
Regulation of cagA-Helicobacter on gastric PIM2 expression in gastric cancer
Abstract
BACKGROUND:
The association between infection with cagA-positive H. pylori and an elevated susceptibility to gastric cancer has been firmly established. PIM2 is known to be overexpressed in various types of cancers; however, the specific mechanism by which cagA influences the regulation of PIM2 expression in gastric cancer remains unidentified at present.
MATERIALS AND METHODS:
A mutant NCTC11637
RESULTS:
Co-culturing gastric cancer cells with NCTC11637 significantly increased PIM2 expression levels (
CONCLUSIONS:
The findings indicate that H. pylori/cagA A could be one of the key factors in regulating PIM2 expression levels, potentially influencing the progression of H. pylori-related Gastric Cancer.
1.Introduction
Gastric cancer (GC) remains a significant public health issue worldwide, despite recent declines in incidence and mortality rates [1]. Risk factors for gastric cancer are older age, low socio-economic status, cigarette smoking, alcohol consumption, familial predisposition, previous gastric surgery, pernicious anaemia, and living in a population at high risk, and Helicobacter pylori (H. pylori) infection is the most well described risk factor for gastric cancer [2]. Chronic inflammation and the pathogenesis of gastric cancer (GC) are posited to be precipitated by the activation of the nuclear transcription factor kappa B (NF
The proviral integration sites for Moloney murine leukemia virus (PIMs) represent a group of serine/threonine kinases that are overexpressed in various hematological malignancies and solid tumors, exerting regulatory effects on cell proliferation and apoptosis [8]. The PIM family comprises