
Golgi phosphoprotein 3 (GOLPH3) promotes endometrial carcinoma cell invasion and migration by regulating the epithelial-mesenchymal transition
Abstract
BACKGROUND:
Golgi phosphoprotein 3 (GOLPH3) is a novel oncogene overexpressed in several human cancers, but specific contributions to endometrial carcinoma (EC) have not been examined. The aims of this study were to evaluate the GOLPH3 expression in EC and investigate its functions in EC cell proliferation, migration, and survival.
METHODS:
The expression levels of GOLPH3 in EC patient samples and EC cell lines (HEC-1A, KLE, RL95-2, and Ishikawa) were examined using qRT-PCR, western blotting and immunohistochemistry. Further, EC cell lines with either ectopic GOLPH3 overexpression or knockdown were established, and the effects on proliferation, apoptosis, invasion, and migration were investigated in vitro using cell viability and transwell assays and in mice following cell injection.
RESULTS:
Compared to adjacent non-cancerous tissues, expression of GOLPH3 was significantly upregulated in EC tissues (
CONCLUSIONS:
The present study provides evidence that GOLPH3 promotes EMT and metastasis of EC cells and predicts the risk of EC progression, highlighting its potential as a therapeutic target for this malignancy.
1.Introduction
Endometrial carcinoma (EC) is the most common malignancy of the female reproductive tract in developed countries [1]. In recent years, EC incidence has increased worldwide, posing a further threat to female health. In many cases the prognosis is satisfying, but 22% of affected women die from the disease. While most cases are diagnosed in the early stages, nearly a third present with regional or distant metastasis. Moreover, despite recent therapeutic advances, metastasizing EC still has a low 5-year survival rate and a high rate of recurrence [2]. The elucidation of molecular pathways underlying tumorigenesis and progression of EC is thus critical for the identification of prognostic factors and the development of diagnostic tests and targeted molecular therapies [3, 4].
Golgi phosphoprotein 3 (GOLPH3) is a highly conserved 34-kDa trans-Golgi matrix membrane protein involved in receptor recycling, glycosylation, and protein trafficking [5, 6, 7]. Recently, GOLPH3 expression was found to correlate with clinical stage, growth, survival, and metastasis of several cancers, including small cell lung cancer [8], breast cancer [7, 9], ovarian epithelial cancer [10], oral tongue cancer [11], liver cancer [12], rectal cancer [13], prostate cancer [14], bladder cancer [15], esophageal squamous cell carcinoma [16], gastric cancer [17, 18], and kidney cancer [19]. It is suggested that GOLPH3 functions as an oncogene in tumorigenesis and migration by activating the mechanistic target of rapamycin (mTOR), Wnt, and nuclear factor
The present study examined the expression of GOLPH3 in tissue samples of EC patients and EC cell lines. We then investigated the possible roles of GOLPH3 in EC cell proliferation, apoptosis, invasion, migration, and epithelial-mesenchymal transition (EMT) by manipulating its expression in EC cell lines in vitro and in animal models.
2.Materials and methods
2.1Design of the study
The expression level of GOLPH3 in EC tissues and EC cell lines were detected by qRT-PCR, western blot and immunohistochemistry to investigate whether GOLPH3 involve in EC development. And In order to analyze the possible roles of GOLPH3 in EC cells, GOLPH3 shRNA and pcDNA3.1 (+)-GOLPH3 plasmid were constructed and transfected into KLE EC cell line and Ishikawa cell line, respectively, the effects of GOLPH3 on EC cell proliferation (by using CCK-8 cell viability assay), migration and invasion (by using transwell assays and Xenograft Tumor model assay) and apoptosis (by using Annexin V FITC apoptosis detection assays) were examined. The levels of EMT-related genes in overexpression and knockdown EC cells were detected by qRT-PCR and western blot to elucidate the mechanism of GOLPH3 involvement in cell migration and invasion.
2.2Patient information and tissue specimens
Thirty EC tissue samples and adjacent non-cancer- ous tissues were randomly collected from EC patients who underwent curative resection from 1/2014 to 12/2015 at the Second Affiliated Hospital of Army Medical University. And endometrial tissue specimen employed in this study was obtained from an endometriotic patient who had no history of endometrial hyperplasia or neoplasia, and had not received any anti-inflammatory or hormonal medications during a period of at least three months before surgery. Study protocols were reviewed and approved by the Ethics Committee of the Second Affiliated Hospital of Army Medical University and conducted according to the principles expressed in the Declaration of Helsinki. Informed written consent was provided by both patients and families before resection surgery.
2.3Tissue chip
The endometrial carcinoma tissue chip was purchased from Shanghai Outdo Biotech Co. Ltd., with the CGT number HUteA060CS01 and the lot number XT15-033. The chip contained 34 endometrial carcinoma tissues and 26 normal endometrial tissues derived from 34 cases, which were fixed in 60 pores.
