
Porphyromonas gingivalis is a Strong Risk Factor for Alzheimer’s Disease
Abstract
Porphyromonas gingivalis (P. gingivalis) is one of the several important bacterial pathogens associated with the sporadic Alzheimer’s disease (AD). Different serotypes are either capsulated or are non-capsulated. It has been demonstrated that P. gingivalis (non-capsulated) can reproduce the neurodegenerative AD-like changes in vitro, and a capsular P. gingivalis (strain W83) could reproduce the cardinal hallmark lesions of AD in a wild-type mouse model. All P. gingivalis forms express proteolytically active proteases that enable cleavage of the amyloid-β protin precursor (AβPP) and tau resulting in the formation of amyloid-β and neurofibrillary tangles. Tau is an established substrate for gingipains, which can cleave tau into various peptides. Some of the P. gingivalis fragmented tau protein peptides contain “VQIINK” and “VQIVYK” hexapeptide motifs which map to the flanking regions of the microtubule binding domains and are also found in paired helical filaments that form NFTs. P. gingivalis can induce peripheral inflammation in periodontitis and can also initiate signaling pathways that activate kinases, which in turn, phosphorylate neuronal tau. Periodontal disease related inflammation has metabolic implications for an individual’s peripheral and brain health as patients suffering from generalized periodontitis often have related co-morbidities and are “at risk” of developing AD. The aim here is to discuss the role of P. gingivalis behind such associations with the backdrop of huge efforts to test P. gingivalis virulence factors clinically (GAIN Trial: Phase 2/3 Study of COR388 in Subjects with AD) with inhibitors, which may lead to an intervention by reducing the pathogenic bacterial load.
ALZHEIMER’S DISEASE AND ITS ASSOCIATION WITH PERIODONTAL DISEASE
The presence of extraneuronal amyloid (Aβ) plaques and intraneuronal neurofibrillary tangles (NFTs) in the brain, together with defined clinical signs of cognitive deficit, form the basis of the diagnostic criteria for AD at autopsy [1, 2]. The origins and the roles of Aβ plaques and NFTs appear quite distinct but together they lead to neurodegeneration. The hippocampus contains abundant intraneuronal NFTs composed of abnormally phosphorylated tau protein. According to Braak staging of AD, the NFTs spread in the brain in a defined distribution, which allows correlations to be made with the clinical stages [3]. Early involvement of tau pathology has been described in the subcortical nuclei such as the locus coeruleus and the pons anatomical areas of the brainstem [3, 4]. This anatomical site of the brain releases norepinephrine in response to emotio-nal and stress related factors. The locus coeruleus also controls the attention and alertness of an individual, so any adverse change in its homeostasis could affect behavior and mood. Notably, the trigeminal ganglion is located adjacent to the locus coeruleus of the brainstem [5]. It follows that the brainstem and the periodontium communicate via the trigeminal nerve because tooth related pain is registered in the brain. In support, the neurons of the trigeminal nerve five are known to be distributed within the periodontal ligament [13]. This provides an important link between periodontitis and the areas of the brain that are affected early in the progression of AD pathology.
Periodontitis is a chronic inflammatory disease which damages the tooth supporting tissues, i.e., gingivae, periodontal ligament, and alveolar bone [6, 7]. Bacteria which can instigate changes in a normally symbiotic microbial community to a dysbiotic state are termed ‘keystone pathogens’. Porphyromonas gingivalis (P. gingivalis) is a Gram negative coccobacillus shaped bacterium which has long been established as a keystone pathogen for periodontitis [6, 7]. This oral commensal becomes pathogenic and can exert its virulence via its endo/exotoxins (surface membrane lipopolysaccharide or LPS, and gingipains) and capsular polysaccharides that allow this bacterium to induce chemokine paralysis of the host and evade immune recognition, even in low abundance. The compromised host innate and adaptive immune responses may be inadequate to control the inflammophilic microbiota and paradoxically, can contribute to connective tissue damage and inflammatory bone loss [8]. Oral microbes can enter the systemic circulation during transient episodes of bacteraemia which can occur with daily oral hygiene activities and dental interventions [9]. Periodontal pathogens including P. gingivalis have been detected at disparate sites such as the atherosclerotic plaque [10] and AD brains [11, 12]. Pertinent to this, it has been demonstrated in AD transgenic mice that extraction of molar teeth generated release of the cytotoxic Aβ, and triggered neurodegeneration in the locus coeruleus via its connection with the trigeminal nerve five pathway connecting the periodontium [13]. This may provide an explanation for the underappreciated concepts such as missing molar teeth and their contribution to neuronal loss in AD [14–17].
This review will cover some salient aspects of P. gingivalis, the keystone periodontal disease pat-hogen, that makes this bacterium “important” as a strong risk factor for developing AD pathophysiology with an emphasis on the pathways that produce hallmark pathology. This is plausible as we now have a better understanding of the P. gingivalis secreted exotoxin known as gingipains that expresses cathepsin B proteolytic enzymatic activity, which enables cleavage of amyloid-β protein precursor (AβPP), to form Aβ plaques. Furthermore, gingipains has the potential to disturb tau homeostasis by hyperphosphorylating serine and threonine residues via inflammatory signaling that activate glycogen synthase kinase-3beta (GSK-3β). In addition, proteolytically active gingipains can hydrolyze tau protein to release fragments [12] with the “VQIINK” and “VQIVYK” hexapeptide motifs that are present in the paired helical filaments (PHF) that constitute the NFT lesion. These are significant recent leaps in scientific advances in order to understand the risk factor involvement of periodontal disease with the development of AD.
WHAT MAKES P. GINGIVALIS A RISK FACTOR FOR AD?
Inflammation is widely thought to contribute to the cognitive decline in AD [18]. However, peripheral episodes of inflammation and/or microbial access to the brain and their impact on triggering cerebral inflammation have largely been ignored. The pathways for microbial access to the brain are many [19] including a leaky blood-brain barrier (BBB). P. gingivalis can trigger the peripheral and cerebral immune responses. Periodontitis can exert its influence indirectly by sustaining peripheral inflammation. This together with defective susceptibility genes that normally help with clearance of waste from the brain, can prime microglial cells into a pro-inflammatory phenotype [20].
Other major risk factors for AD are comorbid in the presence of the Apolipoprotein E allele 4 (APOE ɛ4) susceptibility gene inheritance [21]. One effect is related to the decline of cerebral blood flow across the lifespan of an individual during normal aging but this effect is greater in subjects with APOE ɛ4 susceptibility gene inheritance [22]. Lack of an adequate systemic blood flow to the brain in older individuals and those suffering from dementia harboring the APOE ɛ4 susceptibility gene can precipitate cerebrovascular pathologies such as stroke, small vessel arteriosclerosis, and others [23–25]. In addition, middle aged hypertension and diabetes also increase the risk of AD [26]; this risk may be heightened in the presence of P. gingivalis oral infection due to severe periodontitis, which has metabolic implications for an individual’s peripheral and brain health [27, 28]. Meta-analysis has shown that there is a 250% higher risk for incident dementia in persons with both APOE ɛ4 inheritance and diabetes than those without; and a 35% higher risk for those with APOE ɛ4 alone [29]. Figure 1 illustrates the shared risk factors of periodontal disease and AD, which includes the genetic components, comorbidities, and environmental factors. This further supports the plausibility of P. gingivalis as a risk factor in itself.
