
Sleep and Tau Pathology in Vietnam War Veterans with Preclinical and Prodromal Alzheimer’s Disease
Abstract
Background:
The increasing prevalence of Alzheimer’s disease (AD) and lack of effective medications has led to a need to identify modifiable risk factors as targets for interventions.
Objective:
In this cross-sectional study, we sought to determine whether worse sleep quality is associated with increased pathological tau, and whether this relationship is affected by amyloid pathology.
Methods:
66 male participants underwent Florbetapir (AV45) positron emission tomography (PET) and Flortaucipir (FTP) PET and completed the Pittsburgh Sleep Quality Index questionnaire (PSQI) as part of the Department of Defense Alzheimer’s Disease Neuroimaging Initiative, a multicenter study collecting data from Vietnam War veterans, some of whom have a history of post-traumatic stress disorder, or non-penetrating traumatic brain injury. AV45 PET was used to determine the presence of significant amyloid pathology. We used regression models to determine the effects of amyloid pathology and PSQI on tau deposition in brain regions associated with Braak stages.
Results:
Among the 66 participants, 14 individuals were amyloid positive (21%) and 52 were amyloid negative (79%). In regions associated with Braak stages III-IV, there was a significant interaction of amyloid status on PSQI (β= 0.04, p = 0.003) with higher PSQI correlating with higher FTP SUVr in amyloid-positive individuals only (β= 0.031, p = 0.005).
Conclusion:
Our study found that an AD profile of tau deposition was associated with an interaction between self-reported sleep quality and amyloid pathology such that worse self-reported sleep was related to higher tau in regions usually associated with AD progression, but only in individuals with high cerebral amyloid deposition.
INTRODUCTION
Alzheimer’s disease (AD) is a growing public health crisis due to an aging population and a lack of disease modifying medications. Importantly, it is now possible to identify a preclinical phase of AD, using biomarker identification of elevated amyloid-β (Aβ) deposits and to track the presence and location of pathological tau using positron emission tomography (tau PET). Potentially modifiable risk factors, including disordered sleep, are being increasingly sought as important targets to delay symptom onset and possible clinically detectable memory decline [1].
Poor sleep appears to be both a cause of, and a consequence of, AD [2]. Studies have displayed high prevalence of sleep disorders in people with AD [3–10]. Wide-ranging alterations in sleep physiology have been documented in patients with AD [11–13], and sleep disturbances are associated with the hallmark proteins of AD, Aβ and tau, both in rodents and in humans. These studies have linked subjective and objective measures of sleep quality to Aβ and tau in clinical populations of AD patients using cerebrospinal fluid (CSF) and PET. However, these studies have focused on cognitively impaired and/or demented populations, leaving questions about the relevance of this relationship in the earlier and preclinical stages of AD.
While pathological levels of amyloid are distributed widely in the brain a decade or more prior to symptom onset, pathological tau spreads in a topographically specific manner, with direct links between the regions involved and the cognitive function impaired. Pathological tau begins to accumulate prior to the onset of major cognitive impairment, and thus tau can be detectable in the preclinical stages, as well as in temporal lobe structures during normal aging [14]. Animal studies support the notion that sleep deprivation is related to increases in pathological tau [15] in models without amyloid manipulation, implying that this relationship between sleep and tau deposition may exist independent of amyloid. However, mice bred to be tau-deficient also demonstrate unusual sleep patterns [16], suggesting that the relationship between tau and sleep may be bidirectional. In a human cohort with no or mild cognitive impairment (MCI), about half of whom were amyloid negative (defined by an absence of cerebral amyloid deposition above a threshold indicative of AD dementia [17]), slow wave sleep activity (during non–rapid eye movement sleep) was associated with CSF tau elevations but not amyloid levels [18]. Furthermore, other human studies have reported increased CSF tau and amyloid levels as a result of acute sleep disruption [19, 20]. Thus, for now, it is unclear how sleep relates to the amyloid and tau and their combination.
In this retrospective, cross-sectional, observational study, we used reported sleep data as well as PET data on both tau and Aβ imaging collected in the Department of Defense Alzheimer’s Disease Neuroimaging Initiative (DoD-ADNI). While our study was mainly focused on the role of sleep quality in the AD continuum, we secondarily assessed potential differences in tau PET between individuals traumatic brain injury (TBI) and those with post-traumatic stress disorder (PTSD), since PTSD is often characterized by disrupted sleep [21] and TBI has relationships with both sleep [22] and tau [23]. We hypothesized that elevated Pittsburgh Sleep Quality Index (PSQI), indicative of decreased sleep quality, would be associated with higher tau pathology, regardless of PTSD and TBI diagnoses. We further sought to establish if disrupted sleep was dependent on the presence of pathological levels of amyloid in this population.
MATERIALS AND METHODS
Study participants
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). For up-to-date information, see http://www.adni-info.org. The DoD-ADNI is a multisite study of the effects of mild to moderate non-penetrating TBI and PTSD on AD in a cohort of Vietnam war veterans with and without MCI. We examined the relationship between the PSQI, a self-report measure of sleep quality, and flortaucipir (AV1451, or FTP) tau PET in amyloid positive (Aβ+) and amyloid negative (Aβ-) veterans. The DoD-ADNI recruits participants with normal or mildly impaired cognition, some of whom had diagnoses of mild to moderate non-penetrating TBI, PTSD, or both.
