
Curcumin Downregulates GSK3 and Cdk5 in Scopolamine-Induced Alzheimer’s Disease Rats Abrogating Aβ40/42 and Tau Hyperphosphorylation
Abstract
Alzheimer’s disease (AD) is the most common form of dementia. Extracellular amyloid-β (Aβ) aggregation and tau hyperphosphorylation are the key drivers of AD. Glycogen synthase kinase 3 (GSK3) and cyclin dependent kinase 5 (Cdk5) have been known as leading applicants arbitrating abnormal tau hyperphosphorylation. Thus, we evaluated the efficacy and underlying mechanism of action of curcumin in scopolamine-induced AD rats in our study. We found that curcumin-treated AD rats markedly reduced the levels of Aβ40 and Aβ42 in the brain and in the plasma in comparison to untreated AD rats. Moreover, the levels of phosphorylated tau at Ser396 (PHF13), Ser202/Thr205 (AT8), and Aβ40/42 (MOAB2) were decreased significantly in AD rats treated with curcumin. Phospho-GSK3β (Tyr216), the active form of GSK3β, and total GSK3β were significantly decreased in AD rats treated with curcumin. Furthermore, Cdk5 and its activators p35 and p25 were significantly decreased in curcumin-treated AD rats. The reduced levels of Cdk5, p35, p25, and GSK3β in curcumin-treated AD rats may result decreased Aβ aggregation and tau hyperphosphorylation, thus ameliorating AD. Impaired spatial memory and locomotor activity in AD rats were partially reversed by curcumin. Therefore, curcumin, as a natural compound present in turmeric, may be a more effective therapeutic agent in the treatment of AD in humans.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia, characterized by a profound memory loss and contributes to 50–70% of neurodegenerative disease among adults [1]. AD is characterized by the accumulation of senile amyloid-β plaques (Aβ40 and Aβ42) and hyperphosphorylated tau in the medial temporal lobe and in cortical areas of the brain. Currently there are 47 million cases of AD worldwide which are expected to rise to 76 million in 2030 and the treatment cost is $604.00 billion per year [1].
The abnormal hyperphosphorylation of tau is the key driver of neurofibrillary degeneration in AD. In vitro kinetic studies of the binding between hyperphosphorylated tau and normal tau suggested that Ser202/396 and Thr205 are among the critical phosphorylation sites that result in sequestration of hyperphosphorylated tau into microtubule associated proteins and self-aggregation of tau into filaments, which are the hallmarks of AD [2, 3]. The proline-directed serine/threonine kinases such as glycogen synthase kinase 3 (GSK3) and cyclin dependent kinase 5 (Cdk5), have been known as the leading applicants arbitrating abnormal tau hyperphosphorylation [2]. GSK3 consists of the highly homologous GSK3α and GSK3β and is constitutively active in unstimulated cells under normal circumstances. Therefore, the regulation of GSK3 through phosphorylation at specific residues is a key control mechanism for its activity. Phosphorylation at Ser9 and Ser21 are the most common mechanisms of GSK3α/β inactivation. In contrast, autophosphorylation of GSK3β at Tyr216 enhances the stability and the activity of GSK3. In addition, Cdk5 has also been linked to increased tau phosphorylation via its activator p35/p25 in AD [4]. Increased levels of Cdk5 have been found in the brain of AD patients [5]. Although Cdk5 activation via p35 has been associated with the physiological activity of Cdk5, the truncated form p25 abnormally stimulates Cdk5 signaling leading to abnormal phosphorylation of tau [6]. Thus, efforts have been made by several research groups on the development of precise inhibitors for GSK3 and Cdk5 as prospective therapeutic treatments in tauopathies [4]. However, the inhibition of key kinases could lead to potential adverse effects on AD patients. It is unclear whether a brief or sustained inhibition of Cdk5 and GSK3 is necessary to attenuate tau hyperphosphorylation. In addition, the progression rate in affected AD patients is largely unknown since the precise pathological changes in AD [2]. Currently, no permanent curative or protective treatment is available for AD. Thus, it is important to develop new pharmacological compound (s) or to identify the natural compound (s) for the management of AD.
Scopolamine is a muscarinic cholinergic receptor antagonist. It is commonly used for the study of cognitive deficiency in animal models of AD [7–11]. Donepezil is frequently used to treat patients with mild to moderate AD. Hence, this study explored a more effective therapeutic agent in the treatment of AD in humans based on a natural compound present in turmeric.
Curcumin is a scavenger of free radicals and has anti-inflammatory properties [12]. Importantly, curcumin can cross the blood-brain barrier (BBB) due to its lipophilic nature and can bind to amyloid plaques, abrogates behavioral impairment [13], and also reduces acute and chronic inflammations [12], which are all associated with AD pathogenesis. Moreover, curcumin ameliorates defective insulin signaling in scopolamine-induced AD rats [7].
Therefore, in our study, the effect of curcumin on the key driver proteins of AD such as Aβ40, Aβ42, unaggregated oligomeric fibrillary forms of Aβ, and phosphorylated tau (Ser396, Ser202/Thr205) in the brain and in plasma were examined in scopolamine-induced AD-like changes in rats. Our study also evaluated the effects of curcumin on the key regulators of tau phosphorylation such as GSK3α/β, Cdk5, and its activators p35/p25 in the brain of AD rats. Donepezil was used as a reference drug throughout our study.
MATERIALS AND METHODS
Animals
Male Sprague Dawley rat (n = 32) aged 60–70 days weighing 300±20 g were used in our study. They had free access to food (Specialty Feeds, Western Australia) and water, and were maintained under the standard laboratory conditions (alternating light and dark cycles, each for 12 h). The animal handling and experiments were performed in accordance with the institutional guidelines (Reference No: UBD/AVC-RI/1.26) by PAPRSB Institute of Health Sciences Research Ethics Committee and Universiti Brunei Darussalam Research Ethics Committee.
Materials
Scopolamine, donepezil, and curcumin were purchased from Sigma-Aldrich, United States. Protease inhibitor tablets were purchased from Roche Applied Science (Mannheim, Germany). Immunohistochemistry kit (R.T.U. Vectastain Universal Elite ABC Kit and DAB kit) was purchased from Vector Laboratories (CA, USA). Pierce™ BCA Protein Assay Kit and ECL advanced reagent kit were purchased from Thermo Fisher Scientific (MA, USA) and Bio-Rad (CA, USA), respectively. Antibodies PHF13, p-GSK3 (Ser9/Ser21) and p-GSK3β (Tyr216) were purchased from Cell Signaling; AT8 was purchased from Thermo Fisher Scientific; MOAB2 was purchased from LifeSpanBioSciences; GSK3β was purchased from Millipore; Cdk5 and p35/p25 were purchased from Santa Cruz; and β-Actin were purchased from Sigma, respectively. All analytical grade reagents were purchased from Merck, Germany and Sigma Aldrich, USA.
Drug interventional studies
The rats (n = 32) were randomly divided into four different groups with 8 rats for each group as follows: 1) control (saline-treated) (n = 8), 2) scopolamine-induced (n = 8), 3) scopolamine+donepezil-treated (n = 8), and 4) scopolamine+curcumin-treated (n = 8). The control group was administered with 0.9% saline (w/v) for 28 consecutive days. Scopolamine (2.5 mg/kg), was administered to all the groups through the intraperitoneal route as a disease inducer. The treatment groups were given donepezil (5 mg/kg) and curcumin (80 mg/kg) by oral gavage for 28 consecutive days (Table 1). Both curcumin and donepezil were dissolved in saline (vehicle). Our study did not observe any signs of suffering in animal, although oral gavage was an invasive procedure.
