
Inhibition of phase-1 biotransformation and cytostatic effects of diphenyleneiodonium on hepatoblastoma cell line HepG2 and a CYP3A4-overexpressing HepG2 cell clone
Abstract
Cell-based in vitro liver models are an important tool in the development and evaluation of new drugs in pharmacological and toxicological drug assessment. Hepatic microsomal enzyme complexes, consisting of cytochrome P450 oxidoreductase (CPR) and cytochrome P450 monooxygenases (CYPs), play a decisive role in catalysing phase-1 biotransformation of pharmaceuticals and xenobiotics. For a comprehensive understanding of the phase-1 biotransformation of drugs, the availability of well-characterized substances for the targeted modulation of in vitro liver models is essential. In this study, we investigated diphenyleneiodonium (DPI) for its ability to inhibit phase-1 enzyme activity and further its toxicological profile in an in vitro HepG2 cell model with and without recombinant expression of the most important drug metabolization enzyme CYP3A4.
Aim of the study was to identify effective DPI concentrations for CPR/CYP activity modulation and potentially associated dose and time dependent hepatotoxic effects. The cells were treated with DPI doses up to 5,000nM (versus vehicle control) for a maximum of 48 h and subsequently examined for CYP3A4 activity as well as various toxicological relevant parameters such as cell morphology, integrity and viability, intracellular ATP level, and proliferation. Concluding, the experiments revealed a time- and concentration-dependent DPI mediated partial and complete inhibition of CYP3A4 activity in CYP3A4 overexpressing HepG2-cells (HepG2-CYP3A4). Other cell functions, including ATP synthesis and consequently the proliferation were negatively affected in both in vitro cell models. Since neither cell integrity nor cell viability were reduced, the effect of DPI in HepG2 can be assessed as cytostatic rather than cytotoxic.
1Introduction
In humans, the liver is the main organ for the metabolization and elimination of pharmaceuticals and xenobiotics due to the high expression of phase-1 and -2 enzymes in hepatocytes [1]. For this reason, hepatocytes are the subject of intensive research efforts, and in vitro systems based on these cells are often used in the context of drug development, diagnostics and therapeutics, for example to clarify and reduce drug side effects at an early stage [2, 3].
In the context of phase-1 biotransformation, microsomal enzyme complexes in hepatocytes, consisting of cytochrome P450 oxidoreductase (CPR) and cytochrome P450 monooxygenases (CYPs), are essential components for a large number of oxidative metabolic conversions of pharmaceuticals or xenobiotics [4, 5]. Despite the large number of different CYPs expressed in the human organism (57 are known to date), only a few, mostly from CYP families 1, 2, and 3, are responsible for the oxidative metabolization of more than 75%of all clinically approved drugs [2, 3, 6, 7]. The microsomal flavoprotein CPR has a significantly lower diversity compared to CYPs with only one individually expressed polymorphic variant [8–10]. As the obligatory electron donor for CYPs, CPR is essential for the liver-mediated phase-1 metabolism. Further, CPR plays a vital role in both oxidative processes catalysed by several oxygenase enzymes as well as biosynthesis and metabolism of various endogenous substances of the hormone and fat metabolism [9, 11]. During phase-1 biotransformation several successive oxidative reactions take place in which electrons and activated oxygen are transferred to a substrate in an nicotinamide adenine dinucleotide phosphate (NADPH)-dependent process [12, 13]. In detail, two electrons are initially transferred from NADPH to the prosthetic group flavin adenine dinucleotide (FAD) contained in CPR before these are transferred to flavin mononucleotide (FMN), another co-factor of CPR, by means of interflavin electron transfer. Sequential electron transfer follows this via redox cycling to a heme-bearing microsomal CYP, which catalyses the oxidative conversion of a substrate [14–16]. For the prediction of the pharmacokinetics of new drug candidates, including relevant metabolites and hepatotoxicity, a clear understanding of the enzymatic phase-1 and -2 reactions interplay in the liver is crucial. In this context, preclinical drug screening with regard to biotransformation and toxicology is mostly based on physiologically relevant sensitive, reliable and in particular adaptable in vitro metabolism models of human hepatocytes [17–20]. Research into specific scientific issues also involves the availability of substances for targeted modulation. There are plenty of CYP inducers and inhibitors known for targeted phase-1 activity modifications [9]. However, the range of phase-1 modulating agents on only CPR activity level or on both CPR and CYPs is limited. However, such inhibitors are an important tool in drug studies, e.g. to elucidate side reactions that are not catalysed by phase-1 biotransformation or to monitor CPR/CYP-dependent pro-drug activation. In this study, diphenyleneiodonium (DPI) was investigated as an inhibitor candidate for CPR/CYP enzyme activity. In addition, the toxicological profile of DPI was analyzed in an in vitro hepatocyte model based on the human hepatoblastoma cell line HepG2 and a HepG2 cell clone with overexpression of CYP3A4. CYP3A4 was chosen as enzymes of the CYP3A family are involved in the metabolism of more than 50%of human approved drugs and CYP3A4 is the most important representative of the CYP3A family concerning drug metabolism in adult human liver [7, 11, 21].
DPI, a member of diaryliodonium salts, is an aromatic heterocyclic cation. Owing to their electron deficient properties at the iodine center, diaryliodonium salts are frequently used as aromatic electrophiles in aryl transfer processes [22]. Its chemical nature makes DPI a potent inhibitor of flavin bearing oxidoreductases, which are generally an integral element of electron transport chains. DPI have a wide spectrum of known cellular targets including CPR [13, 15, 23], NADPH oxidase (NOX) [24–31], mitochondrial respiratory chain complex I (NADH ubiquinone oxidoreductase) [28, 32–34], and different types of nitric oxide synthase [13, 35]. It is assumed that DPI inhibition is achieved by covalent modification of flavin and/or heme prosthetic groups within enzymes based on radical formation. NADPH-dependent inhibition of CPR by DPI occurs via irreversible modification of reduced FMN, which effectively prevent electron transfer to their physiological targets [13, 15, 36–38]. In these studies, DPI could be shown as an effective CPR inhibitor in recombinant expressed protein isolates, rat and human liver microsomes as well as in several in vitro cell models. Likewise, it was found, that DPI-mediated CPR inhibition prevented electron flow to CYPs, leading to inhibition of theirmonooxygenase activity [13, 39]. In the context of further studies, DPI was also shown to irreversibly modify heme porphyrin in microsomal CYPs. Since both CPR-flavins and the heme in CYPs are a target for DPI, CYP-dependent monooxygenase activity is inhibited at two levels, with CYPs being significantly more sensitive to DPI than CPR [13].