Individuals who had undergone FTP tau PET, florbetapir (AV45) Aβ PET and completed the PSQI were included in these analyses. Amyloid positivity was determined based on the standard threshold of 1.11 cortical summary Standard Uptake Value ratio (SUVr) in each participant’s AV45 scan available via processed data available on the ADNI database (http://adni.loni.usc.edu). From this, we identified all Aβ+ individuals that had FTP PET data available (N = 16). One of these individuals was excluded due to abnormally high FTP SUVr (3.175 standard deviations above the mean), and one was excluded due to missing PSQI data. Of the Aβ- individuals with FTP PET data (n = 60), seven were excluded due to missing or incomplete PSQI data. In the remaining sample there was one woman who was excluded to eliminate potential sex effects [24].
Of the resulting dataset, 7 participants had an MCI diagnosis at the time of their FTP PET scan, 2 of whom were Aβ+ (i.e., prodromal) and 5 of whom were Aβ-. ADNI criteria for MCI were: 1) Memory complaint by subject or study partner that is verified by a study partner; 2) Abnormal memory function documented by scoring below the education adjusted cutoff on the Logical Memory II subscale (Delayed Paragraph Recall) from the Wechsler Memory Scale –Revised (score ≤8 for those with ≥16 years of education; score ≤4 for those with 8–15 years of education; score ≤2 for those with 0–7 years of education); 3) Mini-Mental State Exam score between 24 and 30 (inclusive) (Exceptions may be made for subjects with less than 8 years of education at the discretion of the project director); 4) global CDR score of 0.5, with Memory Box score at least 0.5; and 5) general cognitive and functional performance sufficiently preserved such that a diagnosis of AD could not be made by the site physician at the time of screening. The remaining participants were cognitively unimpaired (CU) and thus represented either preclinical AD if Aβ+ (n = 12) or were considered to be non-AD (n = 47). The Aβ+ group had a mean (SD) Montreal Cognitive Assessment (MoCA) of 24.79 (2.33) while the Aβ- group had a mean (SD) MoCA of 24.04 (3.19). Participant data is summarized in Table 1.
Table 1
Average Age, Years of Education (YOE), Pittsburg Sleep Quality Index (PSQI), and Montreal Cognitive Assessment (MoCA)s by Amyloid Status and Diagnosis
| Characteristic | Aβ+(CU = 12, MCI = 2) | Aβ- (CU = 47, MCI = 5) | p |
| Age (SD) | 72.39 (6.95) | 70.64 (3.80) | 0.21 |
| YOE (SD) | 15.79 (2.67) | 15.09 (2.44) | 0.36 |
| PSQI (SD) | 8.07 (2.56) | 8.25 (3.13) | 0.85 |
| MoCA (SD) | 24.79 (2.33) | 24.04 (3.19) | 0.41 |
| SUVr in Regions Associated with Braak Stages I-II (SD) | 1.28 (0.17) | 1.17 (0.14) | 0.02 |
| SUVr in Regions Associated with Braak Stages III-IV (SD) | 1.39 (0.13) | 1.33 (0.11) | 0.07 |
| SUVr in Regions Associated with Braak Stages V-VI (SD) | 1.45 (0.13) | 1.42 (0.11) | 0.41 |
| Clinical Diagnosis (%) | ANOVA n.s. | ||
| Neither TBI nor PTSD | 6 | 14 | |
| TBI | 2 | 5 | |
| PTSD | 3 | 23 | |
| PTSD &TBI | 3 | 10 |
In the subgroup of participants with a PTSD diagnosis (n = 39), 14 were taking medication for depression, 3 were taking medication for PTSD, and 1 was taking an antipsychotic medication.
All individuals in the DoD-ADNI are veterans. Because of this, for the purposes of this study, there was no possibility for a non-veteran control group.
Imaging data
We downloaded regional summary AV45 SUVr data and partial volume corrected regional summary FTP SUVr data from the LONI database. Methods for the calculation and partial volume correction of these data have been described elsewhere [25, 26]. Given the relatively small sample size, we limited our analyses to regions relevant to each of the Braak stages [14] where tau is expected in each pathological stage of AD. Braak SUVr data from ADNI are grouped into three pairs (I-II, III-IV, and V-VI), so relationships are reported here as such. In the context of this paper, Braak regions refer not to the Braak stage that each participant is in, but rather refers to regions where we would expect to see tau in relation to AD progression. Briefly, the regions covered in stages I-II are entorhinal and hippocampus, III-IV extend to the amygdala and other subcortical regions as well as the cortex of the inferior temporal lobe and parahippocampal gyrus, V-VI extends isocortically beyond the temporal lobe into the frontal, parietal and occipital cortex. For a full listing of regions see the FTP processing methods available on the ADNI database [27].
PSQI
The PSQI is a self-report questionnaire that determines sleep quality by evaluating seven components of sleep: subjective sleep quality, sleep latency, sleep duration, habitual sleep efficiency, sleep disturbance, use of sleeping medications, and daytime dysfunction [28]. We downloaded PSQI data from the ADNI database and calculated a total score for each participant from 0 to 21, with scores higher than 5 suggesting worse sleep quality.