Table 1
Experimental design and drug doses in different groups of rats
| Group No. | Name of groups | Compound and drug administered |
| 1 | Control | Saline (0.9% w/v) i.p. |
| 2 | Disease Control | Scopolamine (2.50 mg/kg) i.p. |
| 3 | Standard Treatment | Scopolamine (2.50 mg/kg) i.p. + Donepezil (5 mg/kg) oral |
| 4 | Test group | Scopolamine (2.50 mg/kg) i.p + Curcumin (80 mg/kg) oral |
Behavioral studies
Rectangular maze test
The maze consisted of a completely enclosed rectangular box with an entry and reward chamber appended at the opposite ends. The box was partitioned with wooden slats into blind passages leaving just twisting corridor leading from the entry to the reward chamber. Transfer latency (the time taken to reach the reward chamber) was recorded beginning from day 1 till day 28. Three readings were taken per animal, mean was calculated and indicated as learning score (transfer latency) for that animal. Lower scores indicated efficient learning while higher scores indicated poor learning in animals [14].
Locomotor activity test
Rats were placed individually in the activity cage for 5 min and their movements were recorded beginning from day1 till day 28. The photo cell count was recorded and the respective decrease or increase in locomotor activity was calculated [14].
Brain tissue collection
After 28 days of treatment, rats were sacrificed using CO2 anesthetization [7, 15]. The brains were collected in cold 1X PBS (pH 7.4) (Bio-Rad, CA, USA). Four brains from each group were used for immunohistochemistry (IHC) and immunoblotting separately.
Isolation of plasma
After 28 days of treatment, 5 ml of blood was collected from each group of rats and was fractionated by centrifugation at 3000× g for 15 min. Supernatants (plasma) were collected and stored frozen at –80°C until further use.
Tissue studies
Immunohistochemistry and scoring
Brains from each group (n = 4) were washed in phosphate buffered saline (pH 7.4), and were subsequently processed. Brain sections from different groups of rats (n = 4 at each time point for each rat) were used for IHC using R.T.U Vectastain Universal Elite® ABC Kit (Vector Laboratories, USA). Tissue sections were incubated with primary antibodies GSK3 (Ser9/21), GSK3β (Tyr216), total GSK3β, Cdk5, and p35/p25 at 4°C overnight. Subsequent procedures were performed according to the manufacturer’s instruction (Vector Universal elite® ABC kits, Vector Laboratories, USA). A series of four representative images per section was collected. IHC analyses were scored according to the intensity of staining and percentage of positive cells stained with respective antibodies. The scoring was done as follows: 0, <5% (negative); 1, 5–25% (weak); 2, 25–50% (moderate); 3, 50–75% (strong), and 4, >75% (very strong) [16]. The analysis (n = 4) was done in triplicate.
Preparation of brain homogenates
Four rat brains (n = 4) from each group were snapped frozen in liquid nitrogen. The weights of the brains were measured. 1X Cell lysis buffer (Cell Signaling Technology) containing protease inhibitor (Roche) was added in a 40% w/v ratio, homogenized (Thermo Scientific) using 3 pulses of 10 s each, with a 5 s interval on ice. Issue lysates were diluted 10-fold in 1X cell lysis buffer and sonicated (Thermo Scientific) to ensure complete lysis in between pulses. The crude brain homogenates were stored at –80°C until further use. Protein concentrations were determined using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific).
ELISA
The levels of Aβ40 and Aβ42 in the plasma (n = 8) and in the brain (n = 4) were determined using Human Aβ (1–40) (N) assay kit (IBL, Gunma, Japan) and Human Aβ (1–42) assay kit (Immuno-biology) (IBL, Gunma, Japan), respectively, according to the manufacturer’s instructions. Absorbance was measured at 450 nm by ELISA (BioTek Instruments Winooski, USA).
Immunoblotting
50 μg of total brain lysate was boiled at 95°C for 10 min in 4X gel-loading buffer. The protein extract was separated by gel electrophoresis, and transferred onto nitrocellulose membrane, blocked, and probed with their respective primary antibodies. The protein bands were visualized by chemiluminescence using Clarity™ Western ECL Substrate (Bio-Rad). Blots were digitally developed using VersaDoc™ imaging system (Bio-Rad). Pre-stained Precision Plus™protein standards (Bio-Rad) were used to estimate the apparent molecular weight of the protein bands. β-actin (Sigma, USA) was used as an internal control in all analyses to ensure comparable protein loading. The phospho-proteins levels were normalized with corresponding total protein.
Coimmunoprecipitation
Brain lysates containing 1 mg of protein were diluted with 1 ml of ice-cold 1X Cell lysis buffer (Cell Signaling Technology) with proteinase inhibitor (Roche). The mixture was rotated with Cdk5 antibody for 4 h at 4°C, lysates were rotated with 40 μl of Protein A/G PLUS-Agarose beads (sc-2003, Santa Cruz) overnight at 4°C. The supernatant was removed by centrifugation 10000× g at 4°C for 10 min. Agarose beads were washed 3 times in 1X Cell lysis buffer. The bound protein was dissociated from the agarose beads by boiling the beads for 5 min in 2× Laemmli sample buffer and immunoblotted for p35 and p25 subunits. Densitometry analysis was performed by using NIH ImageJ software to measure the optical densities of the targeted protein band and normalized to the intensity of the endogenous β-actin level.
Statistical analysis
Data were expressed as mean±standard error (S.E.) and analyzed using SPSS 20.0 statistical software (SPSS Inc., Chicago, IL, USA). The post-hoc test were used after ANOVA analysis. A p value of less than 0.05 was considered significant.
RESULTS
Reduced spatial memory and locomotor activity in scopolamine-induced AD rats and marked reversal by curcumin
The spatial memory test was executed using the rectangular maze (Fig. 1a). The transfer latency was significantly decreased (p < 0.01) in saline-treated rats from day 14 till day 28 compared to other groups of rats, indicating higher efficiency of learning in saline-treated rats. However, transfer latency was significantly increased (p < 0.001) in scopolamine-induced AD rats during this time period, indicating progressive loss of efficiency of learning in scopolamine-induced AD rats. Additionally, transfer latency was progressively decreased (p < 0.001) in either donepezil- or curcumin-treated AD rats during this time period. Importantly, in comparison to donepezil-treated AD rats, the transfer latency was markedly decreased (p < 0.001) in curcumin-treated AD rats (Fig. 1a).
Fig.1
Behavioral studies of animals. (a) Rectangular maze test and (b) locomotor activity test of AD rats and following treatment. The assay was performed in triplicate. Data expressed as mean±S.E. (*p < 0.001, **p < 0.01, and ***p < 0.05 versus saline-treated group; #p < 0.001, ##p < 0.01, and # # #p < 0.05 versus scopolamine-induced AD rats, ∧p < 0.001, ∧∧p < 0.01, and ∧∧∧p < 0.05 versus scopolamine-donepezil rats. n = 8). Cont., control; Scop., scopolamine; Done., donepezil; Curc., curcumin.
