
Gemfibrozil, a Lipid-Lowering Drug, Lowers Amyloid Plaque Pathology and Enhances Memory in a Mouse Model of Alzheimer’s Disease via Peroxisome Proliferator-Activated Receptor α
Abstract
Deposition of extracellular senile plaques containing amyloid-β is one of the major neuropathological characteristics of Alzheimer’s disease (AD). Therefore, targeting amyloid-β dyshomeostasis is an important therapeutic strategy for treatment of AD. In this study, we demonstrate that gemfibrozil, an FDA-approved drug for hyperlipidemia, can lower the amyloid plaque burden in the hippocampus and cortex of the 5XFAD model of AD. Additionally, gemfibrozil reduced microgliosis and astrogliosis associated with plaque in these mice. Administration of gemfibrozil also improved spatial learning and memory of the 5XFAD mice. Finally, we delineate that gemfibrozil requires the transcription factor peroxisome proliferator-activated receptor alpha (PPARα) to exhibit its amyloid lowering and memory enhancing effects in 5XFAD mice. These results highlight a new therapeutic property of gemfibrozil and suggest that this drug may be repurposed for treatment of AD.
INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disorder and at present there is no treatment available to cure or halt the progression of this debilitating disorder. Although AD involves a multifactorial etiology, the amyloid cascade hypothesis is widely established as the underlying model of AD pathophysiology [1]. According to this hypothesis, deposition of amyloid-β (Aβ) is the initiating and driving event in AD pathogenesis and an imbalance between production and clearance of Aβ leads to other pathological events including neurofibrillary tangles, neuronal dysfunction, and dementia [1–3]. Therefore, many strategies for development of novel therapeutics for AD are focused on targeting Aβ dyshomeostasis.
Senile plaques, one of the major neuropathological hallmarks of AD, are formed by abnormal accumulation of Aβ and are broadly classified into two categories, diffuse and dense-core plaques. Diffuse plaques are thioflavin-S-negative, non-neuritic and are frequently observed in the cognitively intact aged people; however, dense-core plaques, present in the brains of AD patients, are composed of fibrillar Aβ and stain positively for Thioflavin-S [4]. These compact-core plaques are associated with dystrophic neurites or neuritic plaques, accumulation of reactive astrocytes and microglia surrounding the plaque [5–7], neuronal degeneration, and synaptic loss [8–10].
Gemfibrozil, marketed as ‘Lopid’, is a FDA-approved drug for hyperlipidemia that activates the nuclear transcription factor peroxisome proliferator-activated receptor-α (PPARα) by binding to the ligand-binding domain of the receptor [11]. In addition to its hypolipidemic actions, gemfibrozil has been reported to regulate numerous cellular pathways. Gemfibrozil has been demonstrated to upregulate the lysosomal enzyme tripeptidyl peptidase-1 (TPP1) via PPARα and thus has therapeutic implications for late infantile neuronal ceroid lipofuscinosis (LINCL) treatment [12]. Additionally, gemfibrozil reduced the storage material burden and enhanced longevity in a LINCL mouse model [13]. Importantly, activation of PPARα by gemfibrozil was shown to induce lysosomal biogenesis in mouse brain cells via transcriptional upregulation of the master regulator TFEB [14]. In AD, PPARα activation by gemfibrozil has been shown to upregulate the expression of the alpha secretase ADAM10 [15]. Given that gemfibrozil can both inhibit the production of Aβ via upregulating ADAM10 and stimulate cellular clearance by inducing lysosomal biogenesis, we explored the role of gemfibrozil on amyloid plaque pathology and memory in the 5XFAD model of AD.
MATERIALS AND METHODS
Reagents
Gemfibrozil was obtained from Sigma Aldrich. Thioflavin-S and all other molecular biology-grade chemicals were purchased from Sigma Aldrich. Primary antibody for β-Amyloid mouse monoclonal (6E10) was obtained from Biolegend (803001) and used at 1 : 500 dilution for immunohistochemistry and 1 : 1000 for immunoblotting. Primary antibody for β-Actin was obtained from Abcam (ab6276) and used at 1 : 10000 dilutions for immunoblotting. Iba1 (Abcam: cat# ab5076; dilution 1 : 1000), GFAP (Dako: cat# IS524; dilution 1 : 2000), PPARα (Santa cruz: cat# sc-398394; dilution 1 : 250), NeuN (Invitrogen: cat# 702022; dilution 1 : 500), and inducible nitric oxide synthase (iNOS) (BD Biosciences: cat# 610432; dilution 1 : 500) antibodies were used at respective dilutions for immunohistochemistry. Secondory antibodies (Alexa-fluor 488 and 647-conjugated) for immunostaining were purchased from Jackson ImmunoResearch. IR-dye-labeled secondary antibodies were purchased from Li-Cor Biosciences.
Animals
Animal maintaining and experiments were in accordance with National Institute of Health guidelines and were approved by the Institutional Animal Care and Use committee (IACUC) of the Rush University of Medical Center, Chicago, IL. Animals exhibiting mild seizures and tremors were fed and watered through animal feeding needles. However, if any mouse came to the moribund stage, it was decapitated after anesthesia with ketamine/xylazine injectables. Conditions for moribund were as follows: Central nervous system disturbance (Head tilt, Seizures, Tremors, Circling, Spasticity, and Paresis); Inability to remain upright; Evidence of muscle atrophy; Chronic diarrhea or constipation; Rough coat and distended abdomen; Spreading area of alopecia caused by disease; Coughing, rales, wheezing and nasal discharge; Distinct jaundice and/or paleness (anemia); Markedly discolored urine, polyuria or anuria; Frank bleeding from any orifice; Persistent self-induced trauma.
5XFAD [B6SJL-Tg(APPSwFlLon,PSEN1 *M146L *L286V)6799 Vas/J] and PPARα null (B6.129S4-Pparatm1Gonz/J) mice were purchased from Jackson Laboratories (Bar Harbor, ME). These animals were inbred and subsequent generations were screened by PCR. PPARα null mice [16] were maintained as homozygous on the C57BL/6J background. 5XFAD mice mimic some of the features of AD including amyloid plaque, neuroinflammation, impairment in memory and learning, etc. [17–19]. 5XFAD mice were also bred with PPARα (–/–) mice to generate 5XFAD/PPARα (–/–) mice as described before [15, 20].
Gemfibrozil treatment
Six-month-old male and female 5XFAD (n = 6 in each group) or 5X/Ppara–/– mice (n = 5 in each group) were orally administered with gemfibrozil (7.5 mg/kg body weight/day) or vehicle (0.1% Methylcellulose) via gavage for 30 days. This dose of gemfbrozil is equivalent to the human adult dose (600 mg/d/adult). Non-transgenic mice (n = 6) from the same background were used as a control. Following treatment, behavioral tests were performed following which mice were sacrificed. Mice were injected with ketamine-xylazine injectables and anesthetized mice were perfused using PBS for 5 min. Hemi-brain was kept in 4% Paraformaldehyde and processed for immunohistochemistry whereas the hippocampus and cortex was dissected from the other half of the brain for biochemical analysis.
Immunoblotting
Western blotting was performed as described in earlier studies [12, 15]. Briefly, for in vitro experiments, cells were washed and scraped in PBS, collected in a microfuge tubes and spun down into a pellet. Next, cells were lysed in RIPA buffer containing protease phosphatase inhibitors and the supernatant was used for measuring the protein concentration using the Bradford method (Bio-Rad). For hippocampal tissue, the weight of the tissue was measured and it was dissolved in CHAPS buffer containing protease and phosphatase inhibitors followed by a spin of 30 min at 15000 rpm. The protein concentration was measured from the supernatant. Next, 15 to 30μg of total protein was mixed with SDS sample buffer and boiled for 5 min. The samples were electrophoresed on custom-made SDS-Polyacrylamide gels (8%, 10%, or 15%) or NuPAGE Novex 4–12% Bis-Tris gels (Invitrogen) using the Tris/Glycine/SDS or the MES running buffer followed by transfer onto nitrocellulose membrane (Bio-Rad) using semi-dry transfer apparatus (Thermo-Pierce). Following transfer, the membrane was blocked using Odyssey blocking buffer (Li-Cor) for 1 h and then incubated with primary antibody overnight under shaking conditions at 4°C. The following day, the membrane was thoroughly washed (3 washes 10 min each) in PBST (PBS + Tween 20) and incubated in IR-dye conjugated secondary antibody at room temperature for 1 h. Following washes in PBST, membranes were visualized in the Odyssey Infrared Imaging System (Li-COR, Lincoln, NE).
Densitometric analysis
Densitometric quantification of the immunoblots was performed using ImageJ (NIH, Bethesda, MD). Target bands were normalized using their respective β-actin loading controls from the same blot. Data are representative of the average fold change relative to the non-transgenic control.
Immunohistochemistry (IHC)
Following gemfibrozil treatment, mice were anesthetized and intracardially perfused with 1X PBS followed by 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. The brains were post fixed in paraformaldehyde overnight at 4°C, and on next day were stored in phosphate buffer containing 30% sucrose at 4°C. Hippocampal sections were cut and saved in serial order at –20°C until immunostained.
IHC using fluorescence microscopy
For immunohistochemistry, hemi brains were incubated with 4% paraformaldehyde (w/v) followed by incubation in 30% sucrose overnight at 4°C. Next, brains were thoroughly washed in PBS and embedded in O.C.T (Tissue Tech) at –80°C and prepared for cryosectioning. Prior to staining, 40μM free floating hippocampal sections were washed thoroughly in PBS. The sections were blocked using 2% BSA in PBSTT (PBS + Triton X-100 + Tween-20) for 1 h. Next, the sections were incubated with primary antibody in 1% PBSTT at 4°C overnight. The following day, sections were washed in PBSTT and incubated with 488 or 647- conjugated secondary antibody for 1 h at room temperature. Following washes in PBSTT, the sections were mounted on glass sides. MFI for immunostained images were measured using NIH image J software. The final MFI was analyzed after subtracting the value with the background signal of respective images.
For thioflavin-S and amyloid β 6E10 colabeling, following primary and secondary antibody incubation for 6E10, sections were incubated in 0.05% thioflavin-S for 5 min. Next, the sections were washed in 50% ethanol and PBS followed by mounting on a glass slide. The samples were visualized under Olympus BX41 fluorescence microscope.
IHC using light microscopy
Free floating sections (thickness; 40μm) were stained using standard immuno-histochemical procedures as described earlier [21, 22]. Briefly, after quenching with 3% hydrogen peroxide (H2O2) and 10% methanol for 5 min sections were pre-incubated in 2% BSA and normal horse serum (NGS; Vector Laboratories, Burlingame, CA) and 0.3% Triton X-100 for 60 min followed by incubation with primary antibodies overnight under cold conditions, followed by incubation for 2 h with the biotinylated secondary antibody (1 : 200, Vector Laboratories) the next day. Vectastain Elite ABC peroxidase kit (Vector Laboratories) was used for visualization using 0.06% diaminobenzidine and H2O2.
