
Tumour Necrosis Factor in Neuroplasticity, Neurogenesis and Alcohol Use Disorder
Abstract
Alcohol use disorder is a pervasive and detrimental condition that involves changes in neuroplasticity and neurogenesis. Alcohol activates the neuroimmune system and alters the inflammatory status of the brain. Tumour necrosis factor (TNF) is a well characterised neuroimmune signal but its involvement in alcohol use disorder is unknown. In this review, we discuss the variable findings of TNF’s effect on neuroplasticity and neurogenesis. Acute ethanol exposure reduces TNF release while chronic alcohol intake generally increases TNF levels. Evidence suggests TNF potentiates excitatory transmission, promotes anxiety during alcohol withdrawal and is involved in drug use in rodents. An association between craving for alcohol and TNF is apparent during withdrawal in humans. While anti-inflammatory therapies show efficacy in reversing neurogenic deficit after alcohol exposure, there is no evidence for TNF’s essential involvement in alcohol’s effect on neurogenesis. Overall, defining TNF’s role in alcohol use disorder is complicated by poor understanding of its variable effects on synaptic transmission and neurogenesis. While TNF may be of relevance during withdrawal, the neuroimmune system likely acts through a larger group of inflammatory cytokines to alter neuroplasticity and neurogenesis. Understanding the individual relevance of TNF in alcohol use disorder awaits a more comprehensive understanding of TNF’s effects within the brain.
INTRODUCTION
The ability to learn, remember and adapt after unique experiences is allowed by neuroplasticity. On a cellular level, neuroplasticity is the flexibility of neurons to adjust their synaptic strength and relationships through genetic, neurochemical and structural mechanisms. The best characterised type of neuroplasticity is described by Hebbian theory, which dictates colloquially that “neurons that fire together wire together” [1]. This theory postulates that recurrent postsynaptic activation associated with presynaptic activation increases the strength of a synapse above a certain basal level [2]. Other forces of synaptic plasticity include homeostatic plasticity, whereby the strength of a synapse is shifted back towards its basal homeostatic level [3]. Through the forces of plasticity, the shape of the brain is dynamically changing to integrate, disintegrate and modulate different synapses within circuits. It has been suggested that the modulation of neuroplasticity by various drugs of abuse is an integral component of the progression of addiction [4, 5].
Alcohol use disorder (AUD) is characterised by behaviours of problematic alcohol use and is a significant burden to individual and public health. It is estimated that AUD affects approximately 12% of the population in the US [6]. Alcohol use is recognised as a leading risk factor for disease burden and has been linked to 60 acute and chronic diseases [7, 8]. Alcohol’s promiscuous action on neural function is thought to drive neural plasticity that manifests as the many symptoms of AUD including addiction, dependence and tolerance [4, 5]. Alcohol’s activation of the neuroimmune system is a highly relevant component of AUD [9–13]. Indeed, altered neuroimmune function is associated with numerous psychiatric disorders including major depressive disorder, bipolar disorder, schizophrenia and drug addiction [13–16]. Experimentally, it has been shown that activation of the immune system results in increased ethanol self-administration in mice and there is support for the idea that alcohol induced neuroinflammation drives further alcohol consumption in a relentless cycle [17, 18].
Tumour necrosis factor (TNF) is a neuroimmune and inflammatory molecule that was first discovered as a factor in the haemorrhagic necrosis of tumours and has associations with numerous pathological states [19]. Despite its association with pathology, it is now known that TNF has numerous beneficial physiological effects in the CNS which can be altered during inflammation or challenge [20, 21]. The neuroimmune system is involved in neuroplasticity and in AUD and these relationships have been the subject of numerous reviews [22–27]. Given that TNF is an early initiator of inflammation in the CNS, and one of a few cytokines that are involved directly in both synaptic plasticity and neuroimmune signalling, its effect on neurotransmission in AUD deserves discussion. Here, we will provide an overview of TNF’s effect on neuroplasticity in normal physiology, followed by its effect on neuroadaptation and neurogenic deficit during AUD.
ETHANOL’S ACTION
Alcohol has many molecular targets that are involved in its rewarding effects. Notably, alcohol’s interactions with numerous synaptic ion channels, signalling molecules, neuropeptide mechanisms and enzymes elicit short and long term effects [4, 5, 28]. Upon repeated exposure, alcohol’s interactions with these synaptic targets are thought to lead to long term changes in synaptic function [5]. While there is still much unknown about how alcohol interacts with its direct synaptic targets to induce neuroplastic changes, this topic has been the focus of previous reviews [4, 28, 29]. Aside from alcohol’s direct targets, alcohol also affects numerous components of the neuroimmune system. The neuroimmune system includes neurons, microglia, astrocytes, oligodendrocytes, peripheral immune cells and vascular and endothelial cells [16, 30]. Signalling between these cells promotes neuroinflammation in response to injury and stress.
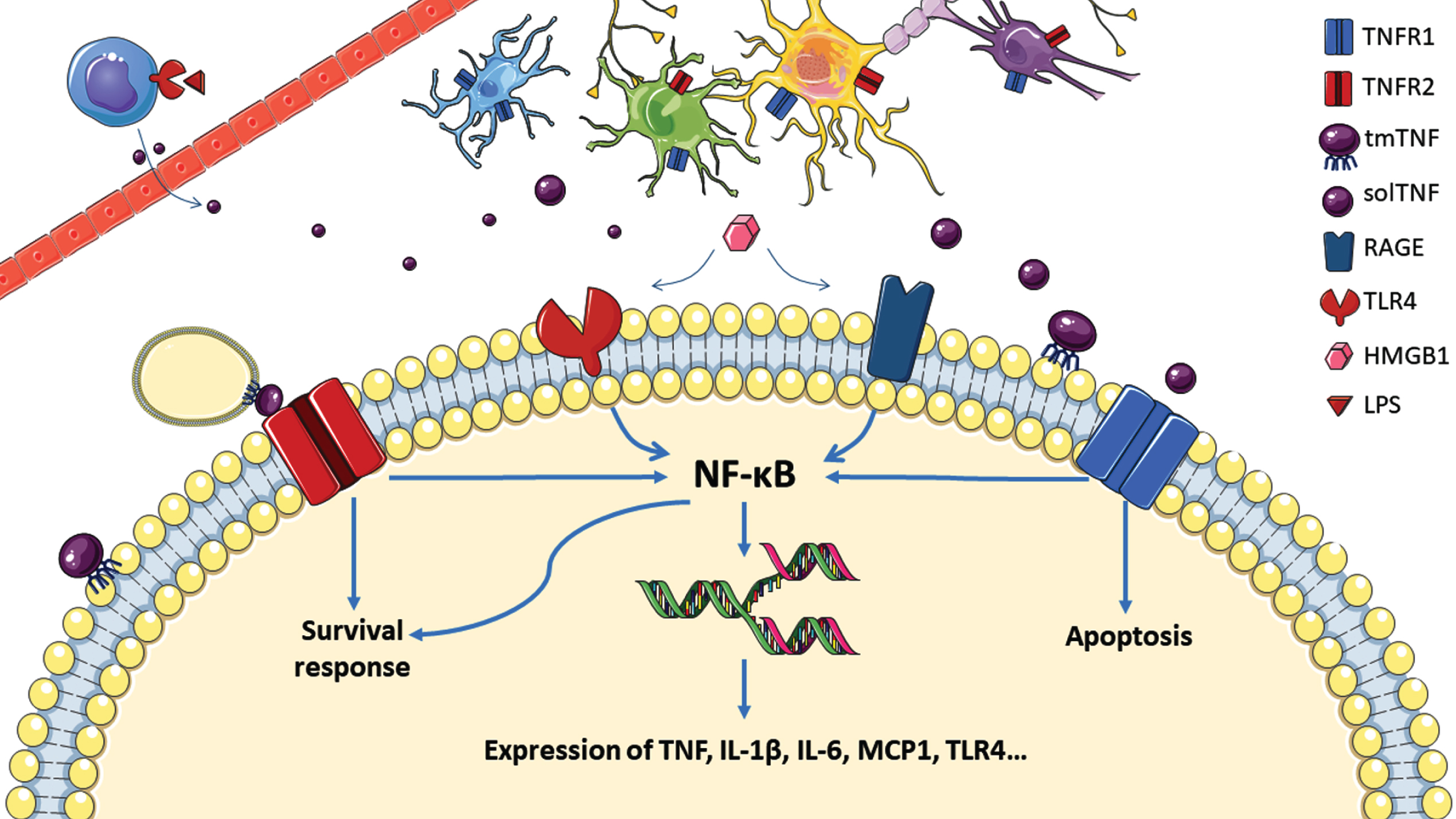
Alcohol consumption can increase intestinal permeability to lipopolysaccharide (LPS or endotoxin), which circulates through the blood and stimulates toll like receptor 4 (TLR4) on peripheral immune cells such as resident macrophages [31–33]. TLR4 activation results in the initiation of cellular cascades that activate nuclear transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) causing the release of inflammatory cytokines, such as TNF, interleukin 1-β and interleukin 6 (IL-6) [34]. Upon alcohol related cell death within the brain, an array of damage associated molecular patterns (DAMPs) are released and stimulate pattern recognition receptors to further the inflammatory response. Of particular significance is the DAMP high motility group box 1 (HMGB-1). HMGB-1 is released actively from neurons and other glial cells upon alcohol exposure but is also passively released after cell death [32, 35]. HMGB-1 has differential effects depending on the status of certain cysteine residues; oxidation of HMGB-1 appears to attenuate its inflammatory effects, while disulphide and all thiol containing HMGB-1 signal for inflammation through TLR4 and receptor for advanced glycation end products (RAGE) respectively [36].