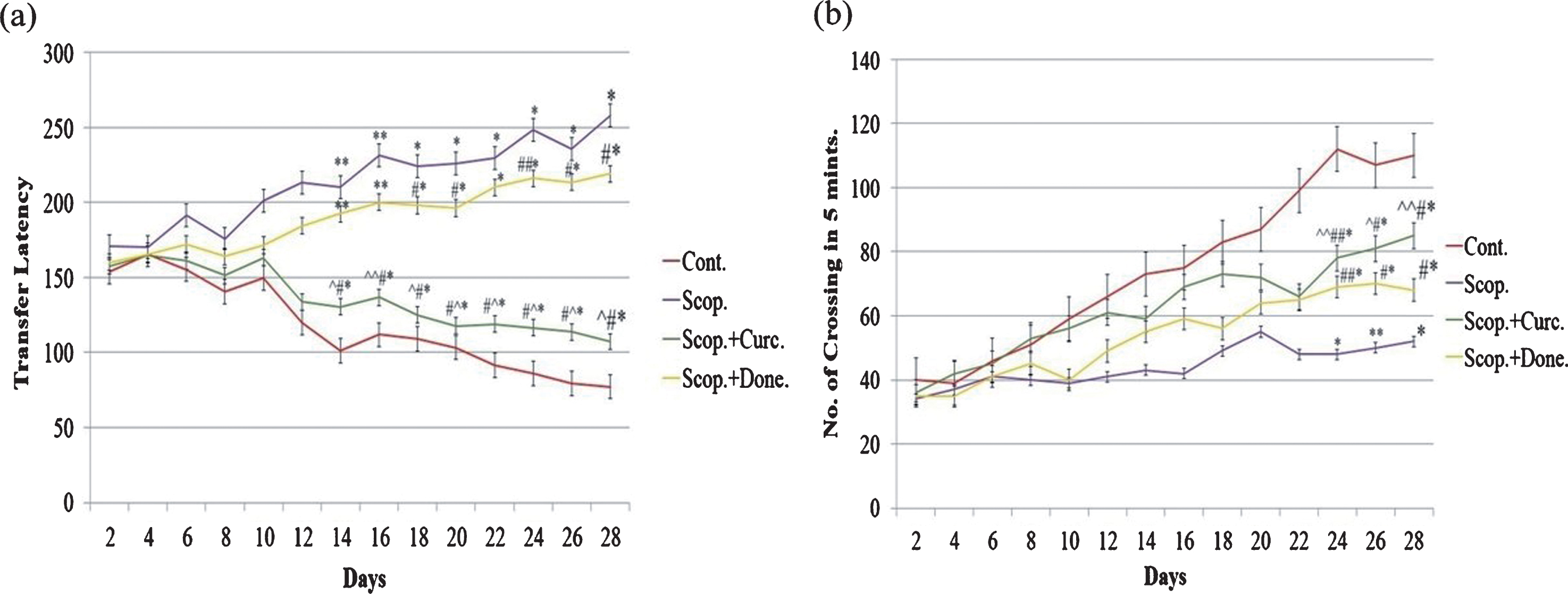
Figure 1b shows the locomotor activity in four groups of AD rats. The locomotor activity was significantly increased (p < 0.001) in saline-treated rats from day 24 till day 28 compared to other groups of rats. However, locomotor activity was significantly decreased (p < 0.001) in scopolamine-induced AD rats during this time period. Moreover, locomotor activity was progressively increased (p < 0.01) in either donepezil- or curcumin-treated AD rats during this time period. Importantly, in comparison to donepezil-treated AD rats, the locomotor activity was markedly increased (p < 0.01) in curcumin-treated AD rats (Fig. 1b).
Increased number of immunostained cells positive for hallmark proteins in scopolamine-induced AD rats’ brains and marked reversal by curcumin
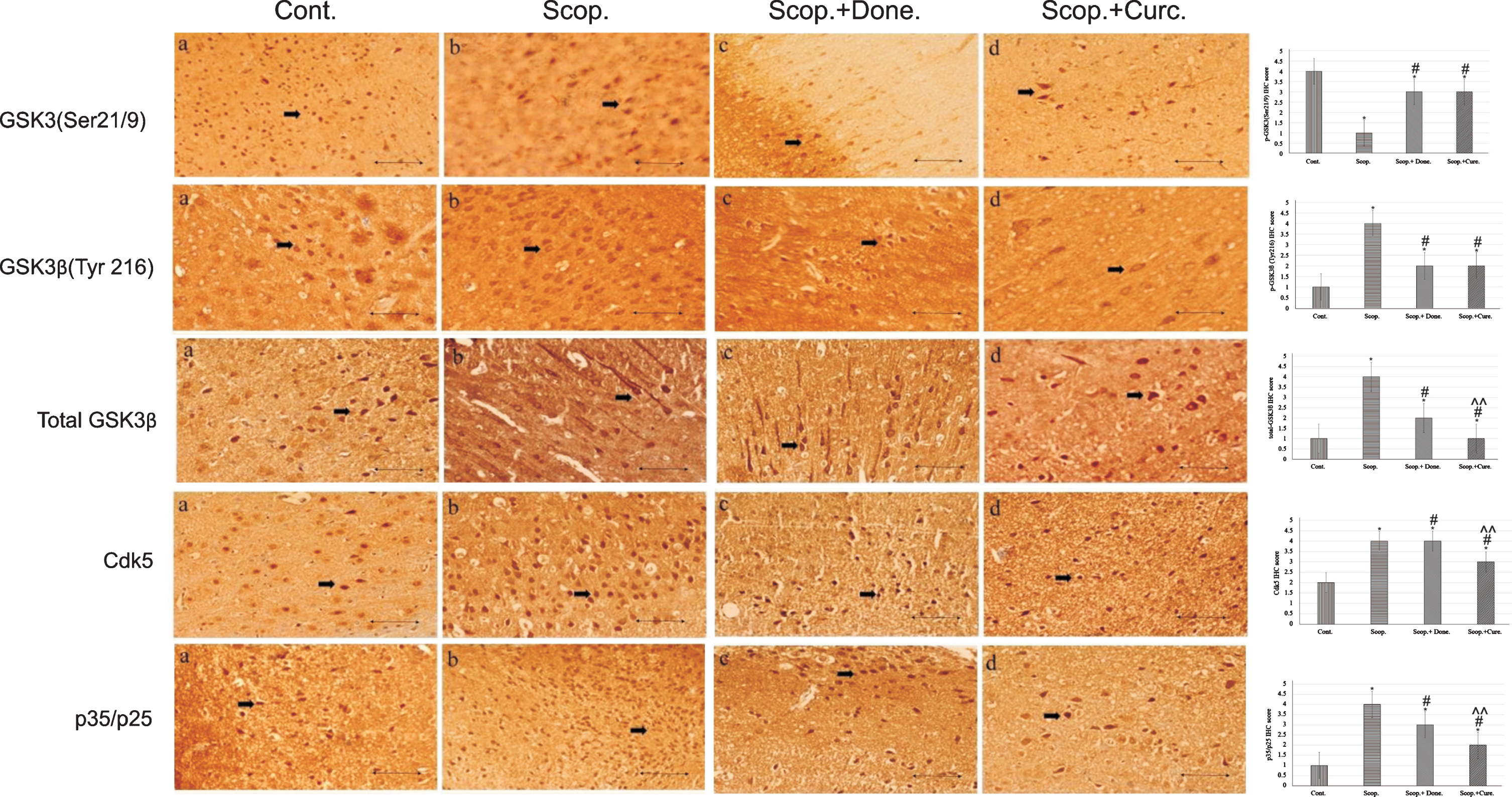
Immunohistochemistry was used to detect the protein expression of GSK3 (Ser9/21), GSK3β (Tyr216), and total GSK3β, Cdk5, and p35/p25 in the cortex region of rat brains (Fig. 2). From the IHC scoring analysis, it was found that the expression of GSK3 (Ser9/21) was significantly decreased; and GSK3β (Tyr216), total GSK3β, Cdk5, and p35/p25 were increased in AD rats compared to saline-treated rats. Treatment with either donepezil or curcumin in AD rats significantly increased the levels of inactive GSK3 (Ser9/21). GSK3β (Tyr216), total GSK3β, Cdk5, and p35/p25 protein expressions were decreased in curcumin and/or donepezil–treated AD rats (Fig. 2).