three isoforms (PIM1, PIM2, and PIM3), which exhibit high conservation across evolutionary lines [8]. While PIM1 and PIM3 are predominantly expressed in hematological and solid tumors, respectively, PIM2 is primarily associated with leukemia [9]. Functionally, PIM2 plays a role in cell apoptosis, proliferation, and differentiation [10], and has been identified as one of the downstream targets of NF
In this study, we explored the impact of H. pylori/cagA on PIM2 expression in gastric cancer cells, using a cagA knockout mutant and a cagA expression vector, uncovering a new mechanism of H. pylori-induced gastric pathogenesis through PIM2 regulation.
2.Materials and methods
2.1Reagents and cell lines
ClonExpress II One Step Cloning Kit, ClonExpress MultiS One Step Cloning Kit, ChamQ Universal SYBR qPCR Master Mix and HiScript® II Q RT SuperMix for qPCR (
2.2Construction of H. pylori cagA knocked-out strain
The upstream and downstream homologous fragments of cagA derived from H. pylori NCTC11637, as well as chloramphenicol resistance gene (cmR) originating from the plasmid pNZ8110, were successfully amplified by PCR. The primers were delineated as follows: P1-F1 sense, 5-CGAGGTCGACGGTATCGATA AGCTTTGCTCGGCAACTGACATCA-3, P2-F1 anti-sense, 5-ATCAATT TTATTAAAGTTCATTGTTTCTC CTTACTATACC-3; P3-F2 sense, 5-CCTAATGACT GGCTTTTATAAAGGATTAAGGAA TACCAAAAAC G-3, P4-F2 anti-sense, 5-GCGGT GGCGGCCGCTC TAGATTCCACTTCTGATCGCAAGC-3; P5-cmR sense, 5-GGTATAG TAAGGAGAAACAATGAACTTT AATAAAATTGAT-3, P6-cmR anti-sense, 5-CGTTTTT GGTATTCCTTAATCCTTTATAAAAGCCAGTCATT AGG-3. PCR conditions included pre-denaturation at 95∘C for 5 min followed by 35 cycles consisting of denaturation at 94∘C for sixty seconds, annealing at 57∘C for fifty