
Machine learning analyses of highly-multiplexed immunofluorescence identifies distinct tumor and stromal cell populations in primary pancreatic tumors1
Abstract
BACKGROUND:
Pancreatic ductal adenocarcinoma (PDAC) is a formidable challenge for patients and clinicians.
OBJECTIVE:
To analyze the distribution of 31 different markers in tumor and stromal portions of the tumor microenvironment (TME) and identify immune cell populations to better understand how neoplastic, non-malignant structural, and immune cells, diversify the TME and influence PDAC progression.
METHODS:
Whole slide imaging (WSI) and cyclic multiplexed-immunofluorescence (MxIF) was used to collect 31 different markers over the course of nine distinctive imaging series of human PDAC samples. Image registration and machine learning algorithms were developed to largely automate an imaging analysis pipeline identifying distinct cell types in the TME.
RESULTS:
A random forest algorithm accurately predicted tumor and stromal-rich areas with 87% accuracy using 31 markers and 77% accuracy using only five markers. Top tumor-predictive markers guided downstream analyses to identify immune populations effectively invading into the tumor, including dendritic cells, CD4+ T cells, and multiple immunoregulatory subtypes.
CONCLUSIONS:
Immunoprofiling of PDAC to identify differential distribution of immune cells in the TME is critical for understanding disease progression, response and/or resistance to treatment, and the development of new treatment strategies.
1.Introduction
Pancreatic cancer is currently the 4
The heterogenous network of structural and immune components that make up the TME result from an oscillating balance of factors that actively combat the tumor or aid its growth [3]. Although some factors facilitating or negating PDAC development and progression have been identified, relatively little is known about the distribution and interactions of distinct immune cell populations within the TME. For example, immunosuppressive cells such as Myeloid Derived
Suppressor Cells (MDSCs) or T and B Regulatory Cells (Tregs and Bregs) can induce exhaustion resulting in inhibition of cancer-killing responses from cytotoxic T lymphocytes (CTLs), natural killer cells (NK), and helper
While traditional histologic stains such as Hematoxylin and Eosin (H&E), trichrome, and mucin stains are commonly used to identify and visualize spatial distributions of cells in cancer, they are usually limited to identification of very few antigens and must be combined with additional immunological labeling to accurately phenotype cells. This has ultimately restricted researchers’ ability to understand the complexity and diversity of cells in the TME [10]. T cells, for example, are identified by CD3 staining. These T cells could be present as cancer promoters including CD4+ Tregs (CD3+, CD4+, FOXP3+), or cancer killing CTLs (CD3+, CD8+, GZMB+) [11, 12]. Furthermore, both cell types can express CD56, which when present on Tregs is associated with poor patient outcome but, when present on cytotoxic T cells increased cancer killing is observed [13, 14]. Additionally, T cells can be exhausted (PD1+), naïve (CD45RA+), or memory (CD62L+) subtypes [14, 15, 16]. Therefore, T cells exist in dozens of states only decipherable by a complex code of co-localized marker-expression profiles. Accurate characterization of putative regulators of the TME therefor requires profiling of several markers to define pro- or anti-tumor cell states.
While new technologies such as single-cell sequencing allow more advanced characterization on a single cell level, many of these techniques require dissociation of the tissue and thus loss of associated spatial data [10]. We used cyclic Multiplex-Immunofluorescence (MxIF) and Whole Slide Imaging (WSI) to characterize the same section of PDAC tissues over nine rounds of IF staining to image 31 different markers. Cyclic multiplexed immunofluorescence is an established technique for re-staining tissue without significant loss to most antigens over the multiplexing process [10, 17]. This allowed us to perform in-depth single-cell-level characterizations and identify complex immune cell subtypes in the TME while preserving spatial information to confirm cell localization in the microenvironment.
Table 1
Patient characteristics
| RAP | Age | Gender | Diagnosis stage | Survival (days) | Tumor grade | Metastatic site (#) | Primary tumor size |
|---|---|---|---|---|---|---|---|
| 116 | 80 | F | IV | 497 | Moderately differentiated | 3 | 5.5 |
| 107 | 83 | F | IV | 269 | Moderately differentiated | 6 | 5.6 cm in autopsy report |
| 89 | 65 | M | IV | 65 | Poorly differentiated | 1 | 4.2 |
| 86 | 64 | M | IV | 64 | Moderately differentiated | 8 | 6.7 |
| 80 | 81 | F | IV | 7 | Moderately differentiated | 1 | 8.0 |
| 8 | 72 | M | IV | 9 | Moderately differentiated | 4 | 3.7 cm in autopsy report |
WSI is data intensive. A single WSI of one patient sample often consists of trillions of pixels representing hundreds-of-thousands of cells. Combining WSI with iterative MxIF exacerbates this problem and introduces new challenges. During cyclic MxIF, each round of staining and imaging must align to accurately visualize marker co-localization. Due to the high resolution of the imaging, minor shifts in the alignment of images across rounds of scanning can disrupt spatial relationships and interfere with cell identification and classification. The size of and number of the images (0.5–1.5 GB per channel, 4 to 5 channels per round) makes manual alignment time prohibitive, if not impossible. To address this issue, we created image registration software that allows us to largely automate the alignment of iteratively stained tissues using nuclear staining (DAPI) as a fiducial marker in all rounds of WSI. Once nuclei are aligned and markers are co-localized, there are a multitude of possible combinations of markers that determine cell type and state. We used machine learning to identify markers that were characteristic of tumor- and stromal-rich areas using a Random Forest Algorithm (RFA).
Random forests are ensemble predictors consisting of a large number of individual decision trees [18]. Each tree in the forest outputs a class prediction (tumor vs. stroma) based on markers expressed in each cell. The final model prediction is determined by voting. RFAs are shown to be robust when encountering noise and resistant to overfitting (memorizing the training data) by using a random selection of features to grow each tree in the forest (fig-tree-snippet). We used the outputs of the RFA to classify cells and perform spatial analyses to determine which cells were capable of invading into the tumor and those remaining trapped within the dense stromal network. Such information is critical for understanding cellular players responsible for modifying and regulating cellular interactions in the TME, uncovering the mechanisms of response and resistance to therapeutic interventions, and discovering novel theragnostic targets.
Table 2
Multiplexed immunofluorescence staining procedure
| Verify Tumor with Surgical Pathologist (serial H&E) | ||||
|---|---|---|---|---|
| Verify Tumor/Stroma Areas with Surgical Pathologist | ||||
| Post-MxIF H&E Staining | ||||
| Cy2 | Cy3 | Cy5 | Cy7 | |
| Round 1 | PRG2 | CD31 | CD141 | CD103 |
| Scan to Verify Quenched Slide (1) | ||||
| Round 2 | SMA | MPO | LOX-1 | |
| Round 3 | CD62L | CD56 | CD8 | Granzyme B |
| Scan to Verify Quenched Slide (2) | ||||
| Round 4 | FOXP3 | CD163 | CD4 | |
| Round 5 | MUC1 | Tryptase | EPCAM | Ki67 |
| Scan to Verify Quenched Slide (3) | ||||
| Round 6 | CCR3 | CD3 | CD19 | CD11b |
| Round 7 | GFAP | IL10 | CXCR3 | CD45RA |
| Scan to Verify Quenched Slide (4) | ||||
| Round 8 | Vimentin | PD1 | FAP | |
| Round 9 | PDL1 | iNOS | ||
2.Materials and methods
2.1Human pancreatic ductal adenocarcinoma (PDAC) specimens
De-identified human tissue samples were collected from consented, end-stage PDAC patients within three-hours of the patient passing. At autopsy, surgical pathologists grossly identified primary and metastatic tumor sites. Tumors, uninvolved tissue immediately adjacent to tumors, and otherwise uninvolved tissue samples were collected from all abdominal organs and stored in formalin (
Consistent with The Code of Ethics of the World Medical Association (Declaration of Helsinki), all human samples were collected from consented patients through the UNMC Pancreatic Rapid Autopsy Program in compliance with Institutional Review Board review (#091-01).