Statistical analysis
We used t-tests to determine if there were differences in age, education, overall PSQI or MoCA, or tau PET uptake within each Braak-combined-region between the Aβ+ and Aβ- groups, and chi square to analyze for differences of frequency in the exposure (PTSD, TBI, PTSD&TBI) categories between the Aβ+ and Aβ- groups. We then conducted three regression models, one for each of the Braak-combined-regions assessing main effects of amyloid status and PSQI on FTP SUVr. In each model, we included the interaction term of interest, amyloid status x PSQI. Each model covaried for age. We corrected for multiple comparisons using the Bonferroni method. In models where the interaction term was significant after correction, we then conducted a further pair of regression models for each of the amyloid-status groups, to understand the interaction. We reran our analyses excluding MCI individuals in order to ensure that our results were not driven by this small group. Finally, given the mixture of exposure to TBI and PTSD in this cohort, we used ANOVA to determine whether FTP SUVr in each of the Braak-combined regions, as well as PSQI, differed between veterans with each kind of exposure. We used pairwise comparisons to investigate significant differences between groups where necessary.
In order to investigate the specific sleep components driving the relationships represented in our study, we repeated our analyses, using the seven components of the PSQI to further understand the relationship between sleep and tau.
Ethics approval and consent
Written informed consent was provided by all study participants in accordance with the Institutional Review Boards of the respective ADNI sites.
RESULTS
