL , Liporace J , Kanner A , Goff SL , Winters L , Fix A ((2001) ) Postmarketing experience with topiramate and cognition. Epilepsia 42: , 1134–40. |
[135] | Szaflarski JP , Allendorfer JB ((2012) ) Topiramate and its effect on fMRI of language in patients with right or left temporal lobe epilepsy. Epilepsy Behav 24: , 74–80. |
[136] | Quon RJ , Mazanec MT , Schmidt SS , Andrew AS , Roth RM , MacKenzie TA , Sajatovic M , Spruill T , Jobst BC ((2020) ) Antiepileptic drug effects on subjective and objective cognition. Epilepsy Behav 104: , 106906. |
[137] | López-Góngora M , Martínez-Domeño A , García C , Escartín A ((2008) ) Effect of levetiracetam on cognitive functions and quality of life: A one-year follow-up study. Epileptic Disord 10: , 297–305. |
[138] | Wheless JW , Ng YT ((2002) ) Levetiracetam in refractory pediatric epilepsy. J Child Neurol 17: , 413–415. |
[139] | Lippa CF , Rosso A , Hepler M , Jenssen S , Pillai J , Irwin D ((2010) ) Levetiracetam: A practical option for seizure management in elderly patients with cognitive impairment. Am J Alzheimers Dis Other Demen 25: , 149–154. |
[140] | Locatelli T , Cursi M , Liberati D , Franceschi M , Comi G ((1998) ) EEG coherence in Alzheimer’s disease. Electroencephalogr Clin Neurophysiol 106: , 229–237. |
[141] | Musaeus CS , Shafi MM , Santarnecchi E , Herman ST , Press DZ ((2017) ) Levetiracetam alters oscillatory connectivity in Alzheimer’s disease. J Alzheimers Dis 58: , 1065–1076. |
[142] | Ricci L , Assenza G , Pulitano P , Simonelli V , Vollero L , Lanzone J , Mecarelli O , Di Lazzaro V , Tombini M ((2021) ) Measuring the effects of first antiepileptic medication in Temporal Lobe Epilepsy: Predictive value of quantitative-EEG analysis. Clin Neurophysiol 132: , 25–35. |
[143] | Dong XX , Wang Y , Qin ZH ((2009) ) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30: , 379–387. |
[144] | Yassa MA , Stark SM , Bakker A , Albert MS , Gallagher M , Stark CEL ((2010) ) High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. Neuroimage 51: , 1242–1252. |
[145] | Bakker A , Krauss GL , Albert MS , Speck CL , Jones LR , Stark CE , Yassa MA , Bassett SS , Shelton AL , Gallagher M ((2012) ) Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74: , 467–474. |
[146] | Cumbo E , Ligori LD ((2010) ) Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer’s disease. Epilepsy Behav 17: , 461–466. |
[147] | Lozupone M , Solfrizzi V , D’Urso F , Di Gioia I , Sardone R , Dibello V , Stallone R , Liguori A , Ciritella C , Daniele A , Bellomo A , Seripa D , Panza F ((2020) ) Anti-amyloid-β protein agents for the treatment of Alzheimer’s disease: An update on emerging drugs. Expert Opin Emerg Drugs 25: , 319–335. |
[148] | Stockburger C , Miano D , Baeumlisberger M , Pallas T , Ar-rey TN , Karas M , Friedland K , Müller WE ((2016) ) A mitochondrial role of SV2a protein in aging and Alzheimer’s disease: Studies with levetiracetam. J Alzheimers Dis 50: , 201–215. |
[149] | Wang W , Zhao F , Ma X , Perry G , Zhu X ((2020) ) Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol Neurodegener 15: , 30. |
[150] | Di Lazzaro V , Oliviero A , Pilato F , Saturno E , Dileone M , Tonali PA , Rossini PM ((2003) ) Motor cortex hyperexcitability to transcranial magnetic stimulation in Alzheimer’s disease: Evidence of impaired glutamatergic neurotransmission? Ann Neurol 53: , 824. |
[151] | Follett PL , Deng W , Dai W , Talos DM , Massillon LJ , Rosenberg PA , Volpe JJ , Jensen FE ((2004) ) Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: A protective role for topiramate. J Neurosci 24: , 4412–440. |
[152] | Nakajima M , Suda S , Sowa K , Sakamoto Y , Nito C , Nishiyama Y , Aoki J , Ueda M , Yokobori S , Yamada M , Yokota H , Okada T , Kimura K ((2018) ) AMPA receptor antagonist perampanel ameliorates post-stroke functional and cognitive impairments. Neuroscience 386: , 256–264. |
[153] | Conroy BP , Black D ((1999) ) Lamotrigine attenuates cortical glutamate release during global cerebral ischemia in pigs on cardiopulmonary bypass. Anesthesiology 90: , 844–854. |
[154] | Tekin S , Aykut-Bingöl C , Tanridaʇ T , Aktan S ((1998) ) Antiglutamatergic therapy in Alzheimer’s disease - effects of lamotrigine. J Neural Transm 105: , 295–303. |
[155] | Cretin B ((2018) ) Pharmacotherapeutic strategies for treating epilepsy in patients with Alzheimer’s disease. Expert Opin Pharmacother 19: , 1201–1209. |
[156] | Rowan AJ , Ramsay RE , Collins JF , Pryor F , Boardman KD , Uthman BM , Spitz M , Frederick T , Towne A , Carter GS , Marks W , Felicetta J , Tomyanovich ML ((2005) ) New onset geriatric epilepsy: A randomized study of gabapentin, lamotrigine, and carbamazepine. Neurology 64: , 1868–1873. |
[157] | Liu J , Wang LN , Wu LY , Wang YP ((2018) ) Treatment of epilepsy for people with Alzheimer’s disease. Cochrane Database Syst Rev 12: , CD011922. |
[158] | Assenza G , Campana C , Formica D , Schena E , Taffoni F , Di Pino G , Di Lazzaro V (2014) Efficacy of cathodal transcranial direct current stimulation in drug-resistant epilepsy: A proof of principle. In 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, EMBC 2014, pp. 530-533. |
[159] | Cantello R , Rossi S , Varrasi C , Ulivelli M , Civardi C , Bartalini S , Vatti G , Cincotta M , Borgheresi A , Zaccara G , Quartarone A , Crupi D , Laganá A , Inghilleri M , Giallonardo AT , Berardelli A , Pacifici L , Ferreri F , Tombini M , Gilio F , Quarato P , Conte A , Manganotti P , Bongiovanni LG , Monaco F , Ferrante D , Rossini PM ((2007) ) Slow repetitive TMS for drug-resistant epilepsy: Clinical and EEG findings of a placebo-controlled trial. Epilepsia 48: , 366–374. |