2.2Multiplexed immunofluorescent (MxIF) staining and whole slide imaging (WSI) of PDAC specimens
MxIF analyses were performed on a tissue section adjacent to the section studied by the pathologist. Tissue sections were deparaffinized and rehydrated using xylene and an alcohol gradient. Antigen retrieval was performed using a microwave heated acidic citrate buffer (20 min, low heat), additional benchtop incubation (20 min), and subsequent blocking with 1% BSA (1 h). Slides were stained with antibodies directly conjugated with fluorophores or by indirect immunofluorescence (Supplementary Table 2). Coverslips were mounted using mounting media with DAPI (Prolong Gold Antifade Mountant, ThermoFisher Scientific) and allowed to dry for 48 h prior to WSI.
Directly conjugated antibodies were either purchased pre-labeled or labeled in-house using a Cy5 or Cy3 labeling kit from Cytiva (Amersham CyDye kits) or lighting-link conjugation kits from Abcam (PE/Cy7
WSI was performed using a Pannoramic 250 whole slide scanner (3D Histech) fitted with a Carl-Zeiss Plan-Apochromat 20
Figure 1.
Representative images from cyclic multiplexed-immunofluorescence (MxIF) labeling of a primary PDAC tumor with image registration. A) Images showing distinct marker distributions in the same area across staining round 3, quenching and re-scanning at the same intensity, and re-staining round 4. Scale bar
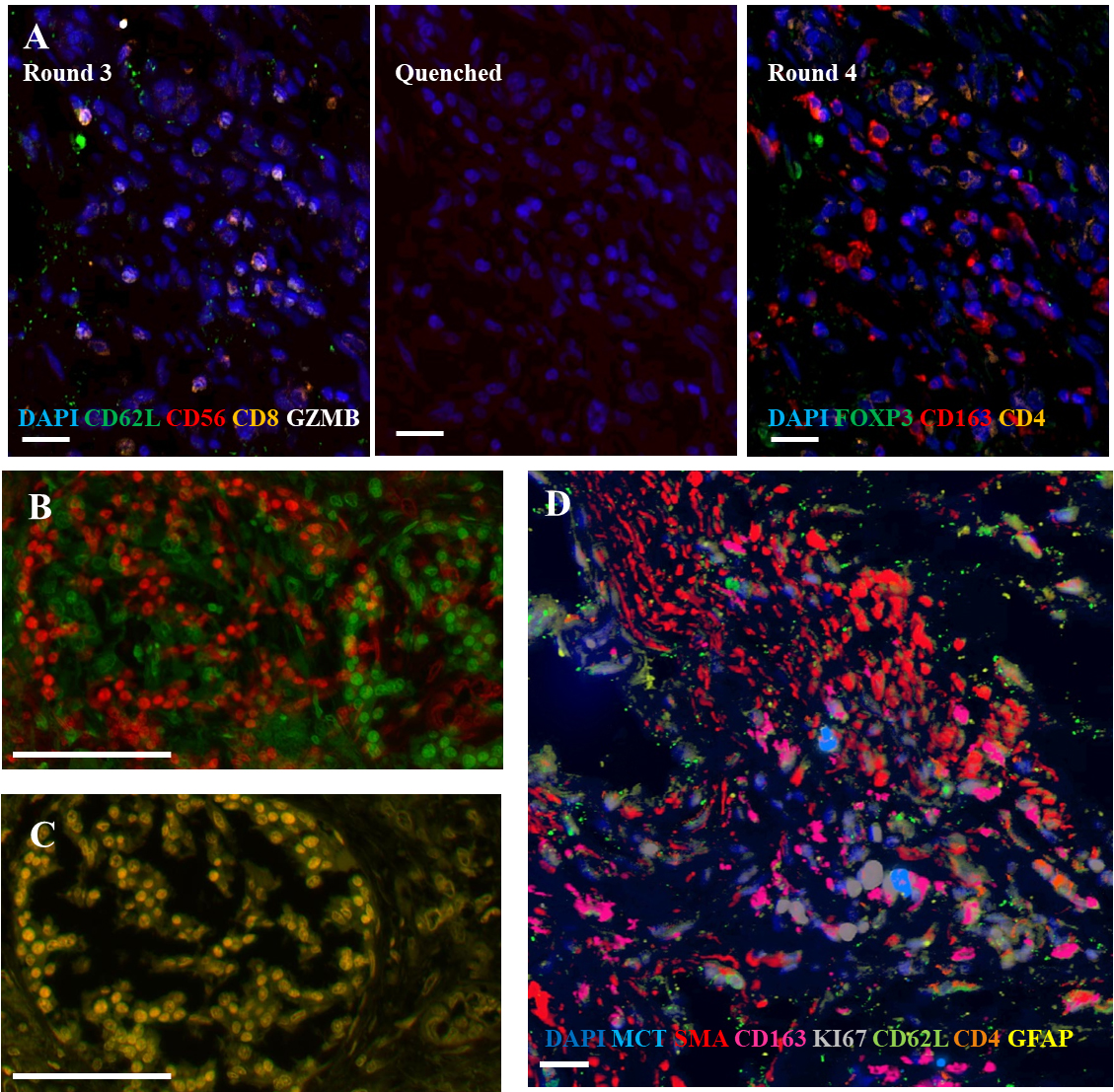
2.3Multiplexed WSI image alignment and pre-processing
WSI images were converted from the proprietary 3DHistech file format (.MXRS), to an 8-bit universal file format (.TIF) for image registration and post processing. Additional information regarding the images and analysis regions are provided in Supplementary Table 1.
Using the above MxIF technique over 30 distinct markers and nine DAPI stainings were aligned. In each of the nine rounds of imaging additional channels align automatically to the corresponding DAPI channel; registration is required to align DAPI frames across rounds. Once DAPI frames are co-registered (see below, Fig. 1B–D), the same translation vector was applied to the associated marker frames bringing all 40 frames into alignment. The code for the registration software is available at GitHub (https://github.com/EDRN/slide-image-registration/releases/tag/v1.0.0).
For alignment of DAPI frames, the first DAPI frame was used as the anchor for subsequent the DAPI frames from all other rounds are registered. The registration method is based on cross-correlation in the frequency domain [18]. The scikit-image implementation of this method uses discrete Fourier transform to achieve arbitrary sub-pixel precision [19]. Frames with either dimension exceeding 30,000 pixels were down-sampled to accommodate larger frames in available memory, making it possible to apply the registration method to entire frames. When conducting cyclic MxIF and iterative WSI, artifacts due to varying staining intensities, tissue damage, or large over-exposed regions can introduce errors in registration [20, 21]. It is therefore important to visually or experimentally verify alignment fidelity. Outputs of the automated alignments were visually inspected and adjusted as needed (see below).
To remain within registration pipeline file size restriction, single channel images were limited to 1.5 GB and some images were cropped to maintain an appropriate file size. A 1.5 GB image still includes tens-of-thousands of cells for analysis. Once registered, successful global alignment was verified visually across all nine rounds of staining using DAPI staining (Fig. 1B and C). Minor misalignments that persisted were adjusted manually in GIMP (GNU Image Manipulation Program) which allowed manipulation of a set of images while preserving spatial consistency. Global alignment was verified visually in GIMP. Files exported from GIMP were converted to .PNG in ImageJ (National Institutes of Health) for background subtraction. We performed further verification of cellular alignment within HALO (High-Plex FL v2.0, see below). Similar analyses could be done visually though at a much larger time expense.
2.4Nuclear segmentation and antigen intensity quantification
Once aligned, nuclei were segmented based on each round of DAPI staining using HALO, a WSI software designed for cellular quantification. Within the HALO analysis platform, the nuclear segmentation algorithm was optimized based on a visual ground truth that could be applied to all images within our dataset. Nuclear contrast, segmentation aggressiveness, and intensity were set at 0.49, 0.795, and 0.05, respectively, using a one-point scale. Abnormally small or large cells were excluded. Segmented nuclei not present in each round of DAPI staining (non-high fidelity) were excluded from further analyses. High-fidelity cells were identified by consistent DAPI co-localization in the same location across all nine rounds of staining. This process excludes cells that go into or out of focus as the
Using HALO, the average staining intensity was measured in the cytoplasmic and nuclear areas of high-fidelity cells. Staining intensity is based on the average number and intensity of pixels within the pre-defined area. The cytoplasmic area, the maximum range around the nucleus classified as the cell area, was set to 1.4
2.5Random forest classifier analysis of marker combinations
We applied a random forest classifier to investigate marker combinations classifying cells in stroma- and tumor-rich areas. Tumor and stroma regions were identified visually as MUC1 high or MUC1 low, respectively, with verification by a surgical pathologist. Normal adjacent or mixed tissue was excluded from analysis. Within our first pass analysis we considered all markers as potential predictors: [‘CCR3’, ‘CD103’, ‘CD11B’, ‘CD141’, ‘CD163’, ‘CD19’, ‘CD3’, ‘CD31’, ‘CD4’, ‘CD45RA’, ‘CD56’, ‘CD62L’, ‘CD8’, ‘CXCR3’, ‘EPCAM’, ‘FAP’, ‘FOXP3’, ‘GFAP’, ‘GZMB’, ‘IL10’, ‘INOS’, ‘KI67’, ‘LOX1’, ‘TRYPTASE’, ‘MPO’, ‘MUC1’, ‘PD1’, ‘PDL1’, ‘PRG2’, ‘SMA’, ‘VIMEN-TIN’]. Upon further analysis we removed markers suspected to be directly expressed on cancer cells (MUC1, Ki67, EPCAM).