Fig.2
Immunohistochemistry images of (i) GSK3α/β(Ser 9/Ser21), (ii) GSK3β(Tyr 216), (iii) total GSK3β, (iv) Cdk5, and (v) p35/p25 in the brain sections (cortex) of rat that were treated with Cont., Scop., Scop.+Done., and Scop.+Curc. The assay (n = 4) was done in triplicate. Lens objective: 40x. Scale bar represents 1 μ. (*p < 0.001, **p < 0.01, and ***p < 0.05 versus saline-treated group; #p < 0.001, ##p < 0.01, and # # #p < 0.05 versus scopolamine-induced AD rats, ∧p < 0.001, ∧∧p < 0.01, and ∧∧∧p < 0.05 versus scopolamine-donepezil rats. n = 4). Cont., control; Scop., scopolamine; Done., donepezil; Curc., curcumin.
Increased levels of Aβ40/42 in the plasma and in the brain of scopolamine-induced AD rats and marked reversal by curcumin
Aβ40 concentrations in the plasma were 96.22±1.27 pg/ml and 329.88±2.13 pg/ml in saline-treated and in scopolamine-induced rats respectively. Aβ40 concentration was decreased to 222.06±6.42 pg/ml in donepezil-treated AD rats. Curcumin treatment in AD rats lowered Aβ40 concentration to 208.02±0.86 pg/ml. In addition, the concentration of Aβ40 was significantly increased (3-folds, p < 0.001) in scopolamine-induced rats compared to saline-treated control rats. Treatment with donepezil and curcumin in AD rats reduced plasma Aβ40 concentration to 1.49-fold (p < 0.001) and 1.59-fold (p < 0.001), respectively, compared to AD rats (Fig. 3a).
Fig.3
ELISA analysis of Aβ40 and Aβ42 content in the plasma (n = 8) and in the brain (n = 4) at each time point for each rat brain. The assay was performed in triplicate. Each value represents the mean±S.E. *p < 0.001, **p < 0.01, and ***p < 0.05 versus saline-treated group; #p < 0.001, ##p < 0.01, and # # #p < 0.05 versus scopolamine-induced AD rats; ∧p < 0.001, ∧∧p < 0.01, and ∧∧∧p < 0.05 versus scopolamine-donepezil rats. Cont., control; Scop., scopolamine; Done., donepezil; Curc., curcumin.
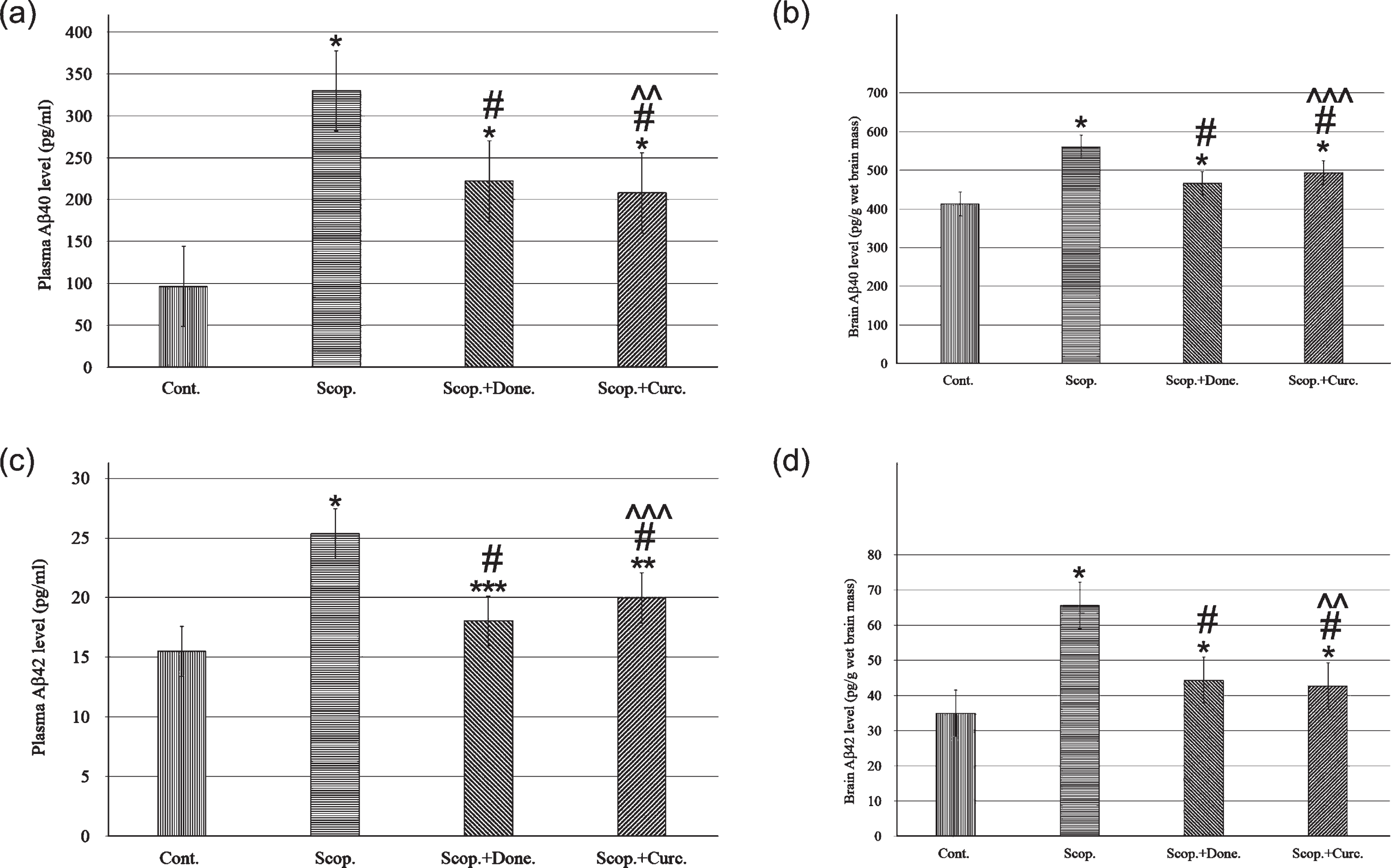
Similarly, Aβ40 concentrations were measured in the brain lysates of all groups. Aβ40 concentration was found to be 413.15±4.78 pg/g wet brain mass and 559.85±3.85 pg/g wet brain mass in saline-treated and in AD rats, respectively. Donepezil treatment in AD rats lowered Aβ40 concentration to 466.37±6.8 pg/g (p < 0.001) wet brain mass. Treatment with curcumin in AD rats lowered Aβ40 concentration to 493.85±2.89 pg/g (p < 0.001) wet brain mass. Taken together, the Aβ40 levels in the brain were significantly increased 1.6-fold (p < 0.001) in AD rats compared to saline-treated control rats. Treatment with curcumin in AD rats decreased Aβ40 levels 1.28-fold (p < 0.001) compared to AD rats (Fig. 3b).
