
Are Porphyromonas gingivalis Outer Membrane Vesicles Microbullets for Sporadic Alzheimer’s Disease Manifestation?
Abstract
Our research into Alzheimer’s disease (AD) focuses on the oral cavity and the brain, from which key evaluations of prospective and retrospective population-based data have shown that chronic periodontal disease existing for ten-years or over doubles the risk for the sporadic form of AD. Furthermore, Porphyromonas gingivalis (P. gingivalis) mono-infections in established periodontal lesions, or introducing its lipopolysachharide (LPS), as demonstrated in in vivo studies, show hallmark pathology inclusive of extracellular amyloid plaques and phospho-tau bound neurofibrillary tangles with AD-like phenotype. Other studies have shown that if periodontitis remains untreated in human AD patients, cognitive decline ensues. This is a bi-directional relationship meaning that the converse is also true; treating periodontal disease in AD patients improves memory. Bacterial cultures and established oral biofilms generate vast numbers of microvesicles and P. gingivalis outer membrane vesicles encase key virulence factors (LPS, gingipains, capsule, fimbriae) as though they are complete destructive “microbullets” when shed in the host. This provides P. gingivalis additional arsenal to manipulate its entry into disparate organs, hijack phagocytosis, destroy tissues, and affect complement related genes while transducing the onset of proinflammatory signaling cascades. The resulting inflammatory mediators may be the cause of disease defining lesions and cognitive decline typical of clinical AD.
INTRODUCTION
Periodontitis is an oral disease presenting with a polymicrobial dysbiosis of the sub-gingival microbiome, which eventually, if untreated, leads to tooth loss. Around 50% of all humans in their middle age (50 years and over) appear to fall victims to periodontitis [1]. The nature of the oral disease is episodic with characteristic recurrent periods of active disease progression followed by periods of quiescence in individuals who are unable to prevent commensals (healthy microbiome) converting into pathogens (pathobiome). However, a plethora of publications provides research-based evidence that an unhygienic oral environment, especially one that encompasses periodontitis, can negatively affect mental health. In the sporadic form of Alzheimer’s disease (AD) there appears to be a sub-population susceptible to Porphyromonas gingivalis (P. gingivalis) infections, because not everyone who develops clinical AD appears to suffer from periodontitis [2]. It follows, therefore, that the risk factor pathway that leads some individuals to AD onset later in life (80 years and over), and development of periodontal disease show an overlap with the preclinical phase of AD (without clinical presentation of disease). Identifying such a group of individuals and focusing on managing periodontal disease at their preclinical AD phase could prevent the incidence of this dementing disease manifesting in the first place.
The bi-directional relationship of periodontal disease and dental hygiene with AD alerts us to the seriousness of a silently incubating risk. This follows that the currently rising numbers of elderly without adequate dental care are falling into the high-risk group of manifesting with AD. Whilst the cause of AD remains unknown, and no imminent treatment via “a pill” looms in the horizon, it is crucial that we recognize all modifiable risk factors and this should include periodontitis, especially in the elderly age group.
Prior to discussing the plethora of literature contribution to the relative importance of periodontitis as a risk factor acceptance, it is necessary to highlight the protective barriers such as the blood-brain barrier (BBB) that keeps the central nervous system (CNS) isolated and protected from aberrant immune challenges (entry of toxins and related cells that detoxify foreign proteins) during early life. The intact BBB also prevents microglial cell activation as the result of systemic antigens such as LPS gaining unrestricted access to the brain parenchyma remains limited. However, this is contrary to our modern understanding of the aging process and the subsequent neurodegenerative disease progression.
PERIODONTAL DISEASE, A RISK FACTOR FOR ALZHEIMER’S DISEASE?
During the course of life, oral infections, of which periodontitis is a typical example, challenge humans by becoming a constant and daily source of bacteraemias [3]. This leads to an increased risk [4] of developing complex diseases and the focus here is on AD. Epidemiological studies (prospective and retrospective) suggest ten-years and over, as a timeline, from the diagnosis of periodontal disease to its impact on AD development [5, 6] and the risk of developing the sporadic form of AD increases twofold [7–9]. Experimentally induced periodontitis in mice supports the fact that this oral disease can be a causal factor in the development of AD in later life [10–17]. Since periodontal disease is modifiable, it follows that there is potential to reduce the unnecessary burden of this debilitating neurodegenerative disease in some individuals through better oral health management and periodontal treatment [18, 19].
There is unanimous agreement between scientists that AD has many risk factors leading to its pathophysiological development. The single greatest risk factor for developing the sporadic form of AD is age (60–65 years onwards). Old age alone is unrelated to conventional medical conditions. This association encompasses immunocompromised individuals whose susceptibility to infection increases and promotes pathobiomes to develop. In periodontitis and related conditions, as discussed by Olsen et al. [20] and Dekita et al. [21], dysbiosis is achieved through manipulation of the host’s adaptive immune system involving cellular immunological responses, specifically of T- and B-cells. However, the common neuropathological characteristics amyloid-beta (Aβ) and hyperphosphorylated tau binding neurofibrillary tangles (NFTs) remain the same irrespective of the course of the AD disease process.
In the amyloidogenic pathway, the amyloid-β protein precursor gives rise to Aβ, a metabolite, which historically has been seen as being neurotoxic through its adverse effect on synapses and memory [22, 23]. However, a newly emerging concept of why Aβ accumulates in the brain, in the first place is gaining support. This concept proposes that Aβ triggers an early innate immune response as an antimicrobial peptide that can trap and kill a broad spectrum of microbes [24–26]. The finding of a wide spectrum of microbes (viruses, bacteria and fungi), in autopsied AD brains, further supports the role of Aβ as an antimicrobial protein [27–34].
LIPOPOLYSACCHARIDE FROM GRAM-NEGATIVE BACTERIA IN AD BRAINS
It has been reported that the AD brains at postmortem had a higher abundance of bacterial lipopolysaccharide (LPS) from Gram-negative bacteria compared to that of non-AD healthy controls that had been matched for age and postmortem interval [35, 36]. Brain scans reveal the presence of Aβ decades before the clinical onset of dementia, implying existence of previous brain infections. Given that AD patients have protracted suffering, which in some cases lasts up to a decade, recurrent infections from the time of clinical diagnosis to death remains a possibility. If Aβ fibrils were the ghost left behind by a previous infection, this would imply that microbial communities within the brain would have shifted over the passage of time, leaving their LPSs behind to degrade slowly. Alternatively, LPS from the gut following dysbiosis and/or shed outer membrane vesicles could add to the existing pool of this endotoxin. Our own research links LPS from the periodontal keystone Gram-negative pathogen P. gingivalis exclusively to AD brains [37]. The definitive link of P. gingivalis LPS, as opposed to LPS from any other bacterium, was assessed by using anti-P. gingivalis clone 1B5 antibody that specifically detects conserved epitopes in lipid A to the O-antigen region [38, 39]. The localization of this endotoxin in AD brains was restricted to glia and the major cerebral vessels [37] (Fig. 1c).