We use scikit-learn implementation to train random forest models [19]. The training of random forest models is supervised, where all marker expression levels are matched with either tumor or stroma class in all data. In each image, there are different numbers of cells detected with the tumor and stroma regions (Supplementary Table 1). To address the effects of imbalanced tumor and stroma cell numbers in predictions, we used the balanced sampling strategy where class weights are adjusted to class frequencies for each tree being trained.
The number of trees and maximum tree depth were determined by monitoring the reduction of out-of-bag error with the addition of each new tree [23]. With all markers as predictors, we found the number of trees to stabilize around 150 trees with max depth of 30, beyond which adding new trees did not reduce the out-of-bag error significantly (Supplementary Fig. 1). While the out-of-bag error is an indicator of the model performance on the test set, cross-validation ensures all samples appear in the training and testing of random forests. We used 5-fold cross-validation with each set repeated ten times to report the average model performance in prediction accuracy.
To measure the importance of specific markers in predicting tumor vs. stromal regions, we used the feature importance scoring of random forest. The internal estimates at each tree node split (fig-single tree snippet) were used in measuring the predictor importance. The code for the RFA can be found at GitHub (https://github.com/EDRN/random-decision-forests/releases/tag/v1.0.0).
2.6Cellular clustering
Once the RFA was trained, top markers were used to identify cell groups based on Louvain clustering, an unsupervised algorithm designed to identify communities with similar features. Louvain clustering removes unintentional visual bias from the clustering method and has been used similarly on other large biological datasets [24, 25]. The Louvain clustering was run on all samples as one concatenated group. The outputs of Louvain clustering were verified with
2.7Neighborhood construction and analysis
Following preprocessing of column structure and centroid calculation from bounding boxes, segmented cell centroids and Louvain-clustering classification were imported into Volumetric Tissue Exploration and Analysis (VTEA) and cellwise neighborhoods were segmented from a nearest-neighbor graph based on distances between cell centroids within 30 microns [26, 27]. VTEA was used to calculate the sum and fraction of the Louvain-cluster defined cell-types in each neighborhood and to export for further analysis in the R environment. Neighborhoods were analyzed in R using a modified neighborhood analysis workflow developed previously [28]. Neighborhoods were first preprocessed to exclude monotypic neighborhoods (one cell type) and then combined across patient samples. Using the total number of cell-types in individual neighborhoods, both Louvain-clustering and projections into
2.8Statistical analyses
Statistical analyses were performed in R and Excel, with
2.9Software and datasets
We have made the registered image sets from these analyses as well as the outputs from HALO following the Z-normalization publicly available at LABCAS (https://doi.org/10.26252/s18j-1h24). Our registration software and RFA are available on GitHub (https:// github.com/EDRN/slide-image-registration/releases/tag /v1.0.0 and https://github.com/EDRN/random-decision-forests/releases/tag/v1.0.0). The neighborhood analysis software is available on GitHub (https://github.com/icbm-iupui/volumetric-tissue-exploration-analysis). R Code for figure generation and analysis is available upon request.
3.Results
3.1Image alignment
Image alignment accounts for slight alterations in slide position and scanning areas that occur when WSI is used over iterative staining rounds. Without correction, image misalignments could confound spatial information and co-localization data for multiplexed datasets. Figure 1C–E shows a successfully aligned area in comparison to pre-alignment images from the first and second round of staining, as well as an example area of high marker intensity post alignment (only subset of markers shown). When comparing segmented areas in the first round to high-fidelity nuclei/cells present throughout the whole staining cycle, we observed an average reduction of 48%. Based on visual inspection this decrease does not appear to be caused by tissue damage or alterations to the Z-depth while scanning but due to over-segmentation in the first round which fails to pass the requirements of high-fidelity cells (nuclear co-localization). There were no significant differences in spurious segmentations between the tumor and stromal tissue (data not shown).
3.2Validation of random forest algorithm
We developed a RFA to identify markers which define the tumor and stroma regions of the TME. Briefly, once slides are processed via MxIF analysis, images are divided into tumor or stromal rich regions (normal tissue and mixed excluded) and the RFA is trained on the presence or absence of markers in the tumor and stromal regions on all high-fidelity cells (Fig. 2). 70% of the cells within the sample were used for training, the other 30% were used for validation. The algorithm was cross-validated with a random selection of the 70:30 split each time to determine accuracy. This was done ten times per run. We tested our algorithm to both exclude and include MUC1, Ki67 and EPCAM, expected to be expressed on tumor cells and therefore an unnecessary bias/factor when seeking novel tumor, stroma predictors.
Figure 2.
Overview of the cyclic MxIF, WSI, and image analysis pipeline. After MxIF processing and pathological validation, images are aligned and classified for tumor-stromal content. Tumor and stromal areas were quantified separately and trained via the RFA. A limited branching decision tree is shown.
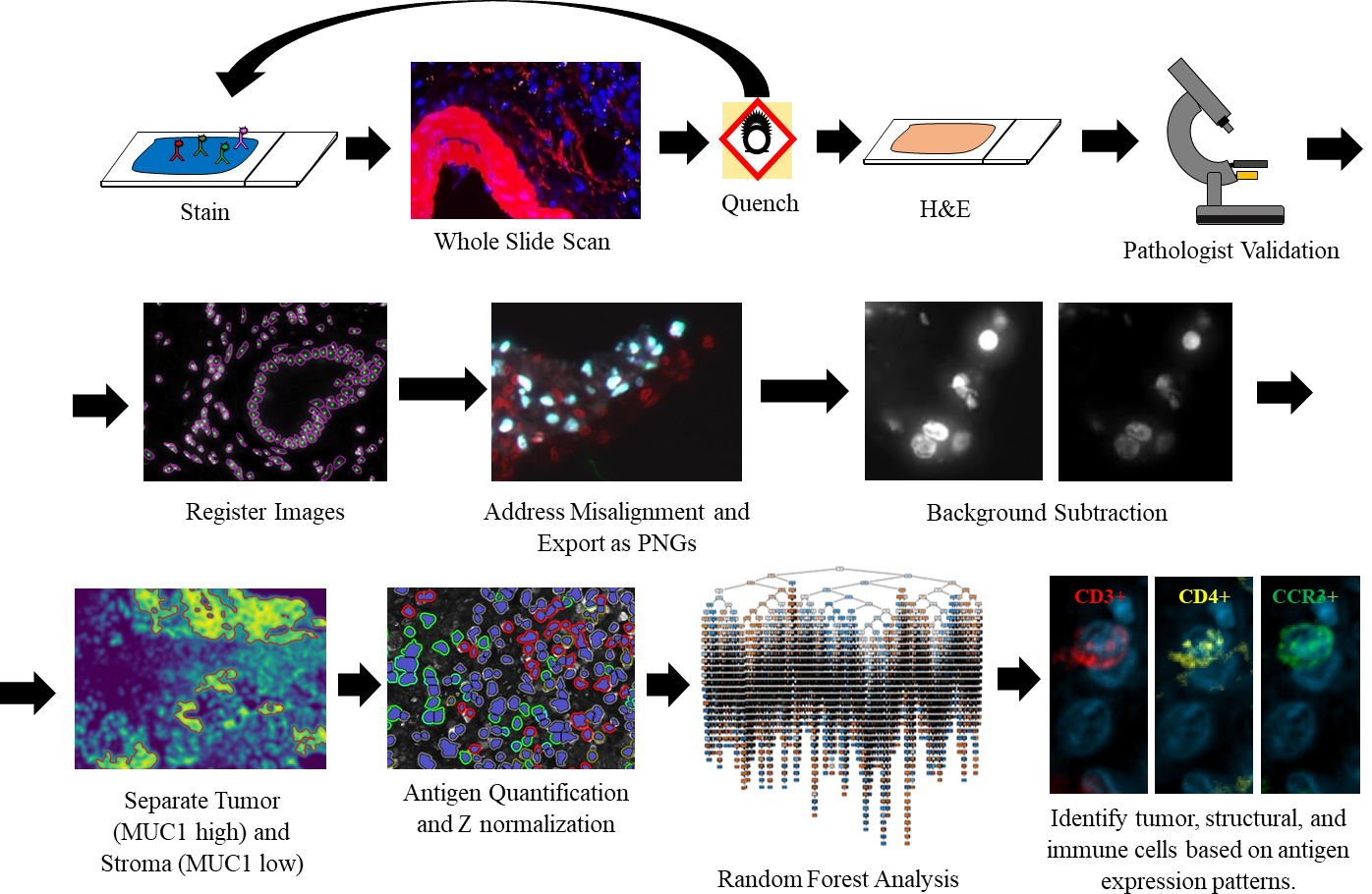
Among individual samples, we had very high accuracy of prediction (average 90.0%
Figure 3.