2.4Chemicals and antibodies
Lipofectamine transfection (Cat. No. L3000-015) and TRIzol (Cat. No. 15596026) reagents were purchased from Life Technologies (Grand Island, NY, USA). Anti-GOLPH3 antibody (Cat. No. ab82377, 1:100) and an Annexin V FITC apoptosis detection kit (Cat. No. ab14085) were purchased from Abcam (Cambridge, MA, USA). Peroxidase-labeled anti-rabbit IgG (H+L) antibody (Cat. No. A0208) was purchased from Beyotime (Shanghai, China). Antibodies against N-cadherin (Cat. No. 4061, 1:500), vimentin (Cat. No. 3932, 1:500), E-cadherin (Cat. No. 3195, 1:500),
2.5Cell lines and cell culture
The human EC cell lines HEC-1A, KLE, RL95-2, and Ishikawa were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). Endometrial stromal cells (ESC) were obtained from the endometrial tissue specimen, Which taken from an endometriotic patient and separated as described by Hou et al. [23]. Cells were cultured at 37
2.6Vectors, retroviral infection, and transfection
For ectopic overexpression, a GOLPH3 expression construct was generated by subcloning PCR-amplified full-length human GOLPH3 cDNA into the pcDNA3.1 (+) plasmid, the empty vector was used as a negative control. For GOLPH3 knockdown, human GOLPH3-targeting short hairpin RNA (shRNA) oligonucleotide sequences were cloned into the pSuper-retro-puro vector. The shRNA sequences were RNAi-1 5’-GCAGCGCCTCATCAAGAAAGT-3’ and RNAi-2 5’-GCATGTTAAGGAAACTCAGCC-3’, a scrambled shRNA (shCtrl) was used as a negative control, the sequence of the scrambled shRNA was 5’-TTCTCCGAACGTGTCACGTTT-3’ (synthesized by Invitrogen). Transfection of plasmids was performed using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, and the transfection efficiency was evaluated by qRT-PCR and Western blotting.
2.7RT-PCR and qRT-PCR
Cells were dissociated with 0.25% trypsin (Invitrogen) and collected for reverse transcription polymerase chained reaction (RT-PCR). Total RNA was isolated using TRIzol reagent (Invitrogen) and cDNA was synthesized using SuperScript II reverse transcriptase. Primers were synthesized by Invitrogen with the assistance of Primer Premier 5.0 (PREMIER Biosoft International, Palo Alto, CA, USA). Sequences used in this study are listed in Table 1. GAPDH was used as the internal control. Quantitative (q)RT-PCR and data collection were performed using an ABI PRISM 7900HT sequence detection system and expression levels analyzed using the comparative threshold cycle (2
Table 1
Primer sequences used for qRT-PCR
| Primer | Sequence (5’-3’) |
|---|---|
| GOLPH3-Forward | GGG CGA CTC CAA GGA AAC |
| GOLPH3-Reverse | CAG CCA CGT AAT CCA GAT GAT |
| Ki67-Forward | CCA CGA GAC GCC TGG TTA CTA T |
| Ki67-Reverse | GCC TCC TGC TCA TGG ATT TCA A |
| N-cadherin-Forward | CCG GTT TCA TTT GAG GGC ACA TGC |
| N-cadherin-Reverse | GCC GTG GCT GTG TTT GAA AGG C |
| Vimentin-Forward | AAC TTA GGG GCG CTC TTG TC |
| Vimentin-Reverse | GGT GGA CGT AGT CAC GTA GC |
| E-cadherin-Forward | TGG GCT GGA CCG AGA GAG TTT C |
| E-cadherin-Reverse | ATC CAG CAC ATC CAC GGT GAC G |
| TCA TTG TGG ACC CCT TGA GC | |
| TTA CGT CCA GCA TTG CCC AT | |
| GAPDH-Forward | GCT GGC GCT GAG TAC GTC GT |
| GAPDH-Reverse | ACG TTG GCA GTG GGG ACA CG |
2.8Western blotting
Cells were washed and treated with radio-immun- oprecipitation assay (RIPA) lysis buffer. Cell lysates were centrifuged and the supernatant proteins mixed with loading buffer and boiled for 10 min. Protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto polyvinylidene difluoride (PVDF) membranes. After blocking in non-fat milk for 1 h at room temperature, the membranes were incubated with primary antibodies for 12 h at 4
2.9Immunohistochemistry
For immunohistochemistry (IHC), the paraffin tissue chip was dehydrated in PBS and underwent antigen retrieval in sodium citrate buffer (10 mM, 0.05% Tween 20, pH 6.0). Then the tissue chip was blocked in 10% normal serum with 1%BSA in TBS for 2 hr at room temperature. The chip was incubated with Anti-GOLPH3 antibody for 12 h at 4
2.10CCK-8 cell viability assay
Cells were seeded onto 96-well plates at 3
2.11Apoptosis assay
Cells were seeded at 1
2.12Invasion and migration assays
Invasion assays were performed using Matrigel-coated transwell inserts (Costar, Manassas, VA, USA) containing polycarbonate filters with 8-
2.13In vivo tumor metastasis
Female Balb/c nude mice (total number
Figure 1.