TNF exists in two forms as transmembrane (tmTNF) and soluble (solTNF) TNF. Upon activation, TNF-α converting enzyme (TACE) cleaves tmTNF into solTNF to be released and have reaching inflammatory effects [21]. The transmembrane form signals through both tumour necrosis factor receptors 1 and 2 (TNFR1 and TNFR2) while solTNF signals solely through TNFR1. Although both receptors are involved in inflammatory responses, TNFR1 activation pathways can lead to cell death while TNFR2 is associated with pro survival responses [21]. Numerous regulatory mechanisms exist to regulate TNF production and curb inflammatory signalling including cyclic AMP, prostaglandins, soluble circulating TNF receptors and anti-inflammatory cytokines [37–40]. Previous reviews provide detailed descriptions of TNFR signalling and regulation [41–43]. During inflammation, released solTNF activates TNFR1 signalling, furthering NF-κB transcription and stimulating activator protein 1 (AP-1) to amplify the inflammatory response in a positive feedback loop [44, 45]. Results in TLR4 knockout mice show that TLR4 signalling is necessary for ethanol induced increases in brain levels of TNF [46]. Despite the importance of TLR4 in ethanol’s effect on TNF, in the context of this paper it is important to note that inactivation of TLR4 does not result in reduced levels of ethanol self-administration in multiple rodent models [47, 48]. Inflammatory cytokines induced by ethanol travel throughout the periphery to promote inflammatory responses and some molecules, including TNF, travel across the blood brain barrier (BBB) to participate in neuroinflammation [11, 49]. In addition to chemokines crossing the BBB, circulating monocytes and macrophages are able to infiltrate the BBB during inflammation (Fig. 1).
Fig. 1
Overview of TNF signalling after alcohol consumption within a neuron. Peripheral immune cells release TNF following binding of lipopolysaccharide (LPS) through toll like receptor 4 (TLR4), which crosses the blood brain barrier (top left) to stimulate TNFR1 signalling and NF-κB translocation to the nucleus for transcription of TNF and other cytokines. High motility group box 1 (HMGB-1) also activates NF-κB through receptor for advanced glycation end products (RAGE) and toll like receptor 4 (TLR4). Microglia (blue), astrocytes (green), neurons (yellow) and oligodendrocytes (purple) are represented here with their respective TNF receptors.
The neuroimmune system differs between the sexes, and is even involved in the masculinisation of the brain during development [50, 51]. It has been shown in animal models that female and male mice display different drinking patterns [52]. While the general trend of an inflammatory response to ethanol is shared between males and females, there are variations in neuroimmune responses and female mice appear more vulnerable to inflammatory effects following binge drinking [53]. Similar sex associated disparities are seen in many aspects of AUD. Generally, men are more likely to develop AUD and consume larger amounts of alcohol while women are more prone to experience AUD related medical problems [54, 55]. An investigation in rats showed that sex had no effect on TNF mRNA levels following exposure to ethanol vapor; however, this review’s bias towards discussing neuroimmune signalling modelled in the male sex remains a shortcoming, which should be kept in mind throughout reading.
TNF IN PHYSIOLOGICAL SYNAPTIC PLASTICITY
TNF is a well known modulator of synaptic plasticity [56–59]. The most popular area of discussion is its ability to potentiate excitatory transmission, which is achieved through numerous mechanisms that will be reviewed throughout this chapter. Firstly, TNF’s ability to increase α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) expression has been a dominant subject of research. Seminal research demonstrated that TNF increases AMPAR surface expression and synaptic strength, with TNF treated slices showing increased frequency of miniature post-synaptic excitatory currents (mEPSCs) [60, 61]. TNF’s activity dependent scaling of synaptic strength in these studies indicates its role as a mediator of homeostatic plasticity [62]. Genetic deletion of TNFR1 suppressed synaptic transmission via AMPAR dependent mechanisms in cortical neurons [63]. Importantly, induction of GLuR2-lacking AMPAR trafficking by TNF allows for enhanced calcium influx which has been shown to contribute to excitotoxicity [64, 65]. In contrast to this mechanism in hippocampal neurons, it has been shown in cortical and motor neurons that TNF can preference the expression of GluR2 containing AMPAR expression, thereby protecting against excessive calcium influx [66]. The reasons for this disparity may include differences in neuron type, which underscores the diverse role of TNF in plasticity [66]. Interestingly, TNF can boost glutamate levels by increasing neuronal glutaminase production, which is involved in the conversion of glutamine to glutamate [67].
An effect on N-methyl-D-aspartate receptor (NMDAR) modulation has also been observed. TNF’s enhancement of hippocampal mEPSC frequency and amplitude was abolished in TNFR1 knockout mice and mice treated with the NMDAR antagonist ifenprodil, with indications for a role of NR2B subunit containing NMDARs in TNFR1 signalling [68]. The involvement of NMDARs in TNF mediated excitatory transmission has been observed elsewhere, with TNF exposure resulting in a quickly observed change in cellular activity that increased the NR1 subunit of NMDAR surface expression and enhanced EPSCs [69, 70].
To further increase neuronal excitability, TNF can decrease inhibitory signalling. TNF was found to cause GABAA receptor endocytosis in hippocampal pyramidal neurons, furthering excitatory transmission at these synapses [60]. Later, the intracellular pathway of this GABAA receptor endocytosis was further characterised in rats and found to be TNFR1 dependent [71]. In contrast, TNF had no effect on reducing the frequency of spontaneous post-synaptic inhibitory currents in rat spinal cord slices [72].
In addition to the neuronal mechanisms discussed above, TNF’s recruitment of glia to enhance excitatory signalling is extremely important, if not critical. For example, TNF induced increases in synaptic strength that were abolished in a model of TNFR1 inactivation were returned with the selective activation of TNFR1 in astrocytes [68]. In particular, TNF’s ability to affect glial glutamate regulation is an important mechanism of TNF excitation [73]. In rat hippocampal entorhinal cortex slices, TNF was shown to dose-dependently decrease uptake of glutamate [74]. Specifically blocking glial glutamate uptake transporters alone resulted in the same NMDAR dependent neurotoxicity as elicited by TNF [74]. TNF has also been shown to downregulate the expression of the excitatory amino acid transporter gene EAAT2, which regulates neuronal glutamate transporters, and has associations with reducing astroglial glutamate transporter 1 [75, 76]. TNF further potentiates excitatory transmission by inducing glial glutamate release. In microglia, TNF activates TNFR1 in an autocrine manner to induce glutamate release through transporters and through gap junction hemi channels [77]. TNF has also been shown to positively regulate astrocytic glutamate release, with microglial-astrocytic communication amplifying release [78]. Furthermore, microglial TNF release potentiates the intrinsic excitability of Purkinje neurons, as seen in a model of LPS induced cerebellar inflammation [79].
In the hippocampus, astrocytic purinergic receptor activation and subsequent calcium influx can induce mEPSCs in adjacent neurons through glutamate release [80]. It was shown in Tnf-/- mice that this effect is abolished due to alterations in the docking and exocytosis of glutamate vesicles, which affects the balance between astrocytic glutamate uptake and release [80]. Investigators noted that low concentrations of TNF appear to be necessary for this mechanism of gliotransmission and that fluctuations in TNF would have subtle, if not greater, effects on synaptic transmission [80]. However, regulatory feedback systems do exist, with glial release of TNF being regulated by detection of extracellular glutamate. Glutamate treated astrocytes transplanted into neuronal cultures released less TNF and elicited less AMPAR expression in neurons, indicating a role in synaptic scaling to protect against excessive excitation [61].
In contrast to TNF’s potentiation of excitatory transmission, studies have demonstrated that TNF application can inhibit components of LTP [81–86]. In the rat hippocampus, it was suggested that TNF’s inhibition of LTP is a biphasic mechanism initially involving p38 mitogen-activated protein kinase in the early stage of inhibition while relying on another mechanism in repressing long term LTP [87]. Later, the same group did similar work and implicated the disruption of intracellular calcium levels related to mGluR5 activation as involved in TNF’s LTP inhibition [88]. Another group showed that TNF in low concentrations enhances LTP expression in mouse pyramidal neurons, an effect which was absent when intracellular calcium stores were depleted [89]. Importantly, this study showed that concentration is critical in what effect TNF will have on synaptic plasticity, as low levels of TNF enhance LTP expression while high levels of TNF impair the establishment of LTP but do not affect previously existing LTP. This effect was able to be reversed with neuronal pretreatment with ryanodine, implicating calcium levels in the effect of TNF on both the inhibition and establishment of LTP. Alterations in calcium transport have also been observed in a study involving old age rats where administration of soluble TNF inactivator Xpro1595 was shown to reverse age related hippocampal LTD and behavioural deficits [90]. The importance of TNF in LTD has been shown elsewhere, with TNFR knockout mice showing no LTD establishment in CA1 neurons [91]. Interestingly, another model of transgenic rats with overexpressed TNF showed increased LTP in CA1 neurons and spatial cognition deficits in behavioural tasks [92]. These results indicate TNF’s negative effect on cognition through modulation of both LTD and LTP.
Notably, there is evidence that directly conflicts with TNF as a booster of synaptic strength. Research by Lewitus and colleagues demonstrates TNF can downregulate synaptic strength in the striatum under certain conditions [93]. In this study, haloperidol treatment resulted in increased AMPA/NMDA ratios in striatal medium spiny neurons (MSNs) in Tnf-/- but not WT mice. In contrast to previous literature, this work illustrates TNF’s ability to dampen synaptic strength rather than increase it under altered function [93, 94]. The authors noted that MSNs are inhibitory cells, and reduced excitatory transmission of these cells would result in reduced overall circuit inhibition [93]. Later, the same group showed that TNF has a protective effect on dopaminergic neurons within the nucleus accumbens in an animal model of cocaine use. It was found that microglial TNF release, regulated by microglia sensing extracellular dopamine through D2 receptors, was able to reduce AMPA/NDMA ratios in D1 expressing MSNs and reduce cocaine sensitisation [95].