Random forest outputs showing contributions of prediction driving markers in all, and those unique to representative long- and short-term survivor PDACs. A) Top 15 tumor-stroma predictors in all PDACs, excluding known cancer markers. B) Top 15 tumor-stroma predictors for long-term survivor (RAP 116), excluding cancer markers. C) Top 15 tumor-stroma predictors in short-term survivor (RAP 80), excluding cancer markers. D) Average marker rankings following jackknife resampling for top 15 markers.
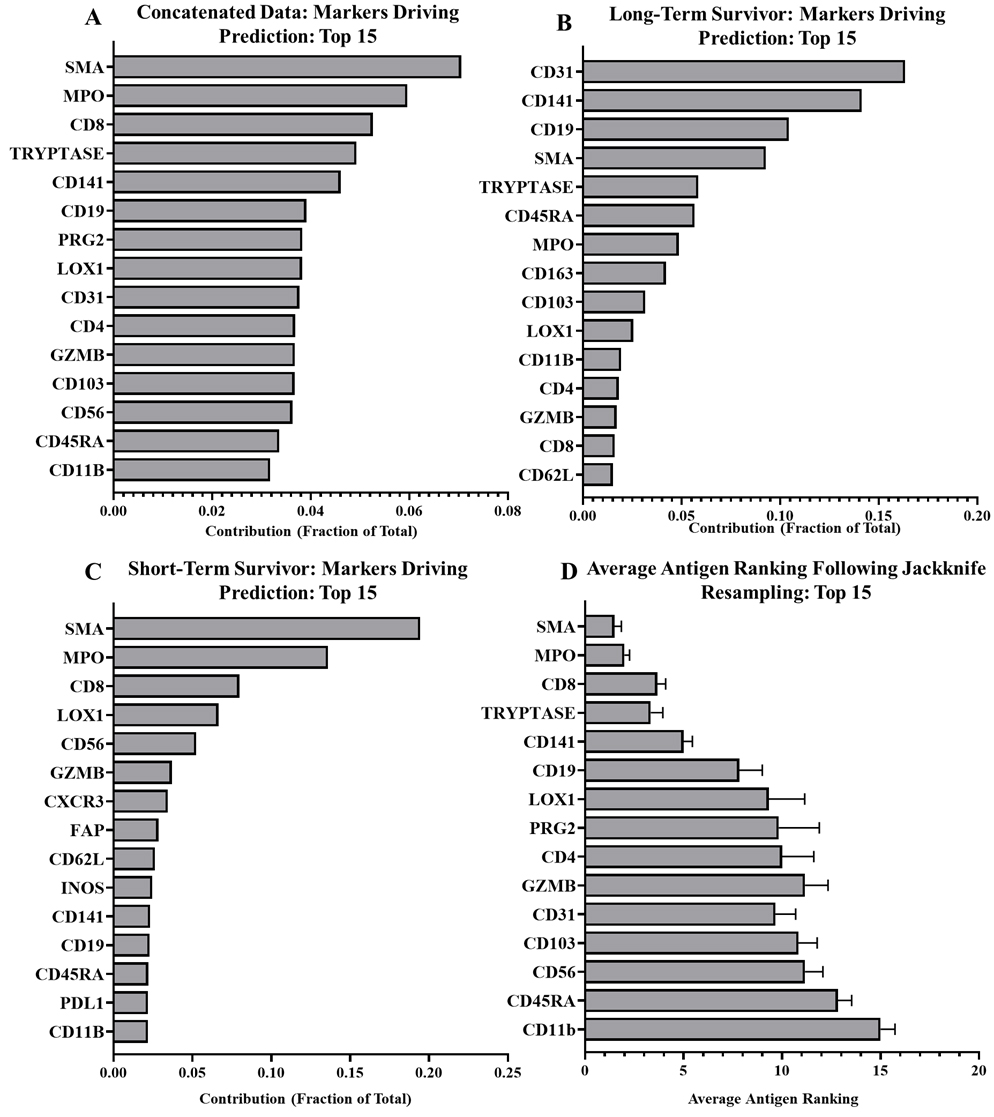
We additionally validated our algorithm by removing individual samples and repeating the RFA to determine if outlier expression was skewing the data set (jackknife resampling). While there were minor fluctuations, markers rarely shifted more than one to two positions in ranking following individual sample removal (Fig. 3D). This suggests the major drivers are particularly stable. Following this jackknife resampling we tested the RFAs produced against the sample that was removed and observed very minimal alterations to the efficacy of the RFA compared to the concatenated data (88.1%
3.3Random forest outputs
Using our RFA, we analyzed six primary pancreatic tumor samples to determine which markers were highly predictive of tumor and stromal regions of the TME. Among all our samples the strongest predictors were smooth muscle actin (SMA), myeloperoxidase (MPO), CD8, Mast Cell Tryptase, and CD141 (Fig. 3A). While we did see remarkable consistency within the marker ranking when jackknife resampling was applied to our patient cohort (Fig. 3D), there is variability between patients. Figure 3 shows the antigen rankings for a long-term and short-term survivor. In the long-term survivor (Fig. 3B), three antigens, CD31, CD141, and CD19 account for 50% of the drivers of prediction. The short-term survivor, matched for gender, age, and stage at diagnosis, presented with a different pattern of tumor-stroma drivers. SMA, MPO, and CD8 were among the highest predictors (Fig. 3C). Compared to the long-term survivor, CD19 (2.2%) and CD31 (1.9%) accounted for a very small prediction portion in the short-term survivor, suggesting patient-specific differences that may be related to disease aggressiveness. Looking at top drivers across patients though, clear consistent patterns emerge with Tryptase and SMA appearing among the top five drivers for five samples, MPO appearing in four, and CD8 and CD141 appearing in three. CD31 appeared twice among the top five drivers while CD56, LOX1, CD19, CD4, CD3, PDL1, CD103 and PRG2 appeared once (Supplementary Table 3).
Figure 4.