In the past, inhibitory effects of DPI were investigated with regard to a potential application in the therapeutic field, i.e. as an antibiotic [29, 40, 41], anti-cancer [31, 42, 43], anti-inflammatory [26, 30] and/or vasodilatory agent [23]. For the analysis of phase-1 biotransformation inhibition, studies were mostly performed in less complex model systems with recombinantly expressed and purified proteins or derived from microsomal fractions in order to clarify size and range of DPI effects and the mechanism of action. Ex vivo and especially in vivo studies are scarcely available. For example, the influence of DPI on CPR-mediated NO formation from glyceryl trinitrate has been investigated both ex vivo in microsomal fractions from rat aorta and in vivo regarding the influence on vasodilation in a rat model [23]. Due to its ability to inhibit phase-1 reactions both at the level of CPR electron transport and CYP monooxygenase activity itself, DPI promises to be an interesting tool for blocking whole biotransformation activity. However, the data available for the application of DPI in more complex in vitro cell models for pharmacological/toxicological biotransformation studies still is limited. Since DPI influences also other physiologically relevant processes such as the mitochondrial respiratory chain, it is of great importance to investigate its effects in a complex in vitro cell model.
Therefore, the aim of our study was to investigate DPI as inhibitor of phase-1 activity via CPR/CYP inhibition in an in vitro hepatocyte model with elevated CYP3A4 activity. The focus was on the elicitation of effective DPI concentrations for CPR/CYP activity manipulation and potentially associated dose- and time-dependent toxic effects on HepG2.
2Methods
2.1Cell culture
Commercially available human hepatocellular carcinoma (HepG2) cells (HB-8065, ATCC, Manassas, VA, USA) as well as genetically modified HepG2 with stable recombinant overexpression of CYP3A4 (HepG2-CYP3A4), generated and kindly provided by the “Molecular Cell Biology” group from the BTU Cottbus-Senftenberg [44], were cultured under standard conditions (37 °C, 5 %CO2) in polystyrene-based tissue culture flasks (SARSTEDT AG & Co. KG, Nümbrecht, Germany) in Dulbecco’s minimal essential medium (D-MEM) supplemented with 10 %fetal bovine serum (FBS) superior, 6 mM L-alanyl-L-glutamine and 49.2 g/L NaHCO3, all purchased from Biochrom GmbH (Berlin, Germany). During standard cell culture the culture medium was replaced every second day. Prior to the inhibition studies with diphenyleneiodonium (DPI), the HepG2-CYP3A4 cell line was post-selected by adding 3μg/mL Blasticidin (AppliChem GmbH, Darmstadt, Germany) to the culture medium over a period of two weeks [45]. No Blasticidin was present in the culture medium during the experiments with DPI. For either cell passaging or experimental seeding, hepatocytes were harvested by trypsin/EDTA treatment (0.05%v/v Trypsin and 0.02%v/v EDTA in water, Biochrom GmbH, Berlin, Germany).
2.2CPR/CYP inhibition studies with diphenyleneiodonium (study design)
The presented study was divided in three consecutive parts. For the assessment of DPI mediated influences on both CYP3A4 monooxygenase activity or toxicological relevant parameters in hepatocytes, HepG2 and HepG2-CYP3A4 cells were seeded in all study parts at a density of 62.500 cells/cm2 into either 96-well or 24-well plates (SARSTEDT AG & Co. KG, Nümbrecht, Germany) 24 h prior to DPI-treatment. The setup of the first study part initially aimed to determine the concentration range of an effective DPI-mediated inhibition of phase-1 biotransformation in the in vitro model system used. For this purpose, HepG2 with recombinant CYP3A4 activity were treated with DPI in a wide concentration range of 2.5–5,000 nM for a short, 30 min period, followed by analysing parameters such as cell morphology and CYP3A4 activity including cell number normalisation via intracellular ATP level. For this purpose, starting from a 1 mM diphenyleneiodonium chloride stock solution in CPR assay buffer (both purchased from BioVision Inc., Milpitas, CA, USA) buffer + 10%DMSO (AppliChem GmbH, Darmstadt, Germany) DPI dilutions (1:10 or 1:100) in cell culture medium were used, by medium change directly before treatment. The vehicle and the untreated parental cell line were always included as controls. Data of monooxygenase activity and intracellular ATP level were generated in triplicates in two independent experiments (n = 6 in sum). Prior and after any DPI treatment, morphological evaluation of the hepatocytes were performed using an Olympus CKX41 inverted microscope (Olympus Corporation, Tokyo, Japan). Pictures were documented in various magnifications in phase-contrast mode. In this part of the study, CYP3A4 activity and intracellular ATP levels were determined directly after DPI treatment as described below (see Section 2.3).
Based on the findings from the first study part, regarding effective DPI concentrations and the DPI-related influence on the intracellular ATP level, as well as anticipating experimental planning for future metabolization studies of substrates/drugs (for which longer conversion times of up to 48 h often are required), the following study parts were performed with an extended setup to elucidate possible time dependent and toxic DPI effects on the HepG2 based in vitro model systems. In the second part of the study, cells were seeded according to the protocol described above in culture vessels suitable for the respective experiments. 24 h after seeding, the cells were treated with different DPI concentrations in the range of 50–5,000 nM over a period of 48 h. In the third part of the study, the cells were treated with higher DPI concentrations of 1,000, 2,500 and 5,000 nM (known to cause effective CPR/CYP inhibition) only for 30 min before switching to DPI-free medium and 48 h cultivation, to investigate a possible recovery of phase-1 activity over time. After 48 h incubation under cell culture conditions, analysis of various parameters including cell morphology, CYP3A4 monooxygenase activity, intracellular ATP, cell integrity, viability and proliferation was performed in the second and third study part with both cell lines as described below.
2.3Determination of CYP3A4 enzyme activity and intracellular ATP level
For the assessment of DPI-induced inhibition of CYP3A4 monooxygenase activity in hepatocytes, HepG2 and HepG2-CYP3A4 cells were analyzed with the P450-Glotrademark CYP3A4 induction/ inhibition assay (Promega, Madison, WI, USA), used according to the manufacturer’s instructions. Briefly, after DPI treatment, cells were incubated with 50μl CYP3A4 substrate Luciferin-IPA diluted in culture medium at 37 °C, 5 vol-%CO2 for 60 min. Subsequently, 25μl of supernatants were transferred into a white-walled 96-well plate (SARSTEDT AG & Co. KG, Nümbrecht, Germany) and an equal volume of luciferin detection reagent was added followed by incubation for 20 min at room temperature in the dark. Luminescence was measured with a FLUOstar Omega microplate reader (Software version: 3.00 R2, BMG LABTECH GmbH, Ortenberg, German), followed by data analysis by MARS Data Analysis Software (Version: 2.41). In addition, the cells and the 25μl substrate solution remaining in the initial 96-well plate were mixed with 25μl ATP reagent solution of the CellTiter-Glo® 2.0 assay (Promega, Madison, WI, USA) and incubated for 10 min in the dark. ATP level was detected by measuring luminescence with the FLUOstar Omega microplate reader to allow normalization to the effective cell number or assessment of DPI mediated influences on the intracellular ATP level.
