
‘Amytrapper’, a Novel Immobilized Sepharose API Matrix, Removes Amyloid-β from Circulation in vitro
Abstract
Alzheimer’s disease (AD) is the most common cause of dementia among elderly patients afflicted by neurodegenerative diseases, caused by the accumulation of amyloid-β (Aβ). Therapeutic interventions in targeting and restricting Aβ production resulted in little or no success. However, recent studies have shown signs of success in validating Aβ as a target. Recombinant Technologies LLC (RTL) has developed and studied its proprietary Amytrap peptide to remove Aβ from circulation which in turn depletes brain Aβ in a clinically relevant mouse model of AD. In the current study, this Amytrap peptide (the active pharmacological ingredient, API) has been linked to sepharose matrix by click chemistry. The derivative namely ‘Amytrapper’ was confirmed to remove Aβ from the surrounding media spiked with Aβ42. Additional testing performed on Amytrapper with sera and plasma containing Aβ42 showed retention of Aβ42 upon increasing concentrations of biotinylated Aβ42 (bio-Aβ42). Specificity of this binding was confirmed via 1) pre-blocking Amytrapper with cold (unbiotinylated) Aβ42 followed by binding experiment with biotinylated Aβ42, 2) 2-dimensional SDS-PAGE analyses on samples harvested before and after the binding experiment, and 3) reconciling the amounts bound to beads and left over in the flow through. The results provide a proof of concept for our proposed prototype design for an Amytrapper device. The results suggest that extracorporeal clearance of Aβ42 by Amytrapper could be a way to manage accumulation of amyloid in AD and thus could become an added mode of therapy for disease modification.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease that has been found to be the fourth most common cause of death. AD affects millions of people worldwide and the risk and number of diagnosed patients continues to increase. Due to increasing life expectancy and the average age, the risks of developing AD are predicted to increase with the number of patients expected to double within the next 20 years [1–3]. Significant accumulation of amyloid-β (Aβ) in the brain is consistently observed in AD along with the accumulation of tau. Research has shown that Aβ accumulation precedes tau, converting tau from normal status to a toxic state. This Aβ-induced cascade results in damage and destruction of synapses mediating memory and cognition [4, 5]. The pathogenesis and trigger in tau pathology of Aβ claims the importance of Aβ for AD therapy.
Concentration of Aβ in the normal brain is tightly regulated through an influx/efflux transport across the blood-brain barrier via the receptor for advanced glycation end products (RAGE) and via the low-density lipoprotein receptor related protein-1 (LRP1), respectively [6–8]. Aβ accumulation progressively forms oligomers, protofibrils, and fibrils that contribute to neuronal death. This observation suggests that prevention of Aβ accumulation may prove beneficial to delay and or prevent the progression of the disease by reducing brain levels. This so-called sink hypothesis-based strategies have been attempted where capturing peripheral Aβ levels reduced brain Aβ levels. Aβ exerts its toxicity at the synapse by binding to the cell surface, thus disrupting functionality of receptors and causing synaptic dysfunction [9, 10]. In addition, accumulation of hyperphosphorylated tau negatively regulates the binding of tau to microtubules, resulting in microtubule destabilization and impaired axonal transport. These observations suggest that Aβ is upstream to tau in the pathological cascade of AD.
Therapeutic interventions have gone to extensive efforts in targeting and restricting Aβ production but with little or no success [11]. One of the early attempts to target Aβ included inhibitors of β-secretase, but due to medicinal chemistry problems, these inhibitors were not found to be successful [12]. γ-secretase, a protease that cleaves amyloid-β protein precursor (AβPP) to Aβ, has been targeted by inhibitors but resulted in undesirable side effects [13]. Advanced novel therapeutic efforts include direct immunization against Aβ or passive immunization with anti-Aβ antibodies. However, these treatments resulted in undesirable effects. The first clinical trial of immunization with pre-aggregated Aβ42 (AN1792) was stopped because of adverse effects resulting in detrimental T-cell mediated brain inflammation [14]. AD patients treated by passive immunization did not show significant improvement in symptoms and exhibited immunological adverse reactions [15]. The hypothesis that Aβ is an attractive target for treating AD has been challenged and its validity questioned based upon these results. Before rejecting the relevancy of Aβ based on these trials, thorough examination of the amyloid theory must be performed before a conclusion on the relevancy of Aβ as a target can be drawn. An important observation favoring Aβ is the promising efficacy of antibodies in a set of patient population. Post-hoc analysis of three separate trials utilizing three different Aβ antibodies (solanezumab, crenezumab, and aducanumab) found a slowing of cognitive decline in mild AD subjects. Among the clinical studies, one antibody in particular, aducanumab (Biogen), showed a cognitive benefit and significant reduction of Aβ in the brains from patients with mild to moderate AD. Thus, the evidence provided by these three promising antibodies endorses Aβ as a viable target for AD treatment.
The use of antibodies in advanced disease stages was found to be unsuitable primarily due to safety issues such as neuro-inflammation. Because patients responded well to Aβ-targeting drugs, a logical inference would be that patients who have developed Aβ plaques with mild cognitive deficits would benefit from the inhibition of Aβ accumulation in the brain. Along with exploring different targets, the influence of Aβ pathology on disease progression at different stages is highly important. The pathogenesis of Aβ has been well understood in early stages, highlighting a need for full evaluation of strategies that inhibit the effect of Aβ and halt disease progression. Thus, researchers are currently revisiting the Aβ hypothesis with a primary focus on anti-Aβ antibodies which may result in beneficial results for AD therapy. Recently, a phase II AD trial has re-emerged with new positive results, after heaps of disappointing attempts previously, where an anti-Aβ protofibril antibody was able to slow clinical symptom decline and reduce accumulation of plaques in the brain [16]. As stated in the article, “The prospect of being able to offer meaningful disease-modifying therapies to individuals suffering from this terrible disease is both exciting and humbling” [16]. The search for alternative candidates for AD therapy requires that the candidate can inhibit or dissociate Aβ aggregation, yet these processes should not induce toxicity as seen with previous candidates such as antibodies or inhibitors of AβPP. One approach would be to screen and develop peptides which prevent aggregation of Aβ or dissociate preformed aggregates as detailed [17].
Different organizations have further derived novel methods in which to reduce circulating Aβ in order to treat patients and modify AD pathology. A plasma-derived therapy for AD has been developed by Grifols with therapeutic Albutein (5% Albumin) to reduce Aβ plasma concentration. The rationale was that circulating albumin binds to 90% plasma Aβ [18] and that this functionality is being reduced or lost in AD patients [19]. Preliminary results revealed that using therapeutic apheresis to replace albumin with Albutein 5% is safe, can mobilize plasma Aβ, and stabilize cognitive abilities [20]. Similarly, Grifols developed intravenous immunoglobulins G (IVIG, Flebogamma DIF) to also mobilize peripheral Aβ [21, 22]. However, hemodialysis or plasma exchange was observed to inadvertently remove biologically important small molecules from the blood, introduce exogenous contaminants or infectious agents or induce allergic reactions in few patients [e.g., Albutein°; 23]. The removal of plasma Aβ could also be achieved by ex vivo adsorption methods or by filtration method. In the adsorption method, molecules that bind to Aβ such as anti-Aβ antibodies, albumin, sLRP1, or other proteins [24] were used as ligand on a solid matrix to remove Aβ from the plasma. Similarly, double filtration method was used to filter out Aβ from the plasma [24]. However, the adsorption methods could be expensive or associated with danger of infection and low effectiveness [23]. Thus, we propose a more specific and less risky apheresis model with no or limited side effects. Conceptually, we aim to generate an Aβ trapper (Amytrapper) by immobilizing the retro-inverso peptide (RI-peptide) in a traditional hemapheresis approach to deplete circulating Aβ from plasma of AD patients. A similar system, Liposorber, is being used successfully to remove lipoproteins from blood plasma.