Mean fluorescence intensity (MFI) calculation
While measuring MFI, a closed polygon tool was used to draw the boundary around GFAP-ir or iNOS-ir cells followed by monitoring MFI from “statistical analysis tool” of the Microsuite V software. The final MFI was analyzed after subtracting the value with the background signal of respective images.
Barnes maze
Barnes maze test was performed as described in previous studies [17, 23] for assessing the spatial memory of the mice. Mice were trained for two days on a 20-hole Barnes maze with only one tunnel containing colored food bait. Mice were food deprived overnight prior to the training and test days to motivate the mice to find the food. During training, each mouse was positioned in the middle of the maze and retained there by a cylindrical 10 cm high start chamber. After 10 s, the cylinder was removed and the mouse was allowed to freely move and explore the maze and find the food baited tunnel on the maze. The maze was always well lit with light to generate sufficient light and heat which motivated the mouse to find the tunnel and escape into the tunnel. The tunnel was positioned under the same hole and was stable within the spatial environment. Following two days of training, mice were given rest for one day and tested on the maze on day 4. On the test day, food-deprived mouse was placed in the middle of the maze and their performance was recorded using the Noldus system. Memory of the mouse was analyzed by latency (time to reach the goal box) and errors (no. of errors made before reaching the goal box). The maze was cleaned thoroughly after each trial.
T maze
T maze for assessing the memory of the mice was performed as described in previous studies [17, 23]. Briefly, mice were trained and habituated on the T maze for two days and were food deprived prior to the training so that they reach for the food reward (placed on the right arm) multiple times during the 10 min training period. During training, mice were placed at the start point for 30 s and allowed to make a forced entry into the right arm which was baited with color food chips while the left arm of the maze was blocked. After entering the right arm, mice were allowed to stay and habituate for 30 to 45 s. On the test day, the left arm was unblocked and food-deprived mouse was placed at the start point allowing it to either enter right arm (positive turn) which had the food bait or left arm (negative turn). The food reward side of the maze was always associated with a visual cue. Mice were allowed to make 15 trials and the numbers of positive and negative turns were recorded. The T maze was thoroughly cleaned after each trial.
Open field test
The open field test was conducted for analyzing the locomotor activity of mice. Mice were placed in the center of a square wooden open field arena (40×40 cm, 30 cm high walls) and allowed to explore freely for 5 min. The movements of the mice were recorded using a camera linked to the Noldus system and EthoVisionXT software. The cumulative duration of movement of the mice were analyzed to assess the general locomotor activity of the mice. The open field was cleaned thoroughly after each trial.
Statistical analysis
Statistical analyses were performed in GraphPad Prism. Data sets were analyzed by one-way ANOVA followed by Tukey’s post hoc test or student’s t-test. Data represented as mean±SEM. A level of p < 0.05 was considered statistically significant ( *p < 0.05, **p < 0.01, and ***p < 0.001).
RESULTS
Gemfibrozil attenuates amyloid plaque load in 5XFAD mice
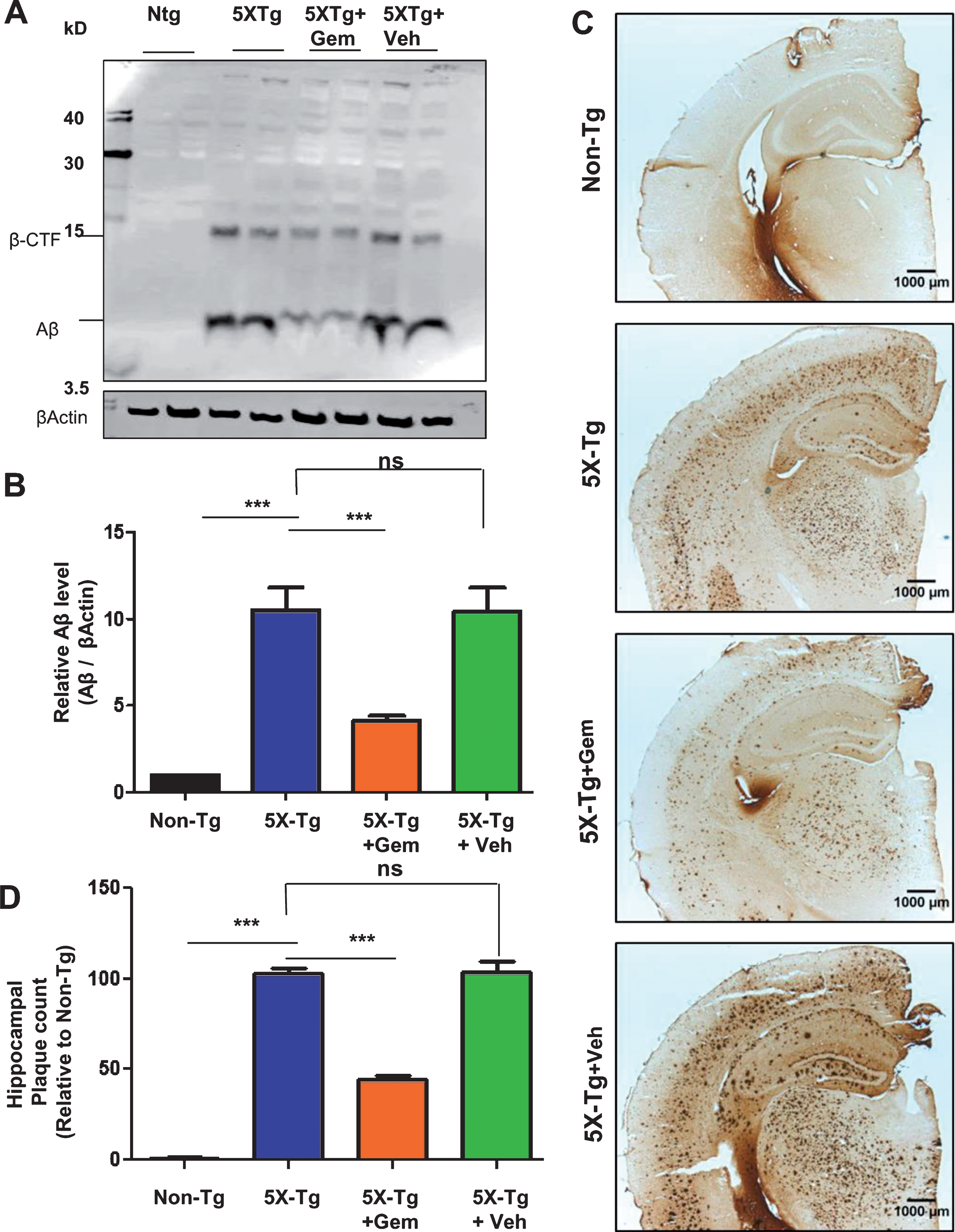
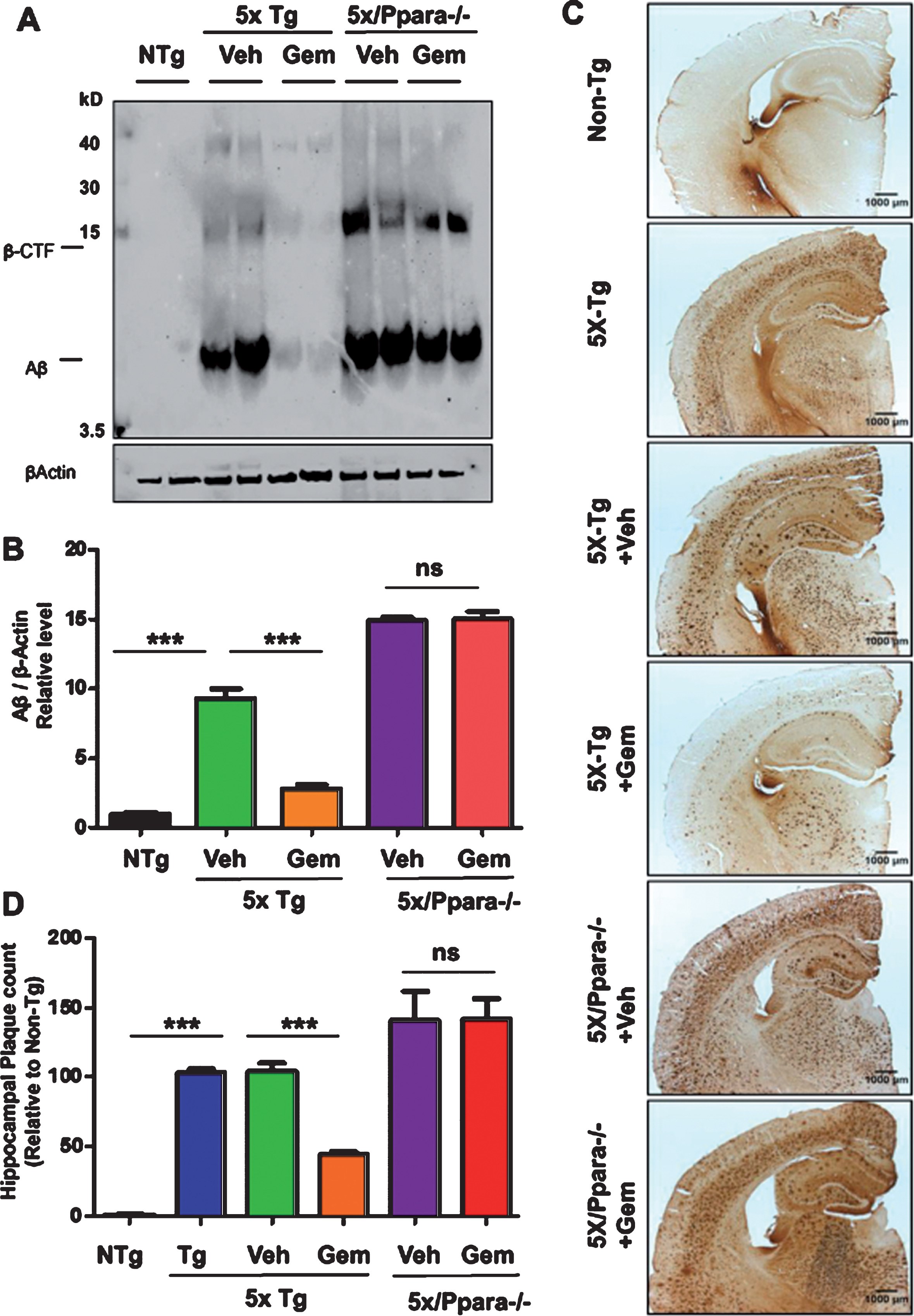
To begin with, we examined the effect of gemfibrozil on plaque deposition in the 5XFAD model of AD. Six-month-old male and female 5XFAD mice were orally treated with 7.5 mg/kg/day Gemfibrozil or vehicle for one month followed by monitoring the Aβ burden in the hippocampus and cortex. We performed double-labeling of hippocampal sections with thioflavin-S, a classical dye that detects the amyloid plaques and 6E10 monoclonal antibody. Data showed that gemfibrozil remarkably lowered the amyloid plaque burden in the hippocampus compared to the transgenic control (Tg) and vehicle treated (Tg + Veh) mice (Fig. 1A–C). Further quantification of the plaque area fraction (% thioflavin S-positive area) in the hippocampus revealed a significant reduction in the gem treated mice but not in the vehicle treated mice (Fig. 1B). Similarly, analysis of the number of thioflavin S-positive puncta in the hippocampus showed that gemfibrozil treated mice had marked decrease relative to the Tg and vehicle treated mice (Fig. 1C). A similar pattern of results was observed in the cortex of these mice where gemfibrozil distinctly attenuated the load of thioflavin S-positive plaques (Fig. 2A, B). Next, we analyzed the levels of Aβ using Aβ monoclonal antibody 6E10 in the hippocampus of these mice. Immunoblotting of hippocampal homogenates showed that gemfibrozil significantly reduced the level of Aβ in hippocampus of 5XFAD mice relative to the transgenic control and vehicle groups (Fig. 3A, B). We further confirmed our results by performing diaminobenzidine staining using Aβ 6E10. Data showed that gemfibrozil markedly reduced the levels of Aβ in the hippocampus of these mice (Fig. 3C, D). Moreover, quantification of the number of amyloid plaques in the hippocampus revealed a significant reduction with gemfibrozil treatment (Fig. 3D). Collectively, these results indicate that oral administration of gemfibrozil attenuates the amyloid plaque pathology in the 5XFAD mice.