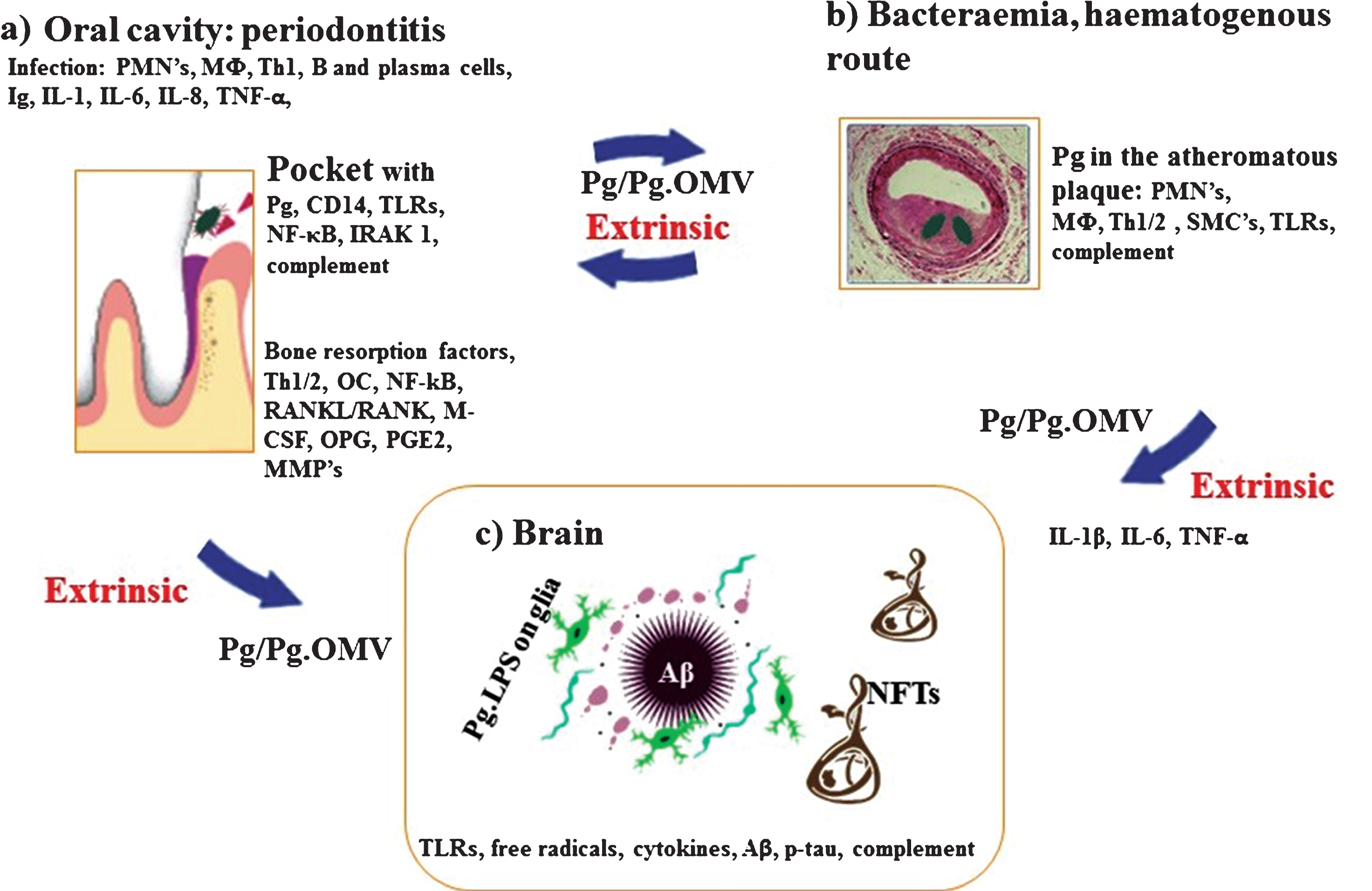
Fig.1
Schematic summary illustrates the extrinsic sources of inflammation involving periodontal and vascular pathologies to the brain including the local inflammation in each co-morbid state. a) Periodontitis: Body’s response is to initiate the innate immune defense mechanism. This results in recruitment of polymorphonuclear neutrophils (PMNs), which P. gingivalis uses as Trojan horses to enter below the gingivae. The host initiates P. gingivalis mediated inflammatory signaling pathways (lipopolysaccharide (LPS) receptor (CD14), and the highly conserved toll like receptors 2 and 4 (TLRs), nuclear factor κB (NF-κB), and the IL-1 receptor-activated kinase 1 (IRAK1). The bacterial proteases and LPS from the outer membrane vesicles (OMVs) directly damage connective tissues (elastin, fibrinogen, collagen) and the host cells also react by up regulating matrix metalloproteinases (MMPs) causing severe loss of host tissues. The humoral and adaptive immune response indicated by B-cells, plasma cells and T-cells (Th2) trigger immunoglobulin release and multiple cytokines (TNF-α, IL-1, IL-6, IL-8) and complement activation. The secretion of PGE2 and other cytokines NF-κB, receptor activator of NF-κB ligand (RANKL/RANK), macrophage colony stimulating factor (M-CSF) and osteoprotegrin (OPG) affect the alveolar bone homeostasis such that osteoclasts (OCs) begin bone resorption. b) Bacteraemias: PMNs and macrophages (MΦs) augment the expression of scavenger receptors in response to either local and/or extrinsic sources of inflammatory mediators and oral bacteraemia. Lipid-laden cells secrete proinflammatory cytokines, with recruitment of Th1/2 and MΦs within the atheromatous plaque where P. gingivalis lurks. Direct innate immune responses and additional cytokines and growth factors cause migration of smooth muscle cells (SMCs) and complement activation. c) Brain: The extrinsic factors from both periodontal disease and the vascular atheroma cause the initial trigger from systemic inflammation to affect the brain. This then leads to intracerebral inflammation (glia activated by P. gingivalis LPS from OMVs) leading to complement activation and emergence of hallmark proteins (Aβ and NFTs) that define Alzheimer’s disease.
In another study, the Escherichia coli (E. coli) K99 strain also appeared to be the source of LPS in AD brains [35]. The striking difference in this report was that LPS co-localized to both amyloid plaques and the vascular amyloid, which accumulates in cerebral amyloid angiopathy in AD [35, 36]. So why did P. gingivalis LPS not bind to Aβ deposits in the Poole et al. [37] investigation? One explanation is that E. coli K99 carries the curli protein gene, which transcribes bacterial functional amyloids on the outer membrane of several prokaryotes; something that P. gingivalis lacks [40]. Therefore, LPS from E. coli K99 binding amyloid in AD brains is to be expected.