Fig. 1
The keystone periodontal pathogen, P. gingivalis (P.g), can enter the bloodstream during episodes of transient bacteremia and gain access to the brain via multiple routes including a leaky blood-brain barrier. The microbial invasion triggers the cerebral immune response resulting in neuroinflammation and in the formation of the two diagnostic lesions of AD. Periodontitis can exert its influence indirectly by sustaining peripheral inflammation (blue dotted arrow). This can affect glial cells by priming them into a pro-inflammatory phenotype. In addition, this could also overload and overwhelm the clearing of toxic neuropeptides from the central nervous system (CNS). The potential causal relationship of periodontitis and AD is further supported by the shared risk factors.
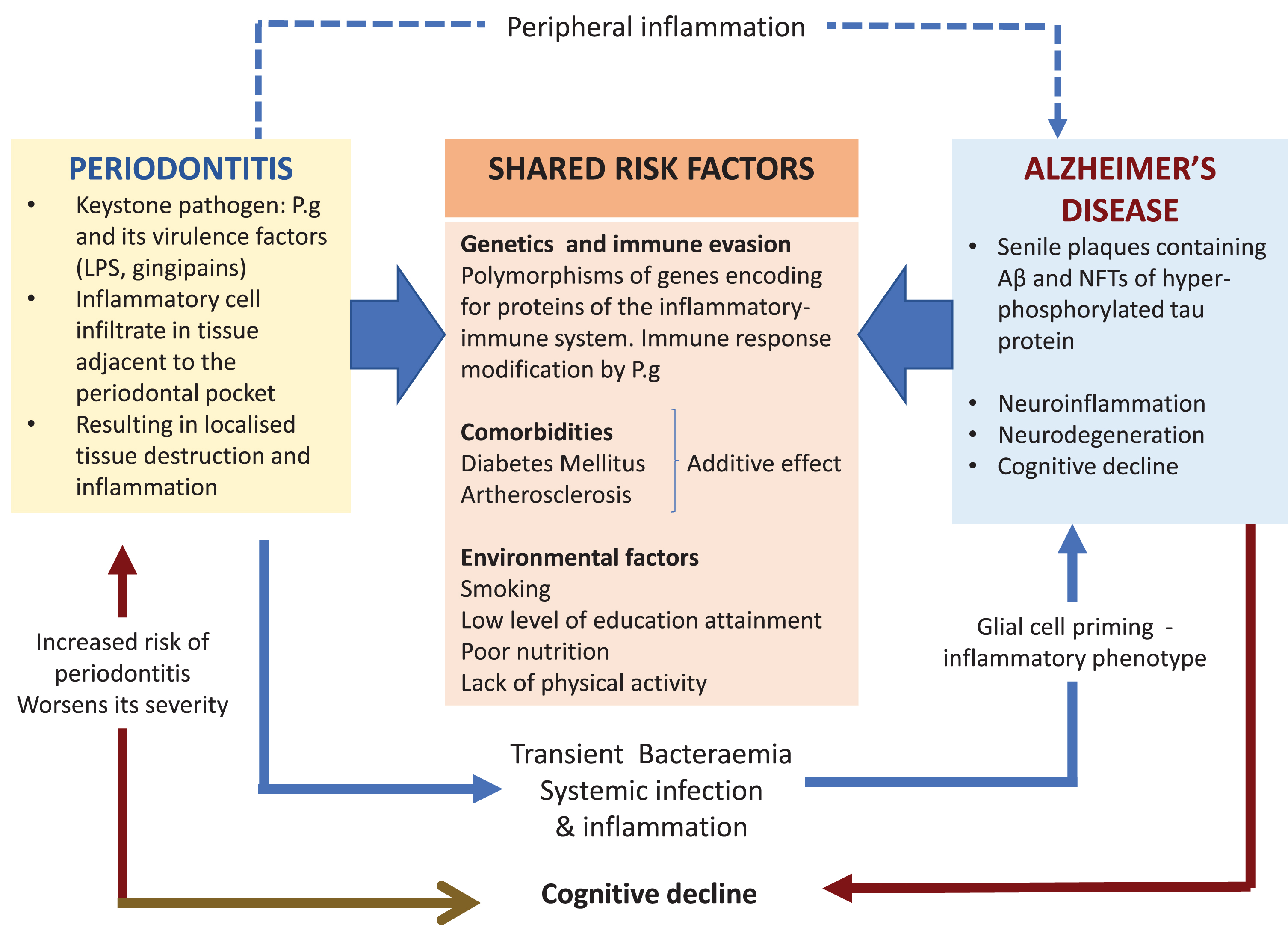
In addition, the BBB function becomes inadequate during normal aging and in cognitively impaired individuals carrying the APOE ɛ4 genetic susceptibility [30–32]. A disrupted BBB during aging and dementia [33] can potentially facilitate the passage of P. gingivalis from the systemic circulation into the brain. The scientific rationale for this statement comes from observations made from P. gingivalis mono-infected apolipoprotein null mice models of periodontitis for AD pathophysiology. P. gingivalis from the oral cavity accessed the brain and the BBB appeared significantly damaged [34–36]. A mechanistic pathway for bacterial entry into the brain is suggested to be via proteolytically active gingipains (the extracellular cysteine proteinases of P. gingivalis strain W83) breaking down the epithelial transmembrane proteins, E-cadherin, β1 integrin, and occludin, thereby disrupting the tight junctions between capillary endothelial cells. This increases the permeability of the BBB [37], and in addition, systemic proinflammatory cytokines also can potentially disrupt the BBB [38]. It is assumed that when tight junction integrity is sufficiently disturbed, passage of bacteria into the brain is likely to take place. In supporting the adverse effects of the cytokine on the BBB integrity, Vernal et al. [39] have reported that capsular serotype K1 and K2 P. gingivalis strains outside the central nervous system induce an enhanced secretion of pro-inflammatory interleukin cytokines (IL)-1β, IL-4, IL-6, IL-10, IL-17, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α in macrophages and dendritic cells, implying capsular P. gingivalis strains have the armory for potentiating BBB permeability through cytokine liberation. Encapsulated bacterial infections, e.g. P. gingivalis W83, harbors an additional virulence factor in the form of the capsular polysaccharide or A-lipopolysaccharide (A-LPS) which is different from the lipopolysaccharide (LPS) of the outer membrane of non-encapsulated bacteria, e.g., P. gingivalis (strain ATCC 33277) (see [40]). The A-LPS in the capsulated P. gingivalis (W83) plays a vital role in this bacterium’s virulence development by inducing proinflammatory cytokine paralysis [41] as it is con-nected to the post-translational additions of Arg-gin-gipains that make the bacterium more virulent [42, 43].
Therefore, a person harboring capsular forms of P. gingivalis could have a higher risk of developing AD, compared with those who carry the non-capsulated bacterium. This may be one reason why not all individuals who suffer from periodontal disease also manifest AD [15, 43]. Prevalence studies among the Dutch population identified K6 serotype (capsular) as the most prevalent (23%) [44], while the K5 serotype (capsular) was frequently observed in 25% of the subgingival isolates examined from Indonesian AD subjects [45].