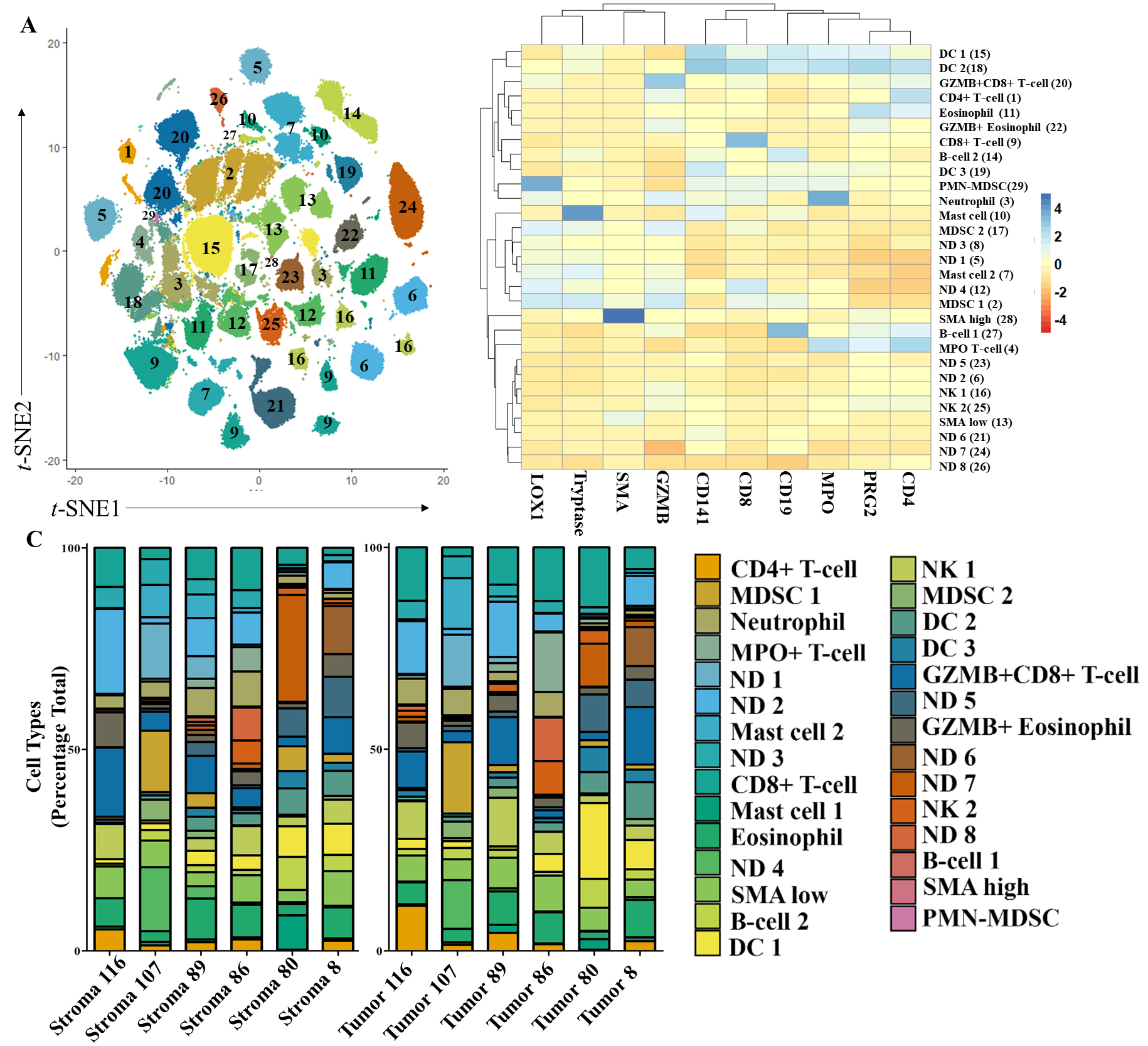
Distribution of tumor-associated immune cells identified in PDAC TMEs using top ten tumor-predicting markers and unsupervised clustering. A)
3.4Identification of tumor- and stroma-specific cell types
To identify discrete cell populations present in tumor- and stroma-rich regions, we performed Louvain clustering using the top ten tumor-and stroma-driving antigens derived from the RFA. While CD31 was in the top ten driving antigens, we determined that more compelling cellular subtypes would be identified using GZMB which had a slightly lower score (0.0009 difference). Louvain clustering with GZMB grouped our dataset into 29 different clusters (Fig. 4A). Using antigen co-expression patterns on the heatmap (Fig. 4B) we were able to identify 21 different cell types in these clusters. Ten driver markers were sufficient for cell identification and allowed us to classify major immune cell subtypes in the TME. We observed expression patterns indicative of most major granulocytes, eosinophils (PRG2+), neutrophils (MPO+), and mast cells (Tryptase), as well as granulocytic-like MDSCs (MPO+, LOX1+, PMN-MDSCs). Of the adaptive immune response regulating cells, we identified clusters of B cells (CD19+) and CD4+ and CD8+ T cells. We also observed high and low levels of SMA expression. While SMA can indicate multiple structural components, high and low expression levels may additionally indicate inflammatory and myofibroblastic cancer associated fibroblasts, respectively [29].
By identifying 21 out 29 groups we were able to account for an average of 72% of cells within the TME. As our classification did not include cancer cells, this represents a sizable portion of cells in the TME. We did not observe significant differences in the number of identified cells between tumor and stroma regions, though a strong trend towards characterization of more cells within the tumor-rich regions was observed (data not shown). Notably, with the exception of the B cell cluster (group 27), all clusters were present all patients (Fig. 4C). While we observed strong trends in CD8+ T cell expression (group 9) increasing in the tumor-rich regions (
Figure 5.
Neighborhood analysis of all samples. A)
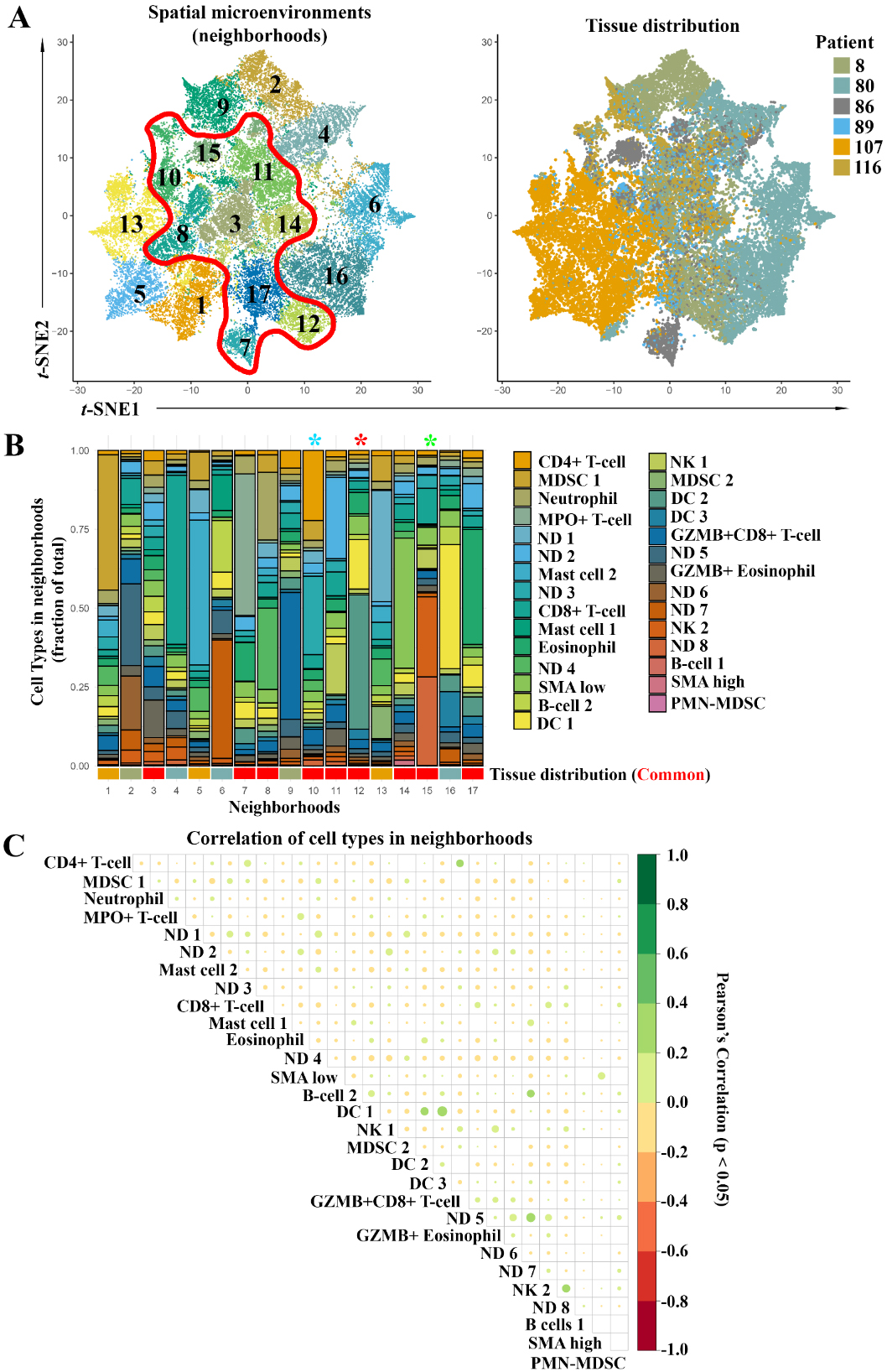
3.5Neighborhood analyses of cells in tumor- and stroma-rich regions
Based on the cell groups identified via Louvain clustering, we performed a neighborhood analysis on all cells within the tissue. We defined neighborhoods cell-wise as cells within 30
Next, we performed a correlation analysis for cell types within our neighborhoods and observed many significant correlations (Fig. 5C). Only significant correlations (
Figure 6.