seconds, and extension at 72∘C also lasting sixty seconds; concluding with a final ten-minute extension step set again to 72∘C. The Clon Express MultiS One Step Cloning Kit facilitated insertion of fragments into plasmid pBluescript IISK (-). The recombinant plasmid underwent transfer into NCTC11637 by electroporation, and subsequently, the mutant strain was selected on a chloramphenicol agar plate. Identification of the mutant strain NCTC1163
2.3Construction of cagA eukaryotic expression vector
The cagA gene fragment was gained via PCR from the genomic DNA of NCTC11637. Then, the plasmid pcDNA3.1 (
2.4Co-culture of H. pylori with GC cells
Initially, H. pylori NCTC11637 was co-cultured with the GC cells, namely HGC27, SGC7901 and AGS, under multiplicity of infection (MOI) ratios of 50:1, 100:1 and 150:1, respectively. Subsequently, the expression levels of PIM2 mRNA within these cells were quantitatively assayed at 24-hour of co-culture initiation. Subsequently, employing NCTC11637 strain as a reference control, the NCTC1163
2.5Cellular transfection
Upon successful identification and confirmation of the recombinant plasmid construct, the gastric cancer cell lines HGC27, SGC7901 and AGS cells were individually subjected to transfection with pcDNA-cagA. Concurrently, a control group was established using the empty vector pcDNA3.1 (
2.6Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted using TRIzol reagent, strictly according to the manufacturer’s protocols provided by the manufacturer. Subsequently, a 20
2.7Western blot