Recombinant Technologies LLC (RTL) has developed and studied Amytrap peptide, a retro inverso peptide that has been shown to sequester Aβ in both in vitro and in vivo studies [Gandbhir O, Sundaram P, unpublished data]. The peptide contains D-amino acids in the reverse sequence of a peptide (Retro-inverso, RI-peptide) that has a high binding affinity for Aβ. RI peptides are generally non-toxic, non-immunogenic, and stable and offer higher resistance to proteolytic degradation [25]. Thus, this prolongs their in vivo half-life in the presence of biological fluids. In addition, non-immune based therapeutics are expected to circumvent the immunological side-effects. We have synthesized multiple RI peptides containing D-amino acids that are capable of binding Aβ. Based on its Aβ binding affinity, Amytrap peptide (RI-peptide) was chosen for incorporation into the proposed Amytrapper extracorporeal system. Amytrap peptide binds to the motif [GXXXG] in Aβ which is responsible for misfolding, oligomerization, and further aggregation. In addition, previous studies of Amytrap peptide (RI-peptide), when conjugated to albumin via PEG linker significantly reduced Aβ burden in the brain and improved cognition in a clinically relevant mouse model of AD following subcutaneous administration [Gandbhir O, Sundaram P, unpublished data].
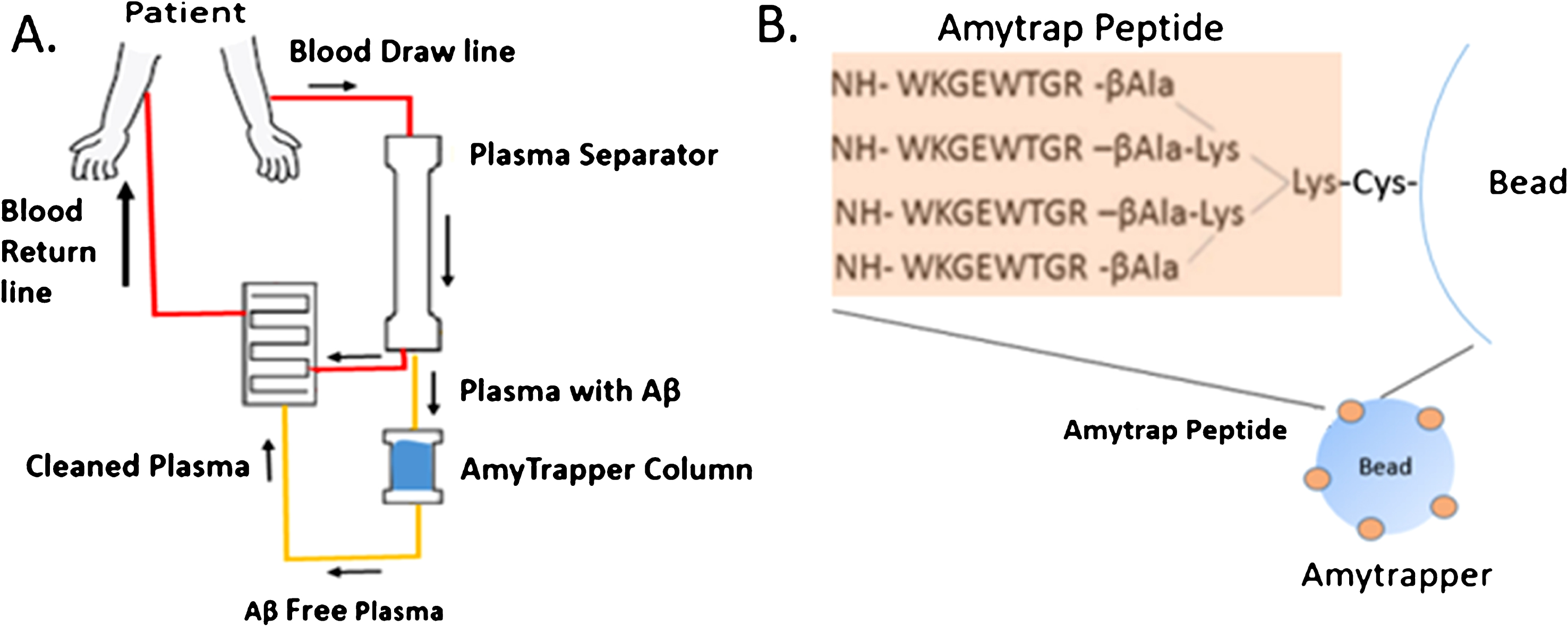
In the present proof-of-concept (POC) study, the target prototype device, Amytrapper, was designed and tested in vitro for functional assessment of binding of its target molecule Aβ. We chose to develop this device based on data obtained with RTLs API, Amytrap peptide, which has yielded promising results that warrant continued examination of this technology. The device would induce a shift in the equilibrium in the CNS through therapeutic apheresis. The design of the device and the structure of the sepharose conjugate are shown in (Fig. 1). Amytrapper would be utilized to treat patients where removal of Aβ is a therapeutic option, thus reducing the Aβ burden within the patient. Amytrapper could turn out to be a robust amyloid removal system to handle the circulating levels of Aβ in the range of 350–400 pg/ml plasma of AD patients. The approximate Aβ binding capacity, based on theoretical estimation, is at least 10 times the circulating levels of Aβ.
Fig.1
A) Flowchart of Amytrapper, a treatment option for Alzheimer’s disease patients. Patient plasma is passed through Amytrapper where API conjugated sepharose beads sequester plasma Aβ returning Aβ-free plasma to the patient. B) The Amytrap peptide, an Aβ binding retro-inverso peptide is conjugated to the bead by a PEG linker.
In the current study, we have designed and developed Amytrapper sepharose beads and demonstrated the effective binding of free Aβ42 to the Amytrapper beads in vitro. We further evaluated the specificity of Amytrapper to its target molecule and analyzed serum samples to evaluate non-specific interactions. Overall, the results show that Amytrapper is functional in binding Aβ42 with high affinity, indicating that Amytrapper is ready to move forward to in vivo studies.
METHODS
Synthesis and characterization of RI-peptide (Amytrap peptide) sepharose matrix
The RI-peptide was coupled to sepharose bead matrix utilizing click-chemistry [26], a chemical reaction with azido-PEG4-NHS ester and maleimide-DBCO. Sepharose beads were washed with distilled water and activated with azido-PEG4-NHS ester overnight at room temperature. RI-peptide (10 mg) was dissolved in potassium phosphate buffer (50 mM pH 6.8) and reacted with maleimide-DBCO (Sigma Aldrich, St. Louis, MO]. This reaction was monitored through high performance liquid chromatography (HPLC) and mass spectrometry (MS) for 1 h. The product of the reaction, RI-peptide-DBCO was reacted with the pegylated beads to produce Amytrapper sepharose beads. The beads were dialyzed against potassium phosphate buffer (50 mM, pH 6.8, 3×20 ml) to remove excess reagent.
Evaluation of the Aβ binding efficiency of RI-peptide sepharose matrix from serum treated with Aβ42
Functional properties of Amytrapper was examined through binding studies. Sepharose Amytrapper beads (100μl) were transferred into a clean column. Five hundred microliters of human serum (Golden West Diagnostics, Temecula, CA) spiked with 32 ng biotinylated Aβ42 (bio-Aβ42) was passed through the beads twice at room temperature. Beads were washed 5 times with 1 ml PBS. At the end of the fifth wash, 20μl of beads from each tube (in triplicate) were transferred to clean tubes and incubated with streptavidin-HRP (1:5,000) for 1 h at room temperature. Probing and detection were performed by ELISA using bio-Aβ42 [27]. Beads were washed 7 times with 1 ml PBS. Two microliters of beads (in duplicate) from each triplicate was transferred into clean tubes and incubated with 50μl TMB substrate (SeraCare, MD) for 5 min and color reaction was stopped with 1 N HCl (50μl). Supernatants were transferred to a microplate and read at 450 nm.
Titration of bio-Aβ42 in animal serum and human serum
Titration of bio-Aβ42 through Amytrapper was performed by adding 100μl of Amytrapper beads to a column. Amytrapper beads were incubated with phosphate buffered saline (PBS) or mouse serum (Golden West Diagnostics) spiked with increasing concentrations of bio-Aβ42 (0, 2, 4, 8, 16, or 32 ng). Unconjugated sepharose beads (sepharose minus API) served as negative controls. Binding of bio-Aβ42 was measured by following the ELISA method described earlier. Further titrations were performed with human serum or plasma samples (Golden West Diagnostics), utilizing concentrations of bio-Aβ42 (0, 4, 8, 16 or 32 ng). Amytrapper beads were pre-equilibrated with the human serum or plasma samples prior to passing the spiked samples.