Expression of GOLPH3 is upregulated in endometrial carcinoma (EC). (A) The mRNA level of GOLPH3 and Ki67 in four EC cell lines (HEC-1A, KLE, RL95-2, and Ishikawa) and endometrial stromal cells (ESCs) as measured by qRT-PCR, Ki67 was detected as positive control. (B) The protein level of GOLPH3 in four EC cell lines and ESCs measured by western blot. (C) GOLPH3 mRNA expression in 30 EC tissue samples and normal adjacent tissue (NAT) analyzed by qRT-PCR. (D) GOLPH3 protein expression in six EC tissues and NAT analyzed by western blot. (E) The expression and localization of GOLPH3 in EC tissue chip detected by IHC Magnification 100
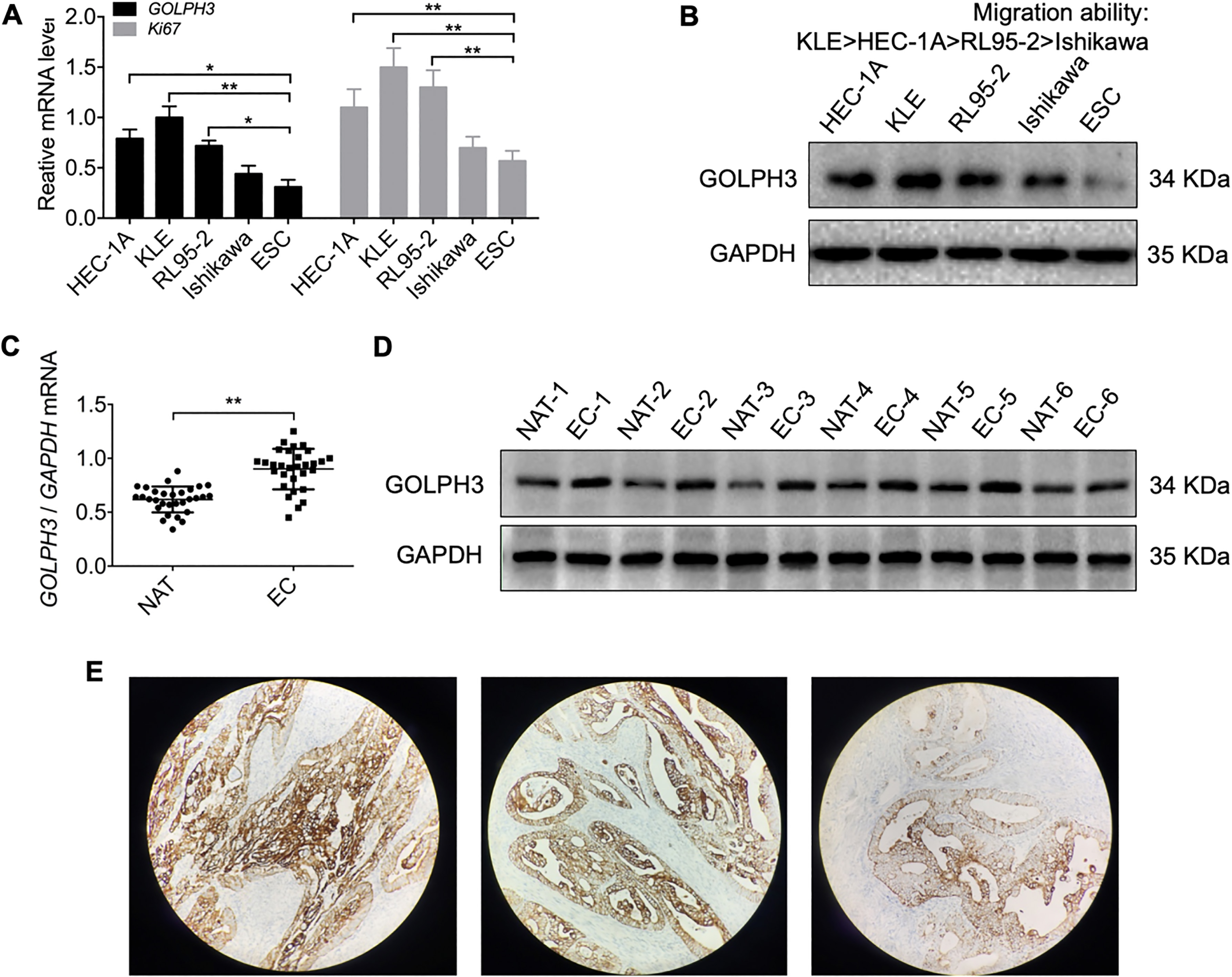
Figure 2.
Downregulation of GOLPH3 in KLE cells reduces cell proliferation, migration, and invasion, and promotes apoptosis in vitro. (A–B) Knockdown of GOLPH3 in KLE cells transfected with shGOLPH3 (KLE-sdGOLPH3 cells) compared to control cells transfected with empty vector was verified by qRT-PCR and western blotting. (C) Serial CCK-8 cell viability assays showing slower proliferation of knockdown cells compared to controls. (D) Downregulation of GOLPH3 also reduced cell migration and invasion in transwell assays. Magnification 100
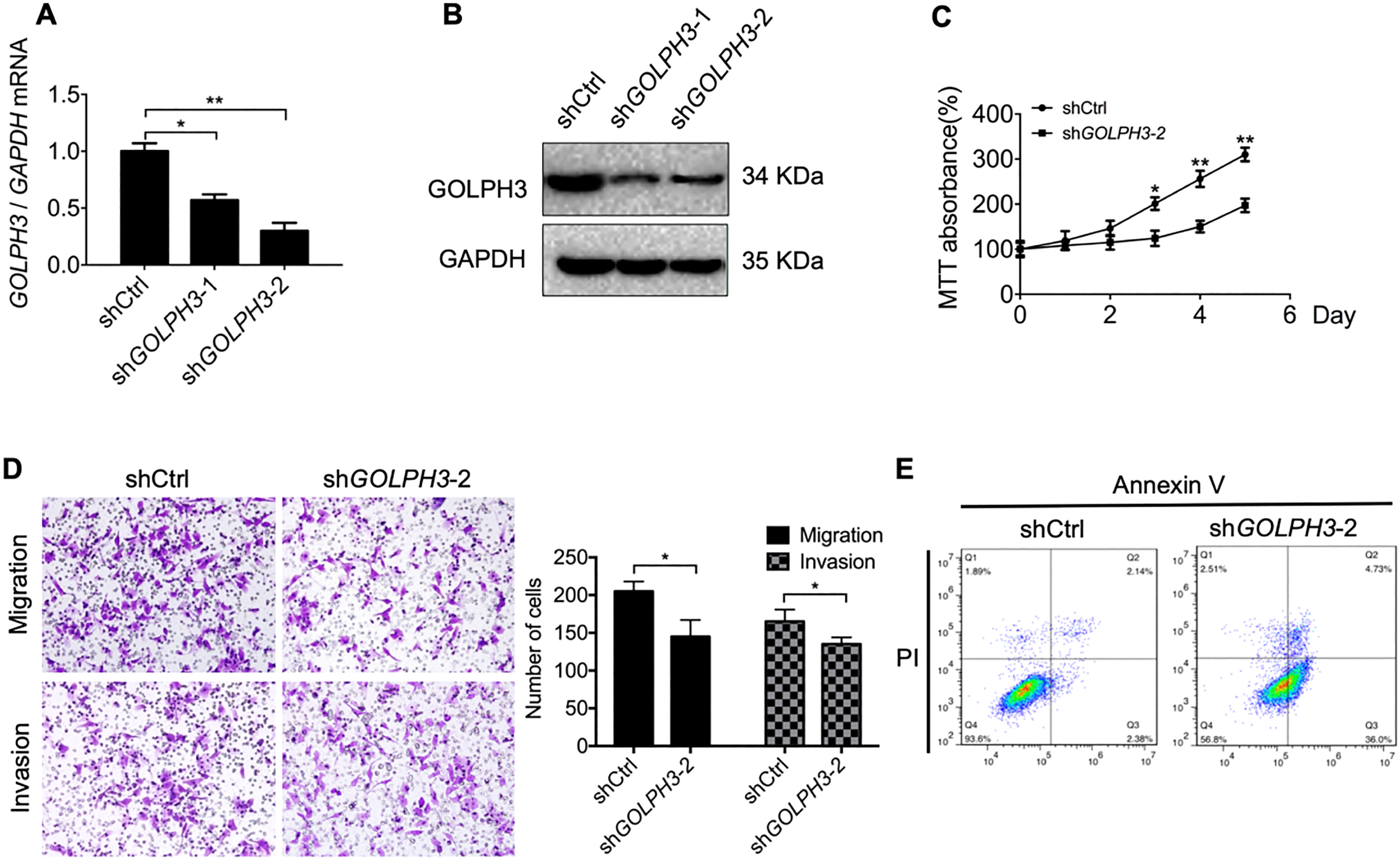
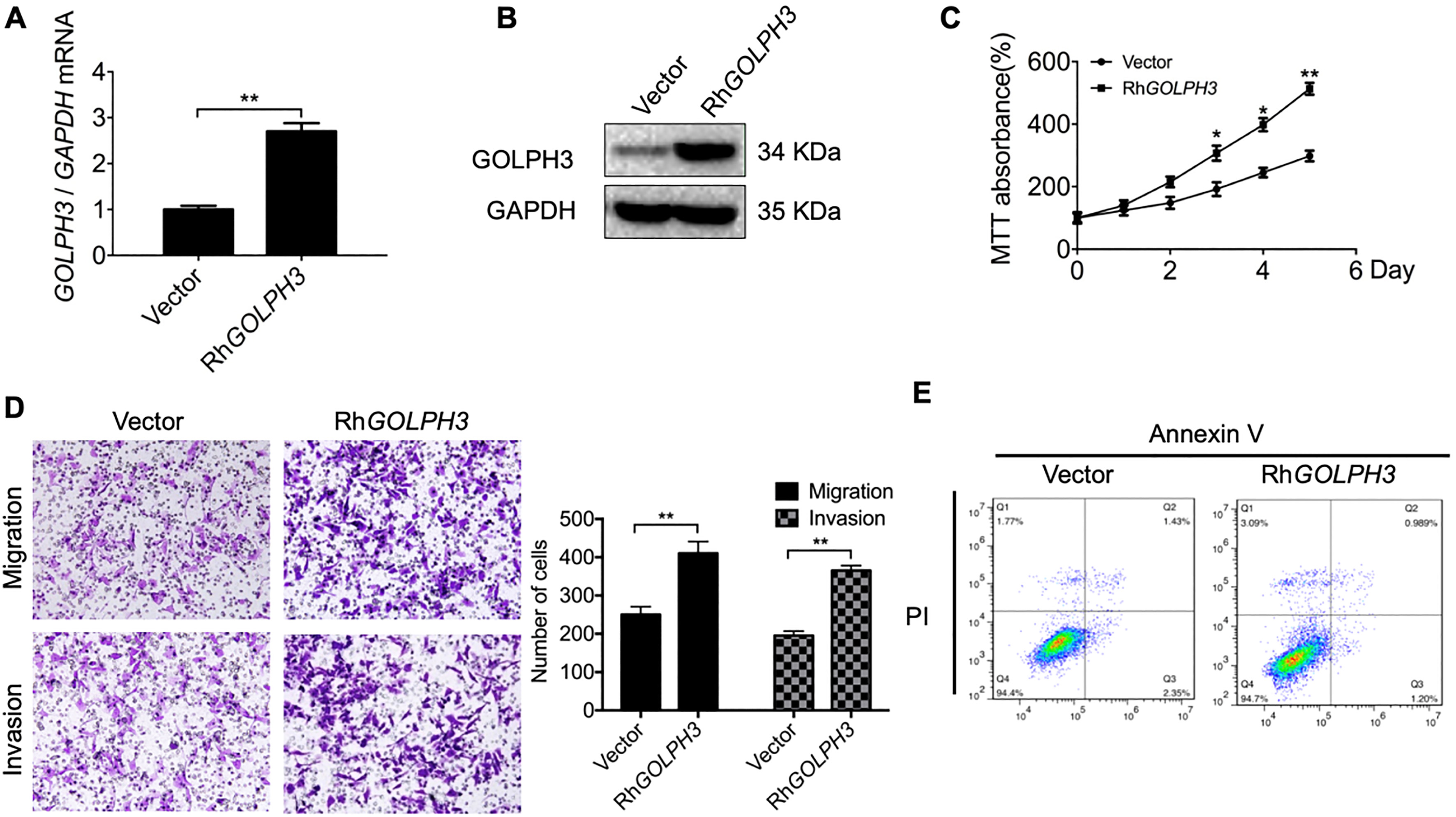
Figure 3.