Sub-Gating of CD4+ and CD8+ T Cells, DCs, and NK cells. A) CD8+ T cell sub-gating using Louvain clustering (
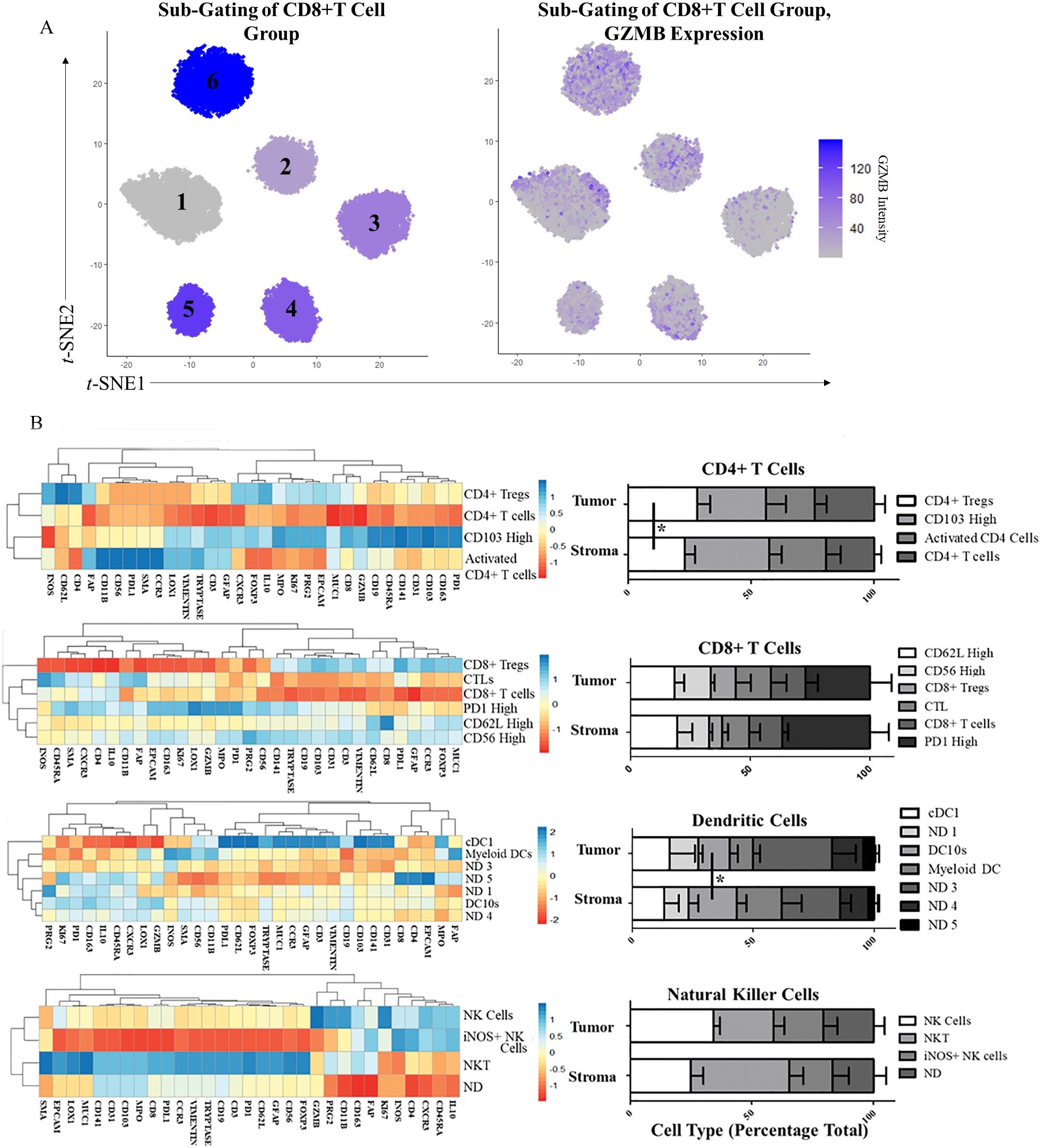
3.6Sub-classification of neighborhood-defining cell types
Following neighborhood classification of our samples, we identified cell types defining features of the neighborhoods that were consistent across all samples. We chose to further investigate CD4+ T cell populations, CD8+ T cell populations, and the DC 2 and NK 2 cell populations, as they made up the largest portion of the DC and cytotoxic neighborhoods, respectively. We repeated the Louvain clustering of these groups independently and were able to identify multiple cell types within each cluster (Example in Fig. 6A). Clustering with only the top ten antigens was sufficient for patterns to emerge from additional antigens uninvolved in the clustering and indicative of other unique cell types.
The CD4+ T cell Louvain clustering identified four groups (Fig. 6B). Using a heatmap of all antigens, we identified FOXP3 high CD4+ Tregs, GZMB high Cytotoxic CD4+ T cells, CD103 high CD4+ T cells, and CD4+ T cells which did not express activation or inactivation factors. We observed a significant increase in CD4+ Tregs in the tumor-rich regions (
3.7Cell type differences correlate with short- and long-term survival
Within our sample cohort we noted patient 116 was a uniquely long-term survivor at 497 days post diagnosis. All other untreated patients diagnosed at stage IV in our RAP cohort survived on average 96 days post diagnosis. We explored the cellular makeup of tumor and stromal regions from this long-term survivor (RAP 116) and compared those finding to a female patient of a similar age (RAP 80), also diagnosed in stage IV, who survived only seven days post diagnosis.
Figure 7.
Differential expression of tumor-associated immune cells in short-term (ST) and long-term (LT) PDAC survivors. Tumor- and stroma-specific cells samples sub-gated for DC 2 (A), CD4+ T cells (B), CD8+ T cells (C), and NK 2 cells (D).