Specificity of Amytrapper column
Amytrapper’s specificity for its target molecule, Aβ42, was analyzed by passing untagged Aβ42 through Amytrapper beads and then passing bio-Aβ42 through the same beads. 100μl of Amytrapper sepharose beads were transferred into clean columns. Five hundred microliters of human serum with excess (100 ng) unlabeled Aβ42 was passed through the column twice to saturate the binding sites. Then another 500μl aliquots of human serum containing increasing concentrations of bio-Aβ42 (0, 4, 8, 16, or 32 ng) was passed through the beads, and the experiment was performed as previously stated.
Prototype Amytrapper column
Sepharose beads conjugated to RI-peptide was prepared in columns (Boca Scientific, Westwood, MA). The column was prepared by packing Amytrapper sepharose beads and control beads into separate 1 ml columns. Binding of bio-Aβ42 was performed as previously described. One hundred microliters of Amytrapper beads was transferred into clean columns. Five hundred microliters of mouse serum, was aliquoted into separate tubes and spiked with increasing concentrations of bio-Aβ42 (0, 4, 8, 16, or 32 ng) which was passed through the column twice at room temperature. Amytrapper sepharose beads were analyzed as described earlier.
Two-dimensional electrophoresis to rule out inadvertent retention of other plasma proteins
Two-dimensional (2D) gel electrophoresis and analysis was performed on two serum samples (500μg of protein at a volume of 150μl) before and after passing through Amytrapper column matrix. Serum was spiked with 16 ng of bio-Aβ42 prior to the passage. 2D gel analysis was performed by Kendrick Labs LLC (Madison, WI), an expert in 2D electrophoresis and computer based comparative analysis [28, 29]. At the end of the 2D run, the gel image was analyzed for any difference between the pre and post run samples by Progenesis Same Spots Software and Progenesis PG240 Software (TotalLab, Newcastle upon Tyne, UK).
RESULTS
Recombinant Technologies has been pioneering the development of non-immune, peptide-based therapeutics to treat AD for over a decade [27, 30]. One such compound, Amytrap, has been successful in binding Aβ42 both in vitro and in vivo [Gandbhir O, Sundaram P, unpublished data]. The present study was taken up to evaluate a novel device called Amytrapper, containing the API (RI-peptide) and coupled to sepharose matrix, which was designed to bind and remove Aβ in AD through apheresis. In a set of pilot experiments, a prototype Amytrapper matrix was tested within buffer, sera or plasma (animal or human) spiked with known amounts of Aβ42 to confirm that the device would bind Aβ42 specifically.
Synthesis of RI-peptide (Amytrap peptide) sepharose matrix
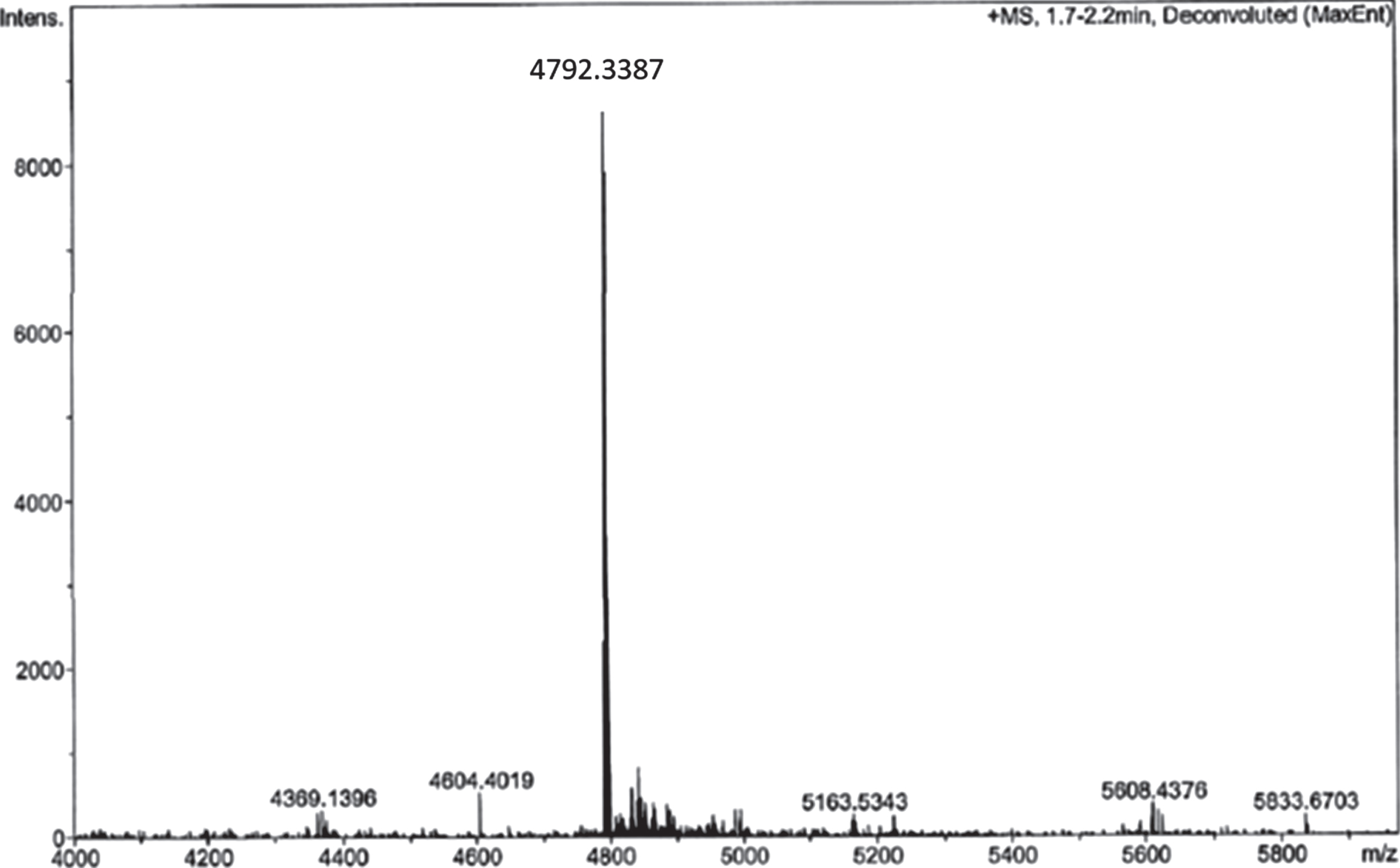
Coupling of RI-peptide to sepharose was performed by click-chemistry, a multi-step reaction to synthesize Amytrapper. To ensure the purity of the intermediate RI-peptide-DBCO, reactions were monitored through high performance liquid chromatography (HPLC) and mass spectrometry (MS) for 1 h. The results of this reaction are depicted in the MS analysis in (Fig. 2). The single peak seen in (Fig. 2) represents the RI-peptide-DBCO intermediate. RI-peptide-DBCO was reacted with the pegylated beads to produce Amytrapper sepharose beads. Total protein concentration of the sepharose beads was estimated to be 0.2 mg/ml for packed beads. This represents the peptide concentration in the beads.
Fig.2
Analysis of RI-peptide-DBCO reaction by ESI-MS analysis of reaction mixture after 1 h.