P. GINGIVALIS GINGIPAINS: THE MOST CRITICAL VIRULENCE FACTOR
P. gingivalis produces gingipains R (RgpA, RgpB) and gingipain K (Kgp). Gingipains are classified as collagenases and trypsin-like cysteine proteinases [46], and are secreted by all strains (with/without capsule) of P. gingivalis [47, 48]. Together they degrade a variety of proteins involved in the immune response [49–52]. Here, our interest is in the trypsin-like cysteine proteinases, secreted by all strains of P. gingivalis [47, 48], which specifically cleave peptides at Arginine-Xaa and Lysine-Xaa (Xaa = any amino acid) from the C-terminus. The Arg-specific proteolytic activity is encoded by rgpA/B genes while the Lys-specific activity is encoded by the kgp gene. Rgp and Kgp are enzymes responsible for the trypsin-like activity associated with P. gingivalis and hence are important in the context of tau NFTs [47].
Recent studies have postulated that tau protein (associated with microtubules) and actin are sub-strates of Kgp gingipain [13, 53]. Neither Lys-gingipain nor Arg-gingipain are inhibited by internal protease inhibitors such as cystatins and α-antichymotrypsin and are therefore, able to diffuse into host tissues. Furthermore, gingipains are thermally stable from 0–45°C and over a range of pH fluctuations 5.5 to 10.5 [54]. It is not surprising in view of the sensitivity of tau to trypsin [55], that it is a potential substrate for gingipains in vivo [12]. This exciting finding is potentially a major breakthrough for therapy based on low molecular weight small-molecules designed to block the toxic effects of the different types of gingipains secreted by P. gingivalis, which are currently in Phase III clinical trials. An aspect of the gingipains inhibitor COR388 based therapy is provided elsewhere [56].
P. GINGIVALIS AND ITS ASSOCIATION WITH NEURODEGENERATION
AD is a neurodegenerative disease and hence neuronal loss is pivotal to the disease process. To this end, Goto et al. [13], performed surgical extractions of molar teeth in their triple transgenic AD (3xTg-AD) mice and uncovered a neurodegenerative pathway involving the trigeminal distribution within the periodontal ligament to the locus coeruleus. From the locus coeruleus, the tau pathology subsequently spreads to the hippocampus. The loss of molar teeth in AD patients is predominantly associated with periodontitis and in general, tooth loss has been linked to a higher risk of AD [14–17]. P. gingivalis (ATCC 33277) has been reported to cause AD-like neurodegeneration in infected neurons derived from induced pluripotent stem cells (iPSC) in an in vitro culture system with persistent expression of active gingipains, resulting in a 25% loss of neurons over three days [57]. The differentiated iPSC neurons can be maintained for months and offer a time efficient in vitro analysis of measuring neuronal degeneration compared to examining the years of AD-related neurodegenerative processes in humans. Furthermore, P. gingivalis can invade and survive within neurons and generate intraneuronal gingipains, which are proteolytically active, implying the plausibility of direct neurodegeneration associated with NFT lesion formation in AD taking place [57].
P. GINGIVALIS INFECTION AND PERIODONTITIS ARE LINKED TO DETERIORATING MEMORY
The first interventional study with human AD subjects was carried out by Rolim et al. [58] in which they demonstrated a beneficial outcome of dental treatment in AD subjects. The reported benefit to the patients with mild AD was in terms of reduced orofacial pain and an improvement in the mandibular function and periodontal indices. These improvements were maintained until the last evaluation after six months, and were followed by a reduction in the functional cognitive impairment. Several proof of concept studies carried out in either mice with experimentally induced periodontal disease with an oral infection with P. gingivalis, or with the introduction of its LPS have indicated that inflammatory signaling pathways contribute to a clinical phenotype in which impaired learning and memory is observed [59–61]. The inflammatory basis of deteriorating memory is upheld by the results from clinical trials in human AD subjects with periodontitis [62]. For a more comprehensive overview, see Singhrao and Olsen [63].
P. GINGIVALIS AND ITS ASSOCIATION WITH AD DIAGNOSTIC LESION FORMATION
P. gingivalis has the potential to induce inflammation peripherally due to periodontitis [64] and subsequently in the brain via its intracerebral entry or entry of its virulence factors (LPS and gingipains) [65–67]. Gingipains released by P. gingivalis which have similar AβPP-cleaving action as cathepsin B (of the host) [12], interact with the AβPP signaling pathways (amyloid cascade) to release Aβ [60]. In this process, gingipains together with LPS can proteolytically activate kinases such as GSK-3β which subsequently phosphorylates neuronal tau [57, 66].
Up until now, abnormally phosphorylated tau protein has not featured negatively in the pathophysiology of periodontal disease per se. However, Adamowicz et al. [68] implicated the role of GSK-3β in bacterial-induced periodontitis because its inhibition rescued bone loss. Thus, GSK-3β may be influencing phosphorylation of brain tau via immune responses mediated by P. gingivalis, in the Ilievski et al. [66] and Haditsch et al. [57] studies. The introduction of phosphorylated tau is an important point, especially since the intraneuronal cytoskeletal alterations precede the formation of amyloid in AD locus coeruleus [69]. This is interesting when compared with the Tg mouse tooth extractions where cytotoxic Aβ, triggered neurodegeneration in the same brain region (i.e., locus coeruleus) [13].