Total protein was extracted from cells employing RIPA Lysis buffer (ComWin Biotech Co., Ltd., Beijing, China), which was supplemented with a 1% protease inhibitor cocktail (ComWin Biotech Co., Ltd., Beijing, China). The protein concentration was quantified using the BCA Protein assay kit (Thermo Fisher Scientific, Inc.). Equivalent quantities of protein extracts (50
2.8Statistical analysis
All data were analyzed utilizing the SPSS software (version 17.0; SPSS, Inc., Chicago, IL, USA). Quantitative data were presented as the mean
3.Results
3.1The expression of PIM2 at different multiplicity of infection (MOI) levels with H. pylori
Figure 1.
Expression of PIM2 mRNA in GC cells infected with H. pylori at a range of MOIs. (*P< 0.05).
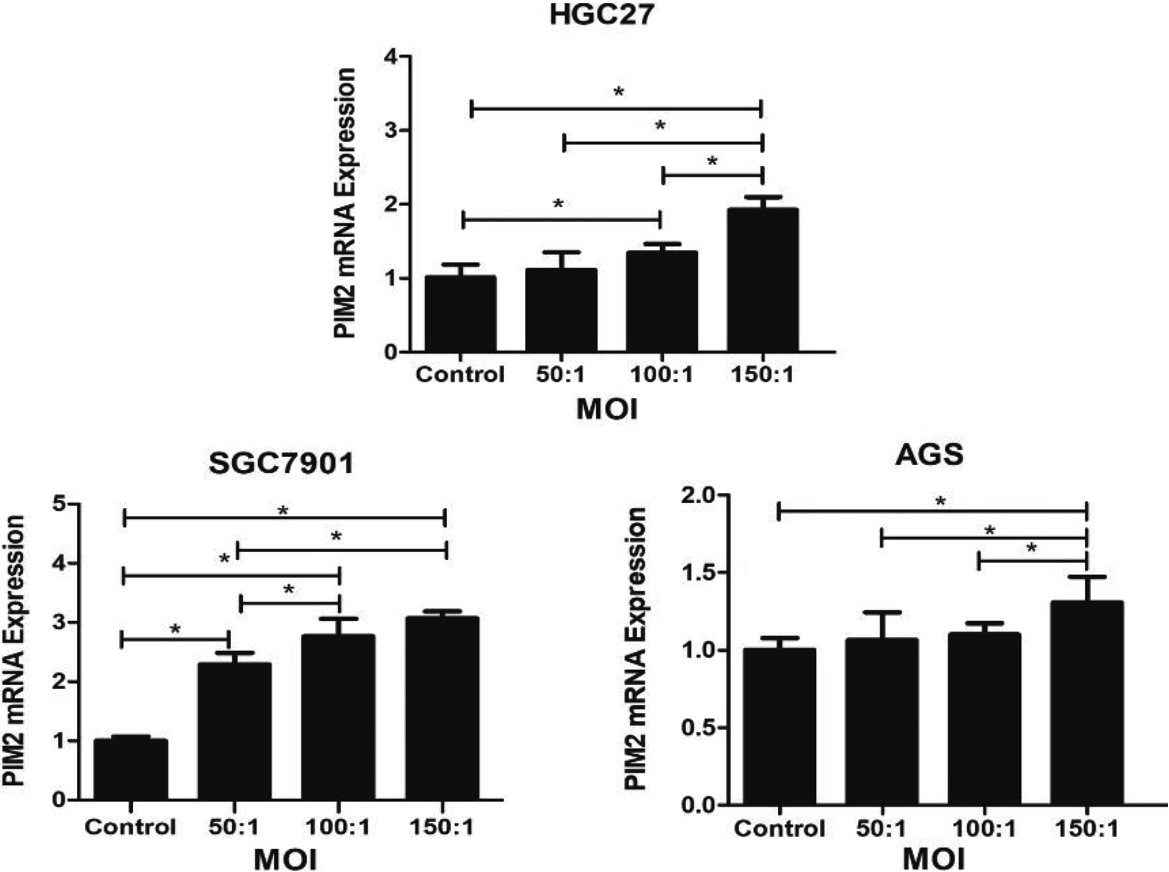
The Fig. 1 demonstrated that the expression levels of PIM2 at varying multiplicities of infection (MOI) with H. pylori. The experimental group with an MOI of 150 exhibited a significant upregulation of PIM2 mRNA expression compared to the control group across three gastric cancer cell lines, HGC27, SGC7901 and AGS (
Figure 2.
The expression of PIM2 mRNA in H. pylori NCTC11637-infected HGC27, SGC7901, and AGS cells at 6 h, 12 h, and 24 h post-infection (*P< 0.05).
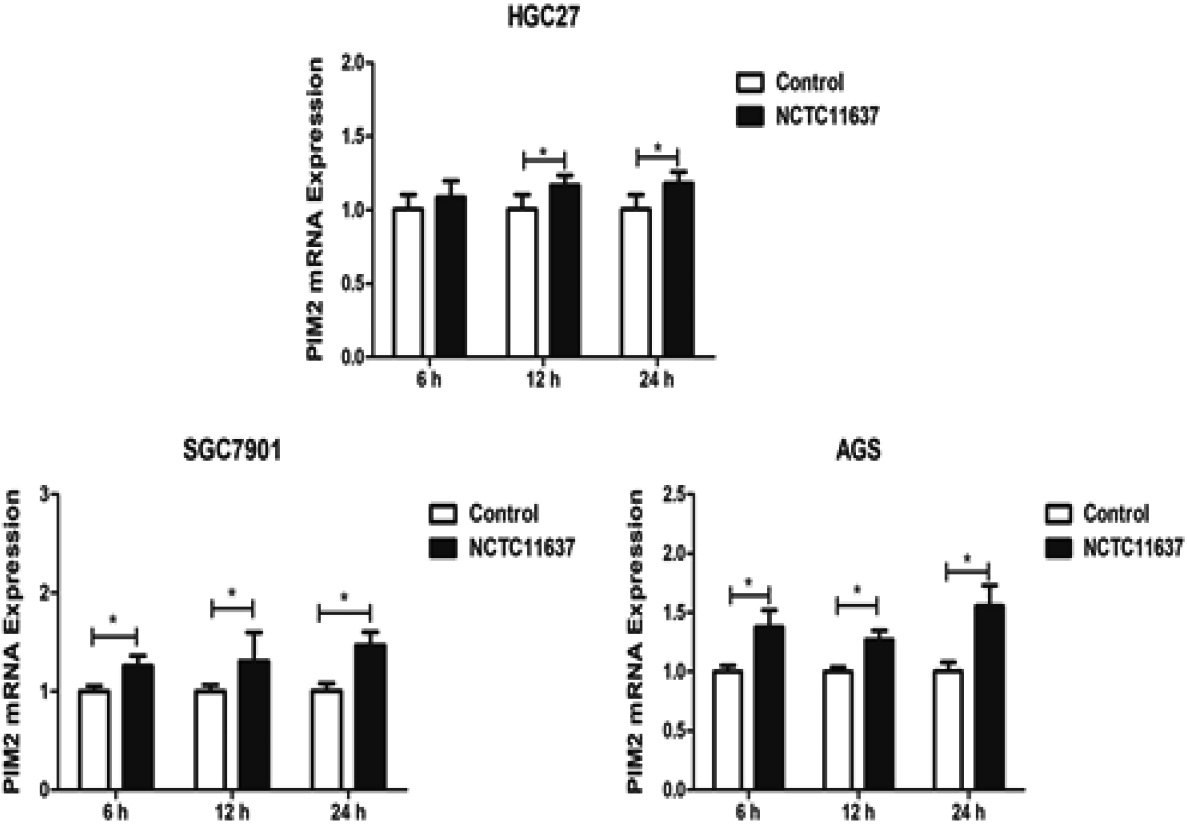
We subsequently investigated the expression levels of PIM2 mRNA in H. pylori GC27, SGC7901, and AGS cells after infection with H. pylori strain NCTC11637 at an MOI of 150:1 (Fig. 2). The results revealed a significant upregulation of PIM2 mRNA expression in HGC27 cells at 12 h (
3.2The expression of PIM2 infected with NCTC11637 and NCTC11637 Δ
Figure 3.
Expression of PIM2 mRNA in the GC cells co-cultured with H. pylori (cagA
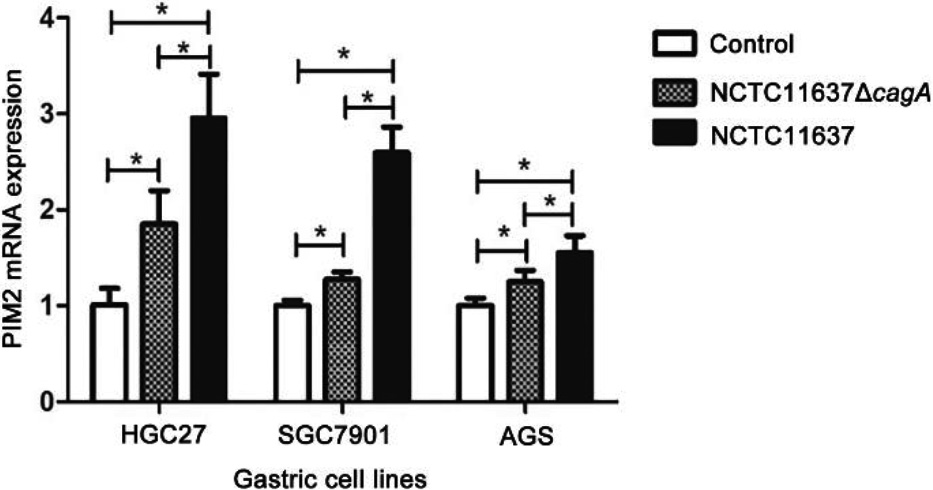
We successfully constructed the H. pylori
3.3The expression of PIM2 infected with the eukaryotic expression vector construction of NCTC11637 cagA
Figure 4.