Fig.1
Gemfibrozil treatment reduces Aβ plaque burden in the hippocampus of 5XFAD mouse model of AD. Six-month-old 5XFAD mice (n = 6) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by (A) double labeling of free-floating hippocampal sections with thioflavin-S (Green) and 6E10 antibody (Red). Scale bar = 1000μm. Characterization of plaques by analyzing (B) Thio-S positive area percentage and (C) Thio-S puncta count. All data are represented as mean±SEM. One way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; *p < 0.05, **p < 0.01, ***p < 0.001.
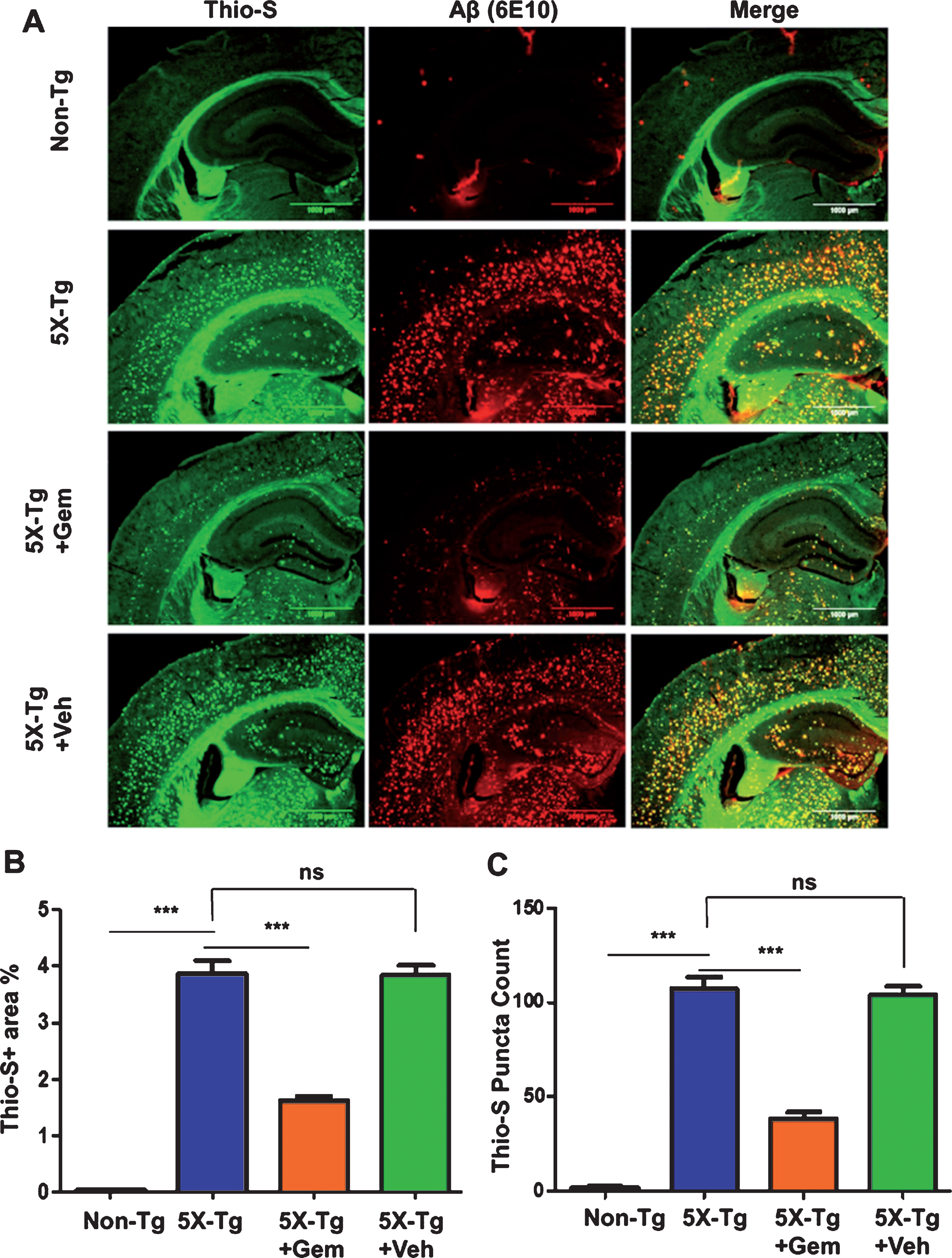
Fig.2
Gemfibrozil lowers amyloid plaque load in cortex of 5XFAD mice. Six-month-old 5XFAD mice (n = 6) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by analyzing the plaque deposition in the cortex by (A) double labeling of free floating sections with thioflavin-S (Green) and 6E10 antibody (Red). Scale bar = 50μm. B) Characterization of plaques by analyzing thio-S positive area percentage. All data are represented as mean±SEM. One way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; *p < 0.05, **p < 0.01, ***p < 0.001.
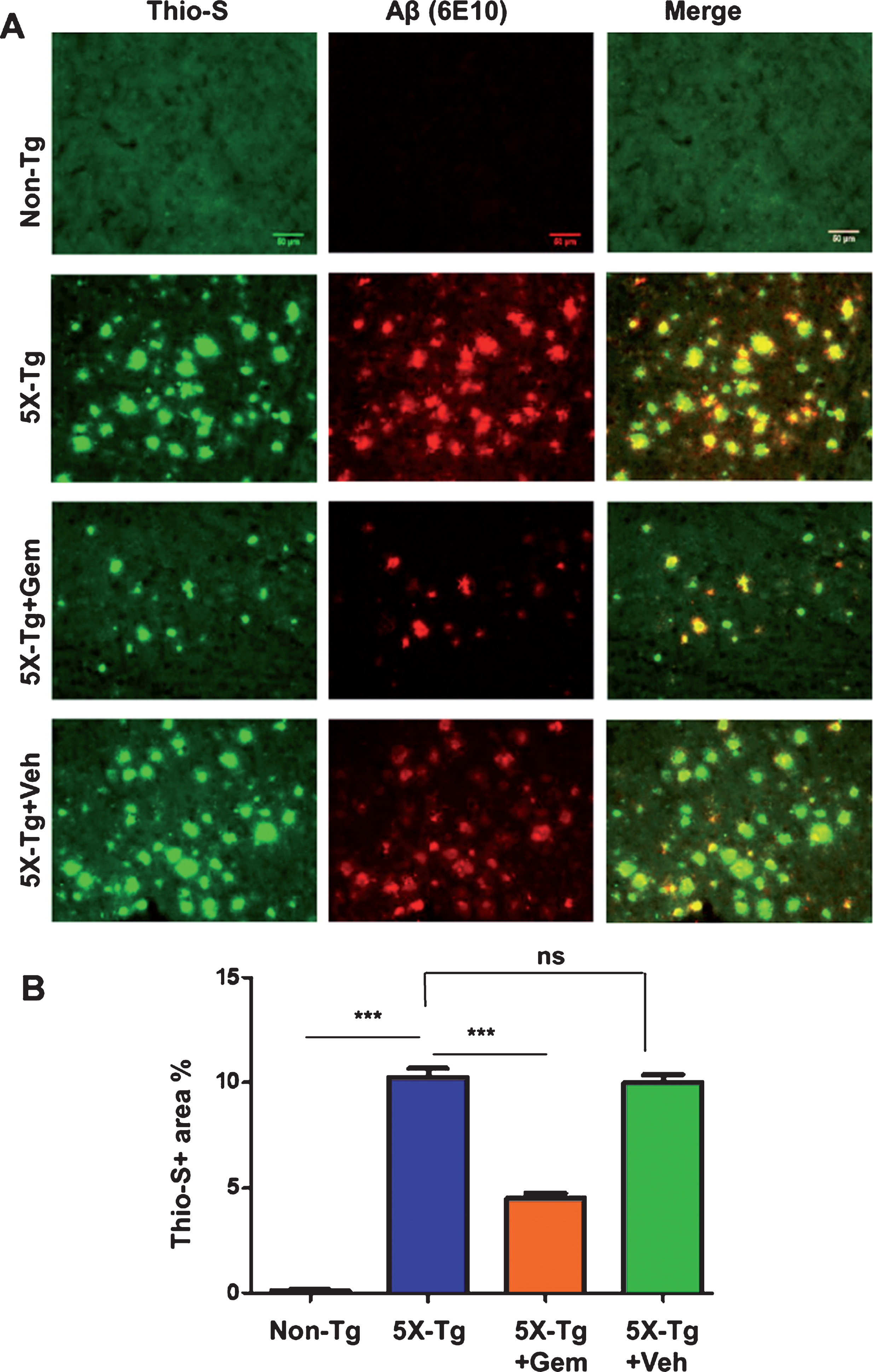
Fig.3
Administration of gemfibrozil reduces amyloid beta levels in hippocampus of 5XFAD mice. Six-month-old 5XFAD mice (n = 6) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by (A) analyzing the Aβ levels in hippocampal homogenates by western blot analysis and (B) densitometric analysis. β-Actin was used as a loading control. C) Diaminobenzidine staining was performed using 6E10 antibody. Scale bar = 1000μm. D) Quantification of number of plaques in the hippocampus relative to nontransgenic. All data are represented as mean±SEM. One way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; *p < 0.05, **p < 0.01, ***p < 0.001.