Plasma Aβ binding efficiency of RI-peptide sepharose matrix
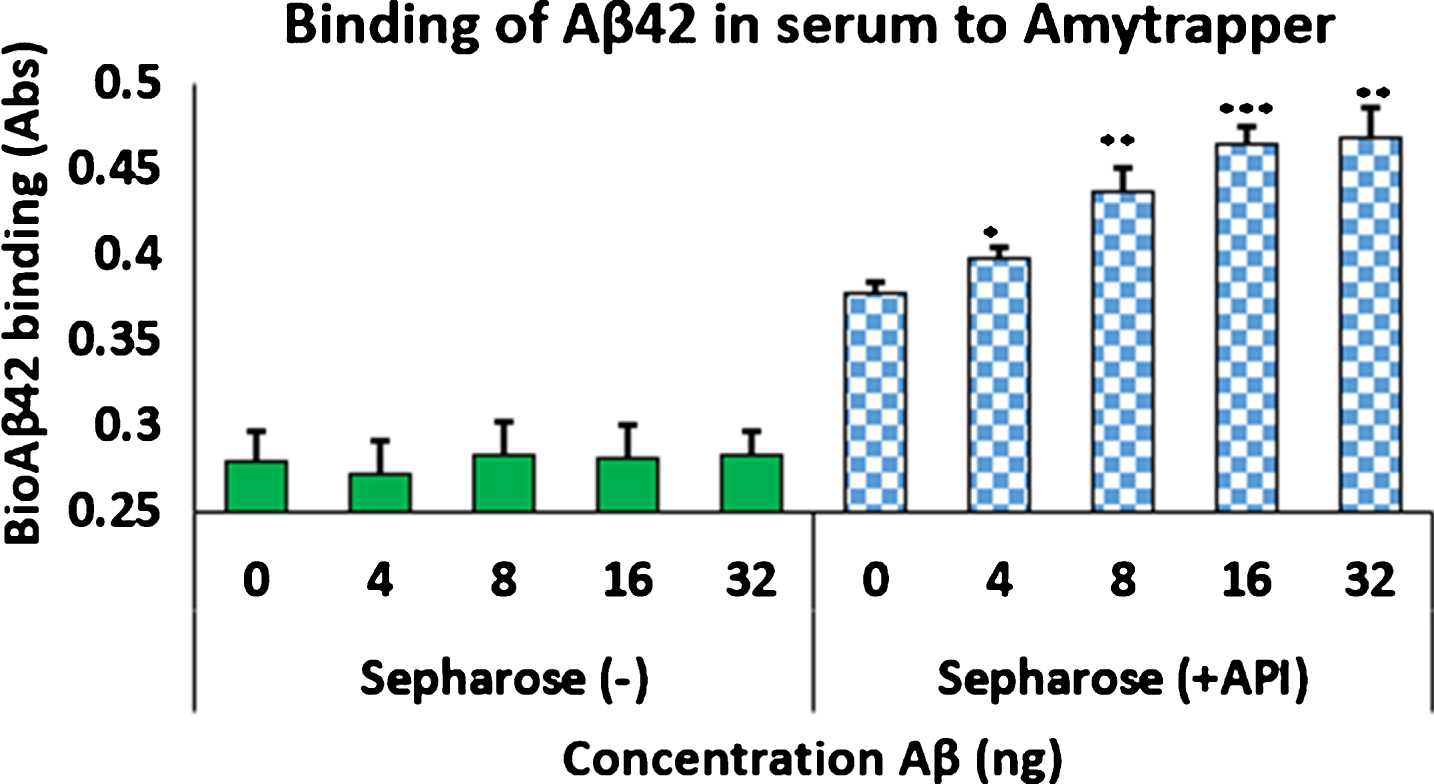
Functional properties of Amytrapper were examined by a developed functional assay. Optimization of Aβ binding was performed in PBS buffer before testing in mouse and human serum (data not shown). Serum binding results detected that there was a significant binding observed from beads treated with serum spiked with bio-Aβ42 (0.6) as compared to non-spiked control serum (Fig. 3). Further, the percent of bound bio-Aβ42 from the spiked serum was found to be 35% of the starting 32 ng concentration. That is 11.5 ng of the 32 ng bio-Aβ42 was found to be bound approximately (Table 1). ELISA was also performed on serum samples before and after passing through sepharose beads. Consequently, there was a 37% reduction in the levels of bio-Aβ42 in the flow through serum samples (Table 1). Thus, the amount of bio-Aβ42 retained on the column agreed with the amount that was reduced in the flow through.
Fig.3
Binding of bio-Aβ42 by Amytrapper (sepharose beads). Values are expressed in absorbance units presented as Mean±SEM from triplicate measurements. Student’s T-test was used to determine significance. *p < 0.001.
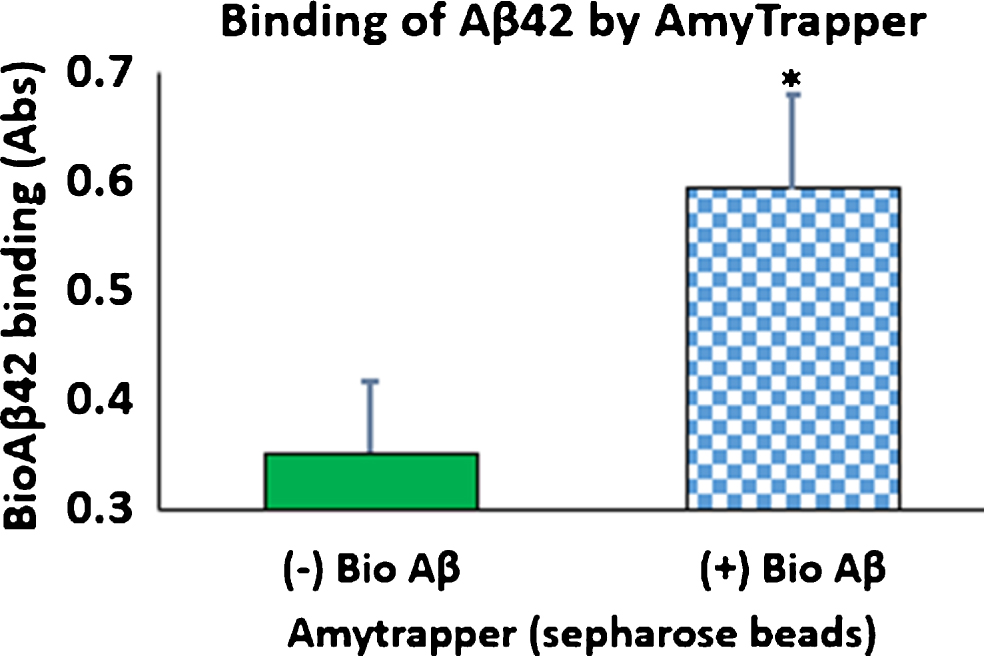
Table 1
Analysis of Aβ in flowthrough and Amytrapper sepharose beads
| Aβ Conc. (ng) | Baseline Pre-incubation (100%) | Post-incubation | Estimated Concentration | |
| Amount in flowthrough | 0 | 0.132 | 0.13 | 0 |
| 32 | 0.235 | 0.152 | 20 | |
| Amount bound to beads | 0 | 17.4 | 17.5 | 0 |
| 32 | 17.6 | 29.665 | 11.5 |
Pre-incubation and Post-incubation totals are given as absorbances at 450 nm. Pre-incubation was taken as 100% and the estimated concentration was calculated. The amount of Aβ bound to the beads and left in the flowthrough after binding was calculated from these absorbances.
Titration of Aβ42 at increased concentrations
Analysis of concentration dependent binding was performed with mouse serum, human serum, or plasma spiked with increasing concentrations of bio-Aβ42 (0, 4, 8, 16, or 32 ng). Unconjugated sepharose beads (Sepharose minus API) served as negative controls. Binding of bio-Aβ42 was measured by following the ELISA method described previously (Evaluation of the plasma Aβ binding efficiency of RI-peptide sepharose matrix). Amytrapper matrix displayed significant concentration dependent binding of bio-Aβ42 at 8, 16, and 32 ng. While binding can be distinguished at 4 ng bio-Aβ42 it was not found to be significant by Student’s t-test (Fig. 4). Low absorbance was detected from Sepharose (–), the unconjugated beads but did not reach absorbances detected from Amytrapper sepharose matrix. Furthermore, Amytrapper sepharose displayed a specific and concentration dependent binding of bio-Aβ42 in human serum (Fig. 5A) and plasma (Fig. 5B). In human serum, concentration dependent binding was found to plateau at 32 ng, while binding in plasma was found to increase at 32 ng. In both cases, binding was calculated to be significant for Amytrapper sepharose beads, while plain sepharose did not have significant binding.
Fig.4
Binding of biotinylated Aβ42 to Amytrapper in mouse serum spiked with bio-Aβ42. Sepharose (–) act as a negative control. Values are expressed in absorbance units presented as Mean±SEM from triplicate measurements. Student’s t-test was used to determine significance. *p < 0.05, **p < 0.01.