Expression of cagA gene in the GC cells transfected with pcDNA-cagA plasmid. (a) (b) (c) CagA mRNA expression in the GC cells, HGC27, AGS and SGC7901, transfected with pcDNA-cagA and pcDNA3.1(
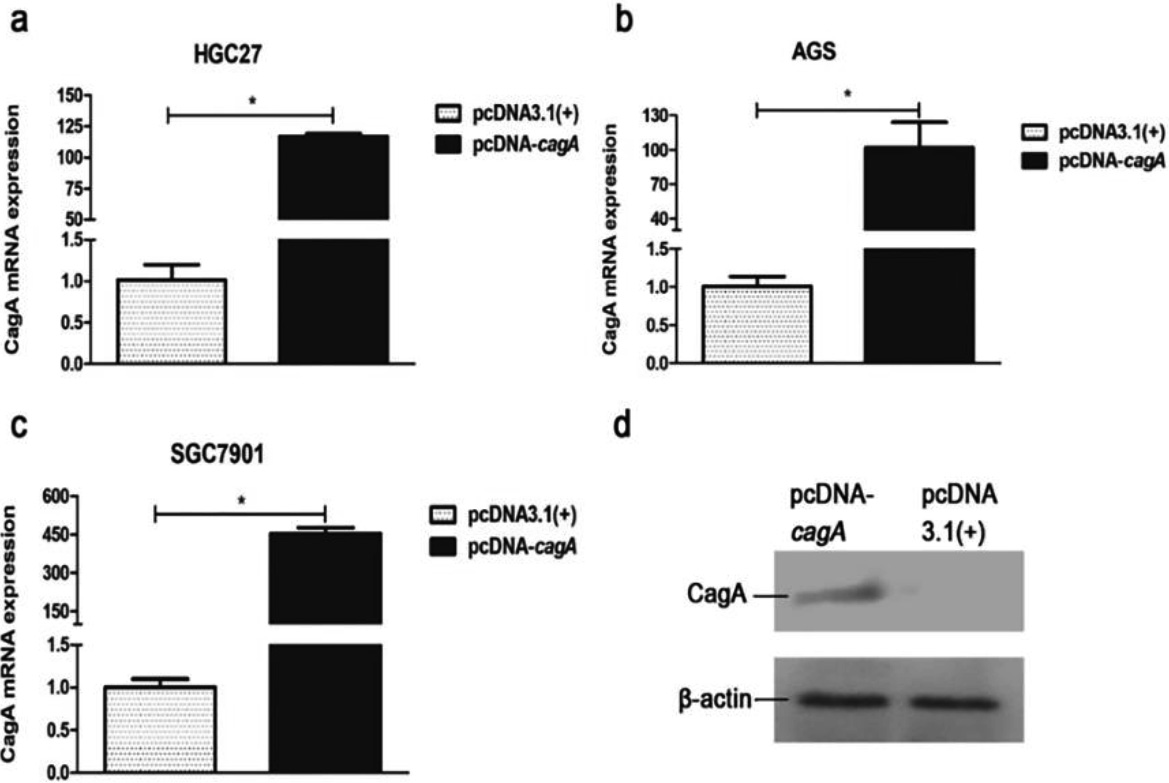
Figure 5.
PIM2 mRNA expression in the GC cells transfected with pcDNA-cagA plasmid. (*P< 0.05).
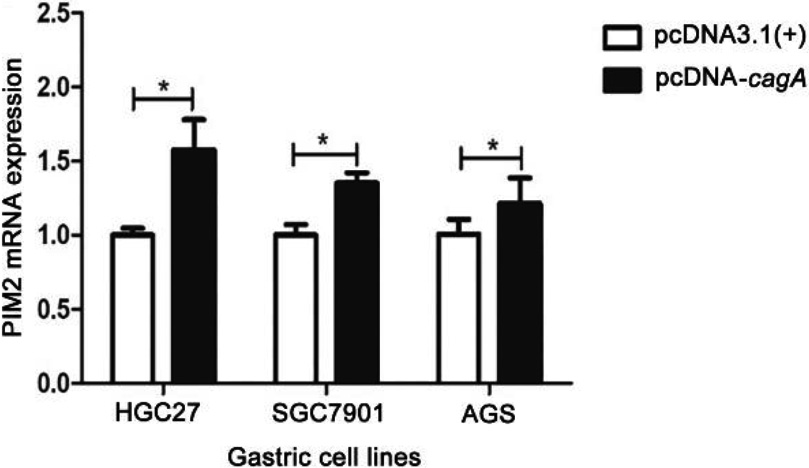
The eukaryotic expression vector pcDNA-cagA was successfully constructed as confirmed by PCR (Fig. S3). Subsequent transfection of HGC27, SGC7901 and AGS cells with pcDNA-cagA showed increased expression of cagA gene as detected by qPCR and Western blot analysis, confirming successful transfection into cells (Fig. 4). The expression levels of PIM2 mRNA in HGC27, SGC7901 and AGS cells transfected with pcDNA-cagA for 48 hours, were significantly higher than control cells treated with pcDNA3.1(
4.Discussion
PIM2 kinase plays an important role as an oncogene in multiple cancers, such as leukemia, liver, lung, myeloma, prostate and breast cancers. PIM2 is largely expressed in both leukemia and solid tumors, and it promotes the transcriptional activation of genes involved in cell survival, cell proliferation, and cell-cycle progression [12]. This study revealed that the MOI and duration of H. pylori infection upregulated the expression of PIM2 in gastric cancer cells. Furthermore, our findings provided additional evidence for the regulatory effect of cagA on PIM2 expression in gastric cancer epithelial cells by demonstrating an increase in PIM2 expression following transfection with cagA.