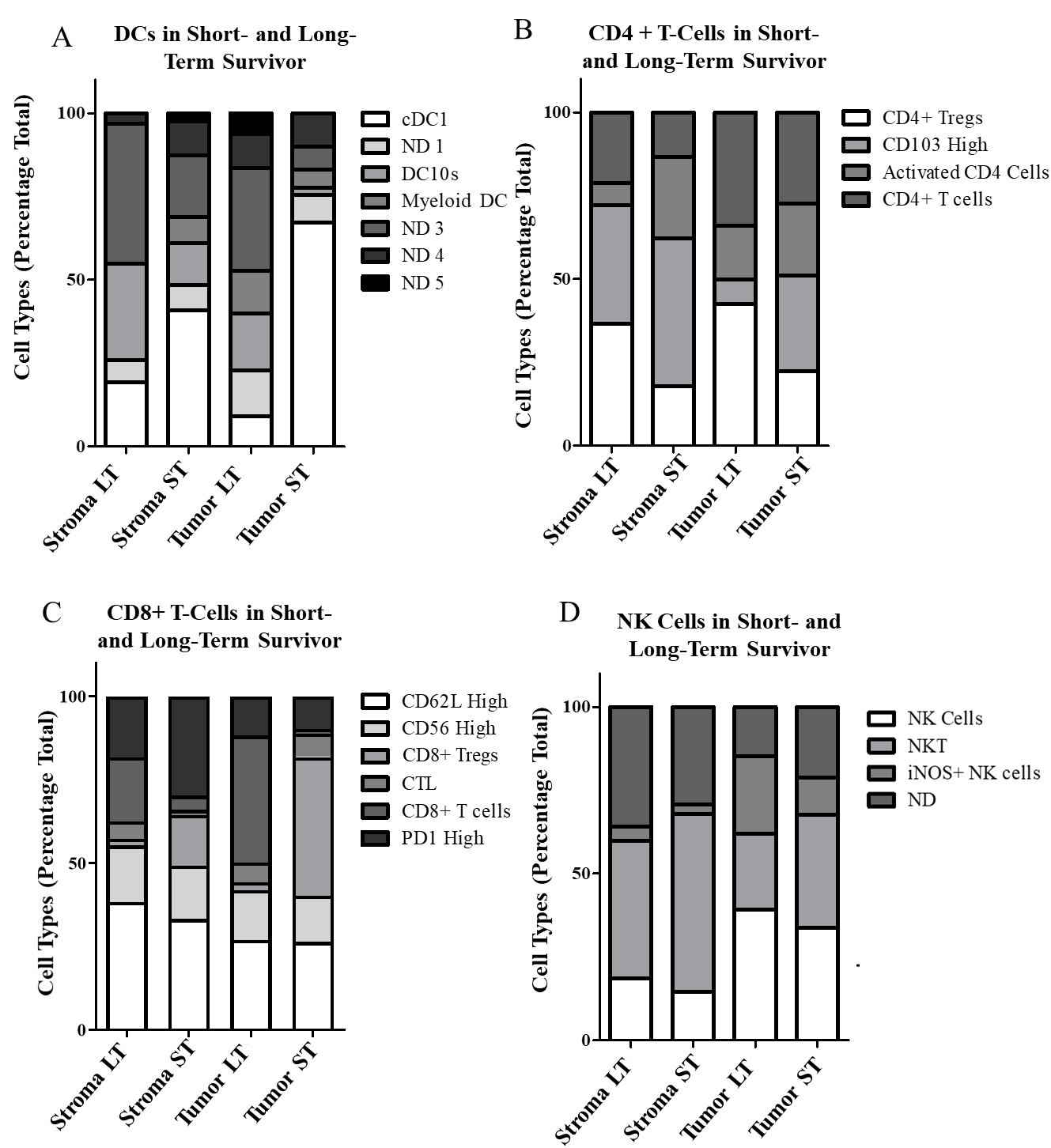
In the DC group, the long-term survivor had the lowest overall levels while patient 80 and 8, another particularly short-term survivor, had the highest levels (Fig. 4B). When sub-gated, most DC high cells in the short-term survivor were identified as cDC1s, while the long-term survivor had higher levels of myeloid like DCs and DC10s (Fig. 7A). The long-term survivor also had the highest levels of CD4+ cells of all patients. Surprisingly, the long-term survivor’s CD4+ cells were predominantly regulatory T cells. This is in contrast to the short-term survivor which had fewer regulatory T cells and higher levels of CD103 high T cells (Fig. 7B). Among CD8+ T cells, the short-term survivor had the highest level in the tumor-rich region but comparatively low levels in the stroma. When we looked at the tumor to stroma ratio of CD8+ expression, short-term survivors (RAP 8, 80), had the lowest ratio, while the long-term survivor was among the highest (Fig. 7C). Finally looking at our NK 2 cell sub-gating it is noteworthy that we saw the highest levels of NK cells within the tumor in the long-term survivor (Fig. 7D).
Looking at the overarching cell groups identified, we observed two B cell groups with an inverse relationships in long- and short-term survivors. In the B cell 1 group the long-term survivor had the largest population while they were conspicuously absent in the short-term survivor. In contrast, the short-term survivor (RAP 80) had the largest population of the B cell 2 group while the short-term survivor (RAP 116) had the smallest (Fig. 4B). Noteworthy from the correlation matrix (Fig. 4C) of the neighborhoods is that the B cell 2 group had a mostly positive correlation with DC cell clusters while the B cell 1 groups had a negative correlation. The CD8+ GZMB+ high clusters also made up a very large portion of the long-term survivor’s TME cells (Fig. 4B). There were no noteworthy features of granulocytic expression in the long-term survivor, though the short-term survivor had the highest level of mast cells of all patients. SMA expression was comparable across all patients.
To better characterize the spatial relationship of the cells we plotted the neighborhoods for all samples (Supplementary Fig. 2). We observed that the largest neighborhoods present in the long-term survivor (RAP 116) were N12, N8, and N7. In Fig. 5B, we can see these neighborhoods characterized as DC rich, granulocytic rich, and neutrophil rich, respectively. Interestingly, the short-term survivor (RAP 80) also has a very pronounced DC rich neighborhoods (N16 and N6) but this DC neighborhood is composed of different clusters of DC cells than those of the long-term survivor’s N12 neighborhood. It is also noteworthy that neighborhoods N16 and N6 have large populations of B cell 2 groups present. The short-term survival patient also had large CD8+ T cell rich neighborhoods (N4). This neighborhood is mostly populated by CD8+ T Cells, with very small populations of CD4+ T cells, NK cells, or B cells in the vicinity which could help orchestrate an adaptive immune response. Together this suggests less of a cytotoxic neighborhood and more of sequestered CD8+ T Cell neighborhood.
4.Discussion
Iterative MxIF and WSI pose exciting new opportunities to delve deeply into the identity and spatial distributions of individual regulatory cells throughout normal and diseased tissues. This type of insight is particularly important for understanding intricate, highly-regulated, and highly-regulatory cellular interactions in discrete regions of complex TMEs. Achieving such understanding also generates an interesting challenge – an overabundance of data. This requires careful handling to ensure accurate analyses and interpretation.
Within our focused dataset we iteratively stained 6 primary tumors from untreated PDAC patients with 31 different markers for immune, stromal, and tumor components. We were able to align the massive image output using a registration software developed herein. Once aligned, we identified high-fidelity cells and quantified antigens on more than a quarter-million tumor or stromal cells. The current studies used HALO (proprietary), though similar analysis can be done using open-source alternatives including ImageJ and CellProfiler [32]. Furthermore, the open-source space for this type of analysis continues to develop. Once antigens were quantified, we developed and used an RFA to focus on markers highly predictive of tumor and stromal TME compartments. Therefore, we were able to determine which cells were effective at passing through the dense stromal network into the tumor to directly interact with cancer cells. Notably, we are to perform this identification without traditional cancer markers.
By choosing to focus on the end-stage patients, as opposed to samples collected from resection, we were able to characterize the tumor at the stage where it became lethal to the patient. While our research team continues to expand our RAPs for pancreatic cancer, untreated patients’ numbers remain relatively low within our program (
Using top tumor-stroma markers, we were able to identify most cells in both TME regions. With further sub-gating we were able to refine the identities of major subsets of prominent cell types across samples. Using only our set of top ten antigens we were able to observe consistent expression patterns for the other 21 antigens in our cohort, some of which indicated specific cell types. This implies some combination of marker intensity variabilities within our top ten cohort are predictive of additional cell types that they are not typically associated with. Future studies will determine if these markers can be used in a simplified labeling panel to robustly characterize the immune cells.
Using sub-gating, we identified multiple, distinct immunosuppressive cell types present at variable levels in the tumor and stroma. Tolerogenic DC10s were significantly increased in the stroma while higher levels of CD4+ Tregs were observed in tumoral regions. DC10s have been shown to activate CD4+ Tregs. Unlike, other DC types requiring direct physical cross-presentation to activate an immune response, DC10s activate Tregs via IL-10 cytokine signaling [33]. So, while we observed a negative spatial correlation between DCs and T cells in our neighborhood analysis, DC10s could still be interacting with Tregs via cytokine signaling. cDC1s are adept at cross presentation and tumor rejection but cross-presentation requires physical interactions with other cell types, while DC10s can cause tolerogenic effects through distant signaling [34]. This could be an avenue of immune suppression in tumors heavily invaded by DCs.
The long-term survivor showed the highest proportion of tumor infiltrating DC10s concomitant with high levels for Tregs. It is possible that the length of survival itself is what causes this immunosuppressive phenotype while the short-term survivors did not have time to develop robust immunosuppression. However, while CD4+ Tregs are well-established as negative indicators for patient survival, the involvement of DC10s in the TME has not been well studied [4]. Though DC10s are known to be tolerogenic and researchers have shown circulating DCs to be correlated to the stage of diagnosis in gastric cancers, similar studies have not been done in the tumor of PDAC patients to determine how they may impact cancer development [35].