Ectopic overexpression of GOLPH3 in Ishikawa cells accelerates proliferation, migration, and invasion while inhibiting apoptosis in vitro. (A–B) GOLPH3 overexpression in Ishikawa-RhGOLPH3 cells compared to control cells was verified by qRT-PCR and western blotting. (C) Serial CCK-8 cell viability assays demonstrating faster proliferation rate of Ishikawa-RhGOLPH3 cells versus controls. (D) Overexpression of GOLPH3 also accelerated cell migration and invasion in transwell assays. Magnification 100
2.14Statistical analysis
All measurement data were presented as means
3.Results
3.1GOLPH3 is highly expressed in endometrial carcinoma (EC) and correlated with distant metastasis
To investigate GOLPH3 involvement in EC, mRNA and protein levels in four EC cell lines were determined by qRT-PCR and western blot, respectively, ki67 was detected as positive control. Results revealed significantly higher expression levels in all lines compared to non-malignant endometrial stromal cells (ESCs) (Fig. 1A and B). We further analyzed the relationship between GOLPH3 expression and metastasis capacity in EC cell lines by transwell migration, and found higher expression in the invasive EC cell lines KLE and HEC-1A than the non-invasive EC cell lines RL95-2 and Ishikawa (Fig. 1A and B). Analysis of 30 EC and matched normal adjacent tissue (NAT) samples by qRT-PCR and 6 EC/NAT samples by western blot revealed significantly higher GOLPH3 mRNA and protein expression in tumor tissue (Fig. 1C and D). Then we carried out IHC to determine the GOLPH3 localization in endometrial carcinoma and the relationship between GOLPH3 expression level and clinical factors of EC. The staining showed that GOLPH3 were overexpressed in the cytoplasm of carcinoma tissues (Fig. 1E). Further, the expression of GOLPH3 was related to the endometrial carcinoma grade. However, no correlation was found between GOLPH3 expression and other clinical factors, such as age, stage and lymph node metastasis (Table 2).
Table 2
The Correlation between GOLPH3 protein and clinic-pathological factors in endometrial carcinoma tissues (
| Factor | GOLPH3 |
| |
|---|---|---|---|
| Positive | Negative | value | |
| Age (years) | 1.000 | ||
| | 8 | 3 | |
| | 17 | 6 | |
| Grade | 0.037 | ||
| I | 3 | 0 | |
| II | 17 | 4 | |
| III | 5 | 5 | |
| Stage | 0.347 | ||
| I | 14 | 4 | |
| II | 6 | 2 | |
| III | 5 | 1 | |
| IV | 0 | 2 | |
| Lymph node metastasis | 0.435 | ||
| Yes | 9 | 5 | |
| No | 16 | 4 | |
3.2Suppression of GOLPH3 expression reduces EC cell proliferation, migration, and invasion while promoting apoptosis
To examine the effects of GOLPH3 on EC cell proliferation, migration, and invasion, the normally invasive KLE EC cell line was transfected with shGOLPH3 or empty vector. KLE cells transfected with shGOLPH3 plasmid displayed significantly lower GOLPH3 expression at the mRNA and protein levels compared to control cells transfected with empty vector (Fig. 2A and B), thus confirming transfection efficiency. In addition, cells transfected with shGOLPH3 demonstrated a slower proliferation rate than cells transfected with the empty vector as assessed by the CCK-8 cell viability assay (Fig. 2C). The migration and invasion capacities of KLE cells transfected with shGOLPH3 were also significantly reduced compared to KLE cells transfected with empty vector as determined by transwell migration assays (
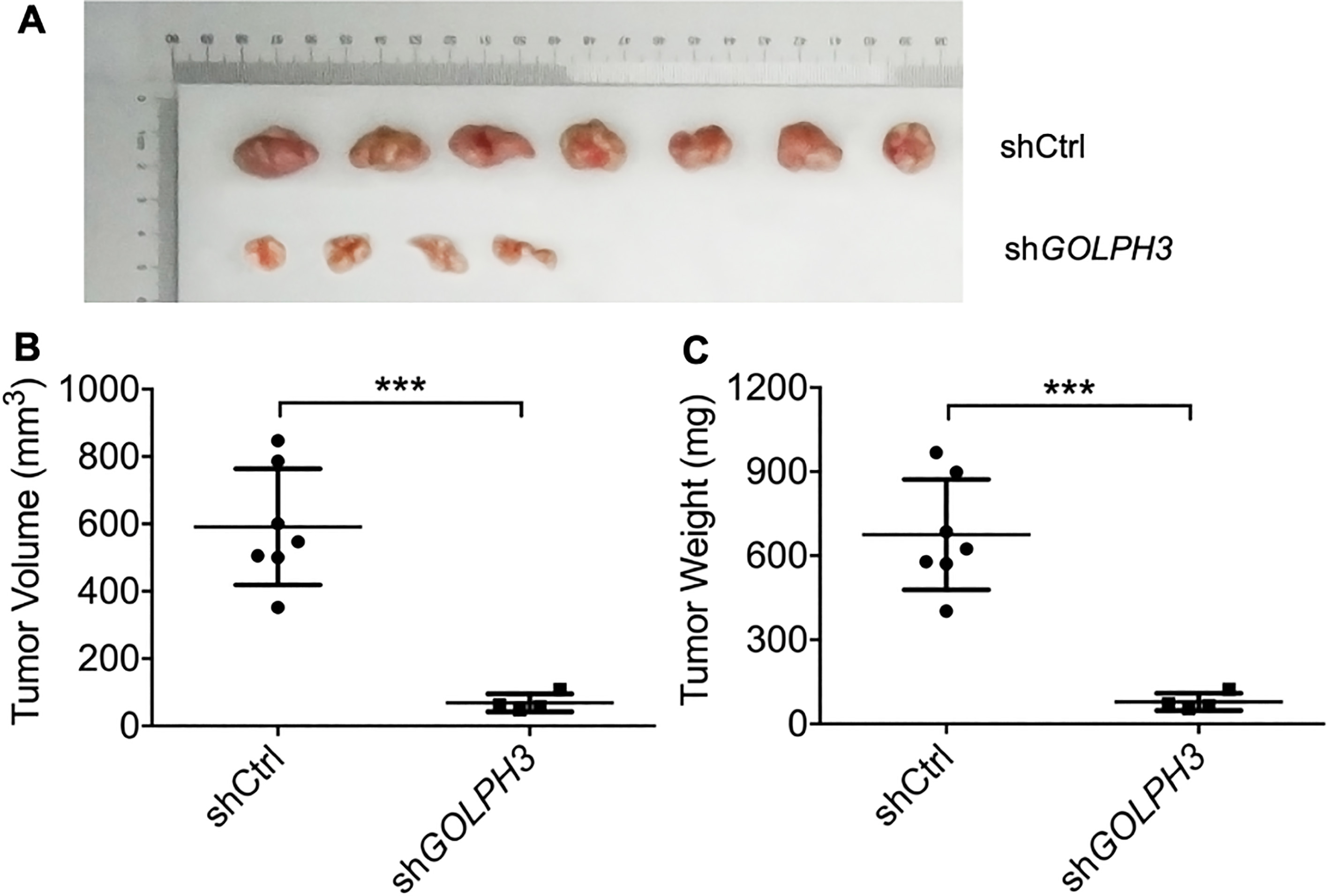
Figure 4.