The Aβ+ group had significantly higher FTP SUVr in regions associated with Braak stages I-II, but not in either of the other Braak-combined regions (Table 1). In the regression models (Table 2), there were no significant main effects of amyloid status or PSQI on FTP SUVr in any of the three Braak-combined regions.
Table 2
Results of Regression Models in the Whole Sample. Bolded p-values indicate significance at corrected α of 0.017
| Whole Sample | |||
| β | t | p | |
| FTP SUVr in Regions Associated with Braak Stages I &II | |||
| Amyloid | –0.1225 | –0.852 | 0.398 |
| PSQI | –0.0006 | –0.097 | 0.923 |
| Amyloid*PSQI | 0.0262 | 1.554 | 0.125 |
| FTP SUVr in Regions Associated with Braak Stages III &IV | |||
| Amyloid | –0.2627 | –2.376 | 0.021 |
| PSQI | –0.0076 | –1.556 | 0.125 |
| Amyloid*PSQI | 0.0400 | 3.084 | 0.003 |
| FTP SUVr in Regions Associated with Braak Stages V &VI | |||
| Amyloid | –0.1623 | –1.515 | 0.135 |
| PSQI | –0.0046 | –0.960 | 0.341 |
| Amyloid*PSQI | 0.0246 | 1.954 | 0.055 |
However, there was a significant interaction of amyloid status on PSQI in regions associated with Braak stage III-IV such that high PSQI was associated with elevated FTP SUVR in Aβ+ participants only (Fig. 1). No such interactions were evident in the other Braak-combined regions. These results held when we excluded MCI individuals.
Fig. 1
Relationship between PSQI and FTP SUVr in Regions Associated with Braak Stages III & IV in Aβ+ and Aβ- groups.
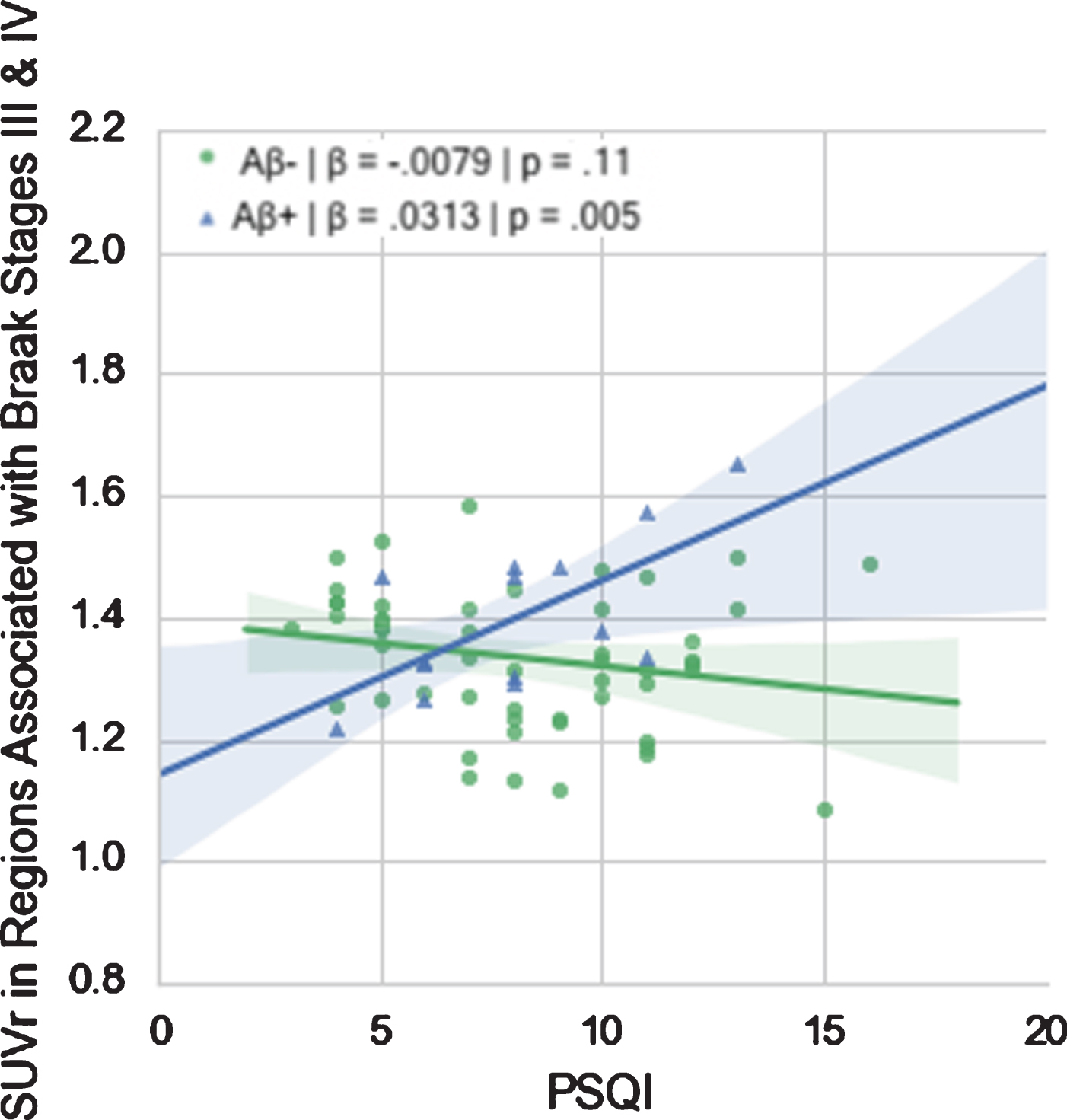
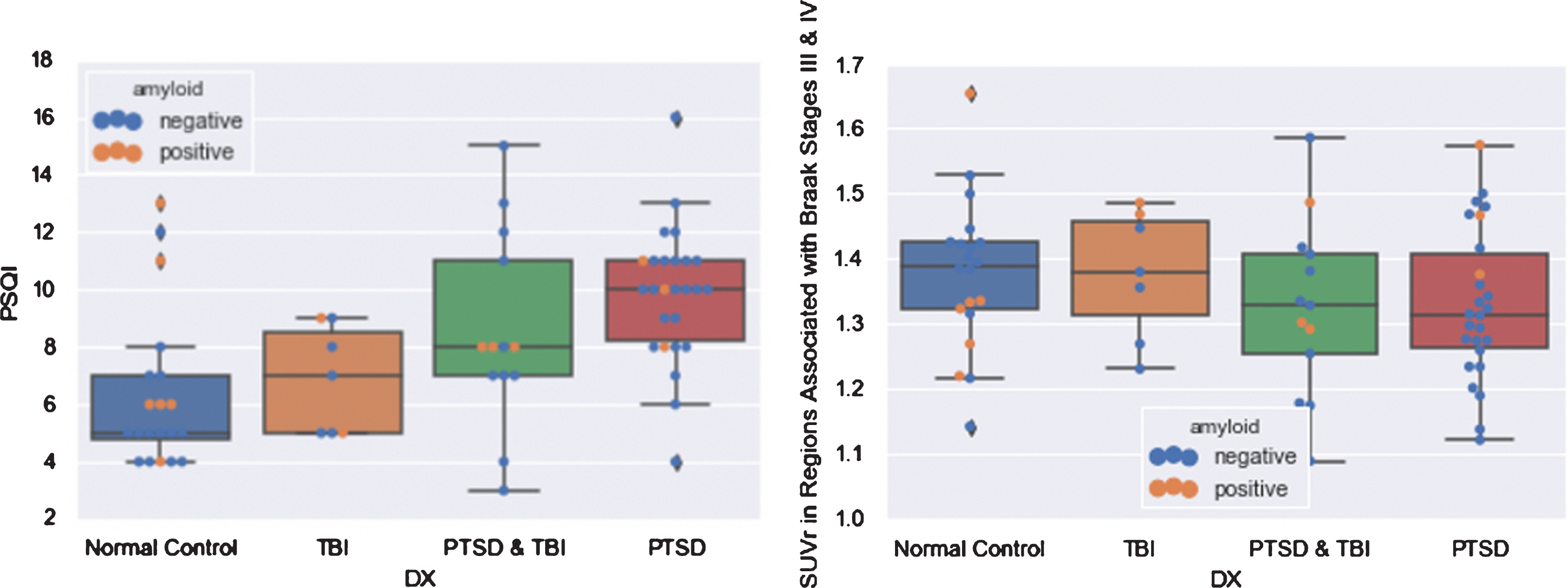
The different diagnoses (PTSD, TBI, both, or neither) did not differ significantly in FTP SUVr in any of the regions investigated, but, as expected, did differ significantly in average PSQI (F = 7.402, p = 0.0003), with higher average PSQI in the PTSD group compared with the normal group (p = 0.001), and the TBI group (p < 0.05), but not the PTSD & TBI group (Fig. 2).