Aβ42 concentrations were also measured in the plasma in all groups of rats. Aβ42 concentrations were 15.51±1.05 pg/ml and 25.35±2.62 pg/ml in saline-treated and in AD rats, respectively. Donepezil treatment in AD rats lowered Aβ42 concentration to 18.05±1.13 pg/ml. Treatment with curcumin in AD rats lowered Aβ42 concentration to 19.99±0.93 pg/ml. Taken together, plasma Aβ42 levels were 1.63-fold higher (p < 0.001) in AD rats compared to saline-treated control rats. Treatment with curcumin in AD rats lowered the Aβ42 levels to 1.27-fold (p < 0.001) compared to AD rats (Fig. 3c).
Similarly, Aβ42 levels in the brain lysate were measured in all groups of rats. The levels were 34.91±1.99 pg/g wet brain mass and 65.62±2.84 pg/g wet brain mass in saline-treated and in AD rats, respectively. Treatment with donepezil, Aβ42 level was decreased to 44.38±1.88 pg/g wet brain mass. Treatment with curcumin in AD rats, decreased the Aβ42 levels to 42.67±1.89 pg/g wet brain mass. Taken together, the levels of Aβ42 in the brain were found to be 1.88-fold higher (p < 0.001) in AD rats compared to saline-treated control rats. Treatment with curcumin in AD rats reduced the Aβ42 levels 1.54-fold (p < 0.001) compared to AD rats (Fig. 3d).
Increased levels of tau phosphorylation (PHF13 and AT8), unaggregated, oligomeric, and fibrillary forms of Aβ40/42 (MOAB-2) in scopolamine-induced AD rats and marked reversal by curcumin
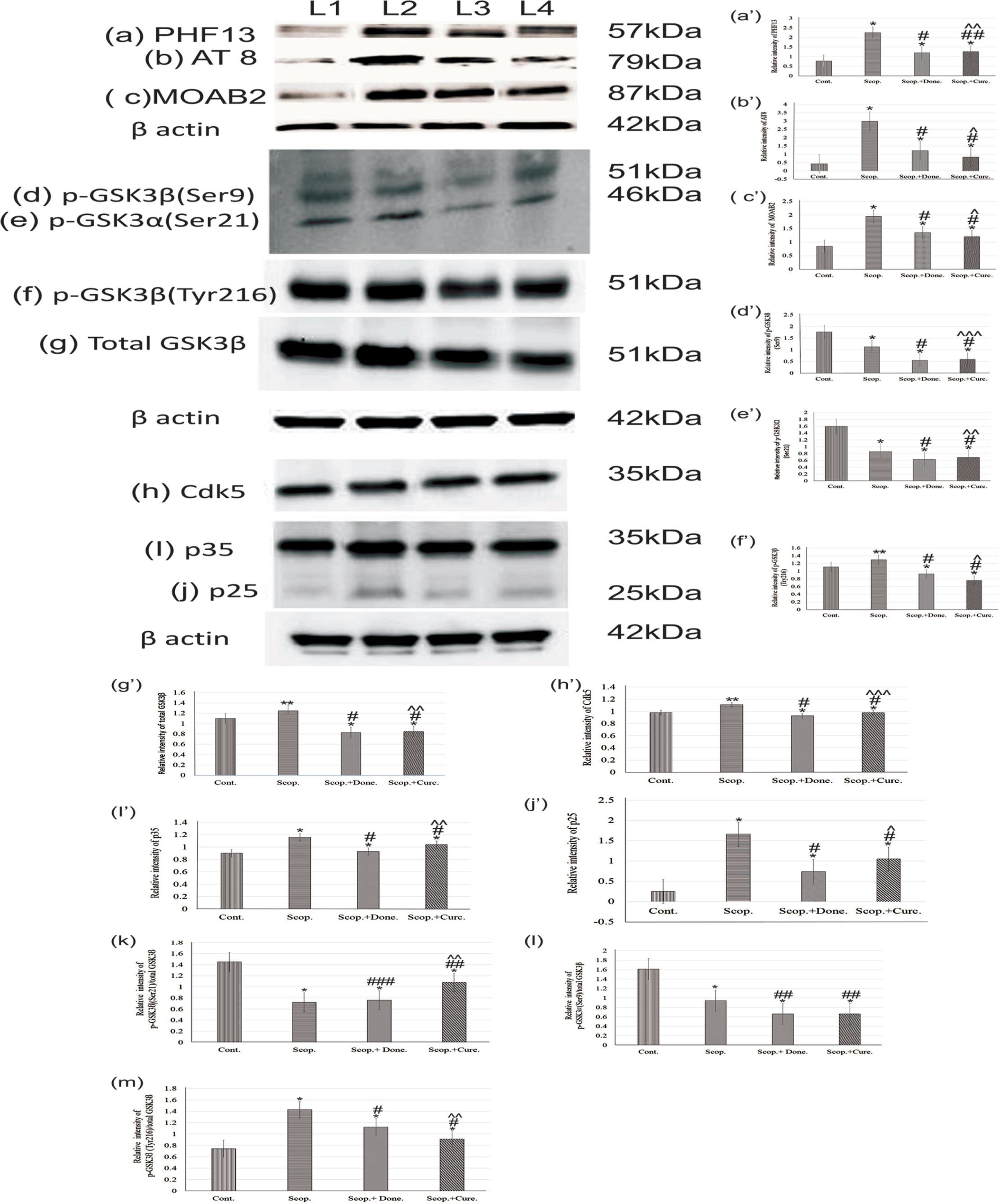
Immunoblotting studies in the brain showed the levels of phosphorylated tau at Ser396 (PHF13) (Fig. 4, a/a’), Ser202/Thr205 (AT8) (Fig. 4, b/b’), unaggregated, oligomeric and fibrillary forms of Aβ40/42 (MOAB2) (Fig.4, c/c’). The levels of phosphorylated tau at Ser396, Ser202/Thr205, and unaggregated oligomeric-fibrillary forms of Aβ40/42 were increased in AD rat brain 2.9-fold (p < 0.001), 6.88-fold (p < 0.001), and 2.67-fold (p < 0.001), respectively, compared to saline-treated rats. Donepezil treatment lowered the levels of tau phosphorylation at sites Ser396, Ser202/Thr205, and unaggregated oligomeric-fibrillary forms of Aβ40/42 1.87-fold (p < 0.001), 2.45-fold (p < 0.001), and 1.44-fold (p < 0.001), respectively, compared to those in AD rat brains. Similarly, curcumin treatment lowered the levels of phosphorylated tau (Ser396, Ser202/Thr205) and unaggregated oligomeric-fibrillary forms of Aβ40/42 4.24-fold (p < 0.001), 3.58-fold (p < 0.001), and 1.6-fold (p < 0.001), respectively, compared to those in AD rats. Thus, curcumin exhibited distinct reduction in the levels of phosphorylated tau (Ser396, Ser202/Thr205) and unaggregated oligomeric-fibrillary forms of Aβ40/42 in AD rats compared to saline-treated control rats (Fig. 4).