Downregulation of GOLPH3 reduces distant EC cell metastasis in vivo. The tumor images (A), tumor volume growth curves (B) and tumor weights (C) 30 days after inoculation of nude mice with GOLPH3 knockdown (KLE-shGOLPH3) EC cells or control cells. All values are the mean
3.3Ectopic overexpression of GOLPH3 accelerates EC cell proliferation, migration, and invasion while inhibiting apoptosis
We transiently transfected pcDNA3.1 (+)-GOLPH3 plasmid into the normally non-invasive Ishikawa cell line, and confirmed transfection efficiency by qRT-PCR and western blot. Compared to Ishikawa cells transfected with empty vector, cells transfected with the pcDNA3.1 (+)-GOLPH3 plasmid displayed significantly greater GOLPH3 expression at both mRNA and protein levels (Fig. 3A and B) as well as accelerated proliferation, migration, and invasion (
Figure 5.
GOLPH3 regulates the transition between epithelial and mesenchymal phenotypes in EC cells. (A–B) Expression levels of epithelial and mesenchymal markers were compared between KLE-shGOLPH3 (knockdown) and control cells by qRT-PCR and western blotting. GOLPH3 knockdown reduced the expression of mesenchymal markers and upregulated epithelial markers (MET). (C–D) Expression levels of epithelial and mesenchymal marker were compared between overexpressing Ishikawa-RhGOLPH3 and control cells by qRT-PCR and western blotting. Ectopic overexpression of GOLPH3 upregulated mesenchymal marker expression and downregulated epithelial marker expression. All values are the mean
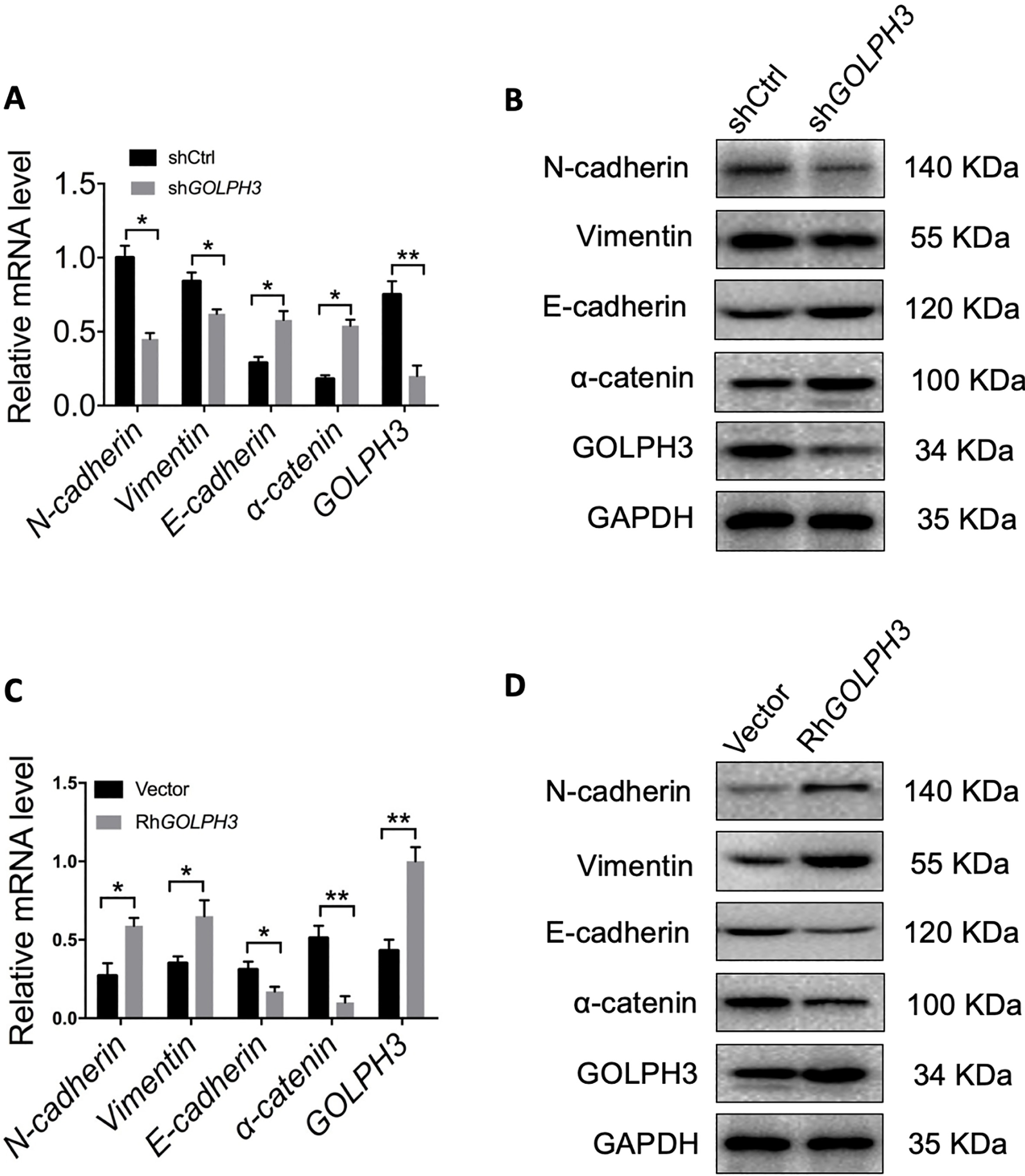
3.4Suppression of GOLPH3 expression reduces EC cell distant metastasis in vivo
We then investigated the functional relevance of GOLPH3 for metastasis in vivo. A suspension of KLE-shGOLPH3 (knockdown) or control KLE cells was injected into nude mice through the tail vein, and animals were sacrificed 30 days later. Knockdown of GOLPH3 significantly decreased the number of mice with distant metastases (4 of 7) compared to the control group (7/7) (Fig. 4A). In addition, tumor volume and tumor weight were significantly lower in the KLE-shGOLPH3 injection group (Fig. 4B and C). Therefore, elevated GOLPH3 expression appears essential for EC cell metastasis in vivo.
3.5GOLPH3 induces epithelial-mesenchymal transition in EC cells
The EMT is critical for cancer cell migration and invasion [24]. To further support the proposal that GOLPH3 facilitates migration and invasion of EC cells, we examined the mRNA and protein levels of EMT-related genes in overexpressing and knockdown EC cells by qRT-PCR and western blotting. As shown in Fig. 5A and B, compared to the control group, knockdown of GOLPH3 in KLE cells inhibited mRNA and protein expression of the mesenchymal markers N-cadherin and vimentin, and stimulated the expression of the epithelial markers E-cadherin and
4.Discussion