Gemfibrozil reduces glial activation in 5XFAD mice
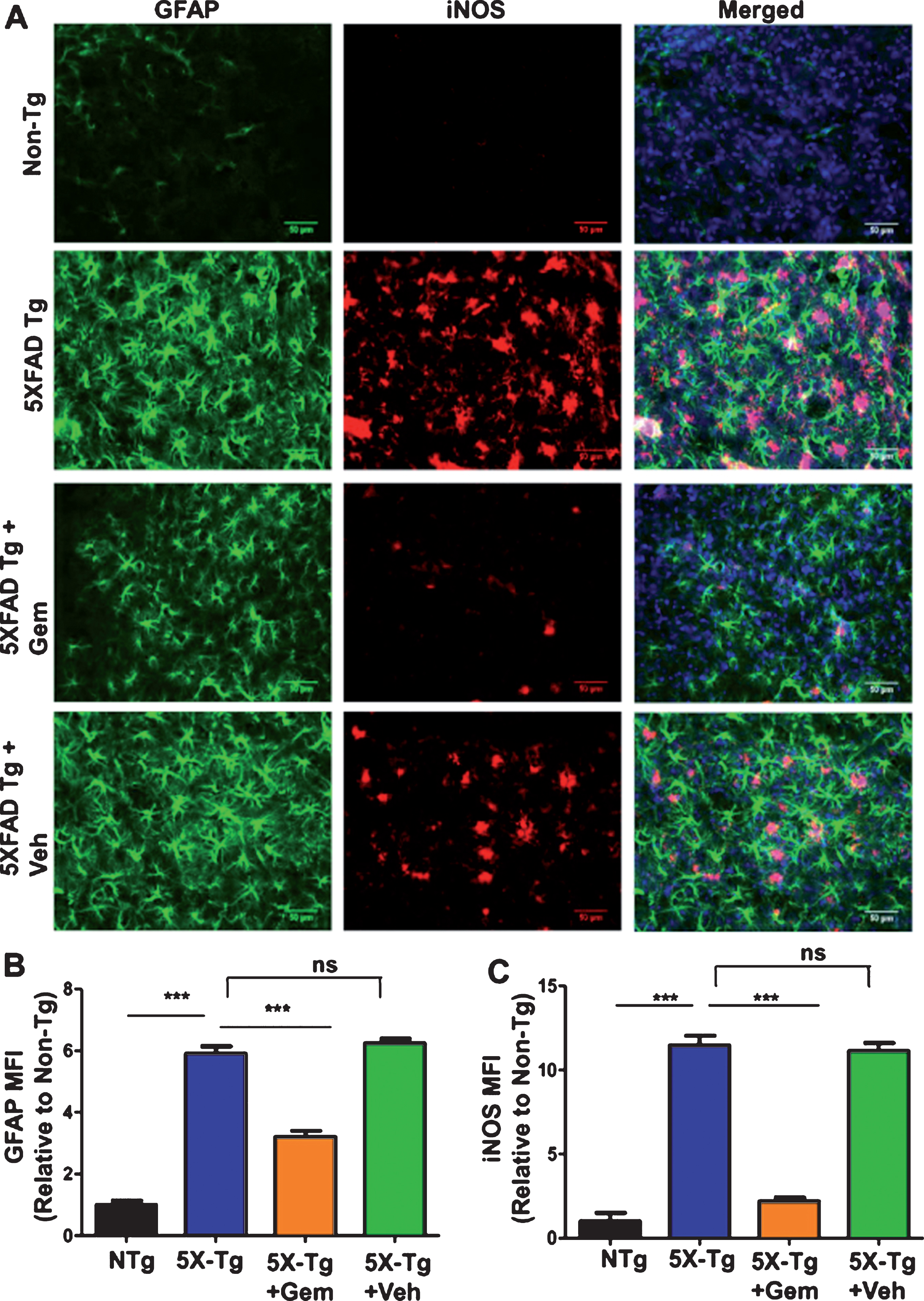
One of the major features of AD is the development of neuroinflammation which contributes to neurodegeneration [24]. The 5XFAD mice exhibit gliosis marked by plaque-surrounding activated microglia and astrocytes starting at 2 months and increasing with age and plaque deposition [25]. Hence, we analyzed the effect of gemfibrozil on microgliosis in the 5XFAD mice by checking microglial marker Iba1 immunoreactivity. Diaminobenzidine staining revealed that gemfibrozil was able to distinctly reduce the microgliosis in different regions of the hippocampus (Dentate gyrus and CA3) (Fig. 4A–D). We further quantified the number of Iba1 positive cells in the DG and CA3 region and observed that gemfibrozil treated mice had significantly reduced number of microglia relative to the untreated transgenic and vehicle treated mice (Fig. 4B, D). In addition to that, we checked the effect of gemfibrozil on microgliosis in the cortex of these mice. Our data showed gemfibrozil lowered microgliosis in the cortex of the 5XFAD mice whereas vehicle did not show any such effect (Fig. 5A, B). Next, we checked the expression of a proinflammatory marker, iNOS in the cortex by double labeling iNOS and Iba1. Our data showed that gemfibrozil treated mice had decreased expression of iNOS in cortex relative to the transgenic control and vehicle groups (Fig. 5C–E). Importantly, Iba1 immunoreactivity was found to be remarkably lowered with gemfibrozil treatment (Fig. 5C, D). Similarly, we observed increased astroglial activation in the cortex of 5XFAD mice as compared to non-Tg mice (Fig. 6A, B). However, gemfibrozil treatment inhibited astroglial activation as evidenced by decreased GFAP immunostaining and reduced iNOS expression (Fig. 6A, B). Together, these results suggest that gemfibrozil reduces the gliosis and neuroinflammation in the hippocampus and cortex of 5XFAD mice.
Fig.4
Gemfibrozil attenuates microgliosis in hippocampus of 5XFAD mice. Six-month-old 5XFAD mice (n = 6) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by monitoring microgliosis by diaminobenzidine staining for IBA1 in the (A) dentate gyrus and (C) CA3 regions of the hippocampus. Scale bar = 50μm. B, D) Quantification of IBA1-positive cells in the DG and CA3 regions respectively. All data are represented as fold change (mean±SEM) with respect to the nontransgenic. One-way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; *p < 0.05, **p < 0.01, ***p < 0.001.
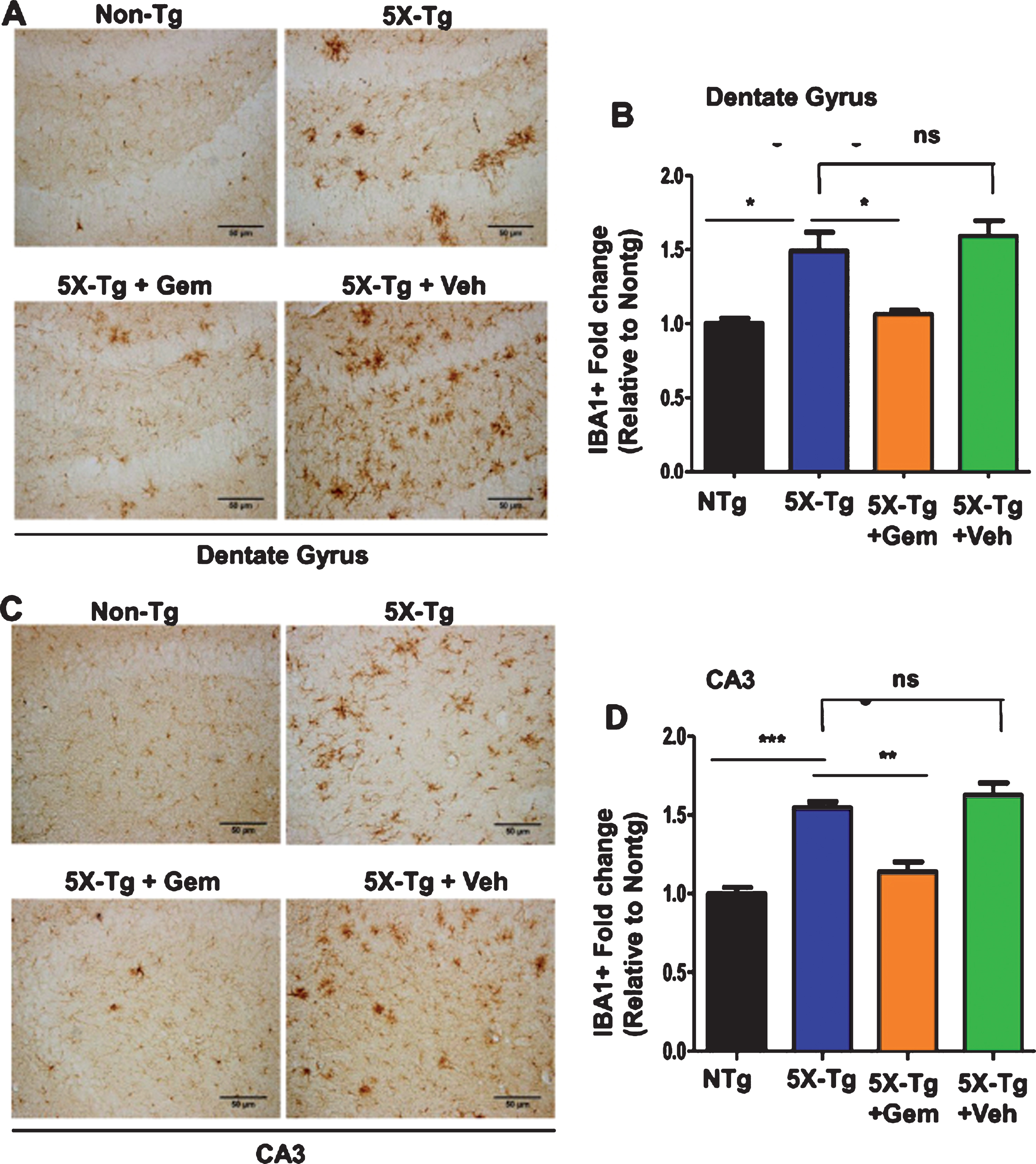
Fig.5
Gemfibrozil reduces microgliosis and iNOS levels in cortex of 5XFAD mice. Six-month-old 5XFAD mice (n = 6) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by monitoring (A) microgliosis by diaminobenzidine staining for IBA1 in the cortex and (B) quantitative analysis of IBA1-positive cells in the cortex; (C–E) Cortical sections were co-labeled with IBA1 and iNOS followed by mean fluorescence intensity analysis for (D) Iba1 and (E) iNOS in the cortex. Scale bar = 50μm. All data are represented as fold change (mean±SEM) with respect to the non-transgenic. One way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; *p < 0.05, **p < 0.01, ***p < 0.001.