Fig. 2
Boxplots of PSQI and FTP SUVr in Regions Associated with Braak Stages III & IV by Diagnosis.
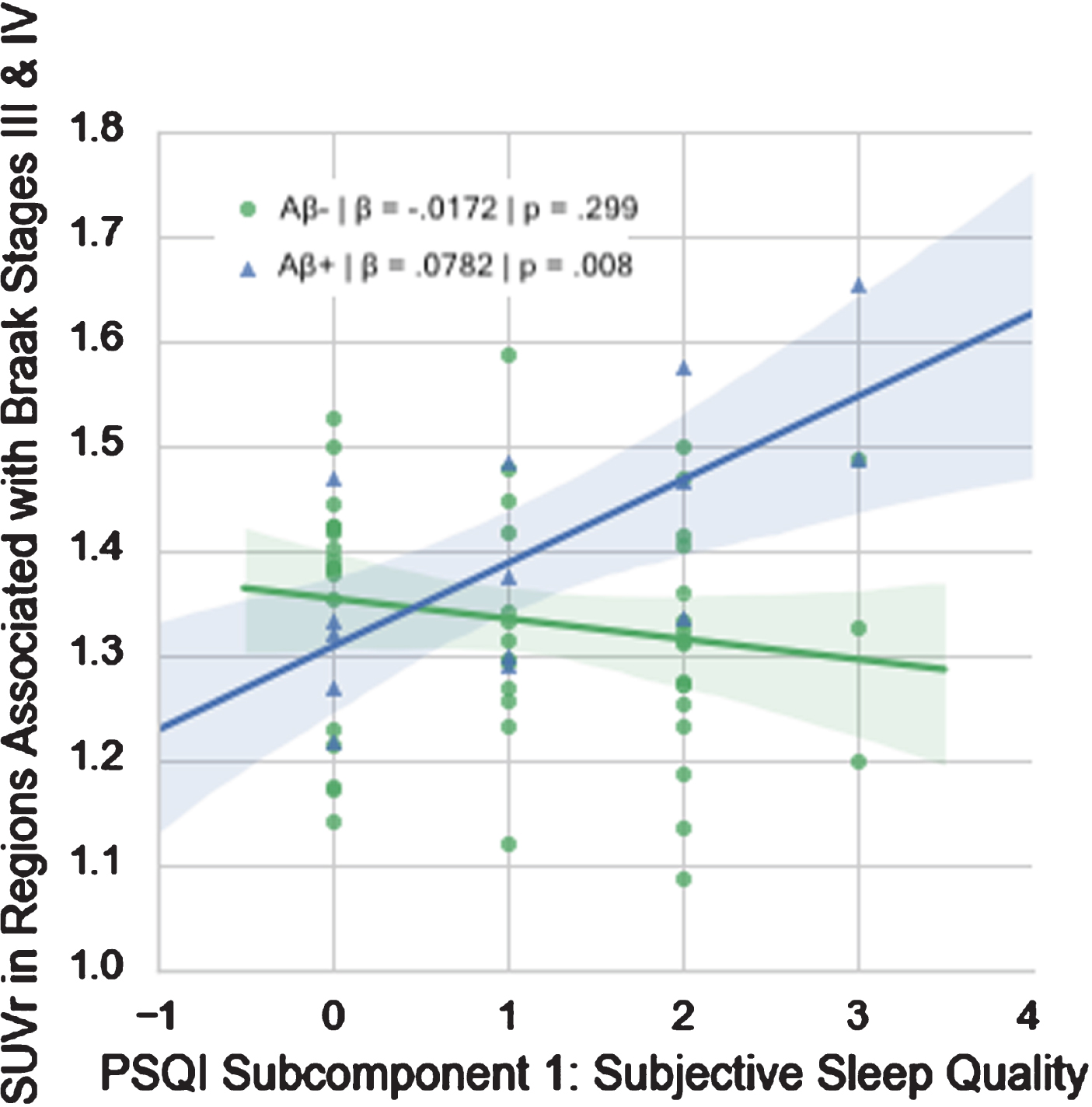
Of the PSQI components, only subjective sleep quality (β= 0.0988, p = 0.003) and sleep efficiency (β= 0.0902, p = 0.013) demonstrated significant interactions with amyloid designation at α= 0.05. Only subjective sleep quality survived multiple comparisons correction (Table 3, Fig. 3).
Table 3
Interactive effect of each PSQI subcomponent with amyloid status on tau SUVr in Regions Associated with Braak Stages III & IV. Bolded p-values indicate significance at corrected α of 0.007. While Habitual Sleep Efficiency was significant in the model, this did not survive correction
| Interaction Term | ||
| β | p | |
| Subjective Sleep Quality | 0.0988 | 0.003 |
| Sleep Latency | 0.0466 | 0.179 |
| Sleep Duration | 0.0591 | 0.077 |
| Habitual Sleep Efficiency | 0.0902 | 0.013 |
| Sleep Disturbances | 0.0250 | 0.670 |
| Use of Sleeping Medication | –0.0667 | 0.126 |
| Daytime Dysfunction | –0.0108 | 0.821 |
Fig. 3
Relationship between PSQI Subcomponent 1 (Subjective Sleep Quality) and FTP SUVr in Regions Associated with Braak Stages III & IV in Aβ+ and Aβ- groups.