Endometrial carcinoma (EC) is the most common malignancy of the female reproductive tract in developed countries and incidence continues to increase [1]. Recent studies indicate that early frequent metastasis that occurs in about one fourth of the high-risk of ECs and is the major reason for poor outcome [25, 26]. Therefore, identifying novel molecules that regulate EC metastasis will aid in the identification of molecular prognostic factors and the development of more efficacious treatment strategies. There is a large body of evidence that induction of EMT confers mobility and invasive capacity to epithelial cancer cells, including EC cells, thereby facilitating the generation of distant metastases [27].
In this study, we show for the first time that GOLPH3 is overexpressed in EC tissues and EC cells compared to adjacent non-cancerous tissues, and GOLPH3 was localized in the cytoplasm of carcinoma tissues and the expression level of GOLPH3 was related to the grade of the tumor. In addition, GOLPH3 overexpression is critical for the enhanced proliferative and invasive properties as well as reduced apoptosis rate characteristic of cancer cells, in vitro and in animal models. Alternatively, downregulation of GOLPH3 in normally aggressive and invasive EC cells inhibited proliferation and metastatic capacity in vitro and in vivo. We also confirmed that GOLPH3 promotes EC cell metastasis by regulating EMT, as EC cells strongly expressing GOLPH3 displayed an EMT phenotype, while downregulation in aggressive EC cells led to mesenchymal-epithelial transition (MET). This study therefore provides important evidence that GOLPH3 overexpression is critical for EC cell tumorigenicity, aggressiveness and metastasis capacity. While EMT is essential for EC cell dissemination and seeding of new tumors at distant sites [28], our results also demonstrate that these GOLPH3-induced processes are reversible by suppression of GOLPH3 expression. Therefore, GOLPH3 may serve as a novel diagnostic and therapeutic target, strongly warranting further investigation.
Previous studies reported that GOLPH3 enhances proliferation and metastasis of various cancer cells by activating the targets of Wnt/
In conclusion, this study demonstrates that relative overexpression of GOLPH3 in EC cells confers an aggressive phenotype characterized by rapid proliferation, strong invasive capacity, and low apoptosis rate. More importantly, these features may be reversed by suppression of GOLPH3 expression, underscoring the potential therapeutic value of strategies targeting GOLPH3 signaling. But unfortunately, we haven’t measured the bioluminescence image of xenografted tumors and intensity, and these results should be confirmed with further clinical studies on human patients.
Conflict of interest
The authors have no conflicts of interest to declare.
Abbreviations
| GOLPH3 | Golgi phosphoprotein 3 |
| EC | endometrial carcinoma |
| ESC | endometrial stromal cell |
| NAT | normal adjacent tissue |
| EMT | epithelial-mesenchymal transition |
| MET | mesenchymal-epithelial transition |
| mTOR | mechanistic target of rapamycin |
|---|---|
| NF- | nuclear factor |
| CCK-8 | Cell Counting Kit-8 |
| RPMI-1640 | Roswell Park Memorial Institute |
| ATCC | American Type Culture Collection |
| DMEM | Dulbecco’s modified Eagle’s |
| medium | |
| FBS | fetal bovine serum |
| RIPA | radio-immunoprecipitation assay |
| PVDF | polyvinylidene difluoride |
| shRNA | short hairpin RNA |
| SD | standard deviation |
| RT-PCR | reverse transcription polymerase |
| chain reaction | |
| IHC | immunohistochemistry |
| SDS-PAGE | sodium dodecyl sulfate polyacry- |
| lamide gel electrophoresis |
References
[1] | P. Morice et al., Endometrial cancer, The Lancet 387: ((2016) ), 1094–1108. |
[2] | K.J. Dedes et al., Emerging therapeutic targets in endometrial cancer, Nature Reviews Clinical Oncology 8: ((2011) ), 261. |
[3] | D. Thanapprapasr and K. Thanapprapasr, Molecular therapy as a future strategy in endometrial cancer, Asian Pacific Journal of Cancer Prevention 14: ((2013) ), 3419–3423. |
[4] | Y.C. Lee, S. Lheureux and A.M. Oza, Treatment strategies for endometrial cancer: current practice and perspective, Current Opinion in Obstetrics and Gynecology 29: ((2017) ), 47–58. |
[5] | H.C. Dippold et al., GOLPH3 Bridges Phosphatidylinositol-4-Phosphate and Actomyosin to Stretch and Shape the Golgi to Promote Budding, Cell 139: ((2009) ), 337–351. |
[6] | C.C. Wu et al., GMx33: a novel family of trans-Golgi proteins identified by proteomics, Traffic 1: ((2000) ), 963–975. |
[7] | S. Tang et al., GOLPH3: a novel biomarker that correlates with poor survival and resistance to chemotherapy in breast cancer, Oncotarget 8: ((2017) ), 105155–105169. |
[8] | W. Tang et al., Overexpression of GOLPH3 is associated with poor survival in Non-small-cell lung cancer, American Journal of Translational Research 8: ((2016) ), 1756. |
[9] | Z. Zeng et al., Overexpression of GOLPH3 promotes proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor, Clinical Cancer Research 18: ((2012) ), 4059–4069. |
[10] | J. Sun et al., GOLPH3 induces epithelial-mesenchymal transition via Wnt/β-catenin signaling pathway in epithelial ovarian cancer, Cancer Medicine 6: ((2017) ), 834–844. |
[11] | H. Li et al., GOLPH3 overexpression correlates with tumor progression and poor prognosis in patients with clinically N0 oral tongue cancer, Journal of Translational Medicine 10: ((2012) ), 168. |
[12] | Q. Li, Y. Ma and W. Xu, High GOLPH3 expression is associated with poor prognosis and invasion of hepatocellular carcinoma, Molecular Medicine Reports 11: ((2015) ), 4315–4320. |
[13] | K. Zhu et al., GOLPH3 overexpression correlates with poor response to neoadjuvant therapy and prognosis in locally advanced rectal cancer, Oncotarget 7: ((2016) ), 68328. |
[14] | X. Hua et al., Increased expression of Golgi phosphoprotein-3 is associated with tumor aggressiveness and poor prognosis of prostate cancer, Diagnostic Pathology 7: ((2012) ), 127. |
[15] | Q. Zhang et al., GOLPH3 is a potential therapeutic target and a prognostic indicatior of poor survival in bladder cancer treated by cystectomy, Oncotarget 6: ((2015) ), 32177–32192. |
[16] | J.H. Wang et al., High Expression of GOLPH3 in Esophageal Squamous Cell Carcinoma Correlates with Poor Prognosis, PLOS ONE 7: ((2012) ), e45622. |
[17] | Y. Liu, Y. Sun and A. Zhao, MicroRNA-134 suppresses cell proliferation in gastric cancer cells via targeting of GOLPH3, Oncology Reports 37: ((2017) ), 2441–2448. |
[18] | J. Peng et al., Mechanisms of GOLPH3 Associated with the Progression of Gastric Cancer: A Preliminary Study, PLOS ONE 9: ((2014) ), e107362. |
[19] | Y. Xue et al., GOLPH3 is a novel marker of poor prognosis and a potential therapeutic target in human renal cell carcinoma, British Journal of Cancer 110: ((2014) ), 2250–2260. |
[20] | K.L. Scott et al., GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer, Nature 459: ((2009) ), 1085–1090. |
[21] | T. Dai et al., Golgi phosphoprotein 3 (GOLPH3) promotes hepatocellular carcinoma cell aggressiveness by activating the NF-κB pathway, Journal of Pathology 235: ((2015) ), 490–501. |
[22] | C.Z. Qiu et al., Correlation of GOLPH3 Gene with Wnt Signaling Pathway in Human Colon Cancer Cells, Journal of Cancer 7: ((2016) ), 928–934. |
[23] | Z. Hou et al., Role of interleukin-1 receptor type II in the pathogenesis of endometriosis, Fertility & Sterility 89: ((2008) ), 42–51. |
[24] | S. Heerboth et al., EMT and tumor metastasis, Clinical & Translational Medicine 4: ((2015) ), 6. |
[25] | N. Colombo et al., ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer, Ann Oncol 27: ((2016) ), 16–41. |
[26] | J.P. Thiery et al., Epithelial-mesenchymal transitions in development and disease, Cell 139: ((2009) ), 871–890. |
[27] | S. Floor et al., Cancer cells in epithelial-to-mesenchymal transition and tumor-propagating-cancer stem cells: Distinct, overlapping or same populations, Oncogene 30: ((2011) ), 4609–4621. |
[28] | A. Makker and M.M. Goel, Tumor progression, metastasis, and modulators of epithelial-mesenchymal transition in endometrioid endometrial carcinoma: an update, Endocr Relat Cancer 23: ((2016) ), R85–R111. |