Observations from pure bacterial cultures and examination of biofilms using electron microscopy demonstrate that bacteria release vast numbers of outer membrane vesicles [41]. These vesicles not only contain LPS, but also a number of other bacterial pathogen associated molecular patterns (PAMPs), which upon entry to the brain have the potential to trigger proinflammatory signalling pathways via toll like receptor 2 and 4 (TLR2/4). This will lead to secretion of proinflammatory cytokines (TNF-α, IL-1β, IL-6, IL-8) via a variety of antigen presenting cell types [42]. A property of Aβ hallmark protein, as an antimicrobial peptide, is to promote complement activation [25, 26], presumably for its phagocytosis by microglia. P. gingivalis oral infections also contribute to cytokine release and to the development of AD hallmark lesions, as demonstrated in vivo by models of induced periodontitis [10, 11], together with the AD-like phenotype [11–13, 17]. All of these elements together suggest that inflammation appears to negatively impact on memory, causing the cognitive decline that is typical of clinical AD.
P. gingivalis AND Pg. MICROBULLETS
A working hypothesis is that the dysbiosis in the sub-gingival niche, which is responsible for periodontal disease [43], may also cause AD through dispersal of the planktonic sub-gingival biofilm and the many potent PAMPs (LPS, gingipains, capsule, fimbriae) of P. gingivalis within Pg. microbullets [44]. Therefore, Pg. microbullets could act as stand-alone units of multiple virulence factors that may be recognized by the host’s pattern recognition receptors (PRRs) for signal transduction pathways following an invasion of the host tissues by microbial attack. The Pg. microbullets, because of their size (nanoscale), can reach the brain following perturbed barrier functions (oral epithelial and BBB alone) during periodontitis and AD (Fig. 2).
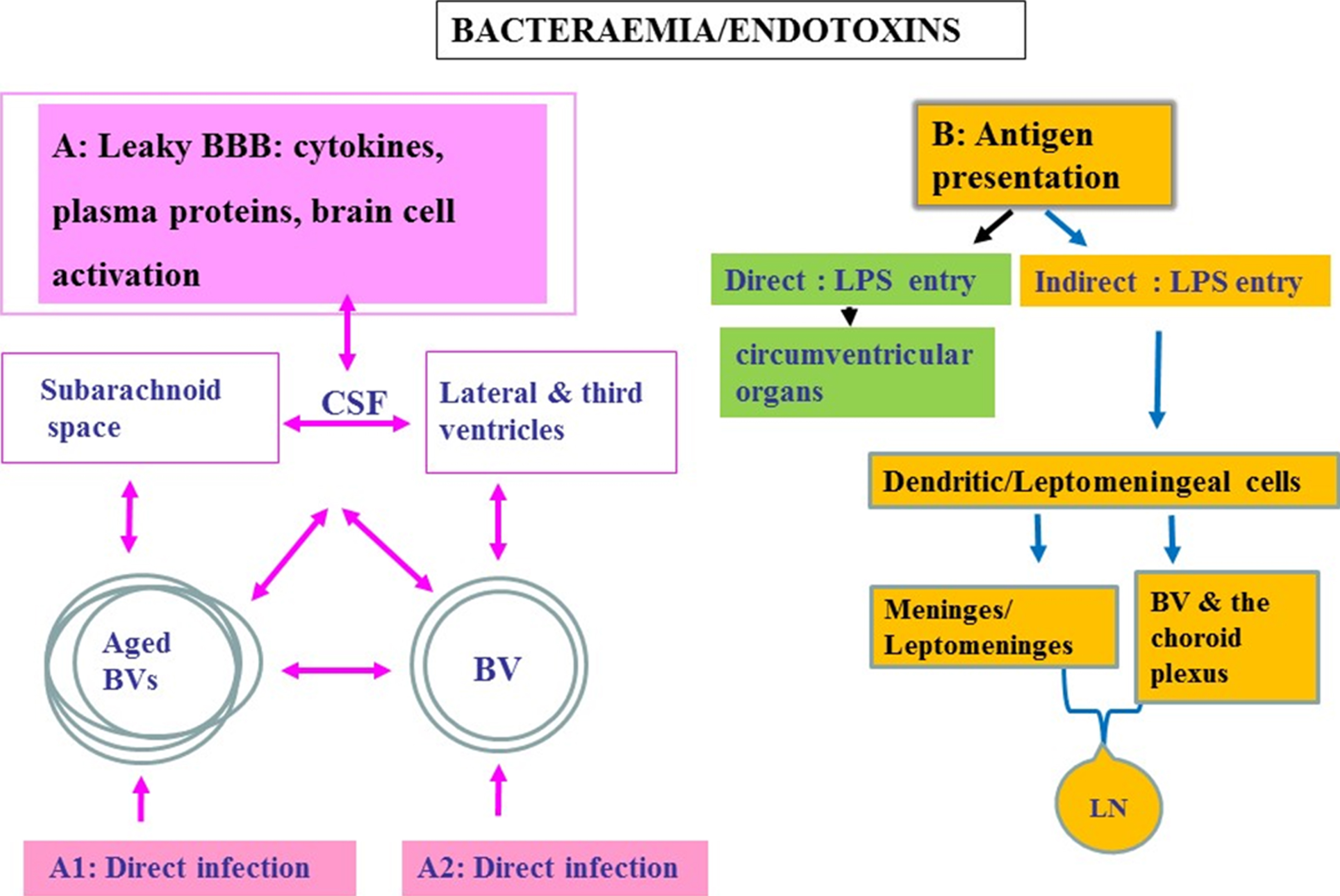
Fig.2
Schematic to show oral pathogen/endotoxin entry direct and/or indirect into the brain following bacteraemias. A) Attenuated P. gingivalis/LPS within the systemic circulation can gain access to the brain via transport across the blood-brain barrier (BBB). Once organisms are present in the ventricular cerebrospinal fluid (CSF), they can potentially invade the subarachnoid space. From the CSF, bacteria cross the ependymal epithelial cell layer lining the ventricle wall for entry into the brain parenchyma. A1 represents an infection encountered in advanced age and A2 in younger age. Bacteria within the systemic circulation can gain access to the perivascular spaces, which in turn connect with the subarachnoid space. The CSF connects to and communicates with the lateral and third ventricles providing access for pathogens to all parts of the CNS. B) Endotoxin transfer directly into the brain via circumventricular organs and indirectly via antigen presentation: The meninges have a well-developed population of dendritic cells and meningeal cells that communicate through lymphatic vessels with the adaptive immune system located in the systemic lymph nodes. Dendritic cells in blood vessels of the choroid plexus and the meninges/leptomeninges house cells expressing the mannose receptor, lipopolysaccharide (LPS) receptor (CD14), and the highly conserved toll like receptors 2 and 4 (TLR 2 and 4).