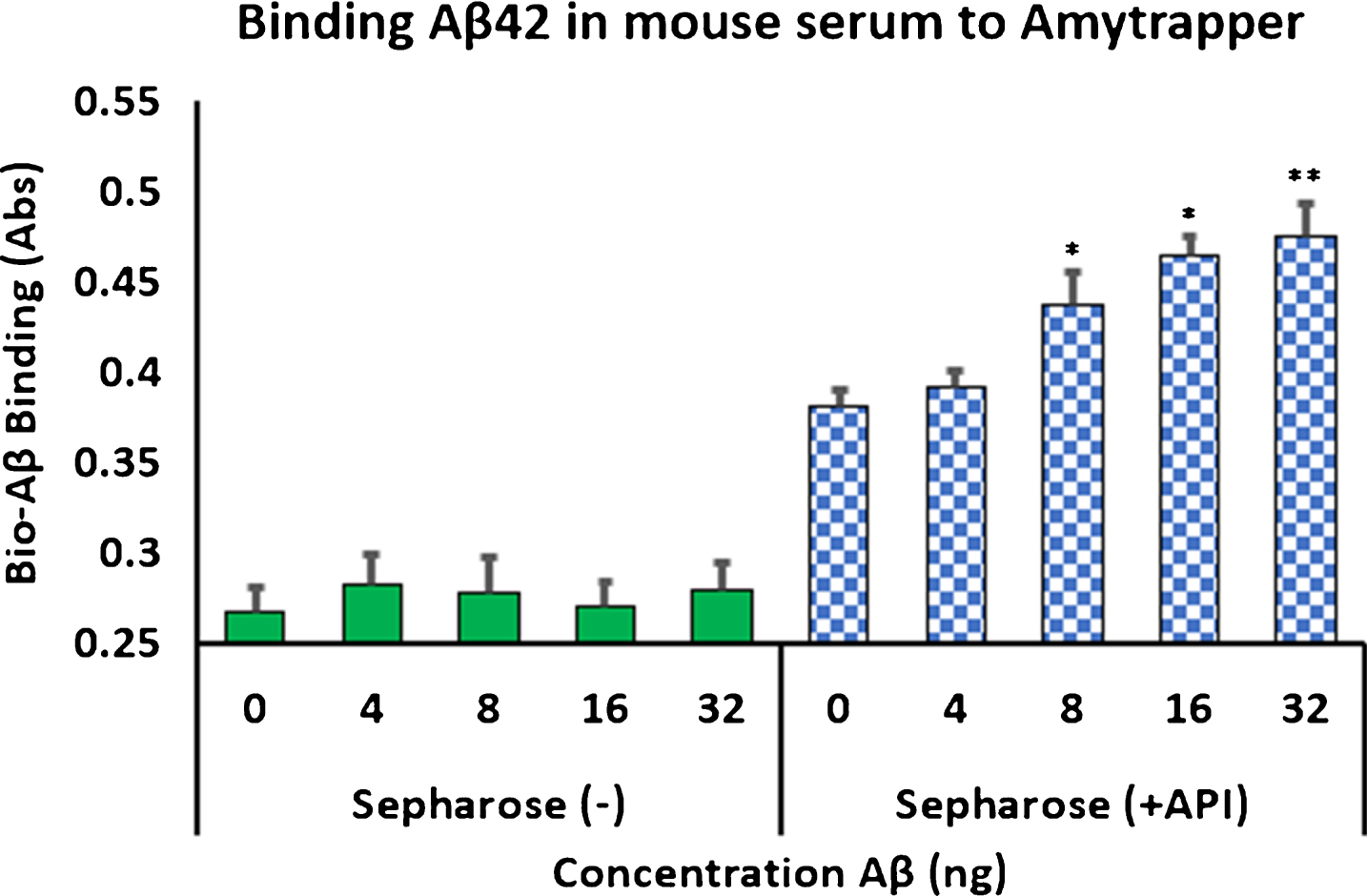
Fig.5
Binding of biotinylated Aβ42 to Amytrapper in (A) human serum spiked with bio-Aβ42 and (B) human plasma spiked with bio-Aβ42. Sepharose (–) act as a negative control. Values are expressed in absorbance units presented as Mean±SEM from triplicate measurements. A) Student’s t-test was used to determine significance. *p < 0.05, **p < 0.01, ***p < 0.001. B) Student’s t-test was used to determine significance. *p < 0.05, **p < 0.01, ***p < 0.001.
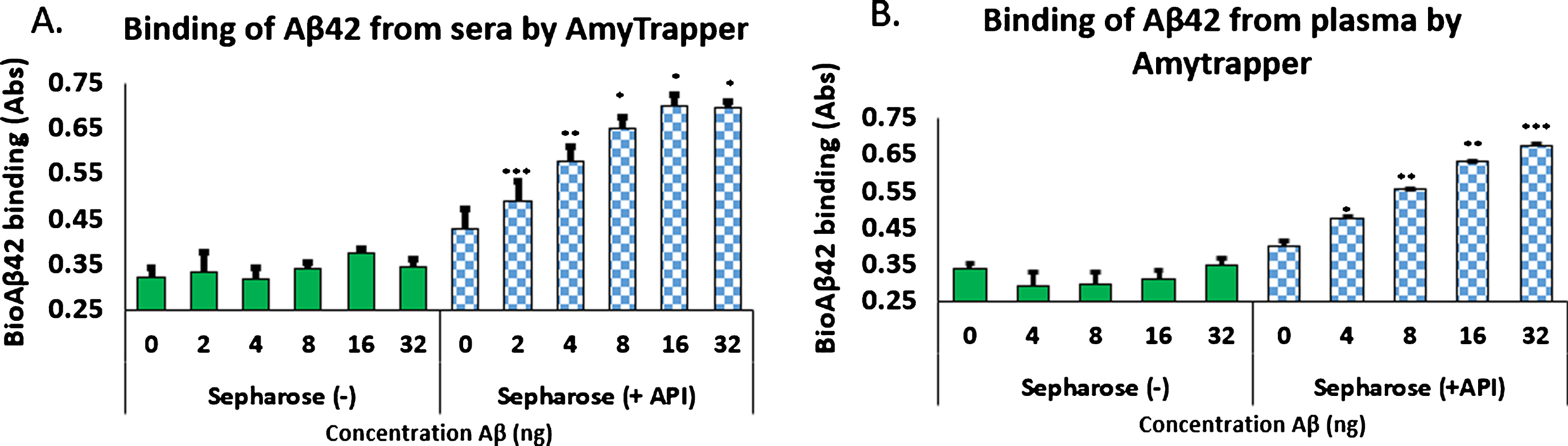
Specificity of Amytrapper
To provide evidence of Amytrapper’s specificity for its target molecule, Aβ42, Amytrapper sepharose beads were saturated with excess unlabeled Aβ42. Then saturated beads were incubated as described in the methods. The results support that Amytrapper presents a high specificity for Aβ42. Beads blocked with unlabeled Aβ42 were calculated to have no difference in binding when compared with the control (0 ng bio-Aβ42) (Fig. 6). Unblocked beads present the same concentration dependent binding that was shown in Fig. 6. This observation further validated the specificity of Amytrapper for Aβ42.
Fig.6
Specificity of Amytrapper for Aβ42. Bio-Aβ42 was allowed to bind to Amytrapper (sepharose beads containing API). Parallel binding was carried out by Amytrapper that was pre-incubated with excess unbiotinylated Aβ42 to block the binding sites. Unblocked and blocked data are represented in the figure. Values are expressed in absorbance units presented as Mean±SEM from triplicate measurements. Student’s t-test was used to determine significance. *p < 0.05.