DISCUSSION
This study investigated the relationship between tau and sleep quality in cognitively intact Vietnam War veterans. We found that worse self-reported sleep quality was associated with higher levels of tau in regions associated with Braak stages III & IV in Aβ+ individuals only. Thus, in these men who have preclinical or prodromal AD, worse subjective sleep quality was associated with more tau. Our post-hoc analyses revealed that, of the PSQI components, Sleep Quality appeared to be driving the overall effect, such that worse subjective sleep quality was associated with higher levels of tau in regions associated with Braak Stages III & IV in Aβ+ individuals only (Fig. 3). While total PSQI and regional FTP SUVr did not differ significantly between the Aβ+ and Aβ- subgroups, no relationship between self-reported sleep and tau was evident in these regions in Aβ- individuals. Given the small group of MCI individuals in our largely preclinical dataset, we reran our regressions excluding these individuals. We found that our results held, suggesting that our results were not driven by the small prodromal subgroup.
Preclinical AD can be defined by presence of pathological levels of amyloid in the absence of significant cognitive or functional loss [29]. Using a more recently developed tau tracer, MK6240, which is known to have less off-target age related binding, Betthauser and colleagues demonstrated that elevated tau PET uptake in combination with elevated amyloid PET uptake was associated with more rapid later cognitive decline than is typical of aging [30]. Thus, we might expect the veterans in this study with the most tau to be more likely to show cognitive decline in their return DoD-ADNI visits.
We limited our regions of interest to those typical of the progression of tau described by Braak and Braak [14]. In this clinic-pathological correlation paper, all samples who were elevated at stages I and II only did not have clinically defined dementia, half of the 10 patients with each of stage III and IV tau had dementia, thus stages III and IV seem to represent a transitional zone between symptoms being evident or not, with stages V and VI representing regions where tau is found on autopsy in those with moderate to severe dementia. We believe that our findings are limited to regions associated with Braak stages III and IV because participants in our sample are relatively early in the progression of the disease, when tau is more limited to these regions and cognition is minimally impacted. The same relationship was not found in regions corresponding to Braak stages I and II, likely due to the fact that patients are too early in the progression of the disease to demonstrate specific AD-related pathology. Tau PET signal related to age and not amyloid status has been reported, but the spatial patterns related to AD are seen much more commonly in Aβ+ participants [31]. Thus, we would expect those in our Aβ+ group with elevations in tau to decline, and we will analyze these longitudinal data once they become available.
Longitudinal and intervention studies are needed to identify whether tau leads to poor sleep, the reverse is true, or if an alternative or bidirectional model exists. Our study was not designed to unravel the causality or directionality in this relationship, but does suggest the possibility of sleep as a modifiable risk factor for AD. Thus, further study of sleep quality in the context of AD, particularly in these preclinical and prodromal periods, is of great importance. The elevation of PSQI in the PTSD cohort points to a potential comorbidity, although we did not see elevated tau in the PTSD group as a whole (Fig. 2). To an extent, this finding is contrary to a Mohamed et al. [32], which reported increased tau deposition in the brain of nondemented war veterans with TBI and/or PTSD decades after their trauma. However, Mohamed et al. focused mainly on topographical differences in tau and did not focus on the possible impact of sleep (PSQI) on tau or amyloid deposition. While overall patterns of tau deposition may differ between diagnostic groups, as reported by Mohamed, in our study, specific deposition in regions associated with AD progression is correlated to an interaction between PSQI and amyloid status, rather than PTSD/TBI diagnosis.
There are several limitations to our study. Primarily, this is a cross-sectional study that, at this time point, precludes a delayed verification of AD diagnosis for cases currently classified as preclinical or prodromal AD. The PSQI is a self-report measure of sleep quality, although a well validated indicator. As a result, our study could not ascertain whether any distinct type of sleep disturbance is driving the relationship with tau. Future research is needed to determine if sleep apnea, insomnia, or other disturbances have a particularly strong relationship with tau, as they have been shown to be associated with increased risk of MCI and AD [33–37]. Furthermore, our study is limited to men, though some research suggests that the relationship between sleep and other biomarkers of AD may be stronger in women [38]. Finally, our study is limited to a very small sample size, which limits our ability to make meaningful conclusions on the role of PTSD and TBI diagnoses in this relationship. Although our study did not find any differences between groups in terms of biomarker results, a control group of non-veterans would be useful to confirm and generalize the results of our study. While sleep quality data do not currently exist in the ADNI study outside of the DoD-ADNI substudy, we hope that these data will become available in the future. Replication and extension of these findings in other datasets and with other methods of assessing sleep quality and tau accumulation in normal aging, preclinical and prodromal AD, and victims of psychological and physical trauma are warranted.
In summary, our results suggest a role of sleep quality in the preclinical and prodromal stages of AD. We found that tau deposition following an AD profile was associated with an interaction between self-reported sleep quality and amyloid status. While our results do not establish directionality in this association, they highlight the potential of sleep quality as a target for future AD research.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Dr. Banks and Murray Andrews are funded in part by the NIA (1R01AG066088) Dr. Brewer directs the UCSD Shiley Marcos Alzheimer’s Disease Center, funded by the NIA (1P30AG062429-0).