Aberrant infections of the brain in the elderly and those suffering from dementias are also documented, with similar inflammatory responses to those linked with P. gingivalis and to its LPS [45, 46]. However, there is no experimental data linking aberrant infections to clinical features in humans, such as sleep pattern disturbances [47, 48], except for a report on the presence of P. gingivalis in mice [49]. Active periodontal disease and reduced glymphatic system functioning, due to disturbed sleep, could impair the brain’s ability to clear microbes, such as P. gingivalis.
BACTERIAL ENTRY INTO THE BRAIN
There are various means of entry for periodontal bacteria to access the brain [32]. However, P. gingivalis and its microbullets predominantly use the circumventricular organs and the hematogenous routes [10, 14, 28, 50] (Fig. 2). Other routes include a permeable BBB or the choroid plexus that is devoid of the BBB function but contains a rich blood supply surrounded by epithelial cells, and the perivascular spaces, which in turn connect with the subarachnoid space. The subarachnoid space contains cerebrospinal fluid (CSF) that connects to and communicates with the lateral and third ventricles providing access for pathogens to all parts of the CNS. Re-absorption of CSF into the venous circulation provides a potential communication with the lymphatic system (Fig. 2B). Once organisms are present in the ventricular CSF, they can potentially invade the subarachnoid space. From the CSF, entry into the brain parenchyma is also possible provided bacteria cross the ependymal epithelial cell layer lining the ventricle wall (Fig. 2A, B). Epithelial cell barrier breakdown is not a challenge for P. gingivalis, as evidenced in periodontal disease. The meninges communicate with the brain through dendritic cells within lymphatic vessels located in the systemic lymph nodes. In vitro studies suggest that the leptomeningeal cells also have the capacity to act as a go between at the interface of peripheral transduction signaling pathways for the innate immune system to convey inflammatory signals to cerebral glia [42]. Dendritic cells express the major histocompatibility complex class II (MHC-class II) antigen on their surface membranes and are also found lying within blood vessels of the choroid plexus (Fig. 2B). In addition, the leptomeningeal cells express TLR 2 and 4, which are able to recognize Gram-negative bacteria [42].
The ependymal cells express regulatory proteins, which protect them against damage from ongoing inflammation, and also express phagocytic receptors, including the mannose receptor and TLRs (additional examples of PRRs), which detect and clear bacteria [51–55]. It seems extremely likely that P. gingivalis and Pg. microbullets hitch ride on erythrocytes (Figs. 1 and 2) [56], firstly by abolishing the anaphylatoxin C5a activity (see review [57]), and then by degrading some of the CR1 molecules [44] that normally facilitate clearance via the spleen. As there is no immune surveillance by cells from the classical adaptive immune system, the resident glia provide a local innate defense mechanism capable of defending the CNS against pathogen entry.
Most importantly, microglia are MHC-class II positive cells capable of antigen presentation as well as expressing CD14 (LPS ligand), TLR 2/4, which recognize PAMPs, as well as a range of complement receptors (CR1, CR3, and CR4), that aid with their phagocytic function [58]. The activated microglia express de novo immune markers, including MHC class II proteins (which allow antigen presentation). They express complement proteins, which destroy pathogens, as well as numerous scavenger and phagocytic receptors (CR1, CR3, CR4, TLR 2 and 4) and pro-inflammatory signaling molecules (C4a, C3a, C5a), which in turn, recruit more activated microglia to the site of tissue injury [54, 58]. Hence, microglia represent “danger” sensing cells within the local CNS microenvironment. Hajishengalis [44] clearly described the ability of P. gingivalis to block some of these receptors to disable phagocytic activity of macrophages.
Microglia become activated, if challenged with systemic LPS, whereby they mount an innate immune defense response to combat the destructive effects of the endotoxin. If exposure to LPS continues, microglia adopt a hypersensitive phenotype. During adoption of this phenotype, an increase in microglial cell synthesis of inflammatory mediators (IL-1β, IL-6, TNF-α, complement factors, TLR 2 and 4, nitric oxide (NO) and reactive oxygen species) is seen [54, 58–63]. This has a negative impact on the severity of neurodegeneration. The increase in NO and TLR 2/4 in AD brains can be explained following entry of P. gingivalis and Pg. microbullets. Hajishengalis [44] described the ability of P. gingivalis to block the inducible NO synthase activity by antagonizing the TLR 2, and by switching over to the cAMP protein kinase (PKA) pathway instead of TLR 4 due to the LPS structure under various hemin deficiencies [39].
The primary function of activated microglia [64] is to provide proinflammatory interleukin classes of cytokines [65], aid phagocytosis of local cellular debris and protect nerve cells. P. gingivalis and its microbullets have the capacity to attenuate phagocytosis by hijacking the opsonophagocytic activity of neutrophils in the gingiva via C3 receptor attenuation and degradation of IgG1 [44, 66]. It is therefore, plausible to suggest that Pg. microbullets, in AD brains, play a role in impairing microglial cell activity related to Aβ phagocytosis. Whole-genome sequencing studies have revealed the Triggering Receptor Expressed on Myeloid cells (TREM), which has a negative impact on microglial phagocytosis of Aβ [67–69]. Not surprisingly, P. gingivalis and its microbullets can cleave at least TREM1 from neutrophils and amplify inflammation [66]. In addition, genome-wide association studies support the role of the complement cascade component genes encoding CR1 and a fluid-phase regulatory protein clusterin, C1s and C9 [70–73] as discussed elsewhere [74]. The fact that P. gingivalis has the capacity to affect all of these genes surely strengthens the causative role of periodontitis, which itself involves complement [75], with AD manifestation (Fig. 1).
Liu et al. [76] provided the first real evidence that infection of microglia with P. gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. The subsequent activation of phosphoinositide 3-kinase/Akt and mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase/ERK pathways stimulated cell migration and an inflammatory response in microglia. This emphasizes the importance of P. gingivalis gingipains in AD pathogenesis, which the Pg. microbullets contain [41, 44].
In the brain, the guardians of host defense are microglia, whilst this function in the gingival sulcus/periodontal pocket is courtesy of neutrophils. P. gingivalis disables neutrophils, and in this way, the bacterium can use it as a vehicle to reach its preferred primary oral niche [44]. Growth of P. gingivalis, within the host, releases vast numbers of Pg. microbullets. As mentioned earlier, Pg. microbullets are loaded with appropriate ammunition (virulence factors). On their dispersal within the body, they reach the AD brain [37], where inflammation (inflammatory cytokines), and concurrent synaptic loss through IL-1β and Aβ accumulation, are present [77].
BRAINS PARA/PERIVASCULAR CLEARANCE PATHWAYS
The brain has many mechanisms for clearance of cellular debris and other solutes. One clearance system relevant to good quality sleep appears to be the glymphatic system, which describes the flow of CSF through the brain parenchyma and the interstitial solutes [78]. During natural sleep, an increase in the interstitial space was detected, which is thought to increase the convective exchange of CSF and interstitial fluid (ISF), which in turn, improved the clearance of waste toxins like Aβ from the brain. The influx of CSF, and the subsequent clearance of ISF, appears to be facilitated by astroglial water (aquaporin 4) channels that line the paravascular pathways.