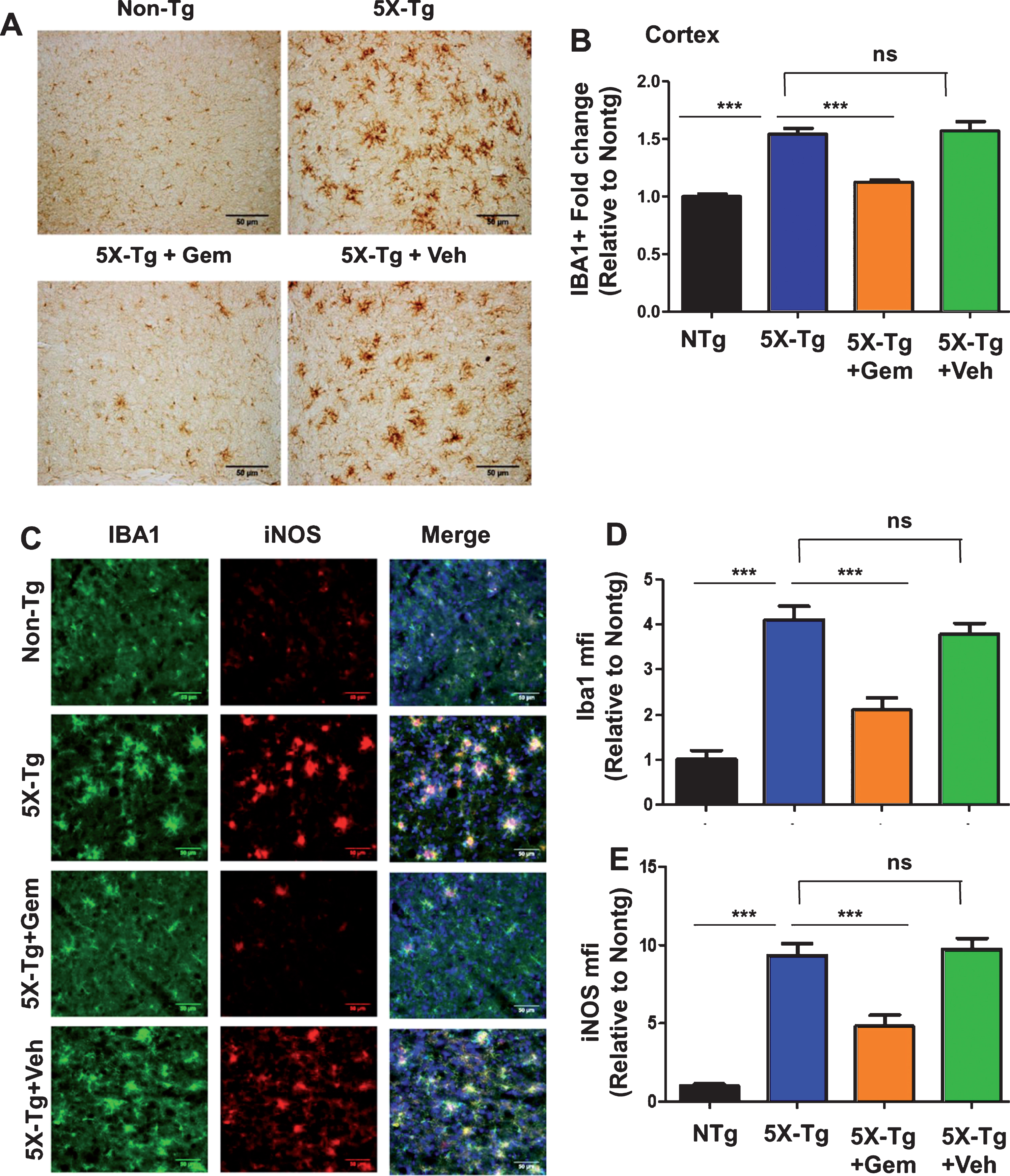
Fig.6
Oral gemfibrozil decreases astroglial activation in the cortex of 5XFAD mice. Six-month-old 5XFAD mice (n = 6) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by double-labeling of cortical sections for GFAP and iNOS (A). Scale bar = 50μm. Mean fluorescence intensity (MFI) of GFAP (B) and iNOS (C) was calculated from two sections (one image/section) of each of six mice per group. All data are represented as fold change (mean±SEM) with respect to the non-transgenic. One way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; ***p < 0.001; ns, not significant.
Gemfibrozil lowers amyloid pathology in a PPARα-dependent manner
Next, we investigated mechanism by which gemfibrozil exhibited plaque-lowering effect in 5XFAD mice. Since gemfibrozil is an agonist of PPARα, we examined the status of PPARα in the hippocampus of 5XFAD mice. The level of PPARα was significantly less in the hippocampus of 5XFAD mice as compared to non-Tg mice (Fig. 7A, B). However, gemfibrozil treatment restored and/or upregulated the level of PPARα in the hippocampus of 5XFAD mice (Fig. 7A, B). Therefore, our next goal was to determine whether gemfibrozil requires PPARα to exhibit amyloid-lowering effects in the 5XFAD mice. To explore the role of PPARα, we treated mice that present the five FAD mutations and are null for PPARα (5XFAD/PPARa–/–) with gemfibrozil or vehicle. We monitored the cerebral amyloid deposition in these cohorts of mice by co-labeling thioflavin-S and Aβ 6E10 antibody. Similar to our previous results, the amount of Aβ plaques in the hippocampus was significantly attenuated following gemfibrozil treatment in the transgenic mice (Fig. 8A–C). Interestingly, in the 5XFAD/PPARa–/– mice, gemfibrozil did not exhibit any effect on hippocampal amyloid plaque deposition (Fig. 8A–C) indicating that gemfibrozil specifically requires PPARα for lowering amyloid plaques. Quantitative analysis of the thioflavin-S area fraction and puncta count in the hippocampus revealed that in contrast to the 5XFAD mice, in the 5XFAD/PPARa–/– mice, there was no significant difference between the vehicle and the gemfibrozil treated mice (Fig. 8B, C). Next, we checked the plaque pathology in the cortex of these mice. A similar pattern of results was observed in the cortex where gemfibrozil reduced plaque deposition in 5XFAD mice, but not in 5XFAD/PPARa–/– mice (Fig. 9A, B). Plaque deposition in the cortex was assessed by the area fraction of thioflavin-S positive plaques which showed that gemfibrozil was not able to lower the plaque pathology in the 5XFAD/PPARa–/– mice (Fig. 9B) further confirming the role of PPARα in gemfibrozil-mediated plaque lowering. Furthermore, we analyzed the Aβ levels in the hippocampal homogenate by immunoblotting using 6E10 antibody. Data showed that unlike the transgenic 5XFAD mice, 5XFAD/PPARa–/– mice did not have any reduction in the level of Aβ (Fig. 10A, B) indicating that gemfibrozil was unable to exhibit in the absence of PPARα. Diaminobenzidine staining using Aβ 6E10 revealed similar results where gemfibrozil attenuated amyloid plaque in the 5XFAD mice, but not in the 5XFAD/PPARa–/– mice (Fig. 10C, D). Taken together, our results indicate that gemfibrozil attenuates amyloid pathology in a PPARα-dependent manner.
Fig.7
Oral gemfibrozil increases the level of PPARα in the hippocampus of 5XFAD mice. Six month old 5XFAD mice (n = 6) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by double-labeling of hippocampal sections for NeuN and PPARα (A). Scale bar = 50μm. Mean fluorescence intensity (MFI) of PPARα (B) was calculated from two sections (one image/section) of each of six mice per group. All data are represented as fold change (mean±SEM) with respect to the non-transgenic. One way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; *p < 0.05, **p < 0.01.
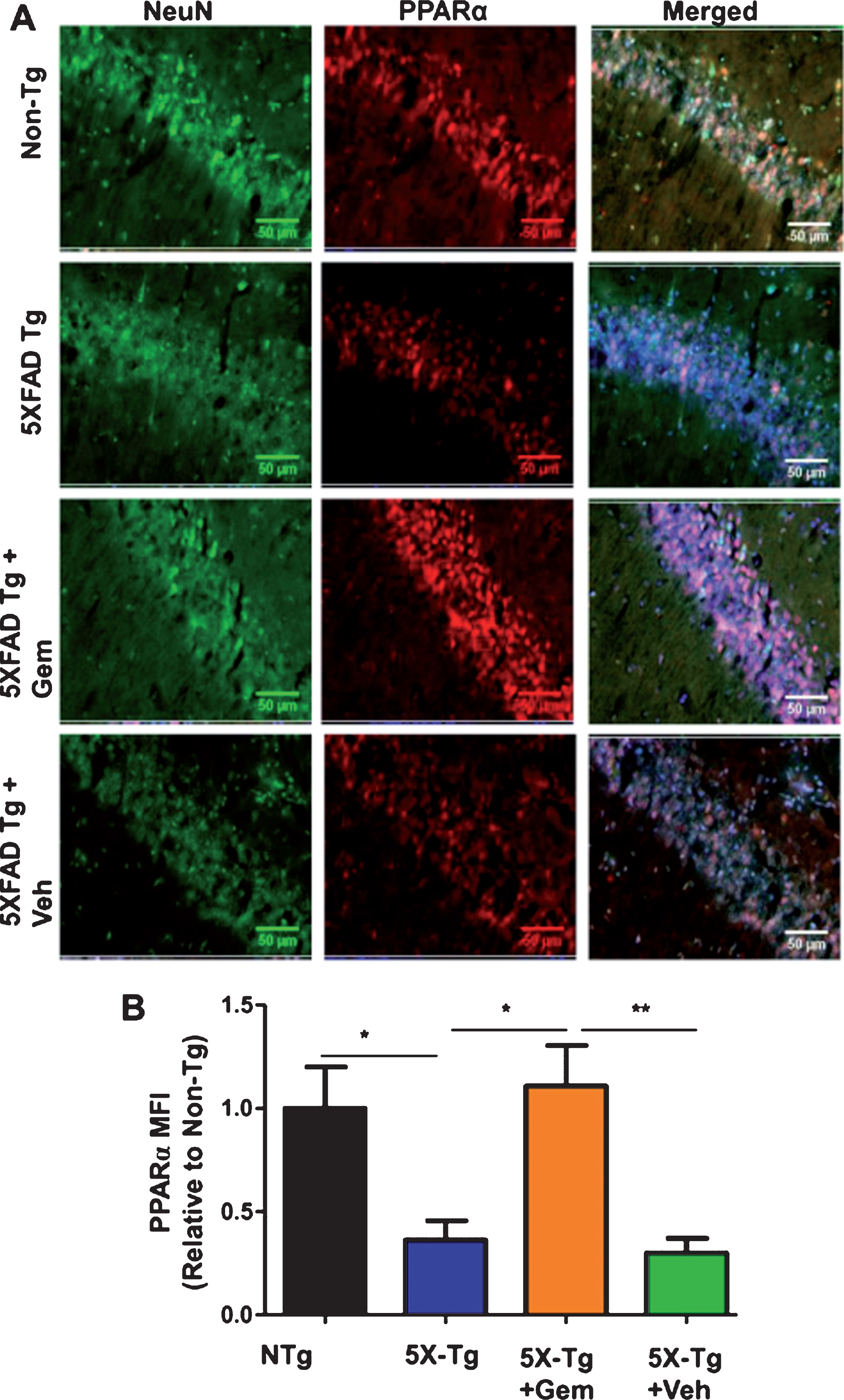
Fig.8
Gemfibrozil attenuates hippocampal amyloid pathology in a PPARα-dependent manner. Six month old 5XFAD (n = 6) and 5XFAD/PPARa–/– mice (n = 5) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by (A) double labeling of free floating hippocampal sections with thioflavin-S (Green) and 6E10 antibody (Red). Scale bar = 1000μm. Characterization of plaques by analyzing (B) Thio-S puncta count and (C) Thio-S positive area percentage. All data are represented as mean±SEM. One way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; *p < 0.05, **p < 0.01, ***p < 0.001.