2.4Determination of cell integrity by LDH assay
To determine a possible concentration and/or time dependent influence of DPI on cell integrity, the amount of lactate dehydrogenase (LDH) released from the cytoplasm into the cell culture supernatant was determined in the second and third study part. For this purpose, the LDH Cytotoxicity Colorimetric Assay Kit II (Biovision GmbH, Ilmenau, Germany) was used according to the manufacturer’s instructions. The experiments were performed in 96-well format (SARSTEDT AG & Co. KG, Nümbrecht, Germany) with both cell lines using triplicates in two independent experiments (n = 6 in sum). The cells were either treated with ascending DPI concentrations (50, 100, 250, 500, 1,000, 2,500, 5,000 nM) for a period of 48 h in the second part of the study or in the third part of the study with higher DPI concentrations for only 30 min (1,000, 2,500, 5,000 nM) before switching to DPI-free medium. After 48 h cultivation, the amount of cell-released LDH in the supernatant was determined. Completely lysed cells (high control), a LDH preparation (positive control) from the kit and a vehicle were always included as controls. High control cell lysis was achieved by adding the cell lysis solution contained in the kit and incubating for 10 minutes under cell culture conditions. After addition of the reagents described in the manual for LDH detection, LDH released from the cells was measured with the FLUOstar Omega microplate reader after 45 minutes of development at OD450 nm (reference: OD650 nm).
2.5Viability and cell density determination by FDA/PI fluorescent staining
DPI-induced changes in proliferation behaviour and cell viability were determined by live-dead staining of the cells with Fluorescein Diacetate (FDA) and Propidium Iodide (PI), both purchased from Sigma Aldrich (St. Louis, MO, US). FDA as a cell-permeant esterase substrate served as a vitality probe, whereby it is hydrolysed into its fluorescent form by intact and metabolically active cells. PI was used to detect dead cells, as it is a DNA-intercalating fluorescent dye that is not cell-permeant. Viability staining was performed in 24 well format (SARSTEDT AG & Co. KG, Nümbrecht, Germany) with both cell lines HepG2 and HepG2-CYP3A4 in two independent experiments with n = 2 wells of each experimental condition. Cells were seeded and treated with DPI analogous to the procedure already described in study design chapter (see Section 2.2). Briefly, for the 48 h treatment in the second part of the study, the cells were exposed to DPI concentrations of 50, 100, 250, 500, 1,000 nM. For the third study part the cells were exposed to higher DPI concentrations (1,000, 2,500, 5,000 nM) for 30 min before switching to DPI-free medium. After 48 h incubation under cell culture conditions, medium was changed and replaced with fresh medium containing FDA (1μg/mL) and PI (2.5μg/mL). The detection of vital/dead cells occurred by means of a LSM800 confocal Laser Scanning Microscope system and ZEN software for picture post processing (Carl Zeiss Microscopy GmbH, Jena, Germany) by taking 3 high resolution pictures of 2×2 tiles (n = 6 in sum from two independent experiments; whole covered area per picture ∼1.5 mm2) from different areas of each well in 10-fold primary magnification. For vitality and proliferation assessment, the cell-covered area was calculated from the pictures by using Image J software (version: 1.53c, National Institutes of Health, Bethesda, MD, USA).
2.6Statistical analysis
For statistical analysis, one-way ANOVA with Turkey’s multiple comparison test was used to calculate differences between groups using Prism 8 software (GraphPad Software, San Diego, CA, USA). Probabilities lower than 0.05 were considered statistically significant.
3Results
3.1Short-term exposure with high-dose DPI completely inhibits CYP3A4 activity and is slightly affecting ATP level
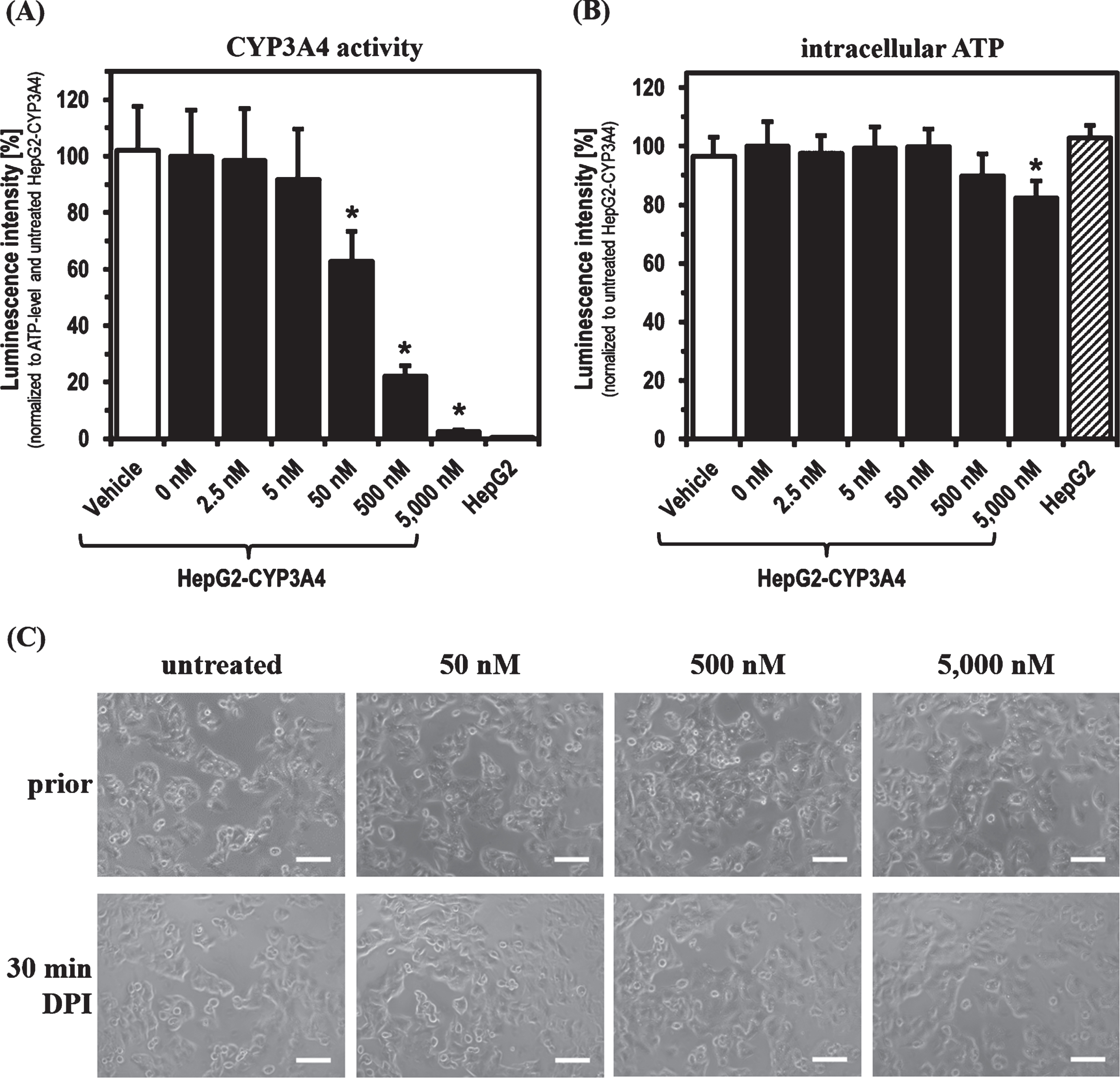
For the experiments with DPI, parental HepG2 and HepG2-CYP3A4 with recombinant CYP3A4 overexpression (described previously [44]) were used as cell models. Initially, the main focus was to determine the DPI concentration range showing an inhibitory effect on phase-1 monooxygenase activity after a 30 min treatment. CYP3A4 activity in the HepG2-CYP3A4 cell line seemed to be slightly decreased already at 5 nM DPI (Fig. 1). Starting with a concentration of 50 nM, a significant reduction of CYP3A4 activity was caused by DPI (p = 0.0004). Treating the cells with DPI concentrations starting from 500 nM, a decrease also in intracellular ATP levels was evident and significant at 5,000 nM DPI (p = 0.0015). In this initial part of the study, the parental cell line HepG2 served as negative control with no detectable CYP3A4 activity. There was no difference in the ATP levels of both cell lines in untreated state. No morphological alterations were observed, when HepG2-CYP3A4 were treated for 30 min with increasing DPI concentrations.