H. pylori colonizes the human gastric mucosa and significantly increases the risk of GC [2]. H. pylori can activate NF
The PIM2 gene is recognized as an oncogene, which plays an essential role in the signal transduction process related to cell proliferation, differentiation, apoptosis, migration, survival and metabolism. It is also transcriptionally regulated by the JAK/STAT and NF
The CagA is s one of the most widely studied H. pylori virulence factor. Previous studies have demonstrated that infection with H. pyloricagA (
As documented, CagA can be injected into target cells. Upon once translocation into the host cytoplasm, CagA may bind to the inner surface of the cell membrane and interact with several host proteins, thereby activating downstream signaling pathways such as NF
The CagA-induced regulation of PIM2 overexpression may represent a critical mechanism underlying the development of GC. Santio et al. demonstrated that in hematopoietic cells, cytokines, hormones or antigens predominantly upregulated PIM2 gene expression mainly via the JAK/STAT signaling pathway [23]. Previous studies has established that STAT protein are regulators of EMT [24]. In breast cancer cells, PIM2 indirectly activates STAT3 protein by upregulating the production of cytokines IL-
Additionally, the present study demonstrated that co-culture of H. pylori and GC cells resulted in the overexpression of PIM2 in the infected GC cells. This effect was observed not only with the H. pylori NCTC11637 (cagA
The present study has limitations. Firstly, the current study was confined to the assessment of PIM2 mRNA expression within gastric cancer cell lines, which may not accurately represent the conditions in the gastric mucosa. The findings, therefore, should be interpreted with caution. Secondly, the study has focused exclusively on the alterations in PIM2 mRNA levels, not accounting for potential changes at the protein level, which may lead to an incomplete understanding of the effects of cagA transfection on the expression and functionality of PIM2 in gastric cancer cells.
5.Conclusions
Our study unveiled that the expression of PIM2 in gastric cancer cells was influenced by both the quantity and duration of H. pylori infection. Furthermore, we pinpointed that cagA of H. pylori played a pivotal role in upregulating PIM2 expression in gastric cancer epithelial cells. Our results further illustrated that transfection of cagA into gastric cancer cells elevated PIM2 expression, providing additional evidence for the regulatory impact of cagA on PIM2 expression in gastric cancer epithelial cells. Considering the oncogenic nature of PIM2, our findings imply that cagA-mediated regulation of PIM2 expression may constitute an important mechanism underlying the pathogenesis of H. pylori infection-induced gastric cancer.
Author contributions
Conception: HW and RZ.
Interpretation or analysis of data: WL.
Preparation of the manuscript: HW, SC.
Revision for important intellectual content: WL, WC, LH.
Supervision: WL and RZ.
Funding
Hainan Provincial Natural Science Foundation of China (820CXTD438) and National Natural Science Foundation of China (82160634) funded this work.
Informed consent statement
Not applicable.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-230351.
Conflict of interest
No conflict of interest.