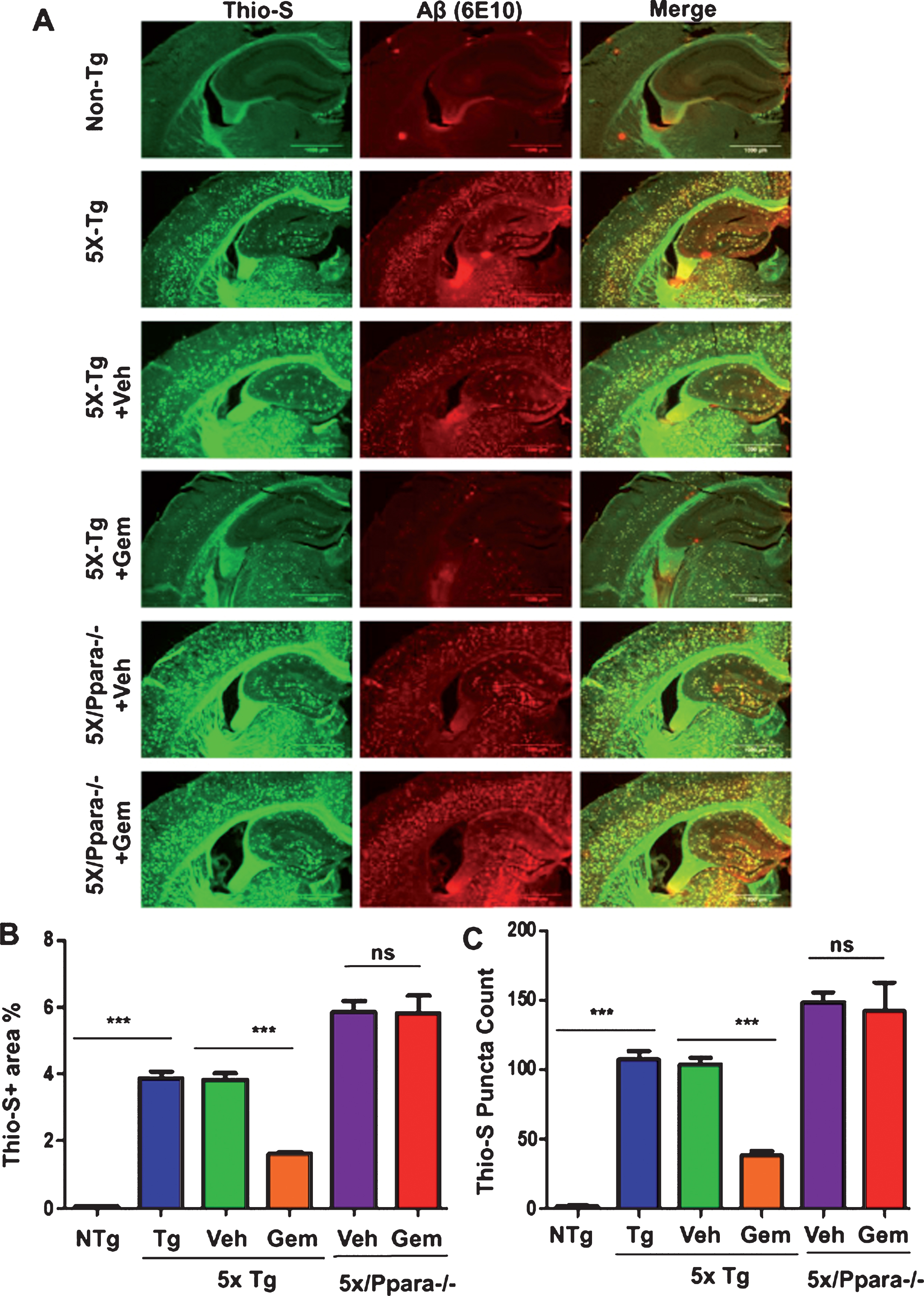
Fig.9
Gemfibrozil reduces cortical amyloid plaque in a PPARα-dependent manner. Six-month-old 5XFAD (n = 6) and 5XFAD/PPARa–/– mice (n = 5) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by (A) double labeling of free floating sections with thioflavin-S (Green) and 6E10 antibody (Red). Scale bar = 50μm. B) Characterization of plaques by analyzing thio-S positive area percentage. All data are represented as mean±SEM. One way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; *p < 0.05, **p < 0.01, ***p < 0.001.
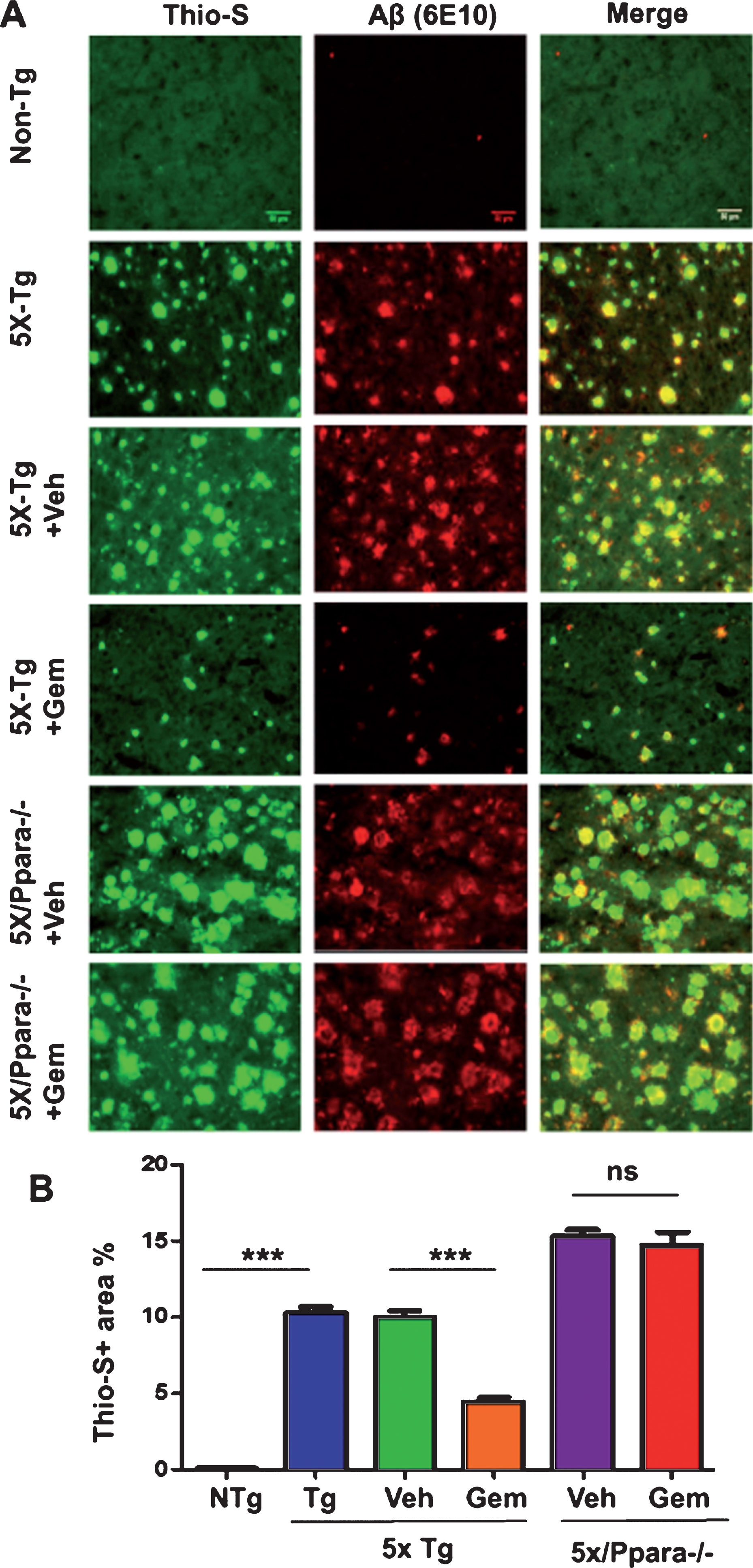
Fig.10
Gemfibrozil reduces amyloid beta levels via PPARα. Six-month-old 5XFAD mice (n = 6) and 5XFAD/PPARa–/– mice (n = 5) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by (A) analyzing the Aβ levels in hippocampal homogenates by western blot analysis using 6E10 antibody and (B) densitometric analysis. β-Actin was used as a loading control. C) Diaminobenzidine staining using 6E10 antibody. Scale bar = 1000μm. D) Quantification of number of plaques in the hippocampus relative to non-transgenic. All data are represented as mean±SEM. One-way ANOVA followed by Tukey’s multiple comparison test was used for statistical analysis; *p < 0.05, **p < 0.01, ***p < 0.001.