Other research using a mouse model of experimental autoimmune encephalomyelitis (EAE) found that microglial TNF was associated with potentiated transmission in the striatum, likely through AMPARs [96]. Anxiety was associated with TNF and altered excitatory transmission in a similar model by the same group [97]. It has been suggested that TNF has region specific effects on AMPAR expression [13]. However, as TNF has displayed opposing effects on synaptic transmission in the same brain region, it is also likely its effect varies based on different conditions, with activity blockade and models of EAE involving the striatum discussed here. As previously mentioned, the concentration of TNF is a critical determinant factor in whether TNF will enhance or inhibit synaptic plasticity; variability in TNF concentration has also been used by others to explain disparate results [89, 98].
Adding to its diverse array of qualifications, TNF has an effect on altering neuronal morphology. Activation of TLR3 signalling following injection of the immunostimulant polyinosinic-polycytidylic acid (poly(I:C)) resulted in TNF dependent remodelling of dendritic spines of pyramidal neurons in the motor cortex [99]. Tnf–/– mice did not show altered dendritic spine dynamics or motor learning deficits following poly (I:C) injection. Interestingly, depletion of microglia did not alter the effects of poly (I:C) administration, indicating that peripheral immune cells are the determinant source of TNF that produce the observed dendritic spine alterations. The importance of peripheral TNF has been demonstrated elsewhere, where increases in peripheral TNF preceded microglial activation and increased dendritic spine turnover in the somatosensory cortex of mice in an EAE model [100]. This is not to discount the importance of microglia in TNF signalling; microglia coordinate numerous TNF effects and have been implicated as important in recruiting peripheral immune cells into the brain in response to peripheral TNF release [101]. Through NF-κB signalling, TNF has also been shown to inhibit neurite outgrowth in mouse neurons in vitro [102].
Importantly, the effect of TNF on excitatory/inhibitory receptor regulation, glial cells, glutamate regulation and intrinsic plasticity means TNF can contribute to vulnerability to glutamate induced excitotoxic cell death [65, 103, 104]. Illustrating this point, treatment with soluble TNFR1 was able to protect against cell death in a model of spinal cord injury in rats [64]. However, TNF does have variable and conflicting effects on synaptic strength, which must be considered when discussing the role of TNF in synaptic plasticity during AUD.
ALCOHOL EXPOSURE AND TNF EXPRESSION
Interestingly, early research found that in vitro cultures of monocytes or rats injected with alcohol showed an abolished or reduced TNF response following immune challenge [105, 106]. This effect was supported using human blood, with ethanol pretreatment reducing TNF levels in response to LPS in vitro [107, 108]. A possible mechanism for alcohol’s ability to acutely diminish TNF levels includes an interaction with TACE function to reduce TNF release [109, 110]. More recent research suggests that LPS tolerance following ethanol exposure in human monocytes is due to alterations in heat shock factor 1 and heat shock protein 70 [111]. In support of ethanol working at a post translational level to inhibit TNF release, increases in TNF mRNA expression are seen in animal models after ethanol administration while protein levels in both the brain and serum remain unaffected [112, 113]. Further supporting alcohol’s role as an immunosuppressive agent, ethanol pretreatment lowers the release of TNF and other cytokines in the brain following traumatic brain injury; similarly, intragastric administration of alcohol in rats for 3 days prior to physical trauma reduces TNF release [114, 115]. Other short term binge models in rats report no significant increases in TNF protein expression in serum, brain or other tissues compared to controls, although other markers of neuroinflammation can be found [116–118]. It should be noted that TNFR and TLR4 pathways are not the only inducers of TNF expression, and alcohol shows minimal effect on TNF release via TLR2 pathways in human monocytes [119].
In contrast to in vivo models and some in vitro models, a range of studies show that both acute and chronic ethanol exposure enhance the expression and release of TNF (Table 1) [46, 120–123]. While there may be multiple mechanisms for TNF release after ethanol consumption, one suggestion involves neuroimmune potentiation after drug use, resulting from the ‘priming’ of microglia which boosts TNF levels following alcohol consumption [10, 13, 123, 124]. It is also likely that the systemic inflammation that can arise in AUD contributes to the enhanced release of TNF after alcohol consumption. This is illustrated in a post-mortem investigation of individuals with AUD, which found the DNA binding patterns of NF-κβ in the prefrontal cortex were modified resulting in inhibition of κβ regulated transcription and upregulation of genes usually repressed by p50 homodimers, which includes TNF [125, 126]. It is clear that differential effects on TNF levels have been observed between in vitro and in vivo results, furthered by disparity between species and brain regions. It is also known that different patterns of alcohol administration can alter neuroimmune gene expression [127]. However, a variable trend of acute alcohol exposure being able to diminish TNF expression with prolonged alcohol exposure increasing TNF expression is observed.
Table 1
Ethanol induced changes in TNF gene expression and protein levels in the brain of rodents
| Ethanol Treatment | Study | Brain region | mRNA levels | Protein levels |
| Mice | ||||
| Acute – 5, 10 or 15 hours of continuous infusion of EtOH (total amount 156 mg/25 g) into the right internal jugular vein | [220] | Whole brain | ↑ | ↑ |
| Acute – 5 g/kg EtOH via intragastric gavage, 25% EtOH (w/v) daily for 10 days | [221] | Whole brain | ↑ | ↑ |
| Acute – 6 g/kg EtOH via intragastric gavage for 10 days | [222] | Hippocampus, cerebellum or cortex | No change | |
| Acute – 5 g/kg EtOH via intragastric gavage daily for 10 days | [223] | Whole brain and serum | ↑ | ↑ |
| Chronic – 5% (v/v) EtOH (36% ethanol-derived calories) or an isocaloric control diet for 5 weeks | [46] | Cerebellum | ↑ | |
| Chronic – 10% EtOH (v/v) in drinking water and solid diet ad libitum for 5 months | [224] | Striatum | ↑ striatum | |
| Rats | ||||
| Acute – 7% EtOH (w/v) for either 15 days ad libitum access or 3 intermittent 5 day binges | [225] | Cortex | ↓acute ↑ chronic | |
| Acute – 2 g/kg EtOH injected intraperitoneally at 3, 9, 15, 18 hours | [226] | Hypothalamus, paraventricular nucleus of the hypothalamus, hippocampus, cerebellum | Hypothalamus =↓ at 3hrs and 9hrs, ↑ 18hrs Hippocampus =↓ 3, 9 and 18hrs Cerebellum =↓ at 3hrs, 9hrs PVN =↓ 3hrs | |
| Acute (single 14 hour exposure), subchronic (1 week intermittent exposure), chronic (6 week intermittent exposure) EtOH vapor | [227] | Basolateral amygdala, nucleus accumbens, and ventral tegmental area | ↑ acute and subchronic in all 3 areas ↑ chronic in the NAC | |
| Acute – 3 g/kg 25% EtOH (v/v) via intragastric gavage every 8 hours for 4 days | [118] | Anterior cerebellar vermis, cingulate cortex, frontal cortex, hippocampus, hypothalamus, striatum | No difference observed | |
| Acute – 25% EtOH (w/v) in drinking water or an isocaloric dextrose diet via intragastric gavage every 8 hours over 4 days | [116] | Hippocampus | No difference observed | |
| Acute – 25% EtOH (w/v) or an isocaloric dextrose diet via intragastric gavage every 8h for 4 days | [117] | Hippocampus and entorhinal cortex | No difference observed | |
| Chronic – 10 weeks voluntary consumption and 4 g/kg EtOH intragastric challenge | [226] | Paraventricular nucleus of the hypothalamus, hippocampus, amygdala | Paraventricular nucleus of the hypothalamus (PVN) = No change Hippocampus =↓ at 3hrs Amygdala = Trend for ↓ at 3 hours | |
| Chronic – 5.0 g/kg via intragastric gavage, 20% EtOH (w/v) on a 2day on/2day off schedule | [228] | Frontal cortex | ↑ |
ALCOHOL’S EFFECT ON TNF IN HUMAN MODELS
As shown in Table 1, there is a general consensus in rodent models that acute ethanol decreases TNF while chronic ethanol treatment increases TNF; however, depending on brain region and alcohol exposure model, gene and protein expression of TNF can still vary [128]. Similar disparities are seen in human studies, shown in Table 2.