The intramural periarterial drainage pathway (IPAD), on the other hand, refers exclusively to the clearance pathway of solutes from the brain parenchyma along arterial smooth muscle cell basement membranes [79, 80]. Recently, a separate periarterial (pial-glial) basement membrane was shown to serve as the pathway for CSF entry into the brain [80]. Bacterial, and/or its LPS, entry into the brain can be via the CSF, or a direct infection in the cerebral tissues (Fig. 2B) and for the LPS via direct/indirect antigen presentation (Fig. 2B). Zhan et al. [35, 36] showed that E. coli K88 LPS co-localized to vascular amyloid accumulates in cerebral amyloid angiopathy. Therefore, LPS clearance via both the IPAD and the glymphatic systems may be taking the same pathway that Aβ uses to exit the brain. Since the CSF eventually drains into the lymph nodes, it seems plausible to suggest that both of these clearance pathways could be involved in removing microbial debris from the brain.
COMPLEMENT SYSTEM AS A CLEARANCE PATHWAY FOR P. gingivalis
The complement cascade, which is an evolutionary important innate immune response system known to combat bacterial infections, plays a role in the pathogenesis of periodontitis, atherosclerosis and AD (Fig. 1a-c), [77]. The complement cascade is mentioned here only in passing because of the plethora of literature that exists already on this subject. However, the need for mentioning this bacterial pathway of clearance comes from the genome-wide association studies having identified susceptibility genes linking this cascade to the pathophysiology of AD. Researchers now have the task to explain how defects in various complement genes are driving neurodegeneration. As mentioned above, P. gingivalis is particularly adept at evading the host’s immune responses and in directing the opsonophagocytic activity of neutrophil function as seen in the periodontium [44, 66]. This implies that organ specific innate immune drivers will play a tissue specific role in the inability to perform clearance of P. gingivalis and its Pg. microbullets [14, 50].
To test the hypothesis of advancing age being the strongest risk factor for developing AD, Ishida et al. [12] and Ding et al. [13], performed functional tests on mice and demonstrated statistically significant outcomes for impaired learning and memory in middle aged P. gingivalis infected mice compared to the younger and middle-aged uninfected mice (wild type and transgenic mice). Wu et al. [11] also supported the AD-like phenotype with the “middle-age” phenomenon in their research where cathepsin B sufficient mice subjected to chronic LPS exposure from P. gingivalis demonstrated intra-neuronal Aβ accumulation. In addition, Wu et al. [11] and Zhang et al. [17] (wild-type C57BL/6 mice) demonstrated that P. gingivalis LPS mediated cytokine release was responsible for cognitive deficit in their respective mouse models. This research provides evidence to support a causal relationship between chronic exposure to a keystone periodontal pathogen and emerging hallmark proteins of AD depositing in areas of pathology together with functional symptoms.
CONCLUSIONS
Based on the evidence presented in this commentary the most likely contribution made by periodontal disease to the development of AD is through the effects of systemic cytokines and/or microbial cell wall molecules such as LPS and microbullets of P. gingivalis on the brain. Alternatively, there may be a negative impact from the oral/gastrointestinal tract dysbiosis. Aβ is not toxic per se, but it is protective as an innate immune protein released by the host in order to trap a wide spectrum of microbes. This suggests that neurodegeneration can result from direct invasion of oral and other microbes to the brain. For the periodontal contribution to AD development, proof of concept has clearly been provided by Ilievski et al. [10], who demonstrated that a chronic oral infection with P. gingvalis with intact virulence factors (LPS) as suggested by Wu et al. [11] play a role in AD hallmark lesion formation and subsequent cognitive deficit as suggested by references [11, 17]. Given that periodontal disease is a peripheral chronic inflammatory condition affecting about 50% of people over 55 years of age, action is definitely required. We call upon health providers and governments to take this research seriously. A ten-year timeline from a diagnosis of periodontal disease gives an individual plenty of time to modify his/her poor oral hygiene habits, and learn improved techniques with the help of dental professionals. Regular dental professional input to treat and manage periodontal disease is vital to reduce the unnecessary high incidence of AD. We call upon global governments to make dental health more widely available to people in their fifties and above.
CONFLICT OF INTEREST
The authors declare no competing interests.
ACKNOWLEDGMENTS
SKS has received a PreViser award from the Oral and Dental Research Trust, 2018, and also acknowledges the continued financial support from the School of Dentistry, University of Central Lancashire, UK.
References
[1] | Eke PI , Dye BA , Wei L , Slade GD , Thornton-Evans GO , Borgnakke WS , Taylor GW , Page RC , Beck JD , Genco RJ ((2015) ) Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol 86: , 611–622. |
[2] | Farhad SZ , Amini S , Khalilian A , Barekatain M , Mafi M , Barekatain M , Rafei E ((2014) ) The effect of chronic periodontitis on serum levels of tumor necrosis factor-alpha in Alzheimer disease. Dent Res J (Isfahan) 11: , 549–552. |
[3] | Olsen I ((2008) ) Update on bacteraemia related to dental procedures. Transfus Apher Sci 39: , 173–178. |
[4] | Tzeng NS , Chung CH , Yeh CB , Huang RY , Yuh DY , Huang SY , Lu RB , Chang HA , Kao YC , Chiang WS , Chou YC , Chien WC ((2016) ) Are chronic periodontitis and gingivitis associated with dementia? A nationwide, retrospective, matched-cohort study in Taiwan. Neuroepidemiology 47: , 82–93. |
[5] | Sparks Stein P , Steffen MJ , Smith C , Jicha G , Ebersole JL , Abner E , Dawson D 3rd. ((2012) ) Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimers Dement 8: , 196–203. |