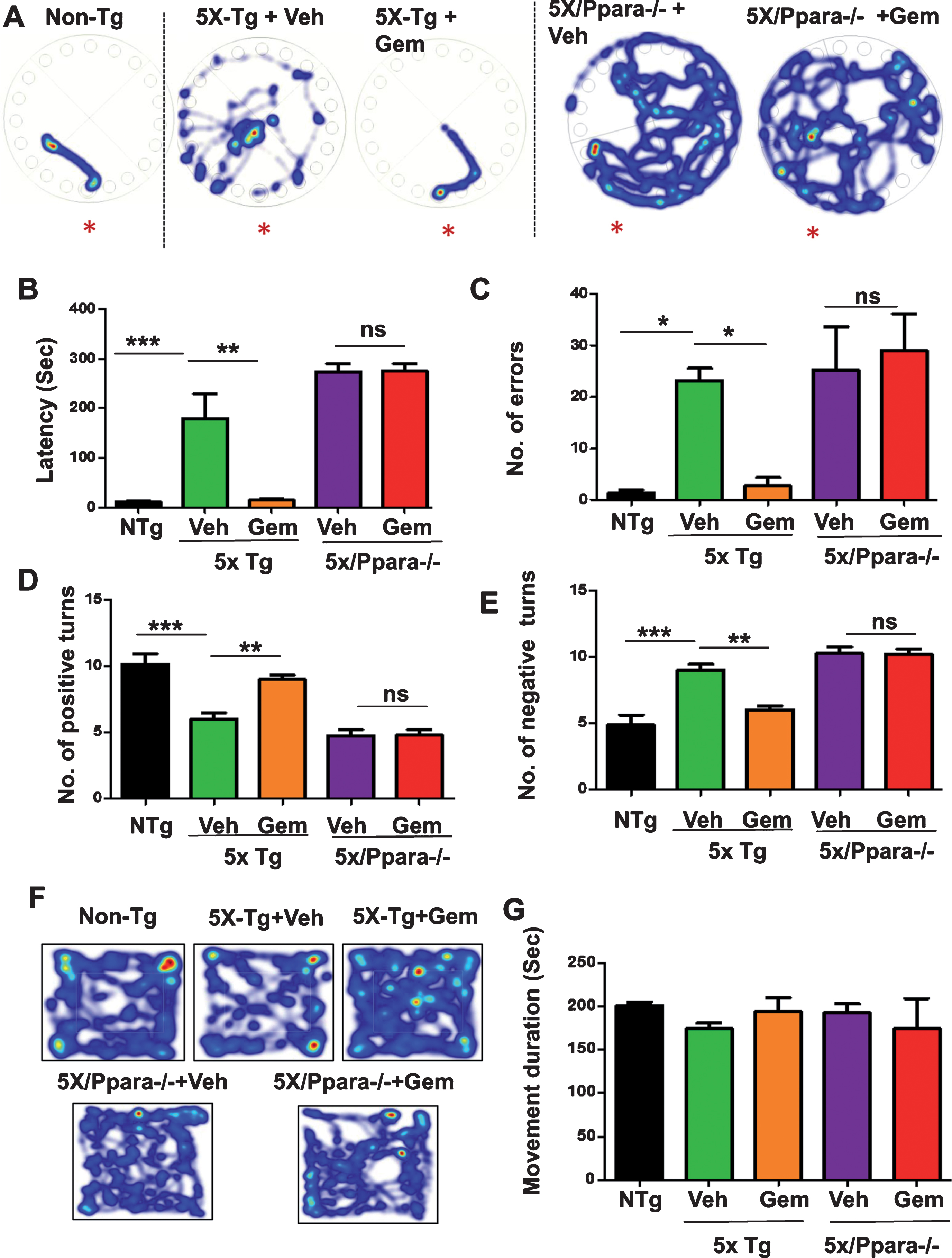
Gemfibrozil improves memory in 5XFAD model via PPARα
Cognitive impairment is the main clinical symptom of AD. Therefore, we analyzed the effect of Gemfibrozil on the memory of 5XFAD mice. Multiple PPARα agonists have been previously established to enhance memory via CREB [26]; however, the role of gemfibrozil on memory improvement has not yet been studied. Following one-month treatment with gemfibrozil or vehicle, the memory of these mice was analyzed by Barnes maze and T-maze tests. In Barnes maze test, gemfibrozil treated mice demonstrated significantly improved memory compared to the vehicle treated mice as shown by less latency (time to reach the goal box) and fewer errors (Fig. 11A–C). Similarly, in T-maze test, gemfibrozil treated mice made significantly more positive turns and less negative turns compared to the vehicle treated mice (Fig. 11D, E) indicating improvement of memory in the gemfibrozil group. We further performed open field test to monitor the locomotor activity of gemfibrozil and vehicle treated mice. Finally, we studied if gemfibrozil-mediated memory improvement is dependent on PPARα by comparing the memory of the 5XFAD and 5XFAD/PPARa–/– mice following gemfibrozil treatment. In this regard, we performed Barnes maze and T maze test which assesses the spatial memory of the mice. Interestingly, we observed that although gemfibrozil improved the memory of 5XFAD mice, in the 5XFAD/PPARa–/– mice, gemfibrozil did not show any effect on the memory (Fig. 11A–C). Vehicle- and gemfibrozil-treated mice did not have any significant difference in latency and errors (Fig. 11A–C). Parallel to our Barnes maze results, in the T maze test, gemfibrozil improved the performance of the 5XFAD, but not the 5XFAD/PPARa–/–, mice as demonstrated by no significant difference in the positive and negative errors in the gemfibrozil and vehicle treated cohorts of 5XFAD/PPARa–/– mice (Fig. 11D-E). These results suggested that under PPARα null conditions, gemfibrozil remains unable to improve the memory in the 5XFAD mice. Next, we analyzed the locomotor functions of these groups of mice by open field test and observed no significant difference in the movement cumulative duration of different groups of mice (Fig. 11F, G) indicating that the improvement in memory is specific and not due to any effect on locomotor activity of these mice. Together, these results demonstrate that gemfibrozil enhances the memory of 5XFAD mice via activation of PPARα.
Fig.11
Gemfibrozil improves memory of 5XFAD mice via PPARα. Six-month-old 5XFAD mice (n = 6) and 5XFAD/PPARa–/– mice (n = 5) were treated with gemfibrozil (7.5 mg/kg/day) or vehicle (0.1% methylcellulose) for 1 month followed by analysis of memory of the mice by (A–C) Barnes maze test; (A) representative heat maps; (B) latency to reach the goal box; (C) no. of errors made before reaching the goal box; (D-E) T maze test demonstrating (D) no. of positive turns and (E) no. of negative turns in T maze. F, G) Open field test for analysis of locomotor function; (F) Representative heat maps from open field test, (G) Velocity of mice in open field test; All data are represented as mean±SEM. Statistical analyses were performed by student’s t test; *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
One of the important therapeutic strategies for AD is to target the amyloid pathology which initiates and drives the disease [1, 2]. In our study, we studied the role of FDA-approved drug, gemfibrozil, on the amyloid plaque pathology in 5XFAD model of AD. Our study shows that administration of gemfibrozil attenuates the amyloid plaque burden and improves memory of 5XFAD mice. Additionally, we demonstrate that gemfibrozil can reduce the microgliosis, a main plaque-associated feature, in the hippocampus and cortex of the 5XFAD mice. We also delineate that gemfibrozil requires PPARα for lowering plaque load and enhancing memory. Our study highlights that gemfibrozil, a lipid-lowering drug, might have therapeutic implications for AD.
The major neuropathological characteristics of AD are the development of extracellular senile plaques containing amyloid beta and formation of intraneuronal neurofibrillary tangles [27]. The amyloid cascade hypothesis describes the deposition of Aβ as the initiating and driving factor in the disease pathogenesis [1–3]. Aβ is derived from AβPP, a neuronal type I transmembrane protein, involved in physiological processes such as neuronal trafficking, neurotrophic pathways, cell adhesion and signaling [28–31]. In the non-amyloidogenic pathway, proteolytic cleavage of AβPP within the Aβ domain by alpha secretase (ADAM 10) precludes Aβ formation [32, 33]. Alternatively, AβPP can be internalized into the early endosomes where it undergoes amyloidogenic processing via sequential cleavage by beta secretase BACE1 and gamma secretase complex to generate Aβ peptide comprised of 40 or 42 residues. Amongst these amyloid species, due to insolubility and a greater propensity to aggregate, Aβ42 is more toxic and abundantly found in the plaques [4, 27, 31, 34]. Our previous study had demonstrated that gemfibrozil upregulates the expression of ADAM10 via activation of PPARα [15]. Therefore, here we explored whether gemfibrozil has any beneficial effect on the amyloid pathology. Our study demonstrates that gemfibrozil reduces the deposition of amyloid plaque in six-month-old male and female 5XFAD mice.
To examine the effect of gemfibrozil on amyloid pathology, we monitored the burden of thioflavin-S and Aβ positive plaques in the hippocampus and cortex following one-month oral treatment with the drug. Thioflavin-S is a classical dye that specifically recognizes the β-pleated sheet conformation in Aβ plaques. While, diffuse plaques can be found in cognitively normal aged people and are thioflavin-S negative, dense core plaques are observed in AD brains and are thioflavin-S positive [4]. In this study, we observed that the 5XFAD mice exhibit profound development of thioflavin-S positive plaque; however, oral administration of gemfibrozil could lower the thioflavin-S positive plaque load in the hippocampus and cortex of the 5XFAD mice. Hence, gemfibrozil might have therapeutic importance for attenuating amyloid plaque in AD.
While examining the underlying mechanism of gemfibrozil-mediated attenuation of amyloid pathology, we observed decrease in PPARα in the hippocampus of 5XFAD mice. Gemfibrozil upregulated PPARα in the hippocampus of 5XFAD mice and required this transcription factor to exhibit its plaque-lowering effect. PPARs are ligand-inducible transcription factors belonging to the class of nuclear hormone receptors that primarily act as lipid sensors [35, 36]. Three different isoforms of PPARs: PPARα, PPARβ/δ, and PPARγ, have distinct tissue expression patterns, physiological functions and ligand specificity [37]. The α isoform, PPARα, is principally involved in regulation of energy homeostasis of the cell through inducing fatty acid and cholesterol metabolism and decreasing serum triglyceride content [35, 38]. Therefore, our study highlights an important crosstalk between lipid lowering and amyloid plaque removal.
Glial activation is a prominent feature of AD and contributes to the neuroinflammation which has been associated with neuronal function impairment, enhanced tau phosphorylation and production of Aβ [42]. Aβ can bind to several receptors on the microglial surface and initiate a cascade of inflammatory functions [43]. Activated microglia and astroglia secerete several proinflammatory cytokines and chemokines, some of which, like IL-1β, TNF-α, IL-6, IL-8, and TGF-β, were observed to have aberrant expression in AD patients [42, 44]. Moreover, polymorphisms in TNF-α, IL-1, and IL-6 are linked with higher risk of AD [45–48]. Given the detrimental effects of glial activation on AD pathology, we examined whether gemfibrozil has any effect on microglial and astroglial activation in 5XFAD mice. Our study shows that oral gemfibrozil treatment reduces both microgliosis and astrogliosis in the hippocampus and cortex of 5XFAD mice. Hence, in addition to reducing the amyloid plaque pathology, gemfibrozil could be beneficial in controlling the neuroinflammation in AD.
Finally, we examined the role of gemfibrozil on behavioral performance of the 5XFAD mice. In addition to its central role in lipid metabolism, mounting evidence suggests that PPARα improves memory via activating the CREB signaling pathway [23, 26, 49]. Therefore, we tested the effect of gemfibrozil on the memory deficit of 5XFAD mice. We observed that gemfibrozil remarkably improved the memory of the 5XFAD mice in Barnes maze and T maze tests. However, in the absence of PPARα, in the 5XFAD/PPARa–/– mice, gemfibrozil did not exhibit any memory-enhancing effect. Thus, our study highlights that gemfibrozil enhances the memory of 5XFAD mice via PPARα.
To summarize, our study identifies a novel role for FDA approved drug, gemfibrozil, in reducing the load of cerebral amyloid plaque in the 5XFAD model of AD. We demonstrate that administration of gemfibrozil lowers amyloid plaque pathology, reduces glial activation and improves memory in 5XFAD mice. We also delineate PPARα activation by gemfibrozil as the underlying mechanism. Therefore, gemfibrozil might be repurposed for lowering Aβ plaques and improving cognitive functions in AD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This study was supported by the Zenith Fellows Award (ZEN–17–438829) from Alzheimer’s Association, a grant (AG050431) from NIH and a merit award (1I01BX003033) from Veteran Affairs.
REFERENCES
[1] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[2] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[3] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[4] | Serrano-Pozo A , Frosch MP , Masliah E , Hyman BT ((2011) ) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1: , a006189. |
[5] | Itagaki S , McGeer PL , Akiyama H , Zhu S , Selkoe D ((1989) ) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24: , 173–182. |
[6] | Vehmas AK , Kawas CH , Stewart WF , Troncoso JC ((2003) ) Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol Aging 24: , 321–331. |
[7] | Pike CJ , Cummings BJ , Cotman CW ((1995) ) Early association of reactive astrocytes with senile plaques in Alzheimer’s disease. Exp Neurol 132: , 172–179. |
[8] | Masliah E , Mallory M , Hansen L , DeTeresa R , Alford M , Terry R ((1994) ) Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett 174: , 67–72. |
[9] | Urbanc B , Cruz L , Le R , Sanders J , Ashe KH , Duff K , Stanley HE , Irizarry MC , Hyman BT ((2002) ) Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc Natl Acad Sci U S A 99: , 13990–13995. |
[10] | Knowles RB , Wyart C , Buldyrev SV , Cruz L , Urbanc B , Hasselmo ME , Stanley HE , Hyman BT ((1999) ) Plaque-induced neurite abnormalities: Implications for disruption of neural networks in Alzheimer’s disease. Proc Natl Acad Sci U S A 96: , 5274–5279. |
[11] | Auwerx J ((1992) ) Regulation of gene expression by fatty acids and fibric acid derivatives: An integrative role for peroxisome proliferator activated receptors. The Belgian Endocrine Society Lecture 1992. Horm Res 38: , 269–277. |
[12] | Ghosh A , Corbett GT , Gonzalez FJ , Pahan K ((2012) ) Gemfibrozil and fenofibrate, Food and Drug Administration-approved lipid-lowering drugs, up-regulate tripeptidyl-peptidase 1 in brain cells via peroxisome proliferator-activated receptor alpha: Implications for late infantile Batten disease therapy. J Biol Chem 287: , 38922–38935. |
[13] | Ghosh A , Rangasamy SB , Modi KK , Pahan K ((2017) ) Gemfibrozil, food and drug administration-approved lipid-lowering drug, increases longevity in mouse model of late infantile neuronal ceroid lipofuscinosis. J Neurochem 141: , 423–435. |
[14] | Ghosh A , Jana M , Modi K , Gonzalez FJ , Sims KB , Berry-Kravis E , Pahan K ((2015) ) Activation of peroxisome proliferator-activated receptor alpha induces lysosomal biogenesis in brain cells: Implications for lysosomal storage disorders. J Biol Chem 290: , 10309–10324. |
[15] | Corbett GT , Gonzalez FJ , Pahan K ((2015) ) Activation of peroxisome proliferator-activated receptor alpha stimulates ADAM10-mediated proteolysis of APP. Proc Natl Acad Sci U S A 112: , 8445–8450. |
[16] | Peters JM , Lee SS , Li W , Ward JM , Gavrilova O , Everett C , Reitman ML , Hudson LD , Gonzalez FJ ((2000) ) Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta). Mol Cell Biol 20: , 5119–5128. |
[17] | Modi KK , Roy A , Brahmachari S , Rangasamy SB , Pahan K ((2015) ) Cinnamon and its metabolite sodium benzoate attenuate the activation of p21rac and protect memory and learning in an animal model of Alzheimer’s disease. PLoS One 10: , e0130398. |
[18] | Patel D , Roy A , Kundu M , Jana M , Luan CH , Gonzalez FJ , Pahan K ((2018) ) Aspirin binds to PPARalpha to stimulate hippocampal plasticity and protect memory. Proc Natl Acad Sci U S A 115: , E7408–E7417. |
[19] | Rangasamy SB , Corbett GT , Roy A , Modi KK , Bennett DA , Mufson EJ , Ghosh S , Pahan K ((2015) ) Intranasal delivery of NEMO-binding domain peptide prevents memory loss in a mouse model of Alzheimer’s disease. J Alzheimers Dis 47: , 385–402. |
[20] | Roy A , Jana M , Kundu M , Corbett GT , Rangaswamy SB , Mishra RK , Luan CH , Gonzalez FJ , Pahan K ((2015) ) HMG-CoA reductase inhibitors bind to PPARalpha to upregulate neurotrophin expression in the brain and improve memory in mice. Cell Metab 22: , 253–265. |
[21] | Ghosh A , Roy A , Liu X , Kordower JH , Mufson EJ , Hartley DM , Ghosh S , Mosley RL , Gendelman HE , Pahan K ((2007) ) Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A 104: , 18754–18759. |
[22] | Ghosh A , Roy A , Matras J , Brahmachari S , Gendelman HE , Pahan K ((2009) ) Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci 29: , 13543–13556. |
[23] | Roy A , Jana M , Corbett GT , Ramaswamy S , Kordower JH , Gonzalez FJ , Pahan K ((2013) ) Regulation of cyclic AMP response element binding and hippocampal plasticity-related genes by peroxisome proliferator-activated receptor alpha. Cell Rep 4: , 724–737. |
[24] | Heppner FL , Ransohoff RM , Becher B ((2015) ) Immune attack: The role of inflammation in Alzheimer disease. Nat Rev Neurosci 16: , 358–372. |
[25] | Oakley H , Cole SL , Logan S , Maus E , Shao P , Craft J , Guillozet-Bongaarts A , Ohno M , Disterhoft J , Van Eldik L , Berry R , Vassar R ((2006) ) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J Neurosci 26: , 10129–10140. |
[26] | Roy A , Kundu M , Jana M , Mishra RK , Yung Y , Luan CH , Gonzalez FJ , Pahan K ((2016) ) Identification and characterization of PPARalpha ligands in the hippocampus. Nat Chem Biol 12: , 1075–1083. |
[27] | LaFerla FM , Green KN , Oddo S ((2007) ) Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci 8: , 499–509. |
[28] | Reinhard C , Hebert SS , De Strooper B ((2005) ) The amyloid-beta precursor protein: Integrating structure with biological function. EMBO J 24: , 3996–4006. |
[29] | Zheng H , Koo EH ((2006) ) The amyloid precursor protein: Beyond amyloid. Mol Neurodegener 1: , 5. |
[30] | Hoareau C , Borrell V , Soriano E , Krebs MO , Prochiantz A , Allinquant B ((2008) ) Amyloid precursor protein cytoplasmic domain antagonizes reelin neurite outgrowth inhibition of hippocampal neurons. Neurobiol Aging 29: , 542–553. |
[31] | Nixon RA ((2007) ) Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 120: , 4081–4091. |
[32] | Asai M , Hattori C , Szabo B , Sasagawa N , Maruyama K , Tanuma S , Ishiura S ((2003) ) Putative function of ADAM9, ADAM10, and ADAM17 as APP alpha-secretase. Biochem Biophys Res Commun 301: , 231–235. |
[33] | Kojro E , Fahrenholz F ((2005) ) The non-amyloidogenic pathway: Structure and function of alpha-secretases. Subcell Biochem 38: , 105–127. |
[34] | Whyte LS , Lau AA , Hemsley KM , Hopwood JJ , Sargeant TJ ((2017) ) Endo-lysosomal and autophagic dysfunction: A driving factor in Alzheimer’s disease? J Neurochem 140: , 703–717. |
[35] | Kummer MP , Heneka MT ((2008) ) PPARs in Alzheimer’s disease. PPAR Res 2008: , 403896. |
[36] | Viswakarma N , Jia Y , Bai L , Vluggens A , Borensztajn J , Xu J , Reddy JK ((2010) ) Coactivators in PPAR-regulated gene expression. PPAR Res 2010: , 250126. |
[37] | Zoete V , Grosdidier A , Michielin O ((2007) ) Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim Biophys Acta 1771: , 915–925. |
[38] | Heneka MT , Landreth GE ((2007) ) PPARs in the brain. Biochim Biophys Acta 1771: , 1031–1045. |
[39] | Kersten S , Wahli W ((2000) ) Peroxisome proliferator activated receptor agonists. EXS 89: , 141–151. |
[40] | Motojima K ((1993) ) Peroxisome proliferator-activated receptor (PPAR): Structure, mechanisms of activation and diverse functions. Cell Struct Funct 18: , 267–277. |
[41] | Roy A , Pahan K ((2009) ) Gemfibrozil, stretching arms beyond lipid lowering. Immunopharmacol Immunotoxicol 31: , 339–351. |
[42] | Solito E , Sastre M ((2012) ) Microglia function in Alzheimer’s disease. Front Pharmacol 3: , 14. |
[43] | Salminen A , Ojala J , Kauppinen A , Kaarniranta K , Suuronen T ((2009) ) Inflammation in Alzheimer’s disease: Amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol 87: , 181–194. |
[44] | Sastre M , Klockgether T , Heneka MT ((2006) ) Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int J Dev Neurosci 24: , 167–176. |
[45] | McCusker SM , Curran MD , Dynan KB , McCullagh CD , Urquhart DD , Middleton D , Patterson CC , McIlroy SP , Passmore AP ((2001) ) Association between polymorphism in regulatory region of gene encoding tumour necrosis factor alpha and risk of Alzheimer’s disease and vascular dementia: A case-control study. Lancet 357: , 436–439. |
[46] | Perry RT , Collins JS , Wiener H , Acton R , Go RC ((2001) ) The role of TNF and its receptors in Alzheimer’s disease. Neurobiol Aging 22: , 873–883. |
[47] | Nicoll JA , Mrak RE , Graham DI , Stewart J , Wilcock G , MacGowan S , Esiri MM , Murray LS , Dewar D , Love S , Moss T , Griffin WS ((2000) ) Association of interleukin-1 gene polymorphisms with Alzheimer’s disease. Ann Neurol 47: , 365–368. |
[48] | Capurso C , Solfrizzi V , D’Introno A , Colacicco AM , Capurso SA , Capurso A , Panza F ((2004) ) Interleukin 6-174 G/C promoter gene polymorphism and sporadic Alzheimer’s disease: Geographic allele and genotype variations in Europe. Exp Gerontol 39: , 1567–1573. |
[49] | Roy A , Pahan K ((2015) ) PPARalpha signaling in the hippocampus: Crosstalk between fat and memory. J Neuroimmune Pharmacol 10: , 30–34. |