References
[1] | Y. Song, X. Liu, W. Cheng, H. Li and D. Zhang, The global, regional and national burden of stomach cancer and its attributable risk factors from 1990 to 2019, Scientific Reports 1: (12) ((2022) ), 11542. |
[2] | X. Guo, B. Yin, C. Wang, H. Huo and Z. Aziziaram, Risk assessment of gastric cancer in the presence of Helicobacter pylori cagA and hopQII genes, Cellular and Molecular Biology 4: (67) ((2022) ), 299–305. |
[3] | N. Ito, H. Tsujimoto, H. Ueno, Q. Xie and N. Shinomiya, Helicobacter pylori-mediated immunity and signaling transduction in gastric cancer, Journal of Clinical Medicine 11: (9) ((2020) ), 3699. |
[4] | Y. Wen, H. Huang, T. Tang, H. Yang, X. Wang, X. Huang et al., AI-2 represses CagA expression and bacterial adhesion, attenuating the Helicobacter pylori-induced inflammatory response of gastric epithelial cells, Helicobacter 2: (26) ((2021) ), e12778. |
[5] | A. Lamb, X.D. Yang, Y.H. Tsang, J.D. Li, H. Higashi, M. Hatakeyama et al., Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination, EMBO Reports 10: (11) ((2009) ), 1242–1249. |
[6] | F. Wang, N. Qu, J. Peng, C. Yue, L. Yuan and Y. Yuan, CagA promotes proliferation and inhibits apoptosis of GES-1 cells by upregulating TRAF1/4-1BB, Molecular Medicine Reports 2: (16) ((2017) ), 1262–1268. |
[7] | S. Chen, G. Duan, R. Zhang and Q. Fan, Helicobacter pylori cytotoxin-associated gene A protein upregulates alpha-enolase expression via Src/MEK/ERK pathway: implication for progression of gastric cancer, International Journal of Oncology 2: (45) ((2014) ), 764–770. |
[8] | N.K, Panchal and E.P Sabina, A serine/threonine protein PIM kinase as a biomarker of cancer and a target for anti-tumor therapy, Life Sciences 15: (255) ((2020) ), 117866. |
[9] | X. Zhang, M. Song, J.K. Kundu, M.H. Lee and Z.Z. Liu, PIM kinase as an executional target in cancer, Journal of Cancer Prevention 3: (23) ((2018) ), 109–116. |
[10] | V. Asati, D.K. Mahapatra and S.K. Bharti, PIM kinase inhibitors: Structural and pharmacological perspectives, European Journal of Medicinal Chemistry 6: (172) ((2019) ), 95–108. |
[11] | J.C. Wang, D.P. Chen, S.X. Lu, J.B. Chen, Y. Wei, X.C. Liu et al., PIM2 expression induced by proinflammatory macrophages suppresses immunotherapy efficacy in hepatocellular carcinoma, Cancer Research 18: (82) ((2022) ), 3307–3320. |
[12] | Y. Wang, J. Xiu, C. Ren and Z. Yu, Protein kinase PIM2: A simple PIM family kinase with complex functions in cancer metabolism and therapeutics, Journal of Cancer 9: (12) ((2021) ), 2570–2581. |
[13] | C. Lu, P. Qiao, Y. Sun, C. Ren and Z. Yu, Positive regulation of PFKFB3 by PIM2 promotes glycolysis and paclitaxel resistance in breast cancer, Clinical and Translational Medicine 4: (11) ((2021) ), e400. |
[14] | X. Li, K. Pan, M. Vieth, M. Gerhard, W. Li and R. Mejias-Luque, JAK-STAT1 signaling pathway is an early response to helicobacter pylori infection and contributes to immune escape and gastric carcinogenesis, International Journal of Molecular Sciences 8: (23) ((2022) ), 4147. |
[15] | P.S. Mary, W. Chaiwangyen, M. Weber, B. Al-Kawlani, R.R. Favaro, U. Jeschke et al., PIM kinases 1, 2 and 3 in intracellular LIF signaling, proliferation and apoptosis in trophoblastic cells, Experimental Cell Research 1: (359) ((2017) ), 275–283. |
[16] | N. Murata-Kamiya and M. Hatakeyama, Helicobacter pylori-induced DNA double-stranded break in the development of gastric cancer, Cancer Science 6: (113) ((2022) ), 1909–1918. |