Use of a RFA to determine pertinent markers for further analyses within a MxIF dataset is not limited to identifying distinguishing factors between the tumor and stroma. With the right training, we can further examine the makeup of the tumor core vs. the leading edge, treated vs. untreated patients, or expand investigations to include a larger subset of long-term vs. short-term survivors or treatment conditions. Many of these questions can be queried within the same tissue sample. One strength of our autopsy program is that samples from multiple primary, metastatic, and unaffected areas in each patient can be used to query organ specificity, tumor heterogeneity, and endless additional factors. While our current version of the RFA is limited to average cell intensities, future versions can extend analyses to look at cell size, shape, and/or other quantifiable morphological features. Looking at other features acquired through the MxIF process, as well as a larger marker set using the methods demonstrated here could significantly increase the data depth obtained from individual samples. Thus, as shown in this paper, the application of comprehensive MxIF labeling and WSI in conjunction with artificial intelligence and machine learning, will enable researchers to use a guided discovery approach to successfully manage and analyze massive immunoprofiling datasets and characterize key regulators in PDAC and other cancers.
Author contributions
Conception: KV, MAH
Interpretation or analysis of data: KV, AA, HJS, BJS, SW
Preparation of the manuscript: KV, AA, HJS, SW
Revision for important intellectual content: HJS, MAH, DC
Supervision: MAH, HJS, DC
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-210308.
Acknowledgments
All samples collected were from consented patients through the University of Nebraska Medical Center Rapid Autopsy Program (supported by P50CA127297, U01CA210240, P30CA36727, and R50CA211462). We would like to thank UNMCs tissue science facilities for sample preparation, the patients and family members who agreed to participate in our RAP program, and the RAP volunteers who provided countless hours of work collecting these tissues.
References
[1] | B.J. Kenner, S.T. Chari, A. Maitra et al., Early detection of pancreatic cancer-a defined future using lessons from other cancers: a white paper, Pancreas 45: ((2016) ), 1073–1079. |
[2] | Surveillance, Epidemiology, and End Results (SEER) Program Populations (1969–2018) [Internet]. National Cancer Institute, DCCPS, Surveillance Research Program. released December (2019) . |
[3] | X. Liu, J. Xu, B. Zhang et al., The reciprocal regulation between host tissue and immune cells in pancreatic ductal adenocarcinoma: new insights and therapeutic implications, Mol Cancer 18: ((2019) ), 184. |
[4] | M. Huber, C.U. Brehm, T.M. Gress et al., The immune microenvironment in pancreatic cancer, Int J Mol Sci 21: ((2020) ). |
[5] | A.N. Hosein, R.A. Brekken and A. Maitra, Pancreatic cancer stroma: an update on therapeutic targeting strategies, Nat Rev Gastroenterol Hepatol 17: ((2020) ), 487–505. |
[6] | S. Barua, P. Fang, A. Sharma et al., Spatial interaction of tumor cells and regulatory T cells correlates with survival in non-small cell lung cancer, Lung Cancer 117: ((2018) ), 73–79. |
[7] | Y. Masugi, T. Abe, A. Ueno et al., Characterization of spatial distribution of tumor-infiltrating CD8(+) T cells refines their prognostic utility for pancreatic cancer survival, Mod Pathol 32: ((2019) ), 1495–1507. |
[8] | J.L. Carstens, P. Correa de Sampaio, D. Yang et al., Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer, Nat Commun 8: ((2017) ), 15095. |
[9] | J. Watt and H.M. Kocher, The desmoplastic stroma of pancreatic cancer is a barrier to immune cell infiltration, Oncoimmunology 2: ((2013) ), e26788. |
[10] | J.R. Lin, B. Izar, S. Wang et al., Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes, Elife 7: ((2018) ). |
[11] | X. Wang, M. Lang, T. Zhao et al., Cancer-FOXP3 directly activated CCL5 to recruit FOXP3(+)Treg cells in pancreatic ductal adenocarcinoma, Oncogene 36: ((2017) ), 3048–3058. |
[12] | A.E. Prizment, R.A. Vierkant, T.C. Smyrk et al., Cytotoxic T Cells and Granzyme B Associated with Improved Colorectal Cancer Survival in a Prospective Cohort of Older Women, Cancer Epidemiol Biomarkers Prev 26: ((2017) ), 622–631. |
[13] | H.H. Van Acker, A. Capsomidis, E.L. Smits and V.F. Van Tendeloo, CD56 in the Immune System: More Than a Marker for Cytotoxicity? Front Immunol 8: ((2017) ), 892. |
[14] | E.I. Buchbinder and A. Desai, CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition, Am J Clin Oncol 39: ((2016) ), 98–106. |
[15] | J. Hang, J. Huang, S. Zhou et al., The clinical implication of CD45RA(+) naive T cells and CD45RO(+) memory T cells in advanced pancreatic cancer: a proxy for tumor biology and outcome prediction, Cancer Med 8: ((2019) ), 1326–1335. |
[16] | D.L. Woodland and J.E. Kohlmeier, Migration, maintenance and recall of memory T cells in peripheral tissues, Nat Rev Immunol 9: ((2009) ), 153–161. |
[17] | M.J. Gerdes, C.J. Sevinsky, A. Sood et al., Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue, Proc Natl Acad Sci USA 110: ((2013) ), 11982–11987. |
[18] | L. Breiman, Random Forests, Machine Learning 45: (1) ((2001) ), 5–32. |
[19] | F.V.G. Pedregosa, A. Gramfort, V. Michel et al., Scikit-learn: Machine Learning in Python., J Mach Learn Res 12: ((2011) ), 2825–30. |
[20] | M. Guizar-Sicairos, S.T. Thurman and J.R. Fienup, Efficient subpixel image registration algorithms, Opt Lett 33: ((2008) ), 156–158. |
[21] | S. van der Walt, J.L. Schonberger, J. Nunez-Iglesias et al., scikit-image: image processing in Python, PeerJ 2: ((2014) ), e453. |
[22] | J.W. Hickey, Y. Tan, G.P. Nolan and Y. Goltsev, Strategies for accurate cell type identification in CODEX multiplexed imaging data, Front Immunol 12: ((2021) ), 727626. |
[23] | T.R.T. Hastie and J. Friedman, The Elements of Statistical Learning, 2 ed, Springer, New York, (2009) . |
[24] | H.W. Jackson, J.R. Fischer, V.R.T. Zanotelli et al., The single-cell pathology landscape of breast cancer, Nature 578: ((2020) ), 615–620. |
[25] | T.E. Bakken, N.L. Jorstad, Q. Hu et al., Comparative cellular analysis of motor cortex in human, marmoset and mouse, Nature 598: ((2021) ), 111–119. |
[26] | M.J. Ferkowicz, S. Winfree, A.R. Sabo et al., Large-scale, three-dimensional tissue cytometry of the human kidney: A complete and accessible pipeline, Lab Invest 101: (5) ((2021) ), 661–76. |
[27] | S. Winfree, S. Khan, R. Micanovic et al., Quantitative three-dimensional tissue cytometry to study kidney tissue and resident immune cells, J Am Soc Nephrol 28: ((2017) ), 2108–2118. |
[28] | B.B. Lake, R. Menon, S. Winfree et al., An atlas of healthy and injured cell states and niches in the human kidney, bioRxiv ((2021) ), 20212007.2028.454201.. |
[29] | C. Han, T. Liu and R. Yin, Biomarkers for cancer-associated fibroblasts, Biomark Res 8: ((2020) ), 64. |
[30] | M. Collin and V. Bigley, Human dendritic cell subsets: an update, Immunology 154: ((2018) ), 3–20. |
[31] | M. Comi, D. Avancini, F. Santoni de Sio et al., Coexpression of CD163 and CD141 identifies human circulating IL-10-producing dendritic cells (DC-10), Cell Mol Immunol 17: ((2020) ), 95–107. |
[32] | Z. Du, J.R. Lin, R. Rashid et al., Qualifying antibodies for image-based immune profiling and multiplexed tissue imaging, Nat Protoc 14: ((2019) ), 2900–2930. |
[33] | M. Comi, G. Amodio and S. Gregori, Interleukin-10-Producing DC-10 Is a Unique Tool to Promote Tolerance Via Antigen-Specific T Regulatory Type 1 Cells, Front Immunol 9: ((2018) ), 682. |
[34] | J.P. Bottcher and C. Reis e Sousa, The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity, Trends Cancer 4: ((2018) ), 784–792. |
[35] | D.P. Xu, W.W. Shi, T.T. Zhang et al., Elevation of HLA-G-expressing DC-10 cells in patients with gastric cancer, Hum Immunol 77: ((2016) ), 800–804. |