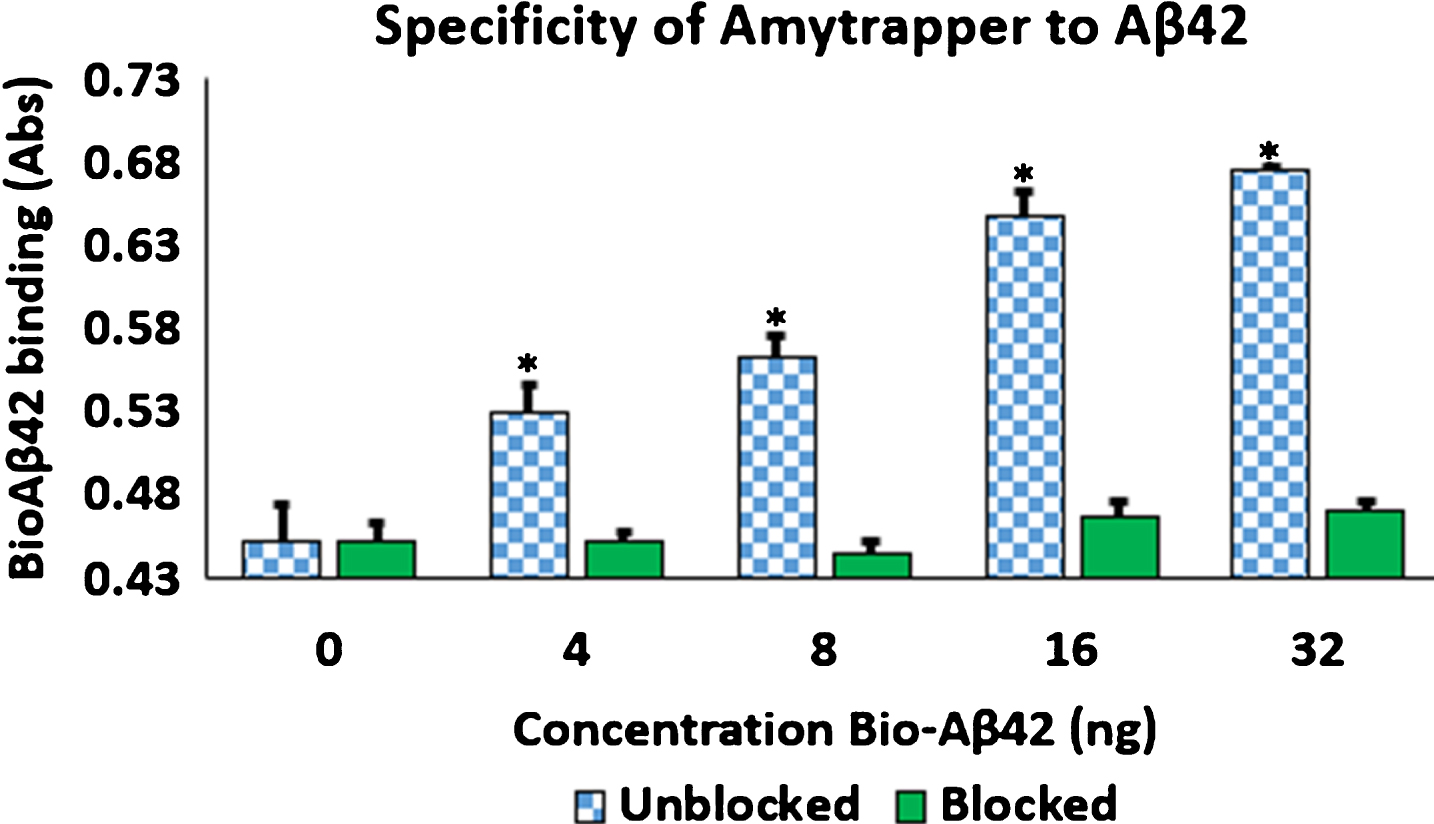
Prototype Amytrapper column
Sepharose beads conjugated to RI-peptide was prepared in columns (Boca Scientific) to confirm proof of concept binding of Aβ42. Amytrapper sepharose beads were analyzed as described previously and were found to maintain concentration dependent binding of bio-Aβ42 was in the prototype matrix reaching a plateau at 32 ng (Fig. 7). Sepharose (–) beads, the negative control, maintained low background binding when compared to Sepharose (+API) beads. Thus, the prototype column was successful in sequestering Aβ42. Independent of the volume of the beads or the amount of Aβ42 that was passed through, the binding efficiency of the Amytrapper matrix was assessed to be 33% of the anticipated theoretical binding. Despite this low efficiency, the binding capacity is sufficient as the circulating levels of amyloid are substantially lower in humans. Varying capacity columns may be generated in the future as needed.
Fig.7
Concentration dependent binding of bio-Aβ42 to Amytrapper. Values are expressed as absorbance and presented as Mean±SEM from triplicate measurements. Student’s T-test was used to determine significance. *p < 0.05, **p < 0.005, ***p < 0.001.
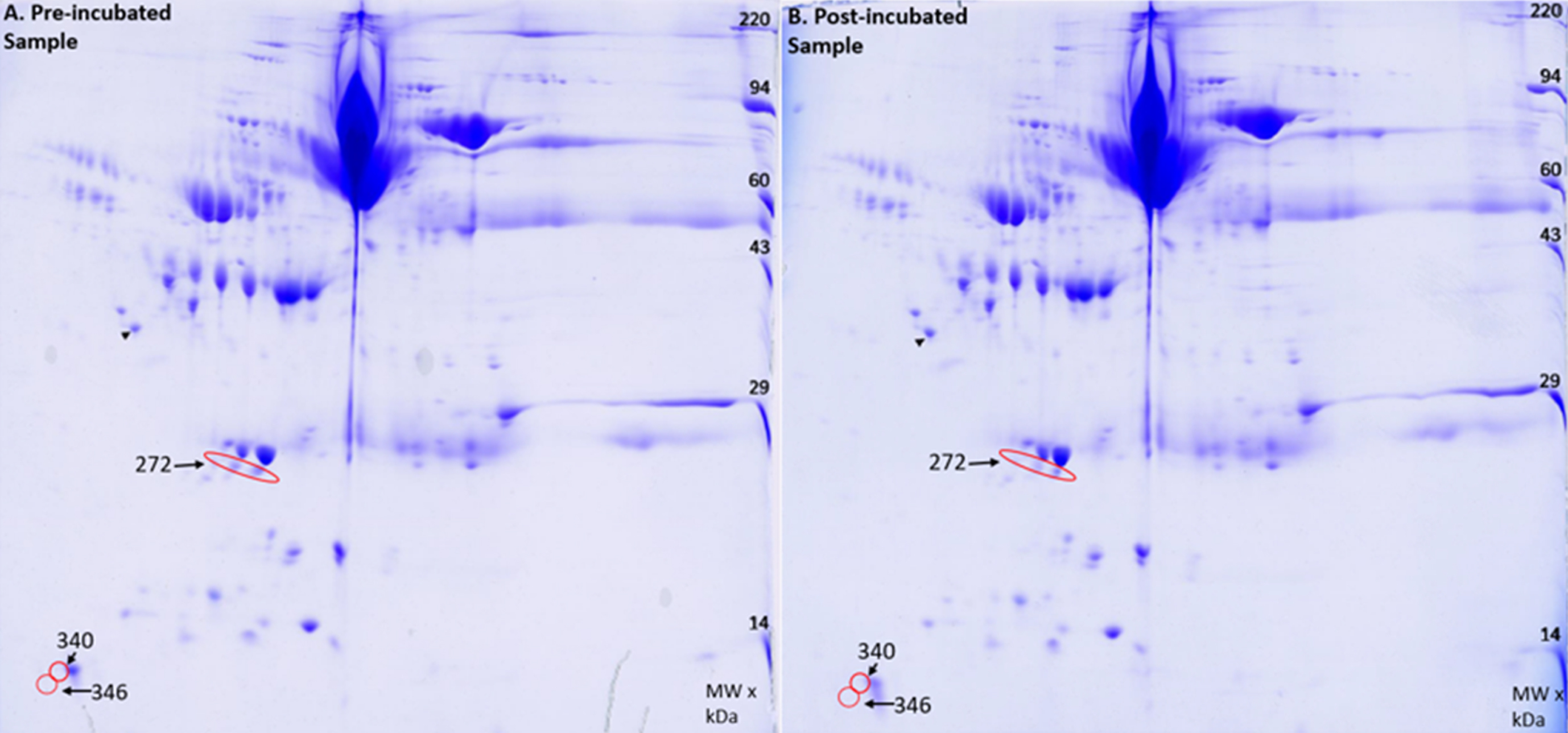
Analysis of native plasma proteins
The Amytrapper device was designed to have plasma pass through it to remove Aβ. Therefore, it was important to know if the sepharose beads would inadvertently bind and retain other plasma proteins from freely passing through the column. To confirm that nonspecific binding and retention did not occur, a 2D gel electrophoresis and analysis was performed on two of the serum samples. A total of 355 spots were analyzed using Kendrick Labs computer software. Of the 355 spots, only 3 spots had a difference in protein of approximately 2% for pre-incubation and post-incubation samples (Fig. 8). This is far smaller than the 33% in amyloid retention as presented in the previous section, thus proving that the retention of amyloid was specific to the API. Further this demonstrates that there was very little retention of plasma proteins. Our future designs will focus on totally eliminating even this small amount of retention.