Table 2
Ethanol induced changes in TNF levels in humans
| Participants | Study | Method | TNF |
| 7 female and 6 male healthy controls, and 9 females and 19 male participants with AUD | [229] | Cerebrospinal fluid levels collected on days 4 and 25 of abstinence | No differences on day 4, ↓ in alcoholics after 25 days |
| 26 female and 28 male healthy controls, and 27 female and 54 male participants with AUD | [230] | Whole blood plasma | ↑ |
| 18 healthy male controls and 30 male participants with AUD | [231] | Serum levels collected during withdrawal at 1, 7 and 14 days | ↑ consistently elevated during the whole period of withdrawal |
| 30 healthy controls and 30 male participants with AUD | [232] | Blood plasma collected during withdrawal at 15–25 hours | ↑ |
| 20 healthy controls and 43 participants with AUD | [233] | Blood sample collected during withdrawal at 12–36 hours | No significant differences |
| 16 healthy controls and 40 participants with AUD | [217] | Blood plasma collected during withdrawal at onset and end of 3 weeks | ↑ onset ↓ decreases during withdrawal but remains increased compared to controls |
| 23 healthy male subjects consuming 4 standard drinks daily for 17 days | [234] | Blood plasma | No differences seen after 17 days |
| 14 healthy controls and 32 participants with AUD. Some participants had AALD | [235] | Blood plasma collected at time of admission (withdrawal around 3 weeks) and 30 days later | No differences in alcoholic patients without clinically apparent liver disease, with alcoholic cirrhosis, or in non-alcoholic healthy controls ↑severe alcoholic hepatitis |
| 459 participants ranging from non-drinkers to heavy drinkers. 137 of these participants were hospitalized for AUD | [236] | Blood plasma collected across a number of hospitals around Spain | ↑ admitted alcoholics ↑ higher TNF levels in alcoholics with liver disease |
| 21 participants with compensated alcoholic liver cirrhosis (ALC) and 23 with decompensated ALC | [237] | Blood sample collected on admission (withdrawal varied) | No difference ↑ decompensated ALC |
| 11 male and 4 female healthy controls, and 11 male and 4 female participants with AUD | [138] | Blood samples collected, peripheral blood mononuclear cells isolated and cultured | No difference was observed with LPS stimulation between groups |
Investigations in humans have shown significantly elevated plasma levels of TNF and IL-6 in alcohol dependent patients compared to healthy controls [129]. Increased plasma TNF was observed at a relatively constant level throughout 14 days of withdrawal and was not associated with self-reported craving or withdrawal symptoms. Keifer and colleagues corroborated this observation, showing a positive association between duration of alcohol abuse but no direct association with self-rated craving during withdrawal [130]. Another study found plasma TNF that was upregulated in alcohol dependent patients, and was able to find a significant and positive association with alcohol craving both at the start and end of a three week alcohol withdrawal period [33]. Importantly, the individuals in this latter study did not have alcohol associated liver disease (AALD), which would be a confounding source of TNF. Increased TNF levels in those with AUD is also supported by Zahr and colleagues [131]. These results highlight an interesting, albeit variable, effect of TNF on withdrawal during AUD.
In contrast, a study of healthy men without AUD showed that the daily consumption of approximately four glasses of whisky for 17 days did not affect plasma TNF levels [132]. In individuals with AUD, levels of TNF within cerebrospinal fluid did not differ significantly to control groups [133]. Another study saw no significant differences in TNF plasma levels between Spanish populations of self-reported alcohol abstainers and light, moderate and heavy drinkers [134]. Similar results were found in AUD patients without AALD and with compensated liver disease in multiple studies [135, 136]. Interestingly, a contrasting study found that elevations in serum TNF predicted decreased long term survival in patients with AALD [137]. Taken together, alcohol related TNF levels in humans show marked differences between studies, even accounting for AALD.
The majority of these studies use subjects who enter clinical settings as patients during withdrawal. Therefore, it is possible that variability in the neuroimmune state of the subject could contribute to incompatible results. Recent studies using positron emission tomography (PET) scans to measure translocator protein (TSPO) binding in withdrawn or abstinent individuals with a history of long term AUD show reduced microglial and astrocytic activation in the striatum and hippocampus compared to controls [138, 139]. Another study showed similar results in total brain TSPO binding, but this difference disappeared when both alcohol dependant patients and healthy controls possessed a TSPO rs6971 polymorphism which confers higher binding affinity for TSPO [140]. The same group later showed that in vitro autoradiography with the TSPO ligand [3H]PK11195 had higher levels of binding than in vivo PET with [11C]PBR28 in the brains of alcohol exposed rats, indicating that observations in the aforementioned human studies may be a result of imaging methodology rather than a true effect [141]. Furthermore, histological results in the brains of humans with alcohol use disorder show region specific increases in microglia immunoreactivity, indicating increased neuroinflammation [142–144]. However, long term alcohol use disorder resulting in a ‘blunting’ of the neuroimmune response, perhaps due to lowered glial density or glial adaptations, remains a possibility.
Additionally, the reliability of serum TNF as an indicator of brain TNF must be considered. While TNF is able to be transported across the BBB, previous work has shown significant increases in TNF within the brain following a 10 day regiment of intragastric ethanol exposure that were not observed in serum [113]. Similarly, other cytokine changes in response to ethanol have previously been restricted to the CNS with no changes in serum levels being detected [145, 146]. Admittedly, these studies were both in mice and it is possible that TNF serum levels may be more representative of brain TNF in the human. However, these results imply the probability that increased TNF within the brain may not be apparent when measuring serum.
TNF’S INVOLVEMENT IN AUD
TNF’s ability to modulate synaptic strength as discussed in this review has implications with drug associated neuroinflammation and neuroplasticity. However, it is difficult to contextualise the neuroplastic effects of TNF in AUD. Firstly, there are contrasting results on the effect of alcohol on TNF levels in those with AUD, and concentration appears to be pivotal in the effect of TNF on neuroplasticity. Secondly, TNF is a single molecule among a family of neuroimmune signals and inflammatory molecules. These cytokines often act in concert in parallel, possibly redundant, pathways that converge on NF-κB transcription and it is therefore difficult to attribute effect to a single immune molecule [62, 147]. Despite this, TNF is in a relatively privileged position of being a potent inflammatory molecule and a modulator of synaptic plasticity and this section will discuss TNF’s role in AUD rather than the collective role of all inflammatory cytokines.
To begin with an unforgiving perspective, ethanol consumption was not significantly decreased in TNFR1 knockout mice [148]. In contrast, interleukin 1 receptor (IL-1R) knockout mice showed significant decreases in ethanol consumption and double knockouts for IL-1R and TNFR1 showed marked decreases in ethanol consumption. However, these results imply that TNF/TNFR1 signalling is not involved in regulating ethanol consumption and do not exclude a contributory effect of TNF on altered neurotransmission, which may affect other aspects of AUD. Demonstrating TNF’s effect on drug related adaptation, TNF has been correlated with alcohol craving in humans, associated with ethanol withdrawal-induced anxiety in mice and is involved in cocaine use in mice [33, 130, 149, 150]. In investigations involving cocaine use, mice subjected to maternal separation show increased levels of TNF gene expression in the nucleus accumbens and the prefrontal cortex in a sex dependent manner, with females experiencing no effect [150]. Importantly, mice who experienced maternal separation showed a place preference for cocaine where their control littermates did not. This preference was abolished after administration of Xpro1595, which blocks solTNF. Although this investigation was not related to AUD, it supports TNF as a regulator of drug induced plasticity and highlights the relationship between stress activated neuroimmune signalling and drug use [13].
A proposed mechanism of brain damage in AUD involves adaptations in GABAergic and glutamatergic signalling leading to higher susceptibility to excitotoxic cell death during withdrawal. Although the effect of alcohol on glutamate signalling is complex, it has been proposed that glutamate excitotoxicity during alcohol use disorder is linked to neurodegeneration [151]. Additionally, while evidence supporting this mechanism in vivo is not clearly demonstrated, drugs used to reduce withdrawal symptoms such as benzodiazepines work by interacting with GABA receptors to reduce excitatory transmission [152, 153]. TNF’s augmentation of excitatory transmission deserves discussion in this context. Theoretically, TNF’s effect on susceptibility to excessive calcium influx, through calcium permeable AMPAR expression, and positive enhancement of glutamate release, should create an optimal environment for excitotoxic cell death [57]. Surprisingly, while there are broad associations, there is a lack of definitive evidence implicating TNF in withdrawal related excitotoxic changes. For example, TNF is known to increase GluR2 subunit containing AMPARs in some models, and the brains of individuals with AUD show increased expression of GluR2 and GluR3 subunit containing AMPARs in the cingulate cortex [66, 154]. However, links such as these are tenuous and do not demonstrate a causative role. Due to the role of TNF in potentiating excitotoxicity, its role in AUD withdrawal warrants investigation. The validity of this topic for further investigation is increased by the observation that TNF administration can manifest increased anxiety related withdrawal symptoms following alcohol withdrawal in mice [149].
In regards to human TNF polymorphisms, a meta-analysis found an association between alcohol dependence and TNF polymorphism in individuals with AALD [155]. However, this relationship was not present in those without AALD and further analysis showed this link was more representative of AALD and AUD rather than TNF polymorphisms. Indeed, associations between TNF and AALD are well documented in some populations [134, 155, 156]. In other populations, no links between TNFR1 or TNFR2 polymorphisms and AALD are found [157]. Therefore, while there is no evidence to support TNF related predisposition to AUD, there is the possibility that TNF polymorphisms could amplify peripheral inflammation, leading to increased TNF levels to exacerbate neurodegeneration and altered neurotransmission. Somewhat nullifying this proposition, a study of common TNF polymorphisms in a Japanese population of alcoholics showed no association between TNF polymorphisms and brain atrophy [158]. An association was found between lymphotoxin, previously known as TNF-β, related alleles and atrophy which is of interest as lymphotoxin signals through both TNFR1 and TNFR2. Polymorphisms involving TNF and brain derived neurotrophic factor (BDNF) genes have been observed to affect spatial memory retention, which is interesting in its own right [159].
Intriguingly, it is possible TNF may play an almost ‘invisible’ role in AUD. In hippocampal rat neurons, it was shown that the presence of a cannabinoid receptor 1 (CB1R) agonist reduced TNF induced AMPAR membrane expression [160]. In alcoholic patients, CB1R availability as measured through PET was lower both during withdrawal and after alcohol consumption compared to healthy controls [161]. Downregulation of CB1R is also observed in animals chronically exposed to ethanol [162, 163]. Therefore, a possible mechanism exists involving changes in neuronal excitability associated with CB1R and TNF in AUD. Notably, changes in the regulation of TNF could result in strengthening or dampening of synaptic transmission independent of changes in TNF brain or serum level.