Fig.4
Immunoblotting and densitometry analysis of (a) PHF 13, (b) AT8, (c) MOAB-2, (d) p-GSK3β (Ser9), (e) p-GSK3α (Ser21), (f) total GSK3β, (g) p-GSK3β (Tyr216), (h) Cdk5, (i) p35, and (j) p25 in brain lysates. The (k) p-GSK3α (Ser21)/total GSK3β (0.51-fold, p < 0.05), (l) p-GSK3β (Ser9)/total GSK3β (0.75-fold, p < 0.05) markedly reduced, and (m) p-GSK3β (Tyr216)/total GSK3β ratio was significantly increased (1.03-fold, p < 0.05) in scopolamine-induced AD rats brain lysate. Immunoprecipitation experiments (n = 4) with brain lysate of different groups rat were carried out using anti-Cdk5 and anti-p35. Analysis was performed using NIH ImageJ software and the values obtained for the individual protein target for each brain sample were normalized against β-actin from the same brain sample. The assay was done in triplicate and ANOVA was used to analyze the differences among groups. Each value represents mean±S.E. for individual rat brain sample (n = 4). Here L1 = Cont., L2 = Scop., L3 = Scop.+Done., and L4 = Scop.+ Curc. *p < 0.001, **p < 0.01, and ***p < 0.05 versus saline-treated group; #p < 0.001, ##p < 0.01, and # # #p < 0.05 versus scopolamine-induced AD rats; ∧p < 0.001, ∧∧p < 0.01, and ∧∧∧p < 0.05 versus scopolamine-donepezil rats. Cont., control; Scop., scopolamine; Done., donepezil; Curc., curcumin.
Increased levels of active GSK3β (Tyr216), Cdk5, and p35/p25 in scopolamine-induced AD rat brains and marked reversals by curcumin
Densitometry analysis showed that the levels of phospho-GSK3β (Ser9) (0.64-fold, p < 0.001) (Fig. 4d/d’) and phospho-GSK3· (Ser21) (0.54-fold, p < 0.001) (Fig. 4e/e’), were significantly decreased in AD rat brain compared to the saline-treated group. Furthermore, the levels of phospho-GSK3β (Tyr216) (1.08-fold, p < 0.05) (Fig. 4f/f’) and total GSK3β (1.09-fold, p < 0.05) (Fig. 4g/g’) were increased in AD rat brain compared to saline-treated rat brain. The levels of GSK3β and phospho-GSK3β (Tyr216) were significantly decreased (p < 0.001) compared to scopolamine-induced rats after treatment with either donepezil or curcumin separately in AD rats. However, the levels of phospho-GSK3α (Ser21) (0.73-fold, p < 0.01) and phospho-GSK3β (Ser9) (0.49-fold, p < 0.01) were not markedly reduced by either donepezil or curcumin (Fig. 4).
Additionally, we examined the protein expression levels of Cdk5 (Fig. 4h/h’), p35 (Fig. 4i/i’), and p25 (Fig. 4j/j’) in the rat brain by co-immunoprecipitation followed by immunoblotting (Fig. 4). Densitometry analysis found that the levels of Cdk5 (1.12-fold, p < 0.01), p35 (1.29-fold, p < 0.001), and p25 (3.0-fold, p < 0.001) were significantly increased in AD rats compared to the saline-treated control rats. However, the levels of Cdk5 (0.89-fold, p < 0.001), p35 (0.89-fold, p < 0.001), and p25 (0.63-fold, p < 0.001) were decreased after treatment with either donepezil or curcumin as compared to untreated AD rats (Fig. 4).
DISCUSSION
Results from numerous studies had shown the cognitive impairments in scopolamine-induced subjects [7–11]. In our study, the effect of scopolamine treatment on spatial memory and locomotor activity test in all groups of rats were assessed by the rectangular maze test and actophotometer (Fig. 1). AD rats exhibited the longest time to complete the maze, thereby indicating a reduced spatial memory performance. In addition, as shown in Fig. 1, impaired spatial memory and locomotor activity in AD rats were markedly improved after treatment with either donepezil or curcumin.
Although the exact roles of Aβ40 and Aβ42 in AD development had not been fully elucidated, it was suggested that reduction in Aβ42 levels could reduce the risk of AD development [17, 18]. Our study showed that the levels of Aβ40 (Fig. 3a, b), Aβ42 (Fig. 3c, d) in the plasma, and tau phosphorylation (Fig. 4) were significantly elevated (p < 0.001) in the brain lysates of scopolamine-induced AD rats, compared to saline-treated control rats. Furthermore, our study showed a reduced memory performance (Fig. 1) in AD rats, which be due to elevated the Aβ aggregation and tau hyperphosphorylation in the brain [19]. Moreover, defective insulin signaling had shown to cause abnormal tau phosphorylation and Aβ40/42 accumulation [20]. Interestingly, we showed that curcumin improved impaired insulin signaling in AD rats [7]. Previous studies [17, 18] showed that the accumulation of Aβ aggregation leads to AD neurodegeneration. In our study, the levels of Aβ40 and Aβ42 (Fig. 3) were increased in the brain and in the plasma. Furthermore, unaggregated-oligomeric and fibrillary forms of Aβ40/42 (Fig. 4c/c’) were also increased in AD rat brains. Moreover, neurofibrillary tangle (NFT) accumulation that contains hyperphosphorylated tau is one of the hallmarks of AD. In our study, tau phosphorylation (Fig. 4) was significantly increased in scopolamine-induced AD rat brains.
Previous studies indicated that GSK3β is a key enzyme that leads to the tau hyperphosphorylation in AD [21–23]. Both GSK3α and GSK3β lead to increased Aβ aggregation and tau hyperphosphorylation [24]. The dysfunctionality of GSK3 is involved in AD [25]. The activity of GSK3 depends on the phosphorylation at the specific sites of GSK3; phospho-GSK3α (Ser21) and phospho-GSK-3β (Ser9) both of which can inhibit the GSK3β activity [26]; whereas phospho-GSK3β (Tyr216) increases the activity of GSK3β [24, 26]. Our study showed a significant reduction of the expression of phospho-GSK3β (Ser9) (p < 0.001) and phospho-GSK3α (Ser21) in AD rats compared to saline-treated AD rats. However, the expression of GSK3β and phospho-GSK3β (Tyr216) were significantly increased (p < 0.01) in AD rats compared to saline-treated rats. In comparison, the expression of phospho-GSK3α (Ser21) was not markedly improved by curcumin in AD rats. However, the levels of phospho-GSK3β (Tyr216) (p < 0.001) and total GSK3β were significantly decreased (p < 0.001) by curcumin in AD rats compared to scopolamine-induced AD rats (Fig. 4). Our study demonstrated that the reduced levels of phospho-GSK3α (Ser21) and phospho-GSK3β (Ser9) (p < 0.001) (inactive forms of GSK3β), and increased levels of phospho-GSK3β (Tyr216) (active form of GSK3β, and the total GSK3β) could possibly explain the increased levels of Aβ40/42 in the brain (Fig. 3) with a concomitant increase of tau phosphorylation (Ser396, Ser202, and Thr205) in AD rats (Fig. 4). The lowered levels of phospho-GSK3β (Tyr216) in AD rats could potentially cause curcumin to lower the levels of Aβ40/42 in AD rats, (Fig. 4), resulting reduced tau phosphorylation and Aβ aggregation in the brain (Fig. 3).