Fig.8
2D gel image of A) Pre-incubated sample and B) the Post-incubated sample. Red circles indicate decreases in protein found in the post incubated sample. The black arrow points to an internal loading control.
DISCUSSION
The goal of the present study was to obtain proof of concept for the Amytrapper device in order to determine if the device would be able to maintain the function of the Amytrap peptide. Amytrapper incorporates a proprietary Amytrap peptide. The present study has yielded promising results that warrant continued examination of this technology. If successful, it would become a viable therapy either as stand alone or in combination with other therapies to treat AD in humans. Amytrapper sepharose matrix was generated through click chemistry, which has been used successfully in our previous studies to link the Amytrap peptide to different scaffolds [Gandbhir O, Sundaram P, unpublished data]. Amytrapper was developed with high purity and yield by linking RTL’s API, Amytrap, to sepharose beads through a PEG linker. Purity was analyzed by HPLC (Fig. 3) and mass spectrometry to produce a pure form of the Amytrap peptide to be conjugated to the sepharose matrix. Click chemistry offers the flexibility to create Amytrapper with higher concentrations of the API, Amytrap peptide. These different concentrations may be useful in future therapies for different severities of AD. Higher concentrations of Amytrapper could be used for treatment in late stages of AD while lower concentrations would be feasible for early stages of the disease.
Our RI-peptide has shown effective binding of Aβ42 in in vitro studies, and further reduced Aβ42 levels from APPswe [Tg2576] mice in a previous study [Gandbhir O, Sundaram P, unpublished]. Amytrap peptide was further found to be safe, non-immunogenic, and non-toxic. RI-peptides have been shown to have high stability as well as have low immunogenicity [25, 31]. This is consistent with other studies utilizing RI-peptides for treatments of various conditions. The benefit of using D-amino acids in developing the Amytrap peptide was that the peptide would be more resistant to proteolytic cleavage. Favorable efficacy of a different D-amino acid peptide in treating AD has previously been reported [25].
Sera samples spiked with bio-Aβ42 were able to flow freely through the beads without mechanical pressure. The Amytrapper sepharose matrix was successfully able to bind bio-Aβ42 that was detectable by the assay described [27]. This test demonstrated that Amytrapper sepharose matrix was capable of binding Aβ42 thus supporting the idea that Amytrapper could be used as an ex vivo device. Thus, Amytrap successfully performed its function in these in vitro studies. Other extracorporeal designs with different bait molecules have proved successful in the past. For instance, a study reported that cellulose bead matrix acts as an adsorption device that targets Aβ42 [32], allowing serum or plasma proteins to freely pass through but sequesters Aβ.
Titrations of Aβ42 was performed in order to determine concentration dependent binding in mouse sera, human sera or human plasma. Removal of Aβ42 from spiked mouse serum was successful at higher concentrations depicting significant differences (Fig. 4) which agrees with other studies utilizing apheresis treatments [20, 33]. Removal from human serum or plasma binding presented even greater success as the presence of Aβ42 was detectable in samples spiked with low concentrations of Aβ42. By decreasing the levels of plasma Aβ42 we expect to see an osmotic effect in the brain levels of Aβ which may alleviate cognitive difficulties as observed with Grifols Albutein plasma treatment [20]. A similar extracorporeal device was capable of performing the same task targeting both Aβ40 and Aβ42 [33] thus corroborating the capabilities of Amytrapper as a potential method for removing Aβ42.
Amytrapper sepharose beads incorporating the API were packed onto a column in order to create a prototype Amytrapper column. The column from Boca Scientific was chosen because they are commercially viable and accepted for this type of applications in proven models. Further analysis of samples passed through the prototype column verified that the Amytrapper would be successful in this commercially viable column (Fig. 7). The columns did not interfere with flow through, or develop blockages, suggesting that this method of plasma apheresis has a high chance of success as a therapeutic option. The POC results obtained so far prove that Amytrapper device would be successful in removing Aβ42 to alleviate Aβ42 toxicity in AD patients. Future testing will require studies with controllable flow rate similar to another study that performed tasks to achieve quick treatment and maximum adsorption of the target molecule [32].
In order to prove that retention of Aβ42 by Amytrapper was specific, binding sites on the Amytrapper sepharose were pre-blocked with excess non-biotinylated Aβ42 followed by binding experiment utilizing bio-Aβ42. If the binding was specific, then the non-biotinylated Aβ42 would block the binding sites and therefore eventual binding of bio Aβ42 would be prevented. As anticipated, untagged Aβ42 prevented binding of bio-Aβ42 to Amytrapper beads, thereby proving that the retention of Aβ42 by Amytrapper was indeed specific to Aβ42 (Fig. 6). Furthermore, unblocked Amytrapper control maintained concentration dependent binding of Aβ42. The specificity of Amytrapper sepharose beads is in agreement with other studies on RI-peptides which have shown evidence that RI-peptides have high selectivity for their target and are thus less likely to cross-react with other proteins [34].
Developing therapeutics for AD has had many successes and road blocks. While many therapeutics are effective in treating AD, they elicit immunogenic responses or neurological responses that can be severe. Amytrapper was designed to address this need without getting inside the body. In order to rule out inadvertent retention of native plasma proteins, we performed 2D SDS-PAGE analyses on serum samples harvested before and after passage through the Amytrapper column. Analysis of the gels suggested negligible loss of serum proteins when compared to the significant amount of Aβ42 (30%) that was bound (Fig. 8). The 30% retention of Aβ compares well with the 30% reduction in the flowthrough, thus accounting for the total amyloid that was used to spike the serum samples. Our 2D SDS-PAGE clearly demonstrated that very small and negligible retention of plasma proteins took place.
In summary, we designed and developed Amytrapper sepharose beads and demonstrated the effective binding of circulating Aβ42 to the Amytrapper prototype in vitro (Fig. 7). We further evaluated the specificity of Amytrapper to its target molecule and analyzed serum samples to evaluate non-specific interactions. Overall, the results show that Amytrapper is functional in binding Aβ42 with high affinity, indicating that it is ready to move forward to in vivo studies. Once animal POC is confirmed, Amytrapper would be used to treat patients where removal of Aβ is a therapeutic option. The binding efficiency of the Amytrapper matrix (containing 100μg/ml equivalent of RI peptide) is estimated to be 30% of the anticipated theoretical binding. However, this level of amyloid binding is more than sufficient as the circulating level of amyloid is substantially lower in humans. Amytrapper could turn out to be a robust amyloid removal system to handle high circulating levels of Aβ in the range of 350–400 pg/ml plasma in AD patients, requiring patients to undergo a treatment regimen where based upon their amyloid levels they may be treated with 2 to 3 ml of packed beads.
CONFLICT OF INTEREST
We declare that there is no actual and potential conflict of interest on this study.
ACKNOWLEDGMENTS
Partial salary support to Dr. Pazhani Sundaram and Omkar Gandbhir was provided by a small business innovative research phase 1 grant # R43 AG0 57327, from National Institute on Aging, National Institutes of Health, USA. Dr. PS the principal investigator and corresponding author thanks NIH for the grant award.