[6] | Chen CK , Wu YT , Chang YC ((2017) ) Association between chronic periodontitis and the risk of Alzheimer’s disease: A retrospective, population-based, matched-cohort study. Alzheimers Res Ther 9: , 56. |
[7] | Gatz M , Mortimer JA , Fratiglioni L , Johansson B , Berg S , Reynolds CA , Pedersen NL ((2006) ) Potentially modifiable risk factors for dementia in identical twins. Alzheimers Dement 2: , 110–117. |
[8] | Stein PS , Desrosiers M , Donegan SJ , Yepes JF , Kryscio RJ ((2007) ) Tooth loss, dementia and neuropathy in the Nun study. J Am Dent Assoc 138: , 1314–1322. |
[9] | Noble J , Scarmeas N , Papapanou P ((2013) ) Poor oral health as a chronic, potentially modifiable dementia risk factor: Review of the literature. Curr Neurol Neurosci Rep 13: , 384. |
[10] | Ilievski V , Zuchowska PK , Green SJ , Toth PT , Ragozzino ME , Le K , Aljewari HW , O'Brien-Simpson NM , Reynolds EC , Watanabe K ((2018) ) Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One 13: , e0204941. |
[11] | Wu Z , Ni J , Liu Y , Teeling JL , Takayama F , Collcutt A , Ibbett P , Nakanishi H ((2017) ) Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun 65: , 350–361. |
[12] | Ishida N , Ishihara Y , Ishida K , Tada H , Funaki-Kato Y , Hagiwara M , Ferdous T , Abdullah M , Mitani A , Michikawa M , Matsushita K ((2017) ) Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech Dis 3: , 15. |
[13] | Ding Y , Ren J , Yu H , Yu W , Zhou Y ((2018) ) Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun Ageing 15: , 6. |
[14] | Poole S , Singhrao SK , Chukkapalli S , Rivera M , Velsko I , Kesavalu L , Crean S ((2015) ) Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE-/- mice brains. J Alzheimers Dis 43: , 67–80. |
[15] | Singhrao SK , Chukkapalli S , Poole S , Velsko I , Crean S , Kesavalu L ((2017) ) Chronic Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE-/- mice brains. J Oral Microbiol 9: , 1270602. |
[16] | Rokad R , Moseley R , Hardy SR , Chukkapalli S , Crean S , Kesavalu L , Singhrao SK ((2017) ) Cerebral oxidative stress and microvasculature defects in TNF-α expressing transgenic and Porphyromonas gingivalis-infected ApoE-/- mice. J Alzheimers Dis 60: , 359–369. |
[17] | Zhang J , Yu C , Zhang X , Chen H , Dong J , Lu W , Song Z , Zhou W ((2018) ) Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signalling pathway in C57BL/6 mice. J Neuroinflammation 15: , 37. |
[18] | Kamer AR , Janal MN , deLeon MJ ((2015) ) Letter to the editor regarding: Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimers Dement (Amst) 1: , 385–386. |
[19] | Harding A , Robinson S , Crean S , Singhrao SK ((2017) ) Can better management of periodontal disease delay the onset and progression of Alzheimer’s disease? J Alzheimers Dis 58: , 337–348. |
[20] | Olsen I , Taubman MA , Singhrao SK ((2016) ) Porphyromonas gingivalis suppresses adaptive immunity in periodontitis, atherosclerosis, and Alzheimer’s disease. J Oral Microbiol 8: , 33029. |
[21] | Dekita M , Wu Z , Ni J , Zhang X , Liu Y , Yan X , Nakanishi H , Takahashi I ((2017) ) Cathepsin S is involved in Th17 differentiation through the upregulation of IL-6 by activating PAR-2 after systemic exposure to lipopolysac- charide from Porphyromonas gingivalis. Front Pharmacol 17: , 470. |
[22] | Lacor PN , Buniel MC , Chang L , Fernandez SJ , Gong Y , Viola KL , Lambert MP , Velasco PT , Bigio EH , Finch CE , Krafft GA , Klein WL ((2004) ) Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci 24: , 10191–10200. |
[23] | Lesne S , Koh MT , Kotilinek L , Kayed R , Glabe CG , Yang A , Gallagher M , Ashe KH ((2006) ) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440: , 352–357. |
[24] | Soscia SJ , Kirby JE , Washicosky KJ , Tucker SM , Ingelsson M , Hyman B , Burton MA , Goldstein LE , Duong S , Tanzi RE , Moir RD ((2010) ) The Alzheimer’s disease-associated amyloid B-protein is an antimicrobial peptide. PLoS One 5: , e9505. |
[25] | Kumar DK , Choi SH , Washicosky KJ , Eimer WA , Tucker S , Ghofrani J , Lefkowitz A , McColl G , Goldstein LE , Tanzi RE , Moir RD ((2016) ) Amyloid-ß peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 8: , 340ra72. |
[26] | Eimer WA , Kumar DKV , Kumar N , Shanmugam N , Rodriguez AS , Mitchell T , Washicosky KJ , Gyorgy B , Breakefield XO , Tanzi RE , Moir RD ((2018) ) Alzheimer’s disease-associated ß-amyloid is rapidly seeded by Her- pesviridae to protect against brain infection. Neuron 99: , 56–63. |
[27] | MacDonald AB , Mirranda JM ((1987) ) Concurrent neocortical borreliosis and Alzheimer’s disease. Hum Pathol 18: , 759–761. |
[28] | Miklossy J ((1993) ) Alzheimer’s disease-a spirochetosis? Neuroreport 4: , 841–848. |
[29] | Pappolla MA , Omar R , Saran B , Andorn A , Suarez M , Pavia C , Weinstein A , Shank D , Davis K , Burgdorfer W ((1989) ) Concurrent neuroborreliosis and Alzheimer’s disease: Analysis of the evidence. Hum Pathol 20: , 753–757. |
[30] | Itzhaki RF , Lin W-R , Shang D , Wilcock GK , Faragher B , Jamieson GA ((1997) ) Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 349: , 241–244. |
[31] | Balin BJ , Gerard HC , Arking EJ , Appelt DM , Branigan PJ , Abrams JT , Whittum-Hudson JA , Hudson AP ((1998) ) Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med Microbiol Immunol 187: , 23–42. |
[32] | Riviere GR , Riviere KH , Smith KS ((2002) ) Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol 17: , 113–118. |
[33] | Kountouras J , Tsolaki M , Gavalas E , Boziki M , Zavos C , Karatzoglou P , Chatzopoulos D , Venizelos I ((2006) ) Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology 66: , 938–940. |
[34] | Carrasco L , Alonso R , Pisa D , Rabano A ((2017) ) Alzheimer’s disease and fungal infection. In Handbook of Infection and Alzheimer’s Disease, Miklossy J, ed. IOS Press, Amsterdam, pp. 281–294. |
[35] | Zhan X , Stamova B , Jin LW , DeCarli C , Phinney B , Sharp FR ((2016) ) Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 87: , 2324–2332. |