Fig. 1
CYP3A4 activity and ATP level after 30 min DPI treatment. Determination of (A) CYP3A4 activity, (B) intracellular ATP level and (C) morphology of HepG2-CYP3A4 after 30 min DPI treatment (Mean±standard deviation; *p < 0.05 compared to untreated cells; n = 6 from two independent experiments; pictures taken by light microscope in phase contrast mode with 10-fold primary magnification; scale: 100μm).
3.2Long-term exposure with DPI inhibits CYP3A4 activity and is affecting ATP levels and proliferation but not cell integrity
Next, we performed DPI treatments of HepG2 and HepG2-CYP3A4 for a longer period (48 h). In addition, we were interested to see if there could be a recovery of CYP3A4 activity as well as intracellular ATP level after short-term DPI treatment. For this, cells were treated with DPI concentrations between 1,000 and 5,000 nM for 30 min followed by 48 h of cultivation in DPI-free culture medium. As before, morphology of DPI-treated cells was analyzed and CYP3A4 activity as well as intracellular ATP level were measured. Moreover, a potential cytotoxic DPI effect on cell integrity was investigated by LDH assay, and the cellular viability status was analyzed with FDA/PI fluorescent staining.
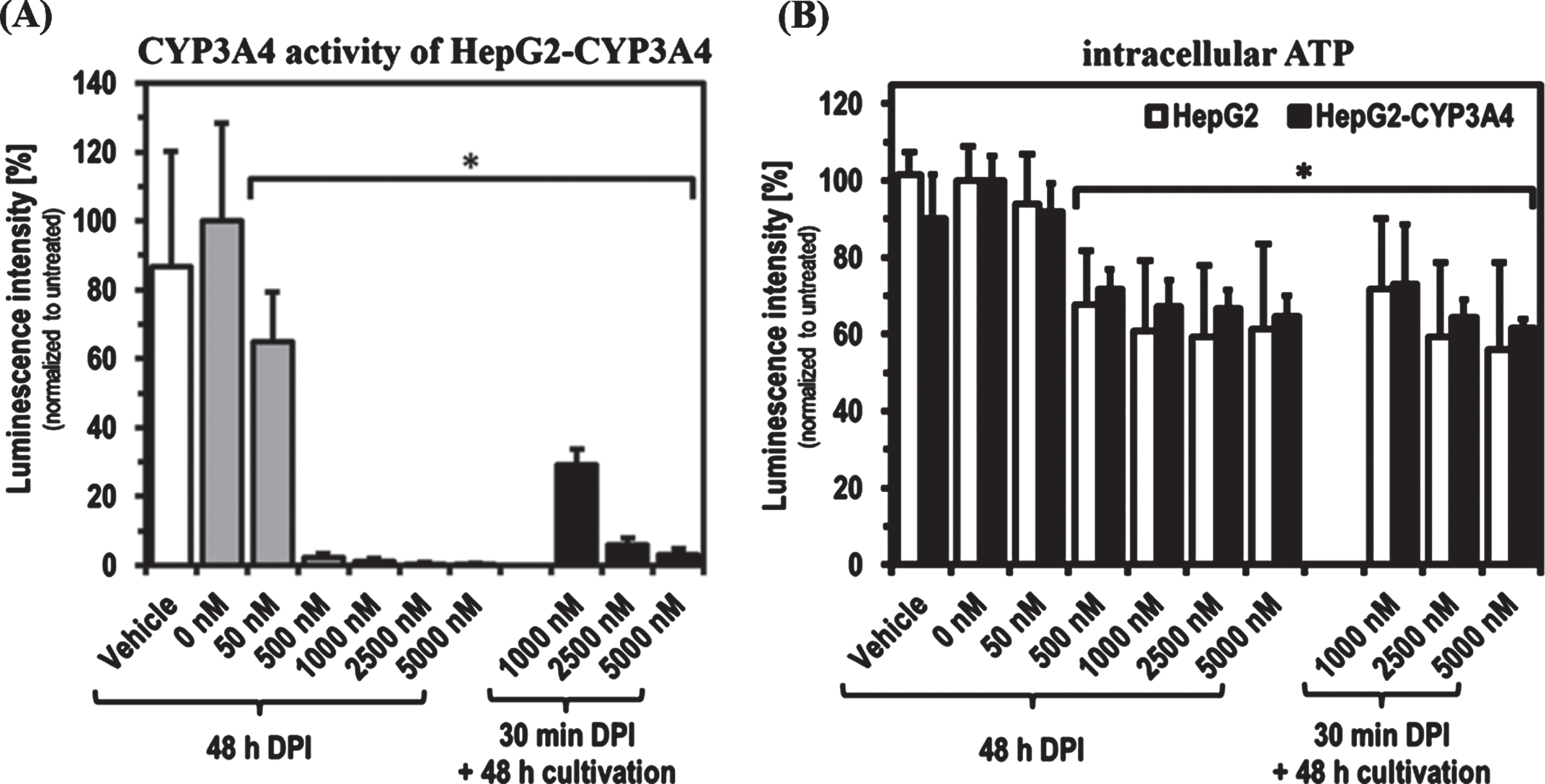
As found with short-term treatments, DPI showed a concentration-dependent inhibitory effect on the CYP3A4 activity of HepG2-CYP3A4 also after 48 h of treatment (Fig. 2). A DPI concentration of 50 nM led to a significant reduction of CYP3A4 activity to about 60%(p = 0.0160). 500 nM was sufficient for an almost complete inhibition of CYP3A4 activity. Recovery experiments showed that HepG2-CYP3A4 cells treated with 1,000 nM DPI for 30 min could reactivate about 30%of CYP3A4 activity when subjected to a 48 h period in DPI-free medium. The recovery capacity was reduced below 10%with 2,500 and 5,000 nM. The intracellular ATP level was significantly reduced by treatment with high DPI concentrations of 1,000 to 5,000 nM. There were no significant differences between a 30 min and a 48 h DPI treatment. Only at 1,000 nM DPI was a tendency towards a slight recovery visible. No significant differences could be detected between both the two setups and the HepG2 cell lines.