While this review highlights TNF’s potentiation of excitatory transmission, there is some evidence that TNF protects against excitotoxicity in cortical and hippocampal neurons [66, 164–166]. Of note, investigators showed that pretreatment of murine cortical neurons with ethanol prevented the neuroprotective effects of TNF against excitotoxic challenge [164]. Additionally, it has been suggested that TNF levels, mediated by drug induced neuroinflammation, and their possibly protective effects against excitatory transmission could drop during withdrawal [13]. However, the sum of human and animal studies taken together imply increases of TNF levels after chronic alcohol consumption. Additionally, some of the studies illustrating neuroprotection involve TNFR2, which is not the primary receptor activated during inflammation induced solTNF release [165].
TNF IN NEUROGENESIS
Neurogenesis describes the generation of new neurons from neural stem cells. Neurogenesis occurs during development and continues throughout adulthood where it is restricted to the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) of the hippocampal dentate gyrus. The neurons born in the SVZ migrate through the rostral migratory stream and become granule and periglomerular neurons within the olfactory bulb whereas neurons born in the SGZ migrate into the granular layer of the dentate gyrus of the hippocampus and become dentate granule cells [167]. The regulation of adult neurogenesis is thought to be involved in memory formation and cognition but is also involved in mood and stress. Recent research has emerged suggesting that inflammation and the immune system itself can influence the way adult neurogenesis occurs in the brain.
While TNF production has been linked to decreased neural stem cell proliferation and shown anti-neurogenic properties, it has demonstrated pro-neurogenic properties in other studies [23, 168–170]. In embryonic neural progenitor cell culture, LPS activated microglia and macrophages inhibited neuronal differentiation or induced neuronal cell death [171, 172]. Treatment with pentoxifylline, an inhibitor of TNF, partially restored neurogenesis [171]. The anti-neurogenic role of TNF has also shown to stimulate differentiation of neuronal progenitor cells into astrocytes [172]. In neuronal spheroid culture, it has been shown that TNF has no effect on differentiation or proliferation and instead induces neuronal progenitor cell migration [168, 173].
The effect of TNF on neurogenesis also depends on whether TNFR1 or TNFR2 signalling is involved. In a TNF receptor knockout model, TNFR1 activation reduced hippocampal neurogenesis in normal conditions and after status epilepticus [169]. Although TNFR2 is known for its neuroprotective effects, it can also induce apoptosis and TNFR2 knockout mouse models show increased proliferation and survival of newborn neurons [169, 174–176]. An investigation using a mouse model of irradiation induced chronic inflammation showed reduced neurogenesis in TNFR2 knockout animals while TNFR1 knockout mice showed little difference compared to controls [177]. This result suggests a role of TNFR2 in promoting neurogenesis after injury. In vivo, TNF knockout models show reduced hippocampal BDNF levels and decreased apical dendrite arborization of CA1 and CA3 pyramidal cells [178]. Another study reported that primary hippocampal neurons in culture reduced neurite outgrowth and dendritic branching in response to TNF [179]. Overall, the proposed mechanism of TNF’s modulation of neurogenesis that is generally accepted involves disruption of I kappa β kinase/ NF-κβ (IKK/NF-κβ) signalling in neural progenitor cells [173]. This process results in an upregulation of Cyclin D1, an important protein for cell cycle progression, which promotes the passage through the G1/S restriction point therefore encouraging proliferation of neural progenitors cells [172, 173]. However, the literature on TNF’s role on neurogenesis varies and further research needs to evaluate the effects of TNF and its receptors on neuronal progenitor cells in neurogenesis.
ALCOHOL’S EFFECT ON TNF AND NEUROGENESIS
Decreased neurogenesis contributes to both anxiety and depression and selective serotonin reuptake inhibitor antidepressants can alleviate and promote hippocampal neurogenesis [180]. Under healthy conditions, stem cells in the brain are under constant stimulation to proliferate, migrate, differentiate and survive. However, pathological conditions like AUD impact these neurogenic stages with differential effects between alcohol intoxication and withdrawal. Ethanol intoxication studies in vivo have shown that the consumption of ethanol decreases neurogenesis through ethanol’s effect on cell proliferation and cell survival [180–185]. However, studies observing abstinence of ethanol after ethanol dependence show increases in neurogenesis [186]. Some studies conflict with the general consensus that ethanol intoxication inhibits neural stem cell proliferation and have observed no effect after 10 days of ethanol consumption, or an increase in proliferation after chronic ethanol exposure [183, 187, 188].
AUD is known to disrupt neurogenesis and hippocampal integrity [182]. Although the exact mechanism is unknown, numerous mechanisms have been proposed. Ethanol’s interaction with neurotransmission may be relevant due to the importance of glutamate and GABA signalling in hippocampal neurogenesis [189]. BDNF is a modulator of neurotransmitters and increases neurogenesis through enhancement of cell birth, survival, and maturation within the hippocampal SGZ [190]. After ethanol consumption, levels of serum BDNF decrease in humans and can return to baseline after alcohol abstinence [191–193]. BDNF levels have also been shown to decrease in rodents in the hippocampus, cortex and hypothalamus [194–196]. Finally, there is considerable evidence suggesting ethanol has significant cellular effects on glia that impact the hippocampal neurogenic niche. Excessive consumption of ethanol results in activation and damage to astrocytes which are primary components of the neural stem cell niche [197–203]. Activation of microglia can also inhibit neurogenesis [204]. Microglial TNF release during neuroinflammation is a key contributor to inflammation induced death of newly formed hippocampal neural progenitor cells in the adult brain after injury [205]. Microglia are activated by ethanol consumption depending on their phenotype and inflammatory environment and the duration, dose, and pattern of alcohol exposure [116, 117, 206–208]. Other cytokines released by microglia such as IL-1β and IL-6 are also thought to contribute to inhibition of neurogenesis [209, 210].
However, limited studies have explored the relationship between alcohol, TNF and adult neurogenesis. Broadly, TNF is usually explored in conjunction with other cytokines and is rarely studied individually. Both LPS and ethanol treated mice have shown to increase proinflammatory cytokines such as TNF, monocyte chemoattractant protein-1 (MCP1) and IL-1β and reduce the proliferation of hippocampal neural progenitors and newly born neuron differentiation in mice [211–213]. However, the role of TNF in neurogenic deficits after alcohol exposure should not be overstated. One study showed reversal of ethanol’s inhibition of neurogenesis by neutralising IL-1β and blocking the IL-1β receptor with an antagonist against IL-1R, an effect that could not be replicated with neutralizing antibodies to TNF or MCP1 [214]. Antioxidant drugs have been tested to rescue the neurogenic deficits elicited after ethanol consumption. Butylated hydroxytoluene (BHT) has been shown to restore ethanol inhibition of neurogenesis by blocking NF-κB induction of proinflammatory genes [215, 216]. Rolipram, a phosphodiesterase-4 inhibitor, has also been tested and induced an increase in neurogenesis after ethanol consumption [214]. The success of these treatments supports a role of inflammatory cytokines in mediating ethanol’s inhibition of neurogenesis but does not highlight TNF as an essential signal in this process.
CONCLUDING REMARKS
TNF has a demonstrated effect on synaptic plasticity and neurogenesis in vitro and in animal models. There is some evidence for TNF having an important role during withdrawal, with associations between alcohol craving and TNF in humans and in animal studies showing a direct effect of TNF on anxiety during withdrawal [149, 217]. This role in anxiety during withdrawal may be important when considering relapse and further studies involving the blocking of TNF during withdrawal may be beneficial. Its possible involvement in excitotoxicity during withdrawal is also an interesting research avenue. Additionally, TNF has prominent involvement in peripheral inflammation during AUD, particularly with AALD, which would exacerbate other neuroimmune signals and their effects [218, 219]. However, there is a lack of definitive evidence for TNF having a clearly defined, unique effect on neuroplasticity and neurogenesis in AUD. Rather, it is likely TNF contributes to neuroimmune signalling within a party of cytokines [32, 62]. Understanding TNF in AUD is also complicated by its differential effects on synaptic strength in different brain regions, which remain to be elucidated. In our opinion, the relevance of TNF in AUD going forward should be decided using animal models of voluntary drinking with inducible knockdowns of TNFR1 or TACE followed by behavioural testing during withdrawal. Furthermore, the roles of TNF should be further investigated to understand its effect on neurotransmission in physiology and pathology.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
This work is supported by the Australian Research Council Future fellowship to S.E.B and National Health and Medical Research funding to S.E.B.