[36] | Zhan X , Stamova B , Sharp FR ((2018) ) Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in Alzheimer’s disease brain: A Review. Front Aging Neurosci 10: , 42. |
[37] | Poole S , Singhrao SK , Kesavalu L , Curtis MA , Crean S ((2013) ) Determining the presence of periodontopathic virulence factors in short-term post-mortem Alzheimer’s disease brain tissue. J Alzheimers Dis 36: , 665–677. |
[38] | Curtis MA , Aduse-Opoku J , Slaney JM , Rangarajan M , Booth V , Cridland J , Shepherd P ((1996) ) Characterization of an adherence and antigenic determinant of the ArgI protease of Porphyromonas gingivalis which is present on multiple gene products. Infect Immun 64: , 2532–2539. |
[39] | Olsen I , Singhrao SK ((2018) ) Importance of heterogeneity in Porhyromonas gingivalis lipopolysaccharide lipid A in tissue specific inflammatory signalling. J Oral Microbiol 10: , 1440128. |
[40] | Pritchard AB , Crean S , Olsen I , Singhrao SK ((2017) ) Periodontitis, microbiomes and their role in Alzheimer’s disease. Front Aging Neurosci 9: , 336. |
[41] | Olsen I , Amano A ((2016) ) Outer membrane vesicles - offensive weapons or good Samaritans? J Oral Microbiol 7: , 27468. |
[42] | Liu Y , Wu Z , Zhang X , Ni J , Yu W , Zhou Y , Nakan- ishi H ((2013) ) Leptomeningeal cells transduce peripheral macrophages inflammatory signal to microglia in reponse to Porphyromonas gingivalis LPS. Mediators Inflamm 2013: , 407562. |
[43] | Hajishengallis G , Darveau RP , Curtis MA ((2012) ) The keystone-pathogen hypothesis. Nat Rev Microbiol 10: , 717–725. |
[44] | Hajishengalis G ((2011) ) Immune evasion strategies of Porphyromonas gingivalis. J Oral Biosci 53: , 233–240. |
[45] | Miklossy J ((2008) ) Chronic inflammation and amyloidogenesis in Alzheimer’s disease -role of spirochetes. J Alzheimers Dis 13: , 381–391. |
[46] | Miklossy J ((2011) ) Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation 8: , 90–106. |
[47] | Musiek ES , Holtzman DM ((2016) ) Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354: , 1004–1008. |
[48] | Cedernaes J , Osorio RS , Varga AW , Kam K , Schioth HB , Benedict C ((2017) ) Candidate mechanisms underlying the association between sleep-wake disruptions and Alzheimer’s disease. Sleep Med Rev 31: , 102–111. |
[49] | Takayama F , Hayashi Y , Wu Z , Liu Y , Nakanishi H ((2016) ) Diurnal dynamic behaviour of microglia in response to infected bacteria through the UDP-P2Y6 receptor system. Sci Rep 6: , 30006. |
[50] | Velsko IM , Chukkapalli SS , Rivera MF , Lee J-Y , Chen H , Zheng D , Bhattacharyya I , Gangula PR , Lucas AR , Kesavalu L ((2014) ) Active invasion of oral and aortic tissues by Porphyromonas gingivalis in mice causally links periodontitis and atherosclerosis. PLoS One 9: , e97811. |
[51] | Stahl PD , Ezekowitz RA ((1998) ) The mannose receptor is a pattern recognition receptor involved in host defense. Curr Opin Immunol 10: , 50–55. |
[52] | McMenamin PG ((1999) ) Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater. J Comp Neurol 405: , 553–562. |
[53] | Burudi EM , Stahl PD , Regnier-Vigouroux A ((1999) ) Identification and functional characterisation of the mannose receptor in astrocytes. Glia 25: , 44–55. |
[54] | Laflamme N , Rivest S ((2001) ) Toll-like receptor 4: The missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J 15: , 155–163. |
[55] | Husemann J , Loike JD , Anankov R , Febbraio M , Silver-stein SC ((2002) ) Scavenger receptors in neurobiology and neuropathology: Their role on microglia and other cells of the nervous system. Glia 40: , 195–205. |
[56] | Bielecka E , Scavenius C , Kantyka T , Jusko M , Mizgalska D ((2014) ) Peptidyl arginine deiminase from Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J Biol Chem 289: , 32481–32487. |
[57] | Olsen I , Singhrao SK , Potempa J ((2018) ) Citrullination as a plausible link to periodontitis, rheumatoid arthritis, atherosclerosis and Alzheimer’s disease. J Oral Microbiol 10: , 1487742. |
[58] | Gasque P ((2004) ) Complement: A unique innate immune sensor for danger signals. Mol Immunol 41: , 1089–1098. |
[59] | Boje KM , Arora PK ((1992) ) Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res 587: , 250–256. |
[60] | Lodge PA , Sriram S ((1996) ) Regulation of microglia activation by TGF-b, IL-10, and CSF-1. J Leukoc Biol 60: , 502–508. |
[61] | Floyd RA ((1999) ) Neuroinflammatory processes are important in neurodegenerative diseases: An hypothesis to explain the increased formation of reactive oxygen and nitrogen species as major factors involved in neurodegenerative disease development. Free Radic Biol Med 26: , 1346–1355. |
[62] | Ye S , Johnson RW ((2001) ) Regulation of interleukin-6 gene expression in brain of aged mice by nuclear factor kappa B. J Neuroimmunol 117: , 87–96. |
[63] | Godbout JP , Chen J , Abraham J , Richwine AF , Berg BM , Kelly KW , Johnson RW ((2005) ) Exaggerated neuroinflammation and sickness behaviour in aged mice after activation of the peripheral innate immune system. FASEB J 19: , 1329–1331. |
[64] | Ransohoff RM , Perry VH ((2009) ) Microglial physiology: Unique stimuli, specialized responses. Annu Rev Immunol 27: , 119–145. |
[65] | Hanisch UK ((2002) ) Microglia as a source and target of cytokines. Glia 40: , 140–155. |
[66] | Olsen I , Hajishengalis G ((2016) ) Major neutrophil functions subverted by Porphyromonas gingivalis . J Oral Microbiol 8: , 30936. |
[67] | Guerreiro R , Wojtas A , Bras J , Carrasquillo M , Rogaeva E , Majounie E , Cruchaga C , Sassi C , Kauwe JS , Younkin S , Hazrati L , Collinge J , Pocock J , Lashley T , Williams J , Lambert JC , Amouyel P , Goate A , Rademakers R , Morgan K , Powell J , St George-Hyslop P , Singleton A , Hardy J ; Alzheimer Genetic Analysis Group ((2013) ) Alzheimer genetic analysis Group TREM2 variants in Alzheimer’s disease. N Engl J Med 368: , 117–127. |
[68] | Jonsson T , Stefansson H , Steinberg S , Jonsdottir I , Jonsson PV , Snaedal J , Bjornsson S , Huttenlocher J , Levey AI , Lah JJ , Rujescu D , Hampel H , Giegling I , Andreassen OA , Engedal K , Ulstein I , Djurovic S , Ibrahim-Verbaas C , Hofman A , Ikram MA , van Duijn CM , Thorsteinsdottir U , Kong A , Stefansson K ((2013) ) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368: , 107–116. |