Cdk5 has also been suggested to directly induce tau phosphorylation at disease-associated sites such as Ser396, Ser202, and Thr205 that might trigger NFT formation as reported previously [28, 29]. Interestingly, Cdk5 has been implicated in the priming of tau at the distinct sites of Ser202, Ser235, and Ser404 for GSK3β [30, 31]. Priming of these sites by Cdk5 and subsequent GSK3β-catalyzed phosphorylation at the adjacent sites resulted in the generation of the disease-associated phospho-epitopes, Ser396, Ser202, and Thr205 [2]. Presumably, the increased levels of p25 in AD rats in our study led to simultaneous hyperactivation of both Cdk5 and GSK3, and these two kinases synergized to induce tau hyperphosphorylation at these critical sites. Several studies indicated that Aβ42 stimulated the cleavage of p35 to p25 [32] and increased the activity of CDK5 [33] (Fig. 4). The increased levels of Cdk5, p35, and p25 observed in our study might be due to increased levels of Aβ40/42 in AD rats. We also observed a reduction in the expression levels of active Cdk5 and its subunits p25/p35 in donepezil- or curcumin-treated AD rats compared to scopolamine-induced AD rats, indicating their protective effects in AD pathogenesis (Fig. 4).
Conclusion
In conclusion, our results suggested that curcumin reduced the levels of GSK3β, Cdk5, p35, and p25. Curcumin also decreased the levels of active GSK3β (Tyr216) resulting decreased tau phosphorylation (Fig. 5). Most importantly, curcumin abrogated Aβ40/42 and tau hyperphosphorylation in AD rats. Moreover, reduced spatial memory and locomotor activity in scopolamine-induced AD-like changes in rats were markedly reversed by curcumin. Thus curcumin, as a natural compound present in turmeric, may be a more effective therapeutic agent in the treatment of AD in humans compared to available pharmacological kinase inhibitors that may have side effects. Further studies should be performed to identify the detailed mechanism of action curcumin in the management of AD.
Fig.5
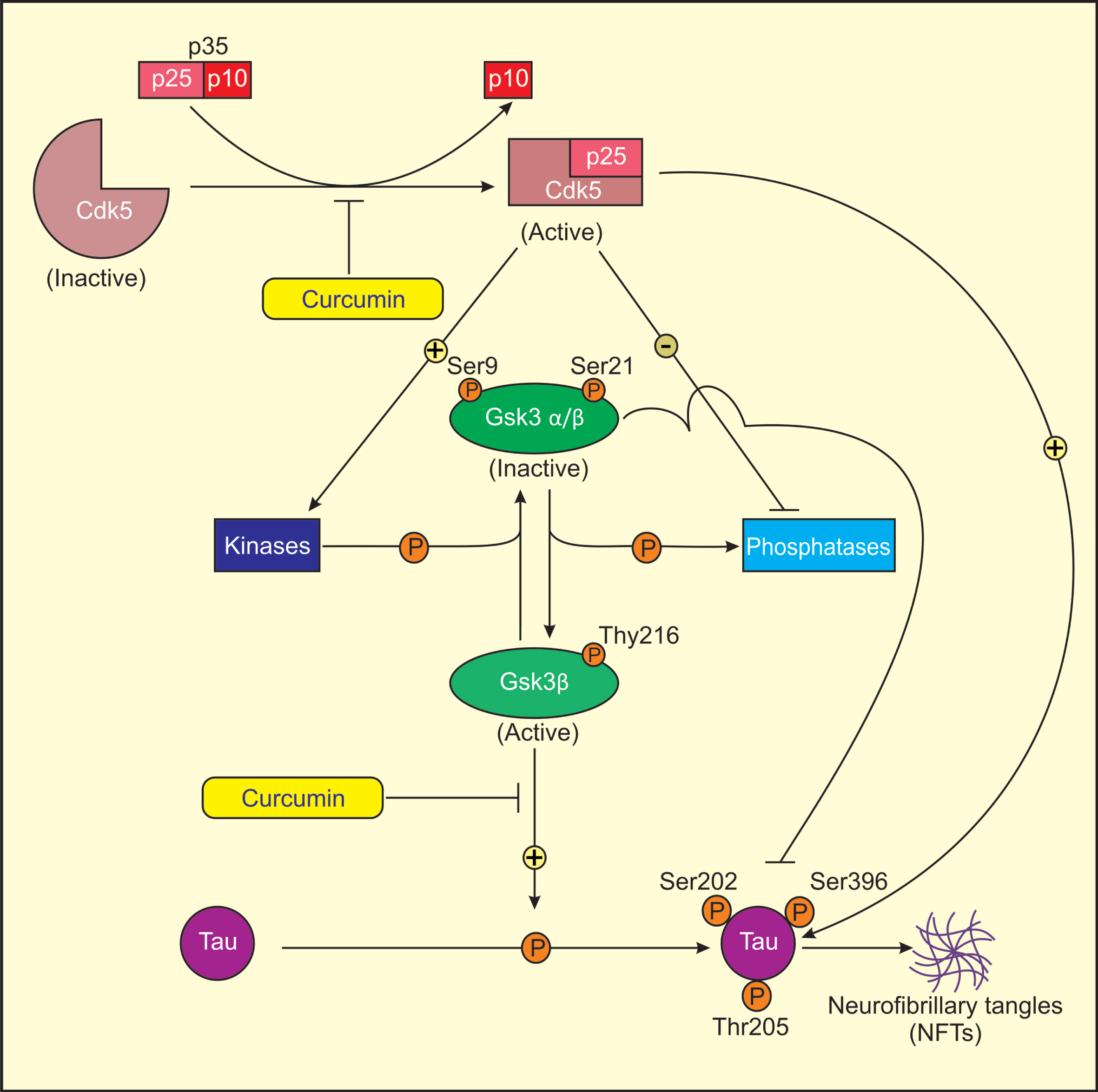
Proposed model for mechanism of action of curcumin in treatment of AD. Cdk5 is activated by its association with p25, as Cdk5-p25 complex is more stable than Cdk5-p35 complex. Activated Cdk5 induces GSK3 phosphorylation (Ser9/21, Tyr216) by direct/indirect regulation of several protein kinases and phosphatases. Ser9/21 phosphorylation of GSK3 renders GSK3 inactive. Activated Cdk5 directly or indirectly induces tau phosphorylation at sites Ser202, Ser396, and Thr205 through activated GSK3 (Tyr216) that results in the formation of NFTs in AD. Curcumin lowers the levels of Cdk5/p25 and active GSK3β (Tyr216) and increases the levels of inactive GSK3β (Ser9/21) in AD rats causing reduced tau hyperphosphorylation.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We thank to Dr. Hjh Fazean Irdayati Binti Hj Idris and Dr. Hjh Hashmet Parveen Ghouse, PAPRSB Institute of Health Sciences, Universiti Brunei Darussalam; for their support and contributions to this study.
This study was funded by the Universiti Brunei Darussalam/ Brunei Research Council-2 (UBD/BRC-2) (Ref: JPKE/DG/83) and Graduate Research Scholarship from Universiti Brunei Darussalam (Ref: UBD/GS/1) ’2015-16.