Fig. 2
CYP3A4 activity and ATP level after 48 h DPI treatment as well as recovery after 30 min DPI treatment. Determination of CYP3A4 activity in HepG2-CYP3A4 (A) and intracellular ATP level in both cell lines (B) after DPI treatment for 48 h as well as for 30 min with following 48 h recovery in DPI-free medium (Mean±standard deviation; *p < 0.05 compared to untreated cells; n = 6 from two independent experiments).
The experiments further revealed that, despite some DPI effects on ATP level, the cell integrity of both cell lines apparently was not negatively affected by DPI at any time (Fig. 3). The release of LDH was even slightly higher in the untreated cells and the vehicle controls (significant in HepG2 for all DPI concentrations). Direct comparison of the two cell lines showed only minor differences. Solely untreated HepG2 and its vehicle control tended to show an increased LDH release compared to HepG2-CYP3A4.
Fig. 3
Cytostatic effect of DPI on HepG2 and HepG2-CYP3A4 cells. Analysis of the HepG2 and HepG2-CYP3A4 cell integrity via LDH release (A), metabolic activity via ATP level (B) and viability via FDA/PI staining (C) (Mean±standard deviation; *p < 0.05 compared to untreated cells; n = 12 pictures from 2 independent experiments; representative cLSM images of cells treated for 48 h with DPI at 10x primary magnification; green = vital cells, red = dead cells; scale: 200μm).
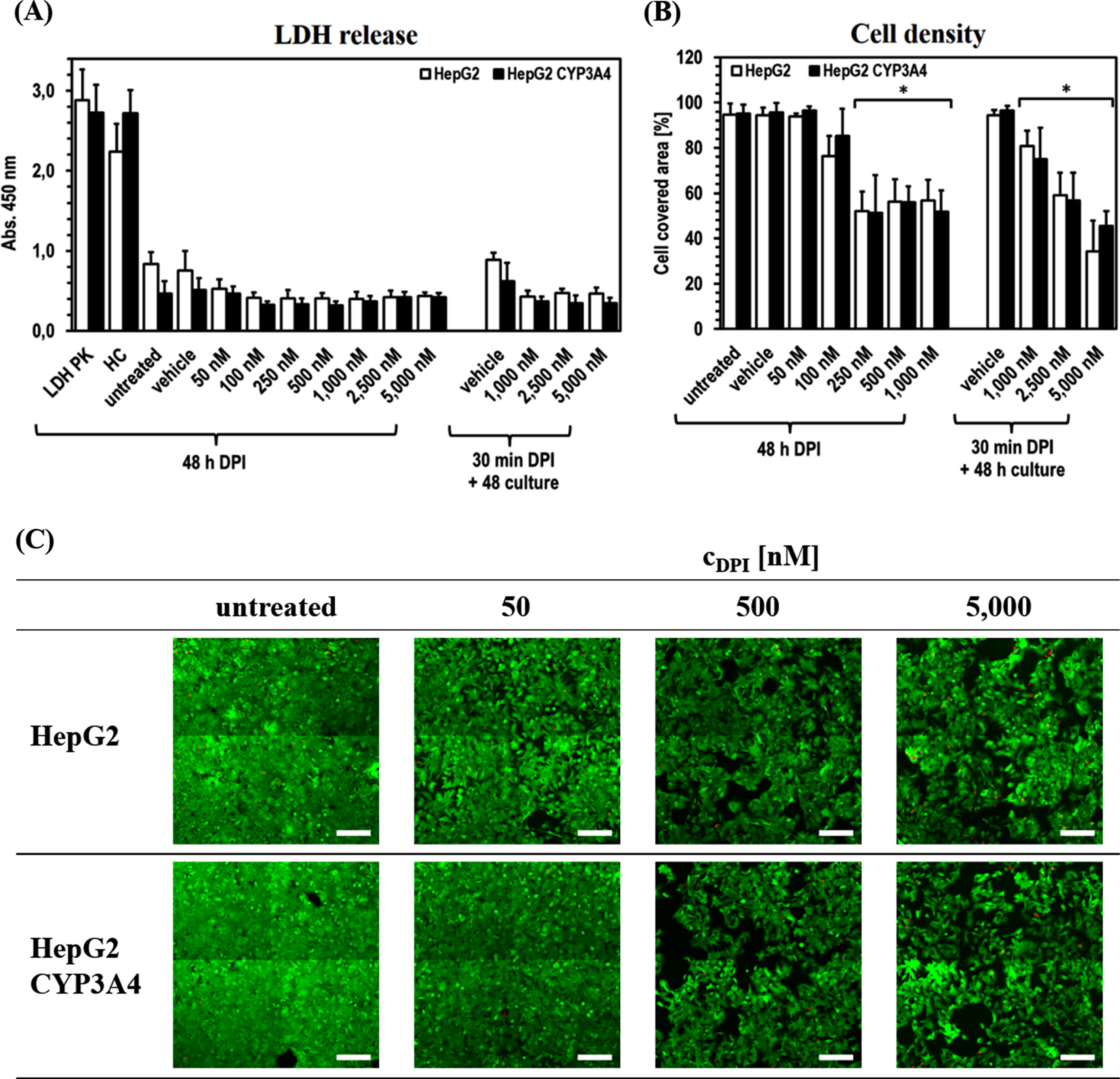
The situation is different for the area covered by vital cells, which was used as a further evaluation parameter. In both cell lines, a comparable reduction of the covered area with increasing DPI concentration was observed. There was a significant difference for the area covered by vital cells to decrease to about 80%after 48 h of treatment with 100 nM DPI (pHepG2100 nM DPI < 0.0001). In HepG2-CYP3A4 only a slight tendency could be observed (pHepG2 CYP3A4100 nM DPI = 0.2710). At higher DPI doses in the range of 250–1,000 nM, a more extensive and in all samples significant reduction of cell density to ∼50%was visible (all p < 0.0001) after 48 h treatment. The recovery experiments with high DPI doses (1,000–5,000 nM) revealed a concentration dependency, whereby higher DPI doses led to lower cell density. Here, 1,000 nM DPI led to a significant reduction of the hepatocyte covered area to about 80%(pHepG2 = 0.0018; pHepG2hboxCYP3A4 < 0.0001). The lowest cell density (∼40%) was observed with 5,000 nM DPI (p < 0.0001 in both cell lines). In none of the experiments, an increased incidence of dead cells caused by DPI could be detected.