[17] | S.Y. Chen, R.G. Zhang and G.C. Duan, Pathogenic mechanisms of the oncoprotein CagA in H. pylori-induced gastric cancer, Oncology Reports 6: (36) ((2016) ), 3087–3094. |
[18] | Z. Ge, Y.L. Zhu, X. Zhong, J.K. Yu and S. Zheng, Discovering differential protein expression caused by CagA-induced ERK pathway activation in AGS cells using the SELDI-ProteinChip platform, World Journal of Gastroenterology 4: (14) ((2008) ), 554–562. |
[19] | R. Lu, Y.G. Zhang and J. Sun, STAT3 activation in infection and infection-associated cancer, Molecular and Cellular Endocrinology 8: (451) ((2017) ), 80–87. |
[20] | I.O. Lee, J.H. Kim, Y.J. Choi, M.H. Pillinger, S.Y. Kim, M.J. Blaser et al., Helicobacter pylori CagA phosphorylation status determines the gp130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells, Journal of Biological Chemistry 21: (285) ((2010) ), 16042–50. |
[21] | A. Sukri, A. Hanafiah, N.R. Kosai, T.M. Mohammed and R. Mohamed, New insight on the role of Helicobacter pylori cagA in the expression of cell surface antigens with important biological functions in gastric carcinogenesis, Helicobacter 5: (27) ((2022) ), e12913. |
[22] | I.S. Sougleri, K.S. Papadakos, M.P. Zadik, M. Mavri-Vavagianni, A.F. Mentis and D.N. Sgouras, Helicobacter pylori CagA protein induces factors involved in the epithelial to mesenchymal transition (EMT) in infected gastric epithelial cells in an EPIYA- phosphorylation-dependent manner, Febs Journal 2: (283) ((2016) ), 206–220. |
[23] | N.M. Santio and P.J. Koskinen, PIM kinases: From survival factors to regulators of cell motility, International Journal of Biochemistry and Cell Biology 12: (93) ((2017) ), 74–85. |
[24] | C.L. Chaffer, J.B. San, E. Lim and R.A. Weinberg, EMT, cell plasticity and metastasis, Cancer and Metastasis Reviews 4: (35) ((2016) ), 645–654. |
[25] | N. Uddin, R.K. Kim, K.C. Yoo, Y.H. Kim, Y.H. Cui, I.G. Kim et al., Persistent activation of STAT3 by PIM2-driven positive feedback loop for epithelial-mesenchymal transition in breast cancer, Cancer Science 6: (106) ((2015) ), 718–725. |
[26] | M. Rokavec, M.G. Oner, H. Li, R. Jackstadt, L. Jiang, D. Lodygin et al., IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis, Journal of Clinical Investigation 4: (124) ((2014) ),1853–1867. |
[27] | C. Cloninger, A. Bernath, T. Bashir, B. Holmes, N. Artinian, T. Ruegg et al., Inhibition of SAPK2/p38 enhances sensitivity to mTORC1 inhibition by blocking IRES-mediated translation initiation in glioblastoma, Molecular Cancer Therapeutics 12: (10) ((2011) ), 2244–2256. |
[28] | R.A. Cairns, I.S. Harris and T.W. Mak, Regulation of cancer cell metabolism, Nature Reviews Cancer 2: (11) ((2011) ), 85–95. |
[29] | G. Yin, Y. Li, M. Yang, X.M. Cen and Q.B. Xie, Pim-2/mTORC1 Pathway Shapes Inflammatory Capacity in Rheumatoid Arthritis Synovial Cells Exposed to Lipid Peroxidations, Biomed Research International 3: (2015) ((2015) ), 240210. |
[30] | M. Liao, F. Hu, Z. Qiu, J. Li, C. Huang, Y. Xu and X. Cheng, Pim-2 kinase inhibits inflammation by suppressing the mTORC1 pathway in atherosclerosis, Aging 18: (13) ((2021) ), 22412–22431. |
[31] | X.H. Zhang, H.L. Yu, F.J. Wang, Y.L. Han and W.L. Yang, Pim-2 Modulates Aerobic Glycolysis and Energy Production during the Development of Colorectal Tumors, International Journal of Medical Sciences 6: (12) ((2015) ), 487–93. |