REFERENCES
[1] | World Alzheimer Report 2015. https://www.alz.co.uk/news/world-alzheimer-report-2015-reveals-global-cost-of-dementia-set-to-reach-usd-1-trillion-by-2018. |
[2] | Plattner F , Angelo M , Giese KP ((2006) ) The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem 281: , 25457–25465. |
[3] | Das TK , Abdul Hamid MRW , Das T , Fatima-Shad K ((2015) ) Potential of glycowithanolides from(Ashwagandha) as therapeutic agents for the treatment of Alzheimer’s disease. World J Pharm Res 4: , 16–38. |
[4] | Chow HM , Guo D , Zhou JC , Zhang GY , Li HF , Herrup K , Zhang J ((2014) ) CDK5 activator protein p25 preferentially binds and activates GSK3β. Proc Natl Acad Sci U S A 111: , 4887–4895. |
[5] | Shah K , Lahiri DK ((2014) ) Cdk5 activity in the brain – multiple paths of regulation. J Cell Sci 127: , 2391–2400. |
[6] | Shukla V , Skuntz S , Pant HC ((2012) ) Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch Med Res 43: , 655–662. |
[7] | Das TK , Chakrabarti SK , Zulkipli IN , Mas RW ((2019) ) Curcumin ameliorates the impaired insulin signaling involved in the pathogenesis of Alzheimer’s disease in rats. J Alzheimers Dis Rep 3: , 59–70. |
[8] | Haider S , Tabassum S , Perveen T ((2016) ) Scopolamine induced greater alterations in neurochemical profile and increased oxidative stress demonstrated a better model of dementia: A comparative study. Brain Res Bull 127: , 234–247. |
[9] | Bihaqi SW , Singh AP , Tiwari M ((2012) ) Supplementation ofattenuates scopolamine-induced increased tau and amyloid precursor protein (APP) expression in rat brain. Indian J Pharmacol 44: , 593–598. |
[10] | Klinkenberg I , Blokland A ((2010) ) The validity of scopolamine as a pharmacological model for cognitive impairment: A review of animal behavioral studies. Neurosci Biobehav Rev 34: , 1307–1350. |
[11] | Newman LA , Gold PE ((2016) ) Attenuation in rats of impairments of memory by scopolamine, a muscarinic receptor antagonist, by mecamylamine, a nicotinic receptor antagonist. Psychopharmacology (Berl) 233: , 925–932. |
[12] | Kumar A , Dora J , Singh A ((2011) ) A review on spice of life curcuma longa (turmeric). Int J Appl Biol Pharm 2: , 371–379. |
[13] | Ono K , Hasegawa K , Naiki H , Yamada MJ ((2010) ) Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta fibrils in vitro. Neurosci Res 75: , 742–750. |
[14] | Goverdhan P1 , Sravanthi A , Mamatha T ((2010) ) Neuroprotective effects of meloxicam and selegiline in scopolamine-induced cognitive impairment and oxidative stress. Int J Alzheimers Dis 2012: , 974013. |
[15] | Boivin GP , Hickman DL , Creamer-Hente MA , Pritchett-Corning KR , Bratcher NA ((2017) ) Review of CO2 as a euthanasia agent for laboratory rats and mice. J Am Assoc Lab Anim Sci 56: , 491–499. |
[16] | Zheng D , Duan H , Wang S , Xu Q , Gan L , Li J , Dong Q ((2018) ) FAK regulates epithelial-mesenchymal transition in adenomyosis. Mol Med Rep 18: , 5461–5472. |
[17] | Das TK , Mas RW , Kaneez FS ((2014) ) Oxidative stress gated by Fenton and Haber Weiss reactions and its association with Alzheimer’s disease. Arch Neurosci 2: , 1–8. |
[18] | Vandal M , White PJ , Tremblay C , St-Amour I , Chevrier G , Emond V , Lefrançois D , Virgili J , Planel E , Giguere Y , Marette A , Calon F ((2014) ) Insulin reverses the high-fat diet–induced increase in brain Aβ and improves memory in an animal model of Alzheimer disease. Diabetes 63: , 4291–301. |
[19] | Lee HK , Kumar P , Fu Q , Rosen KM , Querfurth HW ((2009) ) The insulin/Akt signaling pathway is targeted by intracellular β-amyloid. Mol Biol Cell 20: , 1533–1544. |
[20] | Bedse G , Di Domenico F , Serviddio G , Cassano T ((2015) ) Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front Neurosci 9: , 204.1–13. |
[21] | Wang Y , Zhang JX , Du XX , Zhao L , Tian Q , Zhu LQ , Wang SH , Wang JZ ((2008) ) Temporal correlation of the memory deficit with Alzheimer-like lesions induced by activation of glycogen synthase kinase-3. J Neurochem 106: , 2364–2374. |
[22] | Mendes CT , Mury FB , de Sá Moreira E , Alberto FL , Forlenza OV , Dias-Neto E , Gattaz WF ((2009) ) Lithium reduces Gsk3b mRNA levels: Implications for Alzheimer disease. Eur Arch Psychiatry Clin Neurosci 259: , 16–22. |
[23] | Lesort M , Jope RS , Johnson GV ((1999) ) Insulin transiently increases tau phosphorylation: Involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J Neurochem 72: , 576–584. |
[24] | de Paula VJR , Guimarães FM , Diniz BS , Forlenza OV ((2009) ) Neurobiological pathways to Alzheimer’s disease amyloid-beta, tau protein or both? Dement Neuropsychol 3: , 188–194. |
[25] | Kockeritz L , Doble B , Patel S , Woodgett JR ((2006) ) Glycogen synthase kinase-3—an overview of an over-achieving protein kinase. Curr Drug Targets 7: , 1377–1388. |
[26] | Dajani R , Fraser E , Roe SM , Young N , Good V , Dale TC , Pearl LH ((2001) ) Crystal structure of glycogen synthase kinase 3β: Structural basis for phosphate primed substrate specificity and autoinhibition. Cell 105: , 721–732. |
[27] | Lei P , Ayton S , Bush AI , Adlard PA ((2011) ) GSK-3 in neurodegenerative diseases. Int J Alzheimers Dis 2011: , 189246. |
[28] | Shelton SB , Johnson GV ((2004) ) Cyclin-dependent kinase-5 in neurodegeneration. J Neurochem 89: , 1313–1326. Erratum in: J Neurochem 89, 528, 2004. |
[29] | Crews L , Patrick C , Adame A , Rockenstein E , Masliah E ((2011) ) Modulation of aberrant CDK5 signaling rescues impaired neurogenesis in models of Alzheimer’s disease. Cell Death Dis 2: , 1–13. |
[30] | Li T , Hawkes C , Qureshi HY , Kar S , Paudel HK ((2006) ) Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry 45: , 3134–3145. |
[31] | Li T , Paudel HK ((2006) ) Glycogen synthase kinase 3beta phosphorylates Alzheimer’s disease-specific Ser396 of microtubule-associated protein tau by a sequential mechanism. Biochemistry 45: , 3125–3133. |
[32] | Town T , Zolton J , Shaffner R , Schnell B , Crescentini R , Wu Y , Zeng J , DelleDonne A , Obregon D , Tan J , Mullan M ((2002) ) p35/Cdk5 pathway mediates soluble amyloid-β peptide-induced tau phosphorylation in vitro. J Neurosci Res 69: , 362–372. |
[33] | Otth C , Concha II , Arendt T , Stieler J , Schliebs R , González BC , Maccioni RB ((2002) ) AβPP induces cdk5-dependent tau hyperphosphorylation in transgenic mice Tg2576. J Alzheimers Dis 4: , 417–430. |