P. GINGIVALIS AND THE Aβ PLAQUE LESION
The extraneuronal AD plaque is composed of Aβ forming the basis of the “Amyloid Hypothesis” [70]. Aβ is a cleavage product of AβPP, which is seen in internal vesicular membranes, including the Golgi apparatus and endosomes [63, 71]. Hence, a direct extracellular deposition of soluble/insoluble Aβ as well as an intracellular processed soluble Aβ [63, 72, 73] both contribute to the extracellular fibrillary/insoluble (diffuse and neuritic) plaques in AD brains. The AβPP proteolytic cleavage is completed by the proteolytically active secretase enzymes (α-, β-, and γ-secretases). In rare familial AD cases, however, there is a mutant form of AβPP (Swedish double mutation APP K67ØM, N671L) which has a double mutation at the start of the beta cleavage site of AβPP cleaving enzyme 1 (BACE-1). BACE-1 has been shown to bind this mutated site with much higher affinity than the wtAPP and thus cleaves to form Aβ much more readily [73]. Notably, the enzyme cathepsin B, which is expressed in secretory vesicles within neurons, is shown, conversely, to bind with high affinity to wtAβPP (and not to the Swedish mutant AβPP) and has been suggested as a likely candidate for production of Aβ in the sporadic form of AD [73]. As described earlier, gingipains, the exotoxin of P. gingivalis, also has cathepsin B-like activity [12] and may act to cleave AβPP [73]. Wu et al. [60] demonstrated the host related cathepsin B to interact with AβPP to liberate Aβ. In order to determine the intracellular processing of AβPP by P. gingivalis-LPS (Pg-LPS), Wu et al. [60] challenged the wtAβPP mice and observed that the host’s cathepsin B, together with inflammatory mediators (IL-1β), directed AβPP proteolysis to release soluble amyloid which they interpreted to be Aβ42 species. Why the host’s cathepsin B activity and not the P. gingivalis cathepsin B-like activity are acting here is because we postulate that the highly purified Pg-LPS used by Wu et al. [60] is likely to have been denatured by the purification process leaving the LPS activity intact. Following P. gingivalis infections, an increase in peripheral Aβ1 - 40/42 accumulation within periodontal tissues have been shown in mice models and in the human gingival tissues and human serum thereby potentially adding to the Aβ pool in the AD brain [74–76]. To this end, Zeng et al. [77] identified advanced glycation end products (AGE) as a likely receptor for Aβ1 - 40/42 in cerebral endothelial cells implying AGE products-receptor are a plausible mediator of cerebrovascular-related Aβ accumulation after P. gingivalis infection in their mouse model. This supports the hypothesis that patients harboring the generalized form of periodontal disease may be “at risk” of developing AD via multiple pathways. Key evaluations of prospective and retrospective population-based data have shown that chronic periodontal disease that exists for longer than ten years results in a doubling of the risk for the sporadic form of AD [78–80]. Putting this information in perspective, the 10-year lag allows Aβ plaques to reach a plateau in the human host and this is when the host begins to indicate the earliest stage (mild cognitive impairment or MCI or prodromal) of AD. Following this stage, the pathological cascade of progressive AD would take over. This is interesting because the pieces of the jigsaw puzzle that form the picture of AD is beginning to emerge from reported scientific observations regarding the co-morbidity between periodontal disease and AD in some patients. Periodontal disease can start at any age and the time it takes to become chronic may vary from individual to individual. If that individual was vulnerable to manifesting AD later in life, it should be possible to predict the risk age of that individual from the time that their periodontitis became chronic. Taken together, an early detection of disease means patients may benefit from an earlier medical intervention.
“GINGIPAINS” INTERACTION WITH TAU PROTEIN: WHAT DOES THIS MEAN FOR NEUROFIBRILLARY TANGLES?
The NFTs represent hyperphosphorylated tau proteins binding to microtubules. Hyperphosphorylation of microtubules is abnormal because normal tau becomes insoluble and subsequently aggregates. This also causes the collapse of microtubules into non–membrane-bound masses of abnormal PHF which are found in the perinuclear cytoplasm of specific neurons. These constitute NFTs [81, 82]. Understanding the formation of the NFT lesion due to bacterial interaction is important as autopsied brains from AD cases have confirmed the presence of the following microbes: Actinomycetes [83, 84], P. gingivalis [12, 56], Helicobacter pylori, Chlamydia pneumoniae [85], Herpes Simplex type 1 virus (HSV1) [86], select species of oral/non-oral spirochetes [87], and select fungi [88]. Furthermore, it has been reported that Bacteroides species such as P. gingivalis are more virulent as a result of mixed infection with the Actinobacillus actinomycetemcomitans bacteria facilitating their movement between different organs [89, 90].
Dominy et al. [12] demonstrated tau protein as a substrate for P. gingivalis protease gingipains and as a consequence, the resultant tau protein fragments can be released into the brain’s parenchymal tissues. Depending on their size (Table 1), these extracellular phosphorylated tau fragments may be directly toxic to other neurons. The smaller sized phosphorylated tau fragments may be taken up by other connecting cells at the synaptic clefts during neurotransmitter uptake, thereby facilitating its spread from neuron to neuron and subsequently spreading pathology.
Table 1
Peptides of interest taken from reference 12. Amino acids indicated in green color are putative phosphorylation sites in the tau protein. *indicates peptides of significance to the PHF tau constituting neurofibrillary tangles that bind to microtubule binding domain (MBD)
| Peptide number | Region in Tau N-C termini | Nucleotide/ Peptide |
| 1 | K87–R126 | QAAQPHTEIPEGTTAEEAGIGDTPSLEDEAAGHVTQAR |
| N-terminal projection | ||
| domain Tau(1–165) | ||
| 2 | R211–R221 | TPSLPTPPTR |
| Proline rich domain | ||
| 166-242 | ||
| 3* | K259–K290 | 267HQPGGGKVQIINKKLDLSNVQS184K287 |
| MBD R1/2(274–304) | ||
| 4* | K28–K290 | 275VQIINKKLDLSNVQS184K285 |
| MBD-2 | ||
| R2(274–304) | ||
| 5* | K298–K317 | 298HVPGGGSVQIVYKPVDLSK317 |
| MBD-2/3 | ||
| 305–335 | ||
| 6* | K298–K321 | 298HVPGGGSVQIVYKPVDLSKVTSK321 |
| MBD-2/3 | ||
| 305–335 | ||
| 7* | K294–K317 | 294DNIKHVPGGGSVQIVYKPVDLSK317 |
| MBD-2/3 | ||
| 305–335 | ||
| 8* | K294–K321 | 294DNIKHVPGGGSVQIVYKPVDLSKVTSK321 |
| MBD-2/3 | ||
| 305–335 | ||
| 9 | R406–K438 | HLSNVSST414GS416IDMVDSPQLATLADEVSALAK |
| C-terminal domain (368–441) |
TAU CLEAVAGE BY GINGIPAINS AND ITS INVOLVEMENT IN PAIRED HELICAL FILAMENTS
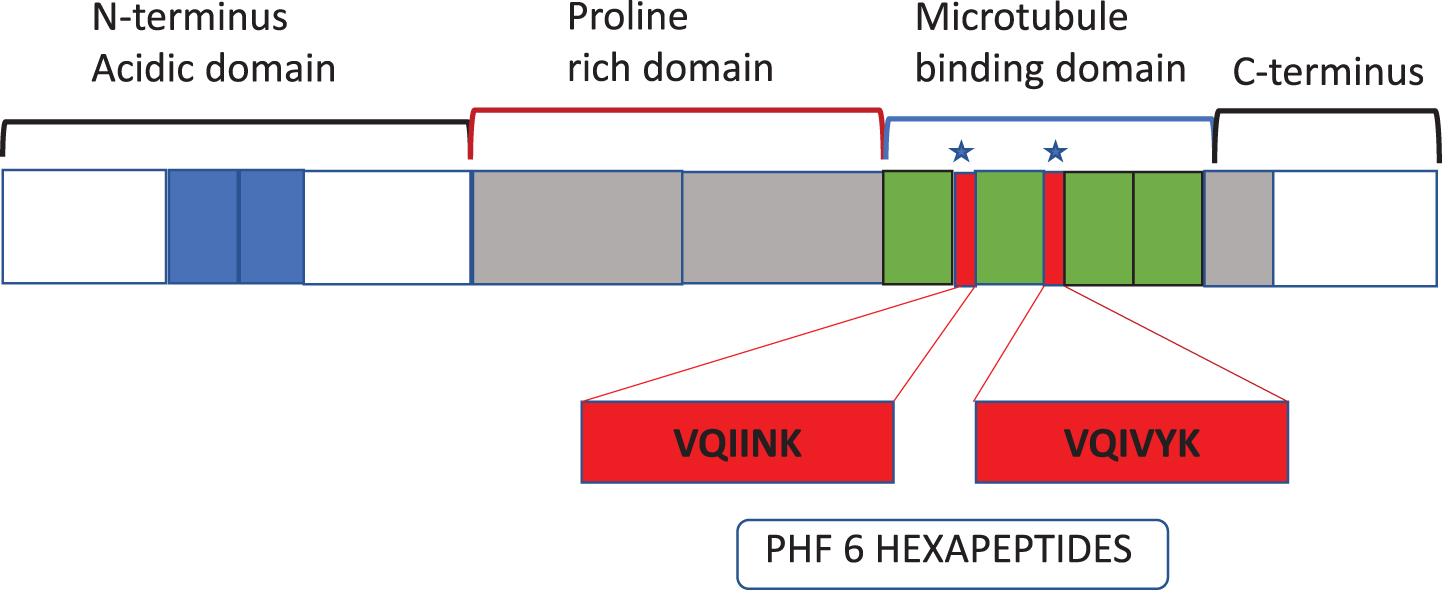
P. gingivalis protease gingipains have been demonstrated to co-localize with microtubules and PHF constituting NFTs in AD brains [12]. As mentio-ned earlier, tau protein can be hydrolyzed by gingipains into several fragments both from the N and C termini [12]. Six out of several fragments have “VQIINK” and “VQIVYK” hexapeptide common motifs that bind to the microtubule binding domains flanking regions, which support the microtubule filaments (Fig. 2, Table 1). This implies that six of the several tau fragments released by gingipains are from the pivotal sites of the functional microtubules and agrees with previously reported observations [12]. Furthermore, PHF filaments also have VQIINK and VQIVYK hexapeptide signatures (see [91]). Therefore, tau cleavage at VQIINK and VQIVYK hexapeptides from the regions of considerable functional significance to microtubules “would collapse” the intact microtubule assembly with the said hexapeptide amino acid signatures within tau and PHFs, constituting the NFT lesions [12, 91]. Further research is needed to clarify the role of gingipains fragmented tau peptides in the pathogenesis of AD.