[69] | Replogle JM , Chan G , White CC , Raj T , Winn PA , Evans DA , Sperling RA , Chibnik LB , Bradshaw EM , Schneider JA , Bennett DA , De Jager PL ((2015) ) A TREM1 variant alters the accumulation of Alzheimer-related amyloid pathology. Ann Neurol 77: , 469–477. |
[70] | Lambert JC , Heath S , Even G , Campion D , Sleegers K , Hiltunen M , Combarros O , Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fiévet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O; European Alzheimer’s Disease Initiative Investigators, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossù P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanché H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alpérovitch A, Lathrop M, Amouyel P ((2009) ) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41: , 1094–1099. |
[71] | Lambert JC , Ibrahim-Verbaas CA , Harold D , Naj AC , Sims R , Bellenguez C , DeStafano AL , Bis JC , Beecham GW , Grenier-Boley B , Russo G , Thorton-Wells TA , Jones N , Smith AV , Chouraki V , Thomas C , Ikram MA , Zelenika D , Vardarajan BN , Kamatani Y , Lin CF , Gerrish A , Schmidt H , Kunkle B , Dunstan ML , Ruiz A , Bihoreau MT , Choi SH , Reitz C , Pasquier F , Cruchaga C , Craig D , Amin N , Berr C , Lopez OL , De Jager PL , Deramecourt V , Johnston JA , Evans D , Lovestone S , Letenneur L , Morón FJ , Rubinsztein DC , Eiriksdottir G , Sleegers K , Goate AM , Fiévet N , Huentelman MW , Gill M , Brown K , Kamboh MI , Keller L , Barberger-Gateau P , McGuiness B , Larson EB , Green R , Myers AJ , Dufouil C , Todd S , Wallon D , Love S , Rogaeva E , Gallacher J , St George-Hyslop P , Clarimon J , Lleo A , Bayer A , Tsuang DW , Yu L , Tsolaki M , Bossù P , Spalletta G , Proitsi P , Collinge J , Sorbi S , Sanchez-Garcia F , Fox NC , Hardy J , Deniz Naranjo MC , Bosco P , Clarke R , Brayne C , Galimberti D , Mancuso M , Matthews F ; European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology, Moebus S , Mecocci P , Del Zompo M , Maier W , Hampel H , Pilotto A , Bullido M , Panza F , Caffarra P , Nacmias B , Gilbert JR , Mayhaus M , Lannefelt L , Hakonarson H , Pichler S , Carrasquillo MM , Ingelsson M , Beekly D , Alvarez V , Zou F , Valladares O , Younkin SG , Coto E , Hamilton-Nelson KL , Gu W , Razquin C , Pastor P , Mateo I , Owen MJ , Faber KM , Jonsson PV , Combarros O , O'Donovan MC , Cantwell LB , Soininen H , Blacker D , Mead S , Mosley TH Jr , Bennett DA , Harris TB , Fratiglioni L , Holmes C , de Bruijn RF , Passmore P , Montine TJ , Bettens K , Rotter JI , Brice A , Morgan K , Foroud TM , Kukull WA , Hannequin D , Powell JF , Nalls MA , Ritchie K , Lunetta KL , Kauwe JS , Boerwinkle E , Riemenschneider M , Boada M , Hiltuenen M , Martin ER , Schmidt R , Rujescu D , Wang LS , Dartigues JF , Mayeux R , Tzourio C , Hofman A , Nöthen MM , Graff C , Psaty BM , Jones L , Haines JL , Holmans PA , Lathrop M , Pericak-Vance MA , Launer LJ , Farrer LA , van Duijn CM , Van Broeckhoven C , Moskvina V , Seshadri S , Williams J , Schellenberg GD , Amouyel P ((2013) ) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45: , 1452–1458. |
[72] | Jones L1 , Holmans PA , Hamshere ML , Harold D , Moskvina V , Ivanov D , Pocklington A , Abraham R , Hollingworth P , Sims R , Gerrish A , Pahwa JS , Jones N , Stretton A , Morgan AR , Lovestone S , Powell J , Proitsi P , Lupton MK , Brayne C , Rubinsztein DC , Gill M , Lawlor B , Lynch A , Morgan K , Brown KS , Passmore PA , Craig D , McGuinness B , Todd S , Holmes C , Mann D , Smith AD , Love S , Kehoe PG , Mead S , Fox N , Rossor M , Collinge J , Maier W , Jessen F , Schürmann B , Heun R , Kölsch H , van den Bussche H , Heuser I , Peters O , Kornhuber J , Wiltfang J , Dichgans M , Frölich L , Hampel H , Hüll M , Rujescu D , Goate AM , Kauwe JS , Cruchaga C , Nowotny P , Morris JC , Mayo K , Livingston G , Bass NJ , Gurling H , McQuillin A , Gwilliam R , Deloukas P , Al-Chalabi A , Shaw CE , Singleton AB , Guerreiro R , Mühleisen TW , Nöthen MM , Moebus S , Jöckel KH , Klopp N , Wichmann HE , Rüther E , Carrasquillo MM , Pankratz VS , Younkin SG , Hardy J , O'Donovan MC , Owen MJ , Williams J ((2010) ) Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One 5: , e13950. |
[73] | International Genomics of Alzheimer’s Disease Consortium (IGAP) ((2015) ) Convergent genetic and expression data implicate immunity in Alzheimer’s disease. Alzheimers Dement 11: , 658–671. |
[74] | Morgan BP ((2018) ) Complement in the pathogenesis of Alzheimer’s disease. Semin Immunopathol 40: , 113–124. |
[75] | Olsen I , Lambris JD , Hajishengallis G ((2017) ) Porphyromonas gingivalis disturbs host-commensal homeostasis by changing complement function. J Oral Microbiol 9: , 1340085. |
[76] | Liu Y , Wu Z , Nakanishi Y , Ni J , Hayashi Y , Takayama F , Zhou Y , Kadowaki T , Nakanishi H ((2017) ) Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci Rep 7: , 11759. Correction: Sci Rep 8: ((2018) ), 10304. |
[77] | Akiyama H , Barger S , Barnum S , Bradt B , Bauer J , Cole GM , Cooper NR , Eikelenboom P , Emmerling M , Fiebich BL , Finch CE , Frautschy S , Griffin WS , Hampel H , Hull M , Landreth G , Lue L , Mrak R , Mackenzie IR , McGeer PL , O'Banion MK , Pachter J , Pasinetti G , Plata-Salaman C , Rogers J , Rydel R , Shen Y , Streit W , Strohmeyer R , Tooyoma I , Van Muiswinkel FL , Veerhuis R , Walker D , Webster S , Wegrzyniak B , Wenk G , Wyss-Coray T ((2000) ) Inflammation and Alzheimer’s disease. Neurobiol Aging 21: , 383–421. |
[78] | Nedergaard M ((2013) ) Neuroscience. Garbage truck of the brain. Science 340: , 1529–1530. |
[79] | Morris AW , Sharp MM , Albargothy NJ , Fernandes R , Hawkes CA , Verma A , Weller RO , Carare RO ((2016) ) Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol 131: , 725–736. |
[80] | Albargothy NJ , Johnston DA , MacGregor-Sharp M , Weller RO , Verma A , Hawkes CA , Carare RO ((2018) ) Convective influx/glymphatic system: Tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol 136: , 139–152. |