REFERENCES
[1] | Brown RE , Milner PM . The legacy of Donald O. Hebb: more than the Hebb Synapse. Nat Rev Neurosci. (2003) ;4: (12):1013–9. |
[2] | Fox K , Stryker M . Integrating Hebbian and homeostatic plasticity: introduction. Philos Trans R Soc Lond B Biol Sci. (2017) ;372: (1715). |
[3] | Turrigiano GG , Nelson SB . Hebb and homeostasis in neuronal plasticity. Current Opinion in Neurobiology. (2000) ;10: (3):358–64. |
[4] | Korpi ER , Hollander B den , Farooq U , Vashchinkina E , Rajkumar R , Nutt DJ , et al. Mechanisms of Action and Persistent Neuroplasticity by Drugs of Abuse. Pharmacol Rev. (2015) ;67: (4):872–1004. |
[5] | Lovinger DM , Kash TL . Mechanisms of Neuroplasticity and Ethanol’s Effects on Plasticity in the Striatum and Bed Nucleus of the Stria Terminalis. Alcohol Res. (2015) ;37: (1):109–24. |
[6] | Grant BF , Chou SP , Saha TD , Pickering RP , Kerridge BT , Ruan WJ , et al. Prevalence of 12-Month Alcohol Use, High-Risk Drinking, and DSM-IV Alcohol Use Disorder in the United States, -to -Results From the National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry. (2017) ;74: (9):911–23. |
[7] | Rehm J , Room R , Graham K , Monteiro M , Gmel G , Sempos CT . The relationship of average volume of alcohol consumption and patterns of drinking to burden of disease: an overview. Addiction. (2003) ;98: (9):1209–28. |
[8] | Litten RZ , Ryan ML , Falk DE , Reilly M , Fertig JB , Koob GF . Heterogeneity of Alcohol Use Disorder: Understanding Mechanisms to Advance Personalized Treatment. Alcoholism: Clinical and Experimental Research. (2015) ;39: (4):579–84. |
[9] | Coppens V , Morrens M , Destoop M , Dom G . The Interplay of Inflammatory Processes and Cognition in Alcohol Use Disorders-A Systematic Review. Front Psychiatry. (2019) ;10: :632. |
[10] | Crews FT , Lawrimore CJ , Walter TJ , Coleman LG . The role of neuroimmune signaling in alcoholism. Neuropharmacology. (2017) ;122: :56–73. |
[11] | Erickson EK , Grantham EK , Warden AS , Harris RA . Neuroimmune signaling in alcohol use disorder. Pharmacology Biochemistry and Behavior. (2019) ;177: :34–60. |
[12] | Mayfield J , Ferguson L , Harris RA . Neuroimmune signaling: a key component of alcohol abuse. Current Opinion in Neurobiology. (2013) ;23: (4):513–20. |
[13] | McGrath AG , Briand LA . A potential role for microglia in stress- and drug-induced plasticity in the nucleus accumbens: A mechanism for stress-induced vulnerability to substance use disorder. Neurosci Biobehav Rev. (2019) ;107: :360–9. |
[14] | Beumer W , Gibney SM , Drexhage RC , Pont-Lezica L , Doorduin J , Klein HC , et al. The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. Journal of Leukocyte Biology. (2012) ;92: (5):959–75. |
[15] | Crews F , Zou J , Qin L . Induction of Innate Immune Genes in Brain Create the Neurobiology of Addiction. Brain Behav Immun. (2011) ;25: (Suppl 1):S4–12. |
[16] | Lacagnina MJ , Rivera PD , Bilbo SD . Glial and Neuroimmune Mechanisms as Critical Modulators of Drug Use and Abuse. Neuropsychopharmacology. (2017) ;42: (1):156–77. |
[17] | Mayfield J , Harris RA . The Neuroimmune Basis of Excessive Alcohol Consumption. Neuropsychopharmacology. (2017) ;42: (1):376. |
[18] | Blednov YA , Benavidez JM , Geil C , Perra S , Morikawa H , Harris RA . Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain, Behavior, and Immunity. (2011) ;25: :S92–105. |
[19] | Vassalli P . The pathophysiology of tumor necrosis factors. Annu Rev Immunol. (1992) ;10: :411–52. |
[20] | Arnett HA , Mason J , Marino M , Suzuki K , Matsushima GK , Ting JP . TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. (2001) ;4: (11):1116–22. |
[21] | Probert L . TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience. (2015) ;302: :2–22. |
[22] | Ben Achour S , Pascual O . Glia: The many ways to modulate synaptic plasticity. Neurochemistry International. (2010) ;57: (4):440–5. |
[23] | Calabrese F , Rossetti AC , Racagni G , Gass P , Riva MA , Molteni R . Brain-derived neurotrophic factor: a bridge between inflammation and neuroplasticity. Front Cell Neurosci. (2014) ;8: :430. |
[24] | Innes S , Pariante CM , Borsini A . Microglial-driven changes in synaptic plasticity: A possible role in major depressive disorder. Psychoneuroendocrinology. (2019) ;102: :236–47. |
[25] | Okun E , Griffioen KJ , Mattson MP . Toll-like receptor signaling in neural plasticity and disease. Trends in Neurosciences. (2011) ;34: (5):269–81. |
[26] | Vezzani A , Viviani B . Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. (2015) ;96: (Pt A):70–82. |
[27] | Wu Y , Dissing-Olesen L , MacVicar BA , Stevens B . Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. (2015) ;36: (10):605–13. |
[28] | McCool BA . Ethanol modulation of synaptic plasticity. Neuropharmacology. (2011) ;61: (7):1097–108. |
[29] | Abrahao KP , Salinas AG , Lovinger DM . Alcohol and the Brain: Neuronal Molecular Targets, Synapses, and Circuits. Neuron. (2017) ;96: (6):1223–38. |
[30] | Peferoen L , Kipp M , van der Valk P , van Noort JM , Amor S . Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology. (2014) ;141: (3):302–13. |
[31] | Bala S , Marcos M , Gattu A , Catalano D , Szabo G . Acute Binge Drinking Increases Serum Endotoxin and Bacterial DNA Levels in Healthy Individuals. PLOS ONE. (2014) ;9: (5):e96864. |
[32] | Crews FT , Vetreno RP . Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology. (2016) ;233: (9):1543–57. |
[33] | Leclercq S , Cani PD , Neyrinck AM , Stärkel P , Jamar F , Mikolajczak M , et al. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav Immun. (2012) ;26: (6):911–8. |
[34] | Lu Y-C , Yeh W-C , Ohashi PS . LPS/TLR4 signal transduction pathway. Cytokine. (2008) ;42: (2):145–51. |
[35] | Wang X , Chu G , Yang Z , Sun Y , Zhou H , Li M , et al. Ethanol directly induced HMGB1 release through NOX2/NLRP1 inflammasome in neuronal cells. Toxicology. (2015) ;334: :104–10. |
[36] | Liu A , Fang H , Dirsch O , Jin H , Dahmen U . Oxidation of HMGB1 Causes Attenuation of Its Pro-Inflammatory Activity and Occurs during Liver Ischemia and Reperfusion. PLOS ONE. (2012) ;7: (4):e35379. |
[37] | Corti A , Ghezzi P . Tumor Necrosis Factor: Methods and Protocols. Springer Science & Business Media; 2004. pp 274. |
[38] | Gérard C , Bruyns C , Marchant A , Abramowicz D , Vandenabeele P , Delvaux A , et al. Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. Journal of Experimental Medicine. (1993) ;177: (2):547–50. |
[39] | McCoy MK , Tansey MG . TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. Journal of Neuroinflammation. (2008) ;5: (1):45. |
[40] | Moss ML , Sklair-Tavron L , Nudelman R . Drug Insight: tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat Rev Rheumatol. (2008) ;4: (6):300–9. |
[41] | Brenner D , Blaser H , Mak TW . Regulation of tumour necrosis factor signalling: live or let die. Nature Reviews Immunology. (2015) ;15: (6):362–74. |
[42] | Liu Z . Molecular mechanism of TNF signaling and beyond. Cell Res. (2005) ;15: (1):24–7. |
[43] | Wallach D , Varfolomeev EE , Malinin NL , Goltsev YV , Kovalenko AV , Boldin MP . Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. (1999) ;17: (1):331–67. |
[44] | Li N , Karin M . Is NF-κB the sensor of oxidative stress? The FASEB Journal. (1999) ;13: (10):1137–43. |
[45] | Natoli G , Costanzo A , Moretti F , Fulco M , Balsano C , Levrero M . Tumor necrosis factor (TNF) receptor 1 signaling downstream of TNF receptor-associated factor 2 nuclear factor κb (NFκB)-inducing kinase requirement for activation of activating protein 1 and NFκB but not of c-Jun n-terminal kinase/stress-activated protein kinase. J Biol Chem. (1997) ;272: (42):26079–82. |
[46] | Lippai D , Bala S , Petrasek J , Csak T , Levin I , Kurt-Jones EA , et al. Alcohol-induced IL-1β in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. Journal of Leukocyte Biology. (2013) ;94: (1):171–82. |
[47] | Harris RA , Bajo M , Bell RL , Blednov YA , Varodayan FP , Truitt JM , et al. Genetic and Pharmacologic Manipulation of TLR4 Has Minimal Impact on Ethanol Consumption in Rodents. J Neurosci. (2017) ;37: (5):1139–55. |
[48] | Blednov YA , Black M , Chernis J , Da Costa A , Mayfield J , Harris RA . Ethanol Consumption in Mice Lacking CD14, TLR2, TLR4, or MyD88. Alcoholism: Clinical and Experimental Research. (2017) ;41: (3):516–30. |
[49] | Gutierrez EG , Banks WA , Kastin AJ . Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. Journal of Neuroimmunology. (1993) ;47: (2):169–76. |
[50] | McCarthy MM , Nugent BM , Lenz KM . Neuroimmunology and neuroepigenetics in the establishment of sex differences in the brain. Nature Reviews Neuroscience. (2017) ;18: (8):471–84. |