Fig. 2
Schematic of tau protein and domain organization to help understand the position of peptides in Table 1 and the 1–4 microtubule binding domains of Tau to microtubules.
P. GINGIVALIS PHOSPHORYLATES TAU PROTEIN
Tau protein binds to microtubules and is prone to hyperphosphorylation. Hyperphosphorylation of tau protein signifies a pathological change, which precedes NFT formation. It is believed that phosphorylated tau proteins accumulate within neurons prior to NFT formation [92]. What causes this shift between bound to free pathological tau in neuronal cells is not clear, but P. gingivalis enzyme activity hydrolyzing this protein as shown by Dominy et al. [12] cannot be ruled out. Haditsch et al. [57] demonstrated an increased tau phosphorylation at two residues (enhanced tau phosphorylation at Thr231 and at Ser396) following P. gingivalis infection in an iPSC differentiated neuronal cell culture model. Furthermore, the capsular serotype K1 P. gingivalis W83 strain has been shown to have the potential to contribute to tau phosphorylation at Ser396 in the in vivo wild-type mouse model [66]. These are accepted phosphorylation sites as evaluated previously by Hanger et al. [93]. However, which kinase may be responsible for phosphorylating Ser396 and Thr231 following P. gingivalis infection is not clear, but GSK-3β is a strong candidate [93]. Recently Liu et al. [94] observed in their gingivalis-infected microglial cell model that phosphoinositide 3-kinase (Pi3K)/ protein kinase B1 (Akt) and mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase/ERK pathways were activated. Our study in which purified P. gingivalis LPS (Pg-LPS) was applied to a neuroblastoma cell line, in vitro cell model also demonstrated that the PI3K/AKT pathways were activated [95]. In addition, Bahar and Singhrao [95] showed GSK-3β and forkhead box class O1 (FOXO1) and NF-kB mRNA expression was also upregulated and that this was dependent on the MyD88 pathway [95]. The importance here is that GSK-3β is one of the enzymes that can phosphorylate tau residues Ser396 and Thr231 [93] and its activation implicates the onset of inflammatory signaling [95] that are known to be involved in AD pathophysiology [96, 97]. This implies that P. gingivalis infection plays an important role in the infected cells where balance of inflammation and inflammation responsive kinases (e.g. GSK-3β) are tipped in favor of phosphorylation, NFT lesion formation, and subsequent pathophysiology of AD.
CONCLUSIONS
This review has applied a pathobiome concept in substantiating the link between P. gingivalis infection and AD lesions. It is clear from the human and proof of concept studies in animal models that whole bacteria and their constituent endo/exotoxins enter the central nervous system. In situ, this bacterium has a range of enzymes that are shown to interact with AβPP and tau, deregulating their structure and intracellular processing, with resultant formation of Aβ and PHF, respectively. We appreciate that other bacterial, fungal, or viral pathogens implicated in AD may follow different pathways towards AD pathophysiology compared to those described here for P. gingivalis.
It is evident that P. gingivalis is a potentially significant etiological agent for AD pathophysiology. While generalized periodontitis develops around middle age, the sporadic form of AD is of much later onset. The lag phase between the two comorbidities could provide us with a mechanistic association between the two diseases and a window of opportunity to inhibit the toxic insults of gingipains in patients with periodontitis, and its downstream inflammatory effects such as systemic inflammation contributing to the risk of developing AD or increasing the severity of dementia by hyperphosphorylating tau. Hence, the clinical “GAIN Trial”: Phase 2/3 Study of COR388 in Subjects with Alzheimer’s disease (ClinicalTrials.gov Identifier: NCT03823404) is key to testing this hypothesis and advancing the management of healthcare landscape in preventing and/or slowing down AD. It is imperative that the oral health component is included as a modifiable risk factor in AD public health messages along with other preventative advice such as keeping active, eating healthily, and exercising.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
SK and SKS in 2017 and again with RW and SKS in 2018 received PreViser awards from the Oral and Dental Research Trust. In addition, SK also acknowledges having received a TC White Young Researcher award (2019).
REFERENCES
[1] | Hyman BT , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Carrillo MC , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Thies B , Trojanowski JQ , Vinters HV , Montine TJ ((2012) ) National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8: , 1–13. |
[2] | Dugger BN , Dickson DW ((2017) ) Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol 9: , a028035. |
[3] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[4] | Braak H , Thal DR , Ghebremedhin E , Del Tredici K ((2011) ) Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 70: , 960–969. |
[5] | Cook IA , Schrader LM , Degiorgio CM , Miller PR , Maremont ER , Leuchter AF ((2013) ) Trigeminal nerve stimulation in major depressive disorder: Acute outcomes in an open pilot study. Epilepsy Behav 28: , 221–226. |
[6] | Socransky SS , Haffajee AD , Cugini MA , Smith C , Kent RL Jr ((1998) ) Microbial complexes in subgingival plaque. J Clin Periodontol 25: , 134–144. |
[7] | Hajishengallis G , Darveau RP , Curtis MA ((2012) ) The keystone-pathogen hypothesis. Nat Rev Microbiol 10: , 717–725. |
[8] | Taubman MA , Kawai T ((2001) ) Involvement of T-lymphocytes in periodontal disease and in direct and indirect induction of bone resorption. Crit Rev Oral Biol Med 12: , 125–135. |
[9] | Parahitiyawa NB , Jin LJ , Leung WK , Yam WC , Samaranayake LP ((2009) ) Microbiology of odontogenic bacteremia: beyond endocarditis. Clin Microbiol Rev 22: , 46–64. |
[10] | Haraszthy VI , Zambon JJ , Trevisan M , Zeid M , Genco RJ ((2000) ) Identification of periodontal pathogens in atheromatous plaques. J Periodontol 71: , 1554–1560. |
[11] | Riviere GR , Riviere KH , Smith KS ((2002) ) Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol 17: , 113–118. |
[12] | Dominy SS , Lynch C , Ermini F , Benedyk M , Marczyk A , Konradi A , Nguyen M , Haditsch U , Raha D , Griffin C , Holsinger LJ , Arastu-Kapur S , Kaba S , Lee A , Ryder MI , Potempa B , Mydel P , Hellvard A , Adamowicz K , Hasturk H , Walker GD , Reynolds EC , Faull RLM , Curtis MA , Dragunow M , Potempa J ((2019) ) Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5: , eaau3333. |
[13] | Goto T , Kuramoto E , Dhar A , Wang RPH , Seki H , Iwai H , Yamanaka A , Matsumoto S-E , Hara H , Michikawa M , Ohyagi Y , Leung WK , Chang RC-C ((2020) ) Neurodegeneration of trigeminal mesencephalic neurons by the tooth loss triggers the progression of Alzheimer’s disease in 3×Tg-AD model mice. J Alzheimers Dis 76: , 1443–1459. |
[14] | Kondo K , Niino M , Shido K ((1994) ) A case-control study of Alzheimer’s disease in Japan-significance of life-styles. Dementia 5: , 314–326. |
[15] | Stein PS , Desrosiers M , Donegan SJ , Yepes JF , Kryscio RJ ((2007) ) Tooth loss, dementia and neuropathology in the Nun Study. J Am Dent Assoc 138: , 1314–1322. |
[16] | Chen J , Ren CJ , Wu L , Xia LY , Shao J , Leng WD , Zeng XT ((2018) ) Tooth loss is associated with increased risk of dementia and with a dose-response relationship. Front Aging Neurosci 10: , 415. |
[17] | Dioguardi M , Gioia GD , Caloro GA , Capocasale G , Zhurakivska K , Troiano G , Russo LL , Muzio LL ((2019) ) The association between tooth loss and Alzheimer’s disease: A systematic review with meta-analysis of case control studies. Dent J 7: , 49. |
[18] | Holmes C , El-Okl M , Williams AL , Cunningham C , Wilcockson D , Perry VHJ ((2003) ) Systemic infection, interleukin 1β and cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 74: , 788–789. |
[19] | Singhrao SK , Harding A ((2020) ) Is Alzheimer’s disease a polymicrobial host microbiome dysbiosis? Expert Rev Anti Infect Ther 18: , 275–277. |
[20] | Olsen I , Singhrao SK ((2020) ) Interaction between genetic factors, Porphyromonasgingivalis and microglia to promote Alzheimer’s disease. J Oral Microbiol 12: , 1820834. |
[21] | Saunders AM , Strittmatter WJ , Schmechel D , George-Hyslop PH , Pericak-Vance MA , Joo SH , Rosi BL , Gusella JF , Crapper-MacLachlan DR , Alberts MJ , et al. ((1993) ) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43: , 1467–1472. |
[22] | Wierenga CE , Clark LR , Dev SI , Shin DD , Jurick SM , Rissman RA , Liu TT , Bondi MW ((2013) ) Interaction of age and APOE genotype on cerebral blood flow at rest. J Alzheimers Dis 34: , 921–935. |
[23] | Tiraboschi P , Hansen LA , Masliah E , Alford M , Thal LJ , Corey-Bloom J ((2004) ) Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 62: , 1977–1983. |
[24] | Yip AG , McKee AC , Green RC , Wells J , Young H , Cupples LA , Farrer LA ((2005) ) APOE, vascular pathology, and the AD brain. Neurology 65: , 259–265. |
[25] | Jin YP , Østbye T , Feightner JW , Hachinski ((2008) ) Joint effect of stroke and APOE E4 on dementia risk: The Canadian Study of Health and Aging. Neurology 70: , 9–16. |
[26] | Xu W , Tan L , Wang HF , Jiang T , Tan MS , Tan L , Zhao QF , Li JQ , Wang J , Yu JT ((2015) ) Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry 86: , 1299–1306. |
[27] | Kulashekar M , Stom SM , Peuler JD ((2018) ) Resveratrol’s potential in the adjunctive management of cardiovascular disease, obesity, diabetes, Alzheimer disease, and cancer. J Am Osteopath Assoc 118: , 596–605. |
[28] | Yang X , Xu S , Qian Y , Xiao Q ((2017) ) Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav Immun 64: , 162–172. |
[29] | Li L , Cavuoto M , Biddiscombe K , Pike KE ((2020) ) Diabetes mellitus increases risk of incident dementia in APOEɛ4 carriers: A meta-analysis. J Alzheimers Dis 74: , 1295–1308. |
[30] | Montagne A , Barnes SR , Sweeney MD , Halliday MR , Sagare AP , Zhao Z , Toga AW , Jacobs RE , Liu CY , Amezcua L , Harrington MG , Chui HC , Law M , Zlokovic BV ((2015) ) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85: , 296–302. |
[31] | Goodall EF , Wang C , Simpson JE , Baker DJ , Drew DR , Heath PR , Saffrey MJ , Romero IA , Wharton SB ((2018) ) Age-associated changes in the blood-brain barrier: Comparative studies in human and mouse. Neuropathol Appl Neurobiol 44: , 328–340. |
[32] | Montagne A , Nation DA , Pa J , Sweeney MD , Toga AW , Zlokovic BV ((2016) ) Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol (Berl) 131: , 687–707. |
[33] | Halliday MR , Rege SV , Ma Q , Zhao Z , Miller CA , Winkler EA , Zlokovic BV ((2016) ) Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 36: , 216–227. |
[34] | Poole S , Singhrao SK , Chukkapalli S , Rivera M , Velsko I , Kesavalu L , Crean S ((2015) ) Active invasion of an oral bacterium and infection-induced complement activation in ApoEnull mice brains. J Alzheimers Dis 43: , 67–80. |
[35] | Singhrao SK , Chukkapalli S , Poole S , Velsko I , Crean SJ , Kesavalu L ((2017) ) Chronic Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE-/- mice brains. J Oral Microbiol 9: , 1270602. |
[36] | Rokad F , Moseley R , Hardy RS , Chukkapalli S , Crean S , Kesavalu L , Singhrao SK ((2017) ) Cerebral oxidative stress and microvasculature defects in TNF-α expressing transgenic and Porphyromonas gingivalis-infected ApoE-/- mice. J Alzheimers Dis 60: , 359–369. |
[37] | Sheets SM , Potempa J , Travis J , Casiano CA , Fletcher HM ((2005) ) Gingipains from Porphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in endothelial cells. Infect Immun 73: , 1543–1552. |
[38] | Lv S , Song HL , Zhou Y , Li LX , Cui W , Wang W , Liu P ((2010) ) Tumour necrosis factor-alpha affects blood-brain barrier permeability and tight junction-associated occludin in acute liver failure. Liver Int 30: , 1198–1210. |
[39] | Vernal R , León R , Silva A , van Winkelhoff AJ , García-Sanz J , Sanz M ((2009) ) Differential cytokine expression by human dendritic cells in response to different porphyromonas gingivalis capsular serotypes. J Clin Periodontol 36: , 823–829. |
[40] | Olsen I , Singhrao SK ((2018) ) Importance of heterogeneity in Porhyromonas gingivalis lipopolysaccharide lipid A in tissue specific inflammatory signalling. J Oral Microbiol 10: , 1440128. |
[41] | Curtis MA , Thickett A , Slaney JM , Rangarajan M , Aduse-Opoku J , Shepherd P , Paramonov N , Hounsell EF ((1999) ) Variable carbohydrate modifications to the catalytic chains of the RgpA and RgpB proteases of Porphyromonas gingivalis W50. Infect Immun 67: , 3816–3823. |
[42] | Zenobia C , Hasturk H , Nguyen D , Van Dyke TE , Kantarci A , Darveau RP ((2014) ) Porphyromonas gingivalis lipid A phosphatase activity is critical for colonization and increasing the commensal load in the rabbit ligature model. Infect Immun 82: , 650–659. |
[43] | Farhad SZ , Amini S , Khalilian A , Barekatain M , Mafi M , Barekatain M , Rafei E ((2014) ) The effect of chronic periodontitis on serum levels of tumor necrosis factor-alpha in Alzheimer disease. Dent Res J (Isfahan) 11: , 549–552. |
[44] | Laine ML , Appelmelk BJ , van Winkelhoff AJ ((1997) ) Prevalence and distribution of six capsular serotypes of Porphyromonas gingivalis in periodontitis patients. J Dent Res 76: , 1840–1844. |
[45] | Van Winkelhoff AJ , Laine ML , Timmerman MF , Van der Weijden GA , Abbas F , Winkel EG , Arief EM , Van der Velden U ((1999) ) Prevalence and serotyping of Porphyromonas gingivalis in an Indonesian population. J Clin Periodontol 26: , 301–305. |
[46] | Smalley JW , Birss AJ , Kay HM , McKee AS , Marsh PD ((1989) ) The distribution of trypsin-like enzyme activity in cultures of a virulent and an avirulent strains of Bacteroides gingivalis W50. Oral Microbiol Immunol 4: , 178–181. |
[47] | Potempa J , Pike R , Travis J ((1995) ) The multiple forms of trypsin-like activity present in various strains of Porphy-romonas gingivalis are due to the presence of either Arg-gingipain or Lys-gingipain. Infect Immun 63: , 1176–1182. |
[48] | Imamura T ((2003) ) The role of gingipains in the pathogenesis of periodontal disease. J Periodontol 74: , 111–118. |
[49] | Fujimura S , Nakamura T ((1987) ) Isolation and characterization of a protease from Bacteroides gingivalis. Infect Immun 55: , 716–720. |
[50] | Ono M , Okuda K , Takazoe I ((1987) ) Purification and characterization of a thiol-protease from Bacteroides gingivalis strain 381. Oral Microbiol Immunol 2: , 77–81. |
[51] | Otsuka M , Endo J , Hinode D , Nagata A , Maehara R , Sato M , Nakamura R ((1987) ) Isolation and characterization of protease from culture supernatant of Bacteroides gingivalis. J Periodont Res 22: , 491–498. |
[52] | Tsutsui H , Kinouchi T , Wakano Y , Ohnishi Y ((1987) ) Purification and characterization of a protease from Bacteroides gingivalis 381. Infect Immun 55: , 420–427. |
[53] | Fontela YC , Kadavath H , Biernat J , Riedel D , Mandelkow E , Zweckstetter M ((2017) ) Multivalent cross-linking of actin filaments and microtubules through the microtubule-associated protein Tau. Nat Commun 8: , 1981. |
[54] | Abe N , Kadowaki T , Okamoto K , Nakayama K , Ohishi M , Yamamoto K ((1998) ) Biochemical and functional properties of lysine-specific cysteine proteinase (Lys-gingipain) as a virulence factor of Porphyromonas gingivalis in periodontal disease. J Biochem 123: , 305–312. |
[55] | Witman GB , Cleveland DW , Weingarten MD , Kirschner MW ((1976) ) Tubulin requires tau for growth onto microtubule initiating sites. Proc Nat Acad Sci U S A 73: , 4070–4074. |
[56] | Ryder MI ((2020) ) Porphyromonas gingivalis and Alzheimer disease: Recent findings and potential therapies. J Periodontol 91: (Suppl 1), S45–S49. |
[57] | Haditsch U , Roth T , Rodriguez L , Hancock S , Cecere T , Nguyen M , Arastu-Kapur S , Broce S , Raha D , Lynch CC , Holsinger LJ , Dominy SS , Ermini F ((2020) ) Alzheimer’s disease-like neurodegeneration in Porphyromonas gingivalis infected neurons with persistent expression of active gingipains. J Alzheimers Dis 75: , 1361–1376. |
[58] | Rolim TS , Fabri GM , Nitrini R , Anghinah R , Teixeira MJ , Siqueira JT , Cesari JA , Siqueira SR ((2014) ) Evaluation of patients with Alzheimer’s disease before and after dental treatment. Arq Neuropsiquiatr 72: , 919–924. |
[59] | Ding Y , Ren J , Yu H , Yu W , Zhou Y ((2018) ) Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun Ageing 15: , 6. |
[60] | Wu Z , Ni J , Liu Y , Teeling JL , Takayama F , Collcutt A , Ibbett P , Nakanishi H ((2017) ) Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun 65: , 350–361. |
[61] | Zhang J , Yu C , Zhang X , Chen H , Dong J , Lu W , Song Z , Zhou W ((2018) ) Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signalling pathway in C57BL/6 mice. J Neuroinflammation 15: , 37. |
[62] | Ide M , Harris M , Stevens A , Sussams R , Hopkins V , Culliford D , Fuller J , Ibbett P , Raybould R , Thomas R , Puenter U , Teeling J , Perry VH , Holmes C ((2016) ) Periodontitis and cognitive decline in Alzheimer’s disease. PLoS One 11: , e0151081. |
[63] | Singhrao SK , Olsen I ((2019) ) Assessing the role of Porphyromonas gingivalis in periodontitis to determine a causative relationship with Alzheimer’s disease. J Oral Microbiol 11: , 1563405. |