REFERENCES
[1] | Alzheimer’s Association ((2006) ) Early onset dementia: A national challenge, a future crisis. Alzheimer’s Association, Washington, DC, https://www.alz.org/media/Documents/early-younger-onset-full-r.pdf |
[2] | Wilson RS , Weir DR , Leurgans SE , Evans DA , Hebert LE , Langa KM , Plassman BL , Small BJ , Bennett DA ((2011) ) Sources of variability in estimates of the prevalence of Alzheimer’s disease in the United States. Alzheimers Dement 7: , 74–79. |
[3] | Alzheimer’s Association ((2012) ) 2012 Alzheimer’s disease facts and figures. Alzheimers Dement 8: , 131–168. |
[4] | Bloom GS ((2014) ) Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71: , 505–508. |
[5] | Hong S , Beja-Glasser VF , Nfonovim BM , Frouin A , Li S , Ramakrishnan S , Merry KM , Shi Q , Rosenthal A , Barres BA , Lemere CA ((2016) ) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: , 712–716. |
[6] | Deane R , Du Yan S , Submamaryan RK , LaRue B , Jovanovic S , Hogg E , Welch D , Manness L , Lin C , Yu J , Zhu H ((2003) ) RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 9: , 907. |
[7] | Deane R , Wu Z , Sagare A , Davis J , Du Yan S , Hamm K , Xu F , Parisi M , LaRue B , Hu HW , Spijkers P ((2004) ) LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron 43: , 333–344. |
[8] | Deane R , Wu Z , Zlokovic BV ((2004) ) RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid β-peptide clearance through transport across the blood–brain barrier. Stroke 35: , 2628–2631. |
[9] | Palop JJ , Mucke L ((2010) ) Amyloid-β–induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat Neurosci 13: , 812. |
[10] | A Massaad C ((2011) ) Neuronal and vascular oxidative stress in Alzheimer’s disease. Curr Neuropharmacol 9: , 662–673. |
[11] | Becker RE , Greig NH , Giacobini E , Schneider LS , Ferruci L ((2014) ) A new roadmap for drug development for Alzheimer’s disease. Nat Rev Drug Discov 13: , 156. |
[12] | Panza F , Solfrizzi V , Frisardi V , Capurso C , D’introno A , Colacicco AM , Vendemiale G , Capurso A , Imbimbo BP ((2009) ) Disease-modifying approach to the treatment of Alzheimer’s disease: From alpha-secretase activators to gamma-secretase inhibitors and modulators. Drugs Aging 26: , 537–555. |
[13] | Crump CJ , Johnson DS , Li YM ((2013) ) Development and mechanism of γ-secretase modulators for Alzheimer’s disease. Biochemistry 52: , 3197–3216. |
[14] | Zotova E , Bharambe V , Cheaveau M , Morgan W , Holmes C , Harris S , Neal JW , Love S , Nicoll JA , Boche D ((2013) ) Inflammatory components in human Alzheimer’s disease and after active amyloid-β42 immunization. Brain 136: , 2677–2696. |
[15] | Salloway S , Sperling R , Fox NC , Blennow K , Klunk W , Raskind M , Sabbagh M , Honig LS , Porsteinsson AP , Ferris S , Reichert M ((2014) ) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370: , 322–333. |
[16] | Kresge N ((2018) ) Biogen surges after positive results in Alzheimer’s Trial. Bloomberg https://www.bloomberg.com/news/articles/2018-07-06/eisai-surges-after-positive-results-in-alzheimer-s-drug-trial?in_source=video_page. |
[17] | Goyal D , Shuaib S , Mann S , Goyal B ((2017) ) Rationally designed peptides and peptidomimetics as inhibitors of amyloid-β (Aβ) aggregation: Potential therapeutics of Alzheimer’s disease. ACS Comb Sci 19: , 55–80. |
[18] | Bohrmann B , Tjernberg L , Kuner P , Poli S , Levet-Trafit B , Näslund J , Grayson R , Huber W , Döbeli H , Nordstedt C ((1999) ) Endogenous proteins controlling amyloid β-peptide polymerization possible implications for β-amyloid formation in the central nervous system and in peripheral tissues. J Biol Chem 274: , 15990–15995. |
[19] | Byun K , Bayarsaikhan E , Kim D , Kim CY , Mook-Jung I , Paek SH , Kim SU , Yamamoto T , Won MH , Song BJ , Park YM ((2012) ) Induction of neuronal death by microglial AGE-albumin: Imlications for Alzheimer’s disease. PloS One 7: , e37917. |
[20] | Boada M , Ramos-Fernández E , Guivernau B , Munoz FJ , Costa M , Ortiz AM , Jorquera JI , Nunez L , Torres M , Paez A ((2016) ) Treatment of Alzheimer disease using combination therapy with plasma exchange and haemapheresis with albumin and intravenous immunoglobulin: Rationale and treatment aroach of the AMBAR (Alzheimer Management by Albumin Replacement) study. Neurología 31: , 473–481. |
[21] | Rovira MB , Núņez L , Lafuente A , Hernandez I , Buendia M , Tarraga L , Roca I , Cuberas G , Torres M , Páez A ((2011) ) Peripheral amyloid-beta mobilization with the intravenous immunoglobulin Flebogamma DIF in Alzheimer’s disease patients. Alzheimers Dement 7: , S456–S457. |
[22] | Costa M , Ortiz AM , Jorquera JI ((2012) ) Therapeutic albumin binding to remove amyloid-β. J Alzheimers Dis 29: , 159–170. |
[23] | Bambauer R , Bambauer C , Lehmann B , Latza R , Schiel R ((2012) ) LDL-apheresis: Technical and clinical aspects. ScientificWorldJournal 2012: , 314283. |
[24] | Agishi T , Kaneko J , Hasuo Y , Hayasaka Y , Sanaka T , Ota K , Amemiya H , Sugino N ((1980) ) Double filtration plasmapheresis with no or minimal amount of blood derivative for substitution. In Plasma Exchange, Plasmapheresis—Plasmaseparation, Sieberth HG, ed. Schattauer, Stuttgart New York, pp. 53–57. |
[25] | Klein AN , Ziehm T , van Groen T , Kadish I , Elfgen A , Tusche M , Thomaier M , Reiss K , Brener O , Gremer L , Kutzsche J ((2017) ) Optimization of D-peptides for Aβ monomer binding specificity enhances their potential to eliminate toxic Aβ oligomers. ACS Chem Neurosci 8: , 1889–1900. |
[26] | Slagle CJ , Thamm DH , Randall EK , Borden MA ((2018) ) Click conjugation of cloaked peptide ligands to microbubbles. Bioconjug Chem 29: , 1534–1543. |
[27] | Sundaram RK , Kasinathan C , Stein S , Sundaram P ((2008) ) Detoxification depot for β-amyloid peptides. Curr Alzheimer Res 5: , 26–32. |
[28] | O’Farrell PH ((1975) ) High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250: , 4007–4021. |
[29] | Burgess-Cassler A , Johansen JJ , Santek DA , Ide JR , Kendrick NC ((1989) ) Computerized quantitative analysis of coomassie-blue-stained serum proteins separated by two-dimensional electrophoresis. Clin Chem 35: , 2297–2304. |
[30] | Sundaram RK , Kasinathan C , Stein S , Sundaram P ((2012) ) Novel detox gel depot sequesters β-amyloid peptides in a mouse model of Alzheimer’s disease. Int J Pept Res Ther 18: , 99–106. |
[31] | Hervé M , Maillére B , Mourier G , Texier C , Leroy S , Ménez A ((1997) ) On the immunogenic properties of retro-inverso peptides. Total retro-inversion of T-cell epitopes causes a loss of binding to MHC II molecules. Mol Immunol 34: , 157–163. |
[32] | Kawaguchi K , Kitaguchi N , Nakai S , Murakami K , Asakura K , Mutoh T , Fujita Y , Sugiyama S ((2010) ) Novel therapeutic approach for Alzheimer’s disease by removing amyloid β protein from the brain with an extracorporeal removal system. J Artif Organs 13: , 31–37. |
[33] | Kawaguchi K , Saigusa A , Yamada S , Gotoh T , Nakai S , Hiki Y , Hasegawa M , Yuzawa Y , Kitaguchi N ((2016) ) Toward the treatment for Alzheimer’s disease: Adsorption is primary mechanism of removing amyloid β protein with hollow-fiber dialyzers of the suitable materials, polysulfone and polymethyl methacrylate. J Artif Organs 19: , 149–158. |
[34] | Cerchietti LC , Yang SN , Shaknovich R , Hatzi K , Polo JM , Chadburn A , Dowdy SF , Melnick A ((2009) ) A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood 113: , 3397–3405. |