[51] | Osborne BF , Turano A , Schwarz JM . Sex differences in the neuroimmune system. Current Opinion in Behavioral Sciences. (2018) ;23: :118–23. |
[52] | Moore CF , Lynch WJ . Alcohol preferring (P) rats as a model for examining sex differences in alcohol use disorder and its treatment. Pharmacology Biochemistry and Behavior. (2015) ;132: :1–9. |
[53] | Pascual M , Montesinos J , Marcos M , Torres J-L , Costa-Alba P , García-García F , et al. Gender differences in the inflammatory cytokine and chemokine profiles induced by binge ethanol drinking in adolescence. Addict Biol. (2017) ;22: (6):1829–41. |
[54] | Erol A , Karpyak VM . Sex and gender-related differences in alcohol use and its consequences: Contemporary knowledge and future research considerations. Drug and Alcohol Dependence. (2015) ;156: :1–13. |
[55] | Wilsnack RW , Vogeltanz ND , Wilsnack SC , Harris TR . Gender differences in alcohol consumption and adverse drinking consequences: cross-cultural patterns. Addiction. (2000) ;95: (2):251–65. |
[56] | Rizzo FR , Musella A , De Vito F , Fresegna D , Bullitta S , Vanni V , et al. Tumor Necrosis Factor and Interleukin-1β Modulate Synaptic Plasticity during Neuroinflammation. Neural Plast. (2018) ;2018: :8430123. |
[57] | Galic MA , Riazi K , Pittman QJ . Cytokines and brain excitability. Frontiers in Neuroendocrinology. (2012) ;33: (1):116–25. |
[58] | O’Connor JJ . Targeting tumour necrosis factor-α in hypoxia and synaptic signalling. Ir J Med Sci. (2013) ;182: (2):157–62. |
[59] | Park KM , Bowers WJ . Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cellular Signalling. (2010) ;22: (7):977–83. |
[60] | Stellwagen D , Beattie EC , Seo JY , Malenka RC . Differential Regulation of AMPA Receptor and GABA Receptor Trafficking by Tumor Necrosis Factor-α. J Neurosci. (2005) ;25: (12):3219–28. |
[61] | Stellwagen D , Malenka RC . Synaptic scaling mediated by glial TNF-α. Nature. (2006) ;440: (7087):1054–9. |
[62] | Konefal SC , Stellwagen D . Tumour necrosis factor-mediated homeostatic synaptic plasticity in behavioural models: testing a role in maternal immune activation. Philosophical Transactions of the Royal Society B: Biological Sciences. (2017) ;372: (1715):20160160. |
[63] | He P , Liu Q , Wu J , Shen Y . Genetic deletion of TNF receptor suppresses excitatory synaptic transmission via reducing AMPA receptor synaptic localization in cortical neurons. FASEB J. (2012) ;26: (1):334–45. |
[64] | Ferguson AR , Christensen RN , Gensel JC , Miller BA , Sun F , Beattie EC , et al. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci. (2008) ;28: (44):11391–400. |
[65] | Leonoudakis D , Zhao P , Beattie EC . Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. (2008) ;28: (9):2119–30. |
[66] | Rainey-Smith SR , Andersson DA , Williams RJ , Rattray M . Tumour necrosis factor alpha induces rapid reduction in AMPA receptor-mediated calcium entry in motor neurones by increasing cell surface expression of the GluR2 subunit: relevance to neurodegeneration. J Neurochem. (2010) ;113: (3):692–703. |
[67] | Ye L , Huang Y , Zhao L , Li Y , Sun L , Zhou Y , et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem. (2013) ;125: (6):897–908. |
[68] | Habbas S , Santello M , Becker D , Stubbe H , Zappia G , Liaudet N , et al. Neuroinflammatory TNFα Impairs Memory via Astrocyte Signaling. Cell. (2015) ;163: (7):1730–41. |
[69] | Weaver-Mikaere L , Gunn AJ , Mitchell MD , Bennet L , Fraser M . LPS and TNF alpha modulate AMPA/NMDA receptor subunit expression and induce PGE2 and glutamate release in preterm fetal ovine mixed glial cultures. Journal of Neuroinflammation. (2013) ;10: (1):916. |
[70] | Wheeler D , Knapp E , Bandaru VVR , Wang Y , Knorr D , Poirier C , et al. TNFα-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J Neurochem. (2009) ;109: (5):1237–49. |
[71] | Pribiag H , Stellwagen D . TNF-α Downregulates Inhibitory Neurotransmission through Protein Phosphatase 1-Dependent Trafficking of GABAA Receptors. J Neurosci. (2013) ;33: (40):15879–93. |
[72] | Kawasaki Y , Zhang L , Cheng J-K , Ji R-R . Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. (2008) ;28: (20):5189–94. |
[73] | Tilleux S , Hermans E . Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. Journal of Neuroscience Research. (2007) ;85: (10):2059–70. |
[74] | Zou JY , Crews FT . TNFα potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NFκB inhibition. Brain Research. (2005) ;1034: (1):11–24. |
[75] | Sitcheran R , Gupta P , Fisher PB , Baldwin AS . Positive and negative regulation of EAAT2 by NF-κB: a role for N-myc in TNFα-controlled repression. EMBO J. (2005) ;24: (3):510–20. |
[76] | Tolosa L , Caraballo-Miralles V , Olmos G , Lladó J . TNF-α potentiates glutamate-induced spinal cord motoneuron death via NF-κB. Mol Cell Neurosci. (2011) ;46: (1):176–86. |
[77] | Takeuchi H , Jin S , Wang J , Zhang G , Kawanokuchi J , Kuno R , et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. (2006) ;281: (30):21362–8. |
[78] | Bezzi P , Domercq M , Brambilla L , Galli R , Schols D , De Clercq E , et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. (2001) ;4: (7):702–10. |
[79] | Yamamoto M , Kim M , Imai H , Itakura Y , Ohtsuki G . Microglia-Triggered Plasticity of Intrinsic Excitability Modulates Psychomotor Behaviors in Acute Cerebellar Inflammation. Cell Reports. (2019) ;28: (11):2923–2938.e8. |
[80] | Santello M , Bezzi P , Volterra A . TNFα Controls Glutamatergic Gliotransmission in the Hippocampal Dentate Gyrus. Neuron. (2011) ;69: (5):988–1001. |
[81] | Cunningham AJ , Murray CA , O’Neill LA , Lynch MA , O’Connor JJ . Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. (1996) ;203: (1):17–20. |
[82] | Tancredi V , D’Arcangelo G , Grassi F , Tarroni P , Palmieri G , Santoni A , et al. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett. (1992) ;146: (2):176–8. |
[83] | Wang D . Tumor Necrosis Factor-Alpha Alters Electrophysiological Properties of Rabbit Hippocampal Neurons. J Alzheimers Dis. (2019) ;68: (3):1257–71. |
[84] | Singh A , Jones OD , Mockett BG , Ohline SM , Abraham WC . Tumor Necrosis Factor-α-Mediated Metaplastic Inhibition of LTP Is Constitutively Engaged in an Alzheimer’s Disease Model. J Neurosci. (2019) ;39: (46):9083–97. |
[85] | Ren W-J , Liu Y , Zhou L-J , Li W , Zhong Y , Pang R-P , et al. Peripheral Nerve Injury Leads to Working Memory Deficits and Dysfunction of the Hippocampus by Upregulation of TNF-α in Rodents. Neuropsychopharmacol. (2011) ;36: (5):979–92. |
[86] | Prieto GA , Tong L , Smith ED , Cotman CW . TNFα and IL-1β but not IL-18 Suppresses Hippocampal Long-Term Potentiation Directly at the Synapse. Neurochem Res. (2019) ;44: (1):49–60. |
[87] | Butler MP , O’Connor JJ , Moynagh PN . Dissection of tumor-necrosis factor-alpha inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early-but not late-phase LTP. Neuroscience. (2004) ;124: (2):319–26. |
[88] | Cumiskey D , Butler MP , Moynagh PN , O’connor JJ . Evidence for a role for the group I metabotropic glutamate receptor in the inhibitory effect of tumor necrosis factor-alpha on long-term potentiation. Brain Res. (2007) ;1136: (1):13–9. |
[89] | Maggio N , Vlachos A . Tumor necrosis factor (TNF) modulates synaptic plasticity in a concentration-dependent manner through intracellular calcium stores. J Mol Med. (2018) ;96: (10):1039–47. |
[90] | Sama DM , Mohmmad Abdul H , Furman JL , Artiushin IA , Szymkowski DE , Scheff SW , et al. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and Ca2+ dysregulation in aged rats. PLoS ONE. (2012) ;7: (5):e38170. |
[91] | Albensi BC , Mattson MP . Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse. (2000) ;35: (2):151–9. |
[92] | Pettigrew LC , Kryscio RJ , Norris CM . The TNFα-Transgenic Rat: Hippocampal Synaptic Integrity, Cognition, Function, and Post-Ischemic Cell Loss. PLOS ONE. (2016) ;11: (5):e0154721. |
[93] | Lewitus GM , Pribiag H , Duseja R , St-Hilaire M , Stellwagen D . An Adaptive Role of TNFα in the Regulation of Striatal Synapses. J Neurosci. (2014) ;34: (18):6146–55. |
[94] | Kaneko M , Stellwagen D , Malenka RC , Stryker MP . Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. (2008) ;58: (5):673–80. |
[95] | Lewitus GM , Konefal SC , Greenhalgh AD , Pribiag H , Augereau K , Stellwagen D . Microglial TNF-α Suppresses Cocaine-Induced Plasticity and Behavioral Sensitization. Neuron. (2016) ;90: (3):483–91. |
[96] | Centonze D , Muzio L , Rossi S , Cavasinni F , De Chiara V , Bergami A , et al. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci. (2009) ;29: (11):3442–52. |