[64] | Olsen I , Lambris JD , Hajishengallis G ((2017) ) Porphyromonas gingivalis disturbs host-commensal homeostasis by changing complement function. J Oral Microbiol 9: , 1340085. |
[65] | Poole S , Singhrao SK , Kesavalu L , Curtis MA , Crean S ((2013) ) Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis 36: , 665–677. |
[66] | Ilievski V , Zuchowska PK , Green SJ , Toth PT , Ragozzino ME , Le K , Aljewari HW , O’Brien-Simpson NM , Reynolds EC , Watanabe K ((2018) ) Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One 13: , e0204941. |
[67] | Olsen I , Singhrao SK ((2019) ) Is there a link between genetic defects in the complement cascade and Porphyromonas gingivalis in Alzheimer’s disease? J Oral Microbiol 12: , 1676486. |
[68] | Adamowicz K , Wang H , Jotwani R , Zeller I , Potempa J , Scott DA ((2012) ) Inhibition of GSK3 abolishes bacterial-induced periodontal bone loss in mice. Mol Med 18: , 1190–1196. |
[69] | Braak H , Del Tredici K ((2004) ) Alzheimer’s disease: Int-raneuronal alterations precede insoluble amyloid-beta formation. Neurobiol Aging 25: , 713–718; discussion 743-746. |
[70] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[71] | Choi RW , Cheng Z , Schekman R ((2012) ) Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc Natl Acad Sci U S A 109: , E2077–2082. |
[72] | Cataldo AM , Barnett JL , Pieroni C , Nixon RA ((1997) ) Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: Neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J Neurosci 17: , 6142–6151. |
[73] | Hook V , Schechter I , Demuth HU , Hook G ((2008) ) Alternative pathways for production of beta-amyloid peptides of Alzheimer’s disease. J Biol Chem 389: , 993–1006. |
[74] | Nie R , Wu Z , Ni J , Zeng F , Yu W , Zhang Y , Kadowaki T , Kashiwazaki H , Teeling JL , Zhou Y ((2019) ) Porphyromonas gingivalis infection induces amyloid-β accumulation in monocytes/macrophages. J Alzheimers Dis 72: , 479–494. |
[75] | Gil-Montoya JA , Barrios R , Santana S , Sanchez-Lara I , Pardo CC , Fornieles-Rubio F , Montes J , Ramirez C , Gonzalez-Moles AM , Burgos JS ((2017) ) Association between periodontitis and amyloid β peptide in elderly people with and without cognitive impairment. J Periodontol 88: , 1051–1058. |
[76] | Leira Y , Iglesias-Rey R , Gómez-Lado N , Aguiar P , Campos F D’Aiuto F , Castillo J , Blanco J , Sobrino T ((2019) ) Porphyromonas gingivalis lipopolysaccharide-induced periodontitis and serum amyloid-beta peptides. Arch Oral Biol 99: , 120–125. |
[77] | Zeng F , Liu Y , Huang W , Qing H , Kadowaki T , Kashiwazaki H , Ni J , Wu Z ((2020) ) Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid β accumulation after Porphyromonas gingivalis infection, J Neurochem, doi: 10.1111/jnc.15096. |
[78] | Chen C-K , Wu Y-T , Chang Y-C ((2017) ) Association between chronic periodontitis and the risk of Alzheimer’s disease: A retrospective, population-based, matched-cohort study. Alzheimers Res Ther 9: , 56. |
[79] | Lin JW , Chang CH , Caffrey JL ((2020) ) Feature article: Examining the association between oral health status and dementia: A nationwide nested case-controlled study. Exp Biol Med (Maywood) 245: , 231–244. |
[80] | Sparks Stein P , Steffen MJ , Smith C , Jicha G , Ebersole JL , Abner E , Dawson D 3rd ((2012) ) Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimers Dement 8: , 196–203. |
[81] | Grundke-Iqbal I , Iqbal K , Quinlan M , Tung YC , Zaidi MS , Wisniewski HM ((1986) ) Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261: , 6084–6089. |
[82] | Goedert M , Klug A , Crowther RA ((2006) ) Tau protein, the paired helical filament and Alzheimer’s disease. J Alzheimers Dis 9: , 195–207. |
[83] | Emery DC , Shoemark DK , Batstone TE , Waterfall CM , Coghill JA , Cerajewska TL , Davies M , West NX , Allen SJ ((2017) ) 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer’s post-mortem brain. Front Aging Neurosci 9: , 195. |
[84] | Siddiqui H , Eribe ERK , Singhrao SK , Olsen I ((2019) ) High throughput sequencing detects gingivitis and periodontal oral bacteria in Alzheimer’s disease autopsy brains. Neuro Res 1: , 3. |
[85] | Balin BJ , Gerard HC , Arking EJ , Appelt DM , Branigan PJ , Abrams JT , Whittum-Hudson JA , Hudson AP ((1998) ) Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med Microbiol Immunol 187: , 23–42. |
[86] | Itzhaki RF , Wozniak MA ((2006) ) Herpes simplex virus type 1, apolipoprotein E and cholesterol: A dangerous liaison in Alzheimer’s disease and other disorders. Prog Lipid Res 45: , 73–90. |
[87] | Miklossy J ((2011) ) Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation 8: , 90. |
[88] | Carrasco L , Alonso R , Pisa D , Rabano A ((2017) ) Alzheimer’s disease and fungal infection. In Handbook of Infection and Alzheimer’s Disease, Miklossy J, ed. IOS Press, Amsterdam, pp. 281-294. |
[89] | Slots J , Bragd L , Wikström M , Dahlén G ((1986) ) The occurrence of Actinobacillus actinomycetemcomitans, Bacteroides gingivalis and Bacteroides intermedius in destructive periodontal disease in adults. J Clin Periodontol 13: , 570–577. |
[90] | Singh PP , Sridharan KB , Bhagi RP , Singla R ((1989) ) Anaerobic infection of the lung and pleural space in tuberculosis. Indian J Chest Dis Allied Sci 31: , 85–89. |
[91] | Barbier P , Zejneli O , Martinho M , Lasorsa A , Belle V , Smet-Nocca C , Tsvetkov PO , Devred F , Landrieu I ((2019) ) Role of tau as a microtubule-associated protein: Structural and functional aspects. Front Aging Neurosci 11: , 204. |
[92] | Brion JP ((1998) ) Neurofibrillary tangles and Alzheimer’s disease. Eur Neurol 40: , 130–140. |
[93] | Hanger DP , Byers HL , Wray S , Leung K-Y , Saxton MJ , Seereeram A , Reynolds CH , Ward MA , Anderton BH ((2007) ) Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem 282: , 23645–23654. |
[94] | Liu Y , Wu Z , Nakanishi Y , Ni J , Hayashi Y , Takayama F , Zhou Y , Kadowak T , Nakanishi H ((2018) ) Author Correction: Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci Rep 8: , 10304. |
[95] | Bahar B , Singhrao SK ((2020) ) An evaluation of the molecular mode of action of trans-resveratrol-resveratrol in the Porphyromonasgingivalis lipopolysaccharide challenged neuronal cell model. Mol Biol Rep, https://doi.org/10.1007/s11033-020-06024-y |
[96] | Liu Y , Liu F , Grundke-Iqbal I , Iqbal K , Gong C-X ((2011) ) Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol 225: , 54–62. |
[97] | Maiese K ((2016) ) Forkhead transcription factors: New considerations for Alzheimer’s disease and dementia. J Transl Sci 2: , 241–247. |