4Discussion
We were interested to evaluate the potential of diphenyleneiodonium (DPI) for the targeted modification of phase-1 monooxygenase activity in cell-based in vitro systems based on previous results from other groups [13, 15, 23, 39]. HepG2 cells as well as recombinant CYP3A4-overexpressing HepG2 cells were used as hepatocyte model systems for functional and toxicological studies [17, 46–50]. HepG2 exhibit in vitro low basal CYP activity and are therefore well suited for recombinant modification with specific CYP activities [44, 51]. In the present study, we investigated DPI concentration- and time-dependent effects both on phase-1 biotransformation and on cell viability. The latter might be detrimental or interfering with HepG2-based in vitro biotransformation studies.
In the first part of the study, we did not find any DPI effects on the cell morphology as analyzed by phase contrast microscopy. However, the strong CYP3A4 enzyme activity in the HepG2-CYP3A4 model could be significantly inhibited by DPI, depending on the concentration. For a relevant inhibition to approximately 20%of the original CYP3A4 activity of the HepG2-CYP3A4 cells, DPI concentrations of at least 500 nM were required. However, there was a negative effect on the intracellular ATP level at higher DPI concentrations detectable, which could have a serious impact on the on the energy balance and metabolism of hepatocytes. The aim of our study was to investigate not only a concentration but also a possible temporal dependence of the DPI effect on phase-1 activity. In addition, toxicological parameters such as cell integrity, viability and proliferation were analyzed to determine to what extent HepG2-CYP3A4 has the ability to regenerate phase-1 activity after a short 30 min DPI treatment and the extent to which toxicologically relevant effects emanate from DPI under these conditions.
With regard to the inhibition of CYP activity, there was no time dependence in the DPI effect when 50 nM was used. After both 30 min and 48 h DPI treatment the residual CYP3A4 activity was ∼60%, when compared to untreated HepG2-CYP3A4. The situation was different at higher DPI concentrations from 500 nM on, where compared to the 30 min treatment (∼20%residual activity) an almost complete inhibition of CYP3A4 activity was achieved after 48 h DPI treatment. Precisely in this concentration range, DPI mediated significant effects on intracellular ATP levels. This means that a substantial inhibition of phase-1 activity by DPI might have a negative impact on ATP synthesis. Higher concentrations of DPI did not further reduce the intracellular ATP level after 48 h of treatment. This could indicate that under the chosen experimental conditions 500 nM DPI was sufficient for maximum inhibition of CYP3A4 activity and the respiratory chain of the in vitro cell system used, and saturation of corresponding DPI targets was achieved. The data collected on cell integrity as well as vitality and cell density provide further insight. In the second and third part of the study, no significant difference between the two cell lines could be detected for any of these parameters, indicating that the genetic modification for recombinant overexpression of CYP3A4 does not significantly affect the DPI mechanism of action or its effect in HepG2. There was a tendency for ATP levels to be slightly increased in HepG2-CYP3A4 compared to the parental cell line, when the cells were treated with higher DPI concentrations. Obviously, cell integrity was not altered even by the highest DPI concentrations used as there was no increase of LDH activity detectable in the cell supernatants. This is in agreement with previous studies in which even higher DPI doses were well tolerated for prolonged periods in various in vitro and in vivo models. DPI was even shown to have anti-inflammatory effects by inhibiting NF-kB mediated free radical formation via NADPH oxidase [26, 29, 30]. The slight reduction in released LDH at higher DPI concentrations in both cell lines correlates with the reduced cell density induced by DPI. In line with that data, the viability of HepG2 and HepG2-CYP3A4 does not seem to be negatively affected by DPI, as no increased occurrence of PI positive cells with increasing DPI concentrations could be determined in any cell line. Nevertheless, an effect on cell viability caused by DPI cannot be completely ruled out, as a part of the dead cells might have been lost due to the medium change immediately before detection in the FDA/PI Assay. However, the results of the LDH assay, in which no increased LDH release could be detected over the 48-hour DPI treatment without medium change, contradict this. An indication that even lower DPI concentrations may be sufficient for the above-mentioned saturation and thus complete inhibition of phase 1 activity is provided by the decreasing cell density with increasing DPI concentrations. The cell density was used as an analytical parameter for the toxicological evaluation of DPI, as no quantification of single cells was possible due to the HepG2 morphology and the high confluence of untreated cells at the end of the incubation period. It was shown that already a 48 h treatment with 250 nM DPI led to maximum detected reduction of cell density to ∼50%compared to untreated cells.
With regard to the detected reduction of the intracellular ATP level after DPI treatment, experimental limitations result in ambiguities in the interpretation of the data. The decreasing intracellular ATP level with increasing DPI concentrations is probably partly due to the lower cell number after DPI treatment. A direct comparison of ATP levels between untreated and treated cells requires a comparable cell number. According to our cell density data, this is no longer given after 48 h treatment at least from 100 nM DPI and in the case of short treatment followed by 48 h cultivation in the third study part at higher DPI concentrations, as the cell density is already substantially lower. Since only the ATP amount in a complete well could be detected after 48 h, it is conceivable that the influence of DPI on the energy metabolism of the individual cell is less than suggested by the detected ATP level per well. However, it was already shown that DPI has an inhibitory influence on complex I of the respiratory chain [42], where the FAD cofactor of the mitochondrial localised NADH-ubiquinone oxidoreductase is a target for DPI [23]. In view of these findings on mitochondrial function in different cell types, as well as the observations from our experiments, it is clear that the ATP synthesis is directly linked to the proliferation [52–54]. The resulting conclusion is, that DPI reduces the ATP level within a short period of time, which has a negative effect on proliferation and results in a reduced cell density after 48 hours.
In our studies, a partial recovery of CYP3A4 activity of up to 30%could also be observed after 48 h of cultivation under DPI-free conditions, following an almost complete inhibition by 30 min treatment with 1,000 nM DPI. These observations do not necessarily contradict findings by others concerning irreversible inhibition of DPI targets [13, 15]. These measurements were made with protein isolates or microsomes. A cell-based system may has the possibility of reproducing enzymes and thus restore enzyme activity over time. At 2,500 and 5,000 nM DPI no recovery could be observed, as both phase-1 residual activity was still reduced below 10%after 30 min treatment followed by DPI-free cultivation as well as ATP levels and cell density were comparable to cells treated for 48 h.
5Conclusion
The objective of the study was to investigate the potential of DPI as an inhibitor of phase-1 monooxygenase activity for in vitro drug and toxicity studies. Based on the HepG2 and HepG2-CYP3A4 in vitro model systems used, the results show that DPI mediated inhibition of phase-1 biotransformation could be achieved. DPI can be used as an inhibitor of CYP3A4 activity at concentrations up to 50 nM without inducing any morphological or toxic effects on the cells. At concentrations > 50 nM, cytostatic effects on HepG2 or HepG2-3A4 are to be expected, so that influences or interactions with activity determinations can not be excluded, which must be taken into account accordingly.
Acknowledgments
This work was funded by grants of the Ministerium für Wirtschaft, Forschung und Kultur (MWFK, state of Brandenburg, Germany) for the Fraunhofer Project Group “Pilzbasierte zellfreie Synthese-Plattformen –PZ-Syn” (project number 22-F241-03-FhG/005/001).