[97] | Haji N , Mandolesi G , Gentile A , Sacchetti L , Fresegna D , Rossi S , et al. TNF-α-mediated anxiety in a mouse model of multiple sclerosis. Exp Neurol. (2012) ;237: (2):296–303. |
[98] | Coleman LG , Crews FT . Innate Immune Signaling and Alcohol Use Disorders. Handb Exp Pharmacol. (2018) ;248: :369–96. |
[99] | Garré JM , Silva HM , Lafaille JJ , Yang G . CX3CR1+ monocytes modulate learning and learning-dependent dendritic spine remodeling via TNF-α . Nature Medicine. (2017) ;23: (6):714–22. |
[100] | Yang G , Parkhurst CN , Hayes S , Gan W-B . Peripheral elevation of TNF-α leads to early synaptic abnormalities in the mouse somatosensory cortex in experimental autoimmune encephalomyelitis. PNAS. (2013) ;110: (25):10306–11. |
[101] | D’Mello C , Le T , Swain MG . Cerebral Microglia Recruit Monocytes into the Brain in Response to Tumor Necrosis Factorα Signaling during Peripheral Organ Inflammation. J Neurosci. (2009) ;29: (7):2089–102. |
[102] | Gutierrez H , O’Keeffe GW , Gavaldà N , Gallagher D , Davies AM . Nuclear Factor κB Signaling Either Stimulates or Inhibits Neurite Growth Depending on the Phosphorylation Status of p65/RelA. J Neurosci. (2008) ;28: (33):8246–56. |
[103] | Gelbard HA , Dzenko KA , DiLoreto D , del Cerro C , del Cerro M , Epstein LG . Neurotoxic effects of tumor necrosis factor alpha in primary human neuronal cultures are mediated by activation of the glutamate AMPA receptor subtype: implications for AIDS neuropathogenesis. Dev Neurosci. (1993) ;15: (6):417–22. |
[104] | Jara JH , Singh BB , Floden AM , Combs CK . Tumor necrosis factor alpha stimulates NMDA receptor activity in mouse cortical neurons resulting in ERK-dependent death. Journal of Neurochemistry. (2007) ;100: (5):1407–20. |
[105] | D’Souza NB , Bagby GJ , Nelson S , Lang CH , Spitzer JJ . Acute Alcohol Infusion Suppresses Endotoxin-induced Serum Tumor Necrosis Factor. Alcoholism: Clinical and Experimental Research. (1989) ;13: (2):295–8. |
[106] | Nelson S , Mason C , Bagby G , Summer W . Alcohol, Tumor Necrosis Factor, and Tuberculosis. Alcoholism: Clinical and Experimental Research. (1995) ;19: (1):17–24. |
[107] | Arbabi S , Garcia I , Bauer GJ , Maier RV . Alcohol (Ethanol) Inhibits IL-8 and TNF: Role of the p38 Pathway. The Journal of Immunology. (1999) ;162: (12):7441–5. |
[108] | Szabo G , Mandrekar P , Girouard L , Catalano D . Regulation of Human Monocyte Functions by Acute Ethanol Treatment: Decreased Tumor Necrosis Factorα, Interleukin-lβ and Elevated Interleukin-10, and Transforming Growth Factor-β Production. Alcoholism: Clinical and Experimental Research. (1996) ;20: (5):900–7. |
[109] | Taıeb J , Delarche C , Ethuin F , Selloum S , Poynard T , Gougerot-Pocidalo M-A , et al. Ethanol-induced inhibition of cytokine release and protein degranulation in human neutrophils. Journal of Leukocyte Biology. (2002) ;72: (6):1142–7. |
[110] | Zhao X-J , Marrero L , Song K , Oliver P , Chin SY , Simon H , et al. Acute Alcohol Inhibits TNF-α Processing in Human Monocytes by Inhibiting TNF/TNF-α-Converting Enzyme Interactions in the Cell Membrane. The Journal of Immunology. (2003) ;170: (6):2923–31. |
[111] | Muralidharan S , Ambade A , Fulham MA , Deshpande J , Catalano D , Mandrekar P . Moderate Alcohol Induces Stress Proteins HSF1 and hsp70 and Inhibits Proinflammatory Cytokines Resulting in Endotoxin Tolerance. The Journal of Immunology. (2014) ;193: (4):1975–87. |
[112] | Baxter-Potter LN , Henricks AM , Berger AL , Bieniasz KV , Lugo JM , McLaughlin RJ . Alcohol vapor exposure differentially impacts mesocorticolimbic cytokine expression in a sex-, region-, and duration-specific manner. Neuroscience. (2017) ;346: :238–46. |
[113] | Qin L , He J , Hanes RN , Pluzarev O , Hong J-S , Crews FT . Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. Journal of Neuroinflammation. (2008) ;5: (1):10. |
[114] | Gottesfeld Z , Moore AN , Dash PK . Acute Ethanol Intake Attenuates Inflammatory Cytokines after Brain Injury in Rats: A Possible Role for Corticosterone. Journal of Neurotrauma. (2002) ;19: (3):317–26. |
[115] | Greiffenstein P , Mathis KW , Stouwe CV , Molina PE . Alcohol binge before trauma/hemorrhage impairs integrity of host defense mechanisms during recovery. Alcohol Clin Exp Res. (2007) ;31: (4):704–15. |
[116] | McClain JA , Morris SA , Deeny MA , Marshall SA , Hayes DM , Kiser ZM , et al. Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain, Behavior, and Immunity. (2011) ;25: :S120–8. |
[117] | Marshall SA , McClain JA , Kelso ML , Hopkins DM , Pauly JR , Nixon K . Microglial activation is not equivalent to neuroinflammation in alcohol-induced neurodegeneration: The importance of microglia phenotype. Neurobiology of Disease. (2013) ;54: :239–51. |
[118] | Zahr NM , Luong R , Sullivan EV , Pfefferbaum A . Measurement of Serum, Liver, and Brain Cytokine Induction, Thiamine Levels, and Hepatopathology in Rats Exposed to a 4-Day Alcohol Binge Protocol. Alcoholism: Clinical and Experimental Research. (2010) ;34: (11):1858–70. |
[119] | Oak S , Mandrekar P , Catalano D , Kodys K , Szabo G . TLR2- and TLR4-mediated signals determine attenuation or augmentation of inflammation by acute alcohol in monocytes. J Immunol. (2006) ;176: (12):7628–35. |
[120] | Kishore R , McMullen MR , Nagy LE . Stabilization of tumor necrosis factor alpha mRNA by chronic ethanol: role of A + U-rich elements and p38 mitogen-activated protein kinase signaling pathway. J Biol Chem. (2001) ;276: (45):41930–7. |
[121] | Kishore R , Hill JR , McMullen MR , Frenkel J , Nagy LE . ERK1/2 and Egr-1 contribute to increased TNF-alpha production in rat Kupffer cells after chronic ethanol feeding. Am J Physiol Gastrointest Liver Physiol. (2002) ;282: (1):G6–15. |
[122] | Pascual M , Baliño P , Aragón CMG , Guerri C . Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: Role of TLR4 and TLR2. Neuropharmacology. (2015) ;89: :352–9. |
[123] | Marshall SA , Geil CR , Nixon K . Prior Binge Ethanol Exposure Potentiates the Microglial Response in a Model of Alcohol-Induced Neurodegeneration. Brain Sciences. (2016) ;6: (2):16. |
[124] | Walter TJ , Crews FT . Microglial depletion alters the brain neuroimmune response to acute binge ethanol withdrawal. Journal of Neuroinflammation. (2017) ;14: (1):86. |
[125] | Ökvist A , Johansson S , Kuzmin A , Bazov I , Merino-Martinez R , Ponomarev I , et al. Neuroadaptations in Human Chronic Alcoholics: Dysregulation of the NF-κB System. PLOS ONE. (2007) ;2: (9):e930. |
[126] | Ziegler-Heitbrock L . The p50-homodimer mechanism in tolerance to LPS. J Endotoxin Res. (2001) ;7: (3):219–22. |
[127] | Gano A , Doremus-Fitzwater TL , Deak T . Sustained alterations in neuroimmune gene expression after daily, but not intermittent, alcohol exposure. Brain Research. (2016) ;1646: :62–72. |
[128] | Roberto M , Patel RR , Bajo M . Ethanol and Cytokines in the Central Nervous System. In: Grant KA, Lovinger DM, editors. The Neuropharmacology of Alcohol. Cham: Springer International Publishing; 2018. pp. 397-431. (Handbook of Experimental Pharmacology). |
[129] | Heberlein A , Käser M , Lichtinghagen R , Rhein M , Lenz B , Kornhuber J , et al. TNF-α and IL-6 serum levels: Neurobiological markers of alcohol consumption in alcohol-dependent patients? Alcohol. (2014) ;48: (7):671–6. |
[130] | Kiefer F , Jahn H , Schick M , Wiedemann K . Alcohol intake, tumour necrosis factor-alpha, leptin and craving: factors of a possibly vicious circle? Alcohol Alcohol. (2002) ;37: (4):401–4. |
[131] | Zahr NM . Peripheral TNFα elevations in abstinent alcoholics are associated with hepatitis C infection. PLOS ONE. (2018) ;13: (2):e0191586. |
[132] | Sierksma A , Patel H , Ouchi N , Kihara S , Funahashi T , Heine RJ , et al. Effect of Moderate Alcohol Consumption on Adiponectin, Tumor Necrosis Factor-α, and Insulin Sensitivity. Diabetes Care. (2004) ;27: (1):184–9. |
[133] | Umhau JC , Schwandt M , Solomon MG , Yuan P , Nugent A , Zarate CA , et al. Cerebrospinal Fluid Monocyte Chemoattractant Protein-1 in Alcoholics: Support for a Neuroinflammatory Model of Chronic Alcoholism. Alcoholism: Clinical and Experimental Research. (2014) ;38: (5):1301–6. |
[134] | Gonzalez-Quintela A , Campos J , Loidi L , Quinteiro C , Perez L-F , Gude F . Serum TNF-α levels in relation to alcohol consumption and common TNF gene polymorphisms. Alcohol. (2008) ;42: (6):513–8. |
[135] | Daniluk J , Szuster-Ciesielska A , Drabko J , Kandefer-Szerszeń M . Serum cytokine levels in alcohol-related liver cirrhosis. Alcohol. (2001) ;23: (1):29–34. |
[136] | Nicolaou