REFERENCES
[1] | Livingston G , Huntley J , Sommerlad A , Ames D , Ballard C , Banerjee S , Brayne C , Burns A , Cohen-Mansfield J , Cooper C , Costafreda SG , Dias A , Fox N , Gitlin LN , Howard R , Kales HC , Kivimäki M , Larson EB , Ogunniyi A , Orgeta V , Ritchie K , Rockwood K , Sampson EL , Samus Q , Schneider LS , Selbæk G , Teri L , Mukadam N ((2020) ) Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396: , 413–446. |
[2] | Mander BA , Winer JR , Jagust WJ , Walker MP ((2016) ) Sleep: a novel mechanistic pathway, biomarker, and treatment target in the pathology of Alzheimer’s disease? Trends Neurosci 39: , 552–566. |
[3] | Carpenter BD , Strauss M , Patterson MB ((1996) ) Sleep disturbances in community-dwelling patients with Alzheimer’s disease. Clin Gerontol 16: , 35–49. |
[4] | Ancoli-Israel S , Klauber MR , Gillin JC , Campbell SS , Hofstetter CR ((1994) ) Sleep in non-institutionalized Alzheimer’s disease patients. Aging (Milano) 6: , 451–458. |
[5] | McCurry SM , Ancoli-Israel S ((2003) ) Sleep dysfunction in Alzheimer’s disease and other dementias. Curr Treat Options Neurol 5: , 261–272. |
[6] | Yesavage JA , Friedman L , Ancoli-Israel S , Bliwise D , Singer C , Vitiello MV , Monjan AA , Lebowitz B ((2003) ) Development of diagnostic criteria for defining sleep disturbance in Alzheimer’s disease. J Geriatr Psychiatry Neurol 16: , 131–139. |
[7] | Leng Y , Redline S , Stone KL , Ancoli-Israel S , Yaffe K ((2019) ) Objective napping, cognitive decline, and risk of cognitive impairment in older men. Alzheimers Dement 15: , 1039–1047. |
[8] | Diem SJ , Blackwell TL , Stone KL , Yaffe K , Tranah G , Cauley JA , Ancoli-Israel S , Redline S , Spira AP , Hillier TA , Ensrud KE ((2016) ) Measures of sleep-wake patterns and risk of mild cognitive impairment or dementia in older women. Am J Geriatr Psychiatry 24: , 248–258. |
[9] | Pat-Horenczyk R , Klauber MR , Shochat T , Ancoli-Israel S ((1998) ) Hourly profiles of sleep and wakefulness in severely versus mild-moderately demented nursing home patients. Aging (Milano) 10: , 308–315. |
[10] | Blackwell T , Yaffe K , Ancoli-Israel S , Schneider JL , Cauley JA , Hillier TA , Fink HA , Stone KL , Study of Osteoporotic Fractures Group ((2006) ) Poor sleep is associated with impaired cognitive function in older women: the study of osteoporotic fractures. J Gerontol A Biol Sci Med Sci 61: , 405–410. |
[11] | Avidan AY ((2006) ) Sleep and neurologic problems in the elderly. Sleep Med Clin 1: , 273–292. |
[12] | Prinz PN , Peskind ER , Vitaliano PP , Raskind MA , Eisdorfer C , Zemcuznikov N , Gerber CJ ((1982) ) Changes in the sleep and waking EEGs of nondemented and demented elderly subjects. J Am Geriatr Soc 30: , 86–93. |
[13] | Petit D , Gagnon J-F , Fantini ML , Ferini-Strambi L , Montplaisir J ((2004) ) Sleep and quantitative EEG in neurodegenerative disorders. J Psychosom Res 56: , 487–496. |
[14] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 82: , 239–259. |
[15] | Rothman SM , Herdener N , Frankola KA , Mughal MR , Mattson MP ((2013) ) Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical Aβ and pTau in a mouse model of Alzheimer’s disease. Brain Res 1529: , 200–208. |
[16] | Cantero JL , Hita-Yañez E , Moreno-Lopez B , Portillo F , Rubio A , Avila J ((2010) ) Tau protein role in sleep-wake cycle. J Alzheimers Dis 21: , 411–421. |
[17] | Fagan AM ((2015) ) What does it mean to be ‘amyloid-positive’? Brain 138: , 514–516. |
[18] | Lucey BP , McCullough A , Landsness EC , Toedebusch CD , McLeland JS , Zaza AM , Fagan AM , McCue L , Xiong C , Morris JC , Benzinger TLS , Holtzman DM ((2019) ) Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci Transl Med 11: , eaau6550. |
[19] | Benedict C , Blennow K , Zetterberg H , Cedernaes J ((2020) ) Effects of acute sleep loss on diurnal plasma dynamics of CNS health biomarkers in young men. Neurology 94: , e1181–e1189. |
[20] | Shokri-Kojori E , Wang G-J , Wiers CE , Demiral SB , Guo M , Kim SW , Lindgren E , Ramirez V , Zehra A , Freeman C , Miller G , Manza P , Srivastava T , De Santi S , Tomasi D , Benveniste H , Volkow ND ((2018) ) β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A 115: , 4483–4488. |