References
[1] | Bell CC , Hendriks DF , Moro SM , Ellis E , Walsh J , Renblom A , et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Scientific Reports. (2016) ;6: (1):1–13. |
[2] | Samer CF , Lorenzini KI , Rollason V , Daali Y , Desmeules JA . Applications of CYP450 testing in the clinical setting. Molecular Diagnosis & Therapy. (2013) ;17: (3):165–84. |
[3] | Lynch T , Price AL . The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. American Family Physician. (2007) ;76: (3):391–6. |
[4] | Guengerich FP . Cytochrome p450 and chemical toxicology. Chemical Research in Toxicology. (2008) ;21: (1):70–83. |
[5] | Zanger UM , Turpeinen M , Klein K , Schwab M . Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Analytical and Bioanalytical Chemistry. (2008) ;392: (6):1093–108. |
[6] | Arimoto R . Computational models for predicting interactions with cytochrome p450 enzyme. Current topics in Medicinal Chemistry. (2006) ;6: (15):1609–18. |
[7] | Shimada T , Yamazaki H , Mimura M , Inui Y , Guengerich FP . Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. Journal of Pharmacology and Experimental Therapeutics. (1994) ;270: (1):414–23. |
[8] | Tee MK , Huang N , Damm I , Miller WL . Transcriptional regulation of the human P450 oxidoreductase gene: hormonal regulation and influence of promoter polymorphisms. Molecular Endocrinology. (2011) ;25: (5):715–31. |
[9] | Zanger UM , Schwab M . Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacology & Therapeutics. (2013) ;138: (1):103–41. |
[10] | Fang Y , Gao N , Tian X , Zhou J , Zhang H-F , Gao J , et al. Effect of P450 oxidoreductase polymorphisms on the metabolic activities of ten cytochrome P450s varied by polymorphic CYP genotypes in human liver microsomes. Cellular Physiology and Biochemistry. (2018) ;47: (4):1604–16. |
[11] | Riddick DS , Ding X , Wolf CR , Porter TD , Pandey AV , Zhang Q-Y , et al. NADPH–cytochrome P450 oxidoreductase: roles in physiology, pharmacology, and toxicology. Drug Metabolism and Disposition. (2013) ;41: (1):12–23. |
[12] | Poulos TL . Intermediates in P450 catalysis. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences. (2005) ;363: (1829):793–806. |
[13] | Szilagyi JT , Mishin V , Heck DE , Jan Y-H , Aleksunes LM , Richardson JR , et al. Selective targeting of heme protein in cytochrome P450 and nitric oxide synthase by diphenyleneiodonium. Toxicological Sciences. (2016) ;151: (1):150–9. |
[14] | Gutierrez A , Paine M , Wolf CR , Scrutton NS , Roberts GC . Relaxation kinetics of cytochrome P450 reductase: internal electron transfer is limited by conformational change and regulated by coenzyme binding. Biochemistry. (2002) ;41: (14):4626–37. |
[15] | Tew DG . Inhibition of cytochrome P450 reductase by the diphenyliodonium cation. Kinetic analysis and covalent modifications. Biochemistry. (1993) ;32: (38):10209–15. |
[16] | Vermilion J , Ballou D , Massey V , Coon M . Separate roles for FMN and FAD in catalysis by liver microsomal NADPH-cytochrome P-450 reductase. Journal of Biological Chemistry. (1981) ;256: (1):266–77. |
[17] | Schulz C , Kammerer S , Küpper J-H . NADPH-cytochrome P450 reductase expression and enzymatic activity in primary-like human hepatocytes and HepG2 cells for in vitro biotransformation studies. Clinical Hemorheology and Microcirculation. (2019) ;73: (1):249–60. |
[18] | Guillouzo A , Guguen-Guillouzo C . Evolving concepts in liver tissue modeling and implications for in vitro toxicology. Expert Opinion on Drug Metabolism & Toxicology. (2008) ;4: (10):1279–94. |
[19] | Lübberstedt M , Müller-Vieira U , Mayer M , Biemel KM , Knöspel F , Knobeloch D , et al. HepaRG human hepatic cell line utility as a surrogate for primary human hepatocytes in drug metabolism assessment in vitro . Journal of Pharmacological and Toxicological Methods. (2011) ;63: (1):59–68. |
[20] | Wu Y , Cheng G , Chen H , Wang J , Wang J , Wang W . IL-17 predicts the effect of TACE combined with apatinib in hepatocellular carcinoma. Clinical Hemorheology and Microcirculation. (2020) (Preprint):1–11. |
[21] | Wrighton SA , Schuetz EG , Thummel KE , Shen DD , Korzekwa KR , Watkins PB . The human CYP3A subfamily: practical considerations. Drug Metabolism Reviews. (2000) ;32: (3-4):339–61. |
[22] | Pacheco-Benichou A , Besson T , Fruit C . Diaryliodoniums Salts as Coupling Partners for Transition-Metal Catalyzed C-and N-Arylation of Heteroarenes. Catalysts. (2020) ;10: (5):483. |
[23] | McGuire JJ , Anderson DJ , McDonald BJ , Narayanasami R , Bennett BM . Inhibition of NADPH-cytochrome P450 reductase and glyceryl trinitrate biotransformation by diphenyleneiodonium sulfate. Biochemical Pharmacology. (1998) ;56: (7):881–93. |
[24] | Cross AR , Jones OT . The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Specific labelling of a component polypeptide of the oxidase. Biochemical Journal. (1986) ;237: (1):111–6. |
[25] | O’Donnell VB , Tew DG , Jones OT , England P . Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochemical Journal. (1993) ;290: (1):41–9. |
[26] | Kono H , Rusyn I , Uesugi T , Yamashina S , Connor HD , Dikalova A , et al. Diphenyleneiodonium sulfate, an NADPH oxidase inhibitor, prevents early alcohol-induced liver injury in the rat. American Journal of Physiology-Gastrointestinal and Liver Physiology.(2001) ;280: (5):G1005–G12. |
[27] | Venkatachalam P , De Toledo S , Pandey B , Tephly L , Carter A , Little J , et al. Regulation of normal cell cycle progression by flavin-containing oxidases. Oncogene. (2008) ;27: (1):20–31. |
[28] | Zavadskis S , Weidinger A , Hanetseder D , Banerjee A , Schneider C , Wolbank S , et al. Effect of Diphenyleneiodonium Chloride on Intracellular Reactive Oxygen Species Metabolism with Emphasis on NADPH Oxidase and Mitochondria in Two Therapeutically Relevant Human Cell Types. Pharmaceutics. (2021) ;13: (1):10. |
[29] | Kim SK , Rho SJ , Kim SH , Kim SY , Song SH , Yoo JY , et al. Protective effects of diphenyleneiodonium, an NADPH oxidase inhibitor, on lipopolysaccharide-induced acute lung injury. Clinical and Experimental Pharmacology and Physiology. (2019) ;46: (2):153–62. |
[30] | Dong W , Li Z , Chen Y , Zhang L , Ye Z , Liang H , et al. NADPH oxidase inhibitor, diphenyleneiodonium prevents necroptosis in HK-2 cells. Biomedical Reports. (2017) ;7: (3):226–30. |
[31] | Piszczatowska K , Przybylska D , Sikora E , Mosieniak G . Inhibition of NADPH Oxidases Activity by Diphenyleneiodonium Chloride as a Mechanism of Senescence Induction in Human Cancer Cells. Antioxidants. (2020) ;9: (12):1248. |
[32] | Majander A , Finel M , Wikström M . Diphenyleneiodonium inhibits reduction of iron-sulfur clusters in the mitochondrial NADH-ubiquinone oxidoreductase (Complex I). Journal of Biological Chemistry. (1994) ;269: (33):21037–42. |
[33] | Holland PC , Clark MG , Bloxham DP , Lardy HA . Mechanism of action of the hypoglycemic agent diphenyleneiodonium. Journal of Biological Chemistry. (1973) ;248: (17):6050–6. |
[34] | Gatley SJ , Stanley H , Sherratt A . A Comparison of the Catalysis of Chloride-Hydroxyl Ion Exchange Across the Inner Membrane of Rat Liver Mitochondria by Several Substituted Diphenyleneiodonium Derivatives and Their Effects on Mitochondrial Oxidations in Chloride-Free and Chloride-Containing Media. Portland Press Ltd.; (1974) . |
[35] | Stuehr DJ , Fasehun OA , Kwon NS , Gross SS , Gonzalez JA , Levi R , et al. Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs 1. The FASEB Journal. (1991) ;5: (1):98–103. |
[36] | Gray JP , Heck DE , Mishin V , Smith PJ , Hong J-Y , Thiruchelvam M , et al. Paraquat increases cyanide-insensitive respiration in murine lung epithelial cells by activating an NAD (P) H: paraquat oxidoreductase: identification of the enzyme as thioredoxin reductase. Journal of Biological Chemistry. (2007) ;282: (11):7939–49. |
[37] | O’Donnell V , Smith G , Jones O . Involvement of phenyl radicals in iodonium inhibition of flavoenzymes. Molecular Pharmacology. (1994) ;46: (4):778–85. |
[38] | Wang Y , Gray JP , Mishin V , Heck DE , Laskin DL , Laskin JD . Role of cytochrome P450 reductase in nitrofurantoin-induced redox cycling and cytotoxicity. Free Radical Biology and Medicine. (2008) ;44: (6):1169–79. |
[39] | Rezende F , Prior K-K , Löwe O , Wittig I , Strecker V , Moll F , et al. Cytochrome P450 enzymes but not NADPH oxidases are the source of the NADPH-dependent lucigenin chemiluminescence in membrane assays. Free Radical Biology and Medicine. (2017) ;102: 57–66. |
[40] | Pandey M , Singh AK , Thakare R , Talwar S , Karaulia P , Dasgupta A , et al. Diphenyleneiodonium chloride (DPIC) displays broad-spectrum bactericidal activity. Scientific Reports. (2017) ;7: (1):1–8. |
[41] | Chung J-W , Kim SY , Park HJ , Chung CS , Lee HW , Lee SM , et al. In vitro activity of diphenyleneiodonium toward multidrug-resistant Helicobacter pylori strains. Gut and Liver. (2017) ;11: (5):648. |
[42] | Ozsvari B , Bonuccelli G , Sanchez-Alvarez R , Foster R , Sotgia F , Lisanti MP . Targeting flavin-containing enzymes eliminates cancer stem cells (CSCs), by inhibiting mitochondrial respiration: vitamin B2 (Riboflavin) in cancer therapy. Aging (Albany NY). (2017) ;9: (12):2610. |
[43] | Rohr-Udilova N , Klinglmüller F , Seif M , Hayden H , Bilban M , Pinter M , et al. Oxidative stress mediates an increased formation of vascular endothelial growth factor in human hepatocarcinoma cells exposed to erlotinib. Oncotarget. (2017) ;8: (34):57109. |
[44] | Herzog N , Katzenberger N , Martin F , Schmidtke K-U . Generation of cytochrome P450 3A4-overexpressing HepG2 cell clones for standardization of hepatocellular testosterone 6β-hydroxylation activity. Journal of Cellular Biotechnology. (2015) ;1: (1):15–26. |
[45] | Geburek I , Rutz L , Gao L , Küpper J-H , These A , Schrenk D . Metabolic Pattern of Hepatotoxic Pyrrolizidine Alkaloids in Liver Cells. Chemical Research in Toxicology. (2021) ;34: (4):1101–13. |
[46] | Bokhari M , Carnachan RJ , Cameron NR , Przyborski SA . Culture of HepG2 liver cells on three dimensional polystyrene scaffolds enhances cell structure and function during toxicological challenge. Journal of Anatomy. (2007) ;211: (4):567–76. |
[47] | Thabrew MI , HUGHES RD , MCFARLANE IG . Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay. Journal of Pharmacy and Pharmacology. (1997) ;49: (11):1132–5. |
[48] | Hussein KH , Park KM , Ghim JH , Yang SR , Woo HM . Three dimensional culture of HepG2 liver cells on a rat decellularized liver matrix for pharmacological studies. Journal of Biomedical Materials Research part B: Applied Biomaterials. (2016) ;104: (2):263–73. |
[49] | Kade S , Herzog N , Schmidtke K-U , Küpper J-H . Chronic ethanol treatment depletes glutathione regeneration capacity in hepatoma cell line HepG2. Journal of Cellular Biotechnology. (2016) ;1: (2):183–90. |
[50] | Zuchowska A , Kwapiszewska K , Chudy M , Dybko A , Brzozka Z . Studies of anticancer drug cytotoxicity based on long-term HepG2 spheroid culture in a microfluidic system. Electrophoresis. (2017) ;38: (8):1206–16. |
[51] | Steinbrecht S , Pfeifer N , Herzog N , Katzenberger N , Schulz C , Kammerer S , et al. HepG2-1A2 C2 and C lentivirus vector-mediated stable and functional overexpression of cytochrome P450 1A2 in human hepatoblastoma cells. Toxicology Letters. (2020) ;319: 155–9. |
[52] | Coppi E , Pugliese AM , Urbani S , Melani A , Cerbai E , Mazzanti B , et al. ATP modulates cell proliferation and elicits two different electrophysiological responses in human mesenchymal stem cells. Stem Cells. (2007) ;25: (7):1840–9. |
[53] | Mandal S , Lindgren AG , Srivastava AS , Clark AT , Banerjee U . Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cells. Stem Cells. (2011) ;29: (3):486–95. |
[54] | Song D , Liu X , Liu R , Yang L , Zuo J , Liu W . Connexin 43 hemichannel regulates H9c2 cell proliferation by modulating intracellular ATP and [Ca2+]. Acta Biochim Biophys Sin. (2010) ;42: (7):472–82. |