[21] | Lamarche LJ , De Koninck J ((2007) ) Sleep disturbance in adults with posttraumatic stress disorder: a review. J Clin Psychiatry 68: , 1257–1270. |
[22] | Viola-Saltzman M , Watson NF ((2012) ) Traumatic brain injury and sleep disorders. Neurol Clin 30: , 1299–1312. |
[23] | Zanier ER , Bertani I , Sammali E , Pischiutta F , Chiaravalloti MA , Vegliante G , Masone A , Corbelli A , Smith DH , Menon DK , Stocchetti N , Fiordaliso F , De Simoni M-G , Stewart W , Chiesa R ((2018) ) Induction of a transmissible tau pathology by traumatic brain injury. Brain 141: , 2685–2699. |
[24] | Digma LA , Madsen JR , Rissman RA , Jacobs DM , Brewer JB , Banks SJ ((2020) ) Women can bear a bigger burden: Ante- and post-mortem evidence for reserve in the face of tau. Brain Commun 2: , fcaa025. |
[25] | Jagust WJ , Landau SM , Shaw LM , Trojanowski JQ , Koeppe RA , Reiman EM , Foster NL , Petersen RC , Weiner MW , Price JC , Mathis CA , Alzheimer’s Disease Neuroimaging Initiative ((2009) ) Relationships between biomarkers in aging and dementia. Neurology 73: , 1193–1199. |
[26] | Baker SL , Maass A , Jagust WJ ((2017) ) Considerations and code for partial volume correcting [18F]-AV-1451 tau PET data. Data Brief 15: , 648–657. |
[27] | Landau S , Jagust W (2016) Flortaucipir (AV-1451) processing methods. Alzheimer’s Disease Neuroimaging Initiative. |
[28] | Buysse DJ , Reynolds CF , Monk TH , Berman SR , Kupfer DJ ((1989) ) The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res 28: , 193–213. |
[29] | Dubois B , Hampel H , Feldman HH , Scheltens P , Aisen P , Andrieu S , Bakardjian H , Benali H , Bertram L , Blennow K , Broich K , Cavedo E , Crutch S , Dartigues J-F , Duyckaerts C , Epelbaum S , Frisoni GB , Gauthier S , Genthon R , Gouw AA , Habert M-O , Holtzman DM , Kivipelto M , Lista S , Molinuevo J-L , O’Bryant SE , Rabinovici GD , Rowe C , Salloway S , Schneider LS , Sperling R , Teichmann M , Carrillo MC , Cummings J , Jack CR ((2016) ) Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement 12: , 292–323. |
[30] | Betthauser TJ , Koscik RL , Jonaitis EM , Allison SL , Cody KA , Erickson CM , Rowley HA , Stone CK , Mueller KD , Clark LR , Carlsson CM , Chin NA , Asthana S , Christian BT , Johnson SC ((2020) ) Amyloid and tau imaging biomarkers explain cognitive decline from late middle-age. Brain 143: , 320–335. |
[31] | Lowe VJ , Bruinsma TJ , Wiste HJ , Min H-K , Weigand SD , Fang P , Senjem ML , Therneau TM , Boeve BF , Josephs KA , Pandey MK , Murray ME , Kantarci K , Jones DT , Vemuri P , Graff-Radford J , Schwarz CG , Machulda MM , Mielke MM , Roberts RO , Knopman DS , Petersen RC , Jack CR ((2019) ) Cross-sectional associations of tau-PET signal with cognition in cognitively unimpaired adults. Neurology 93: , e29–e39. |
[32] | Mohamed AZ , Cumming P , Götz J , Nasrallah F , for the Department of Defense Alzheimer’s Disease Neuroimaging Initiative ((2019) ) Tauopathy in veterans with long-term posttraumatic stress disorder and traumatic brain injury. Eur J Nucl Med Mol Imaging 46: , 1139–1151. |
[33] | Gehrman PR , Martin JL , Shochat T , Nolan S , Corey-Bloom J , Ancoli-Israel S ((2003) ) Sleep-disordered breathing and agitation in institutionalized adults with Alzheimer disease. Am J Geriatr Psychiatry 11: , 426–433. |
[34] | Yaffe K , Laffan AM , Harrison SL , Redline S , Spira AP , Ensrud KE , Ancoli-Israel S , Stone KL ((2011) ) Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 306: , 613–619. |
[35] | Zimmerman ME , Aloia MS ((2012) ) Sleep-disordered breathing and cognition in older adults. Curr Neurol Neurosci Rep 12: , 537–546. |
[36] | Neikrug AB , Ancoli-Israel S ((2010) ) Sleep disturbances in nursing homes. J Nutr Health Aging 14: , 207–211. |
[37] | Elias A , Cummins T , Tyrrell R , Lamb F , Dore V , Williams R , Rosenfeld JV , Hopwood M , Villemagne VL , Rowe CC ((2018) ) Risk of Alzheimer’s disease in obstructive sleep apnea syndrome: amyloid-β and tau imaging. J Alzheimers Dis 66: , 733–741. |
[38] | Kim TH , Carroll JE , An SK , Seeman TE , Namkoong K , Lee E ((2016) ) Associations between actigraphy-assessed sleep, inflammatory markers, and insulin resistance in the Midlife Development in the United States (MIDUS) study. Sleep Med 27-28: , 72–79. |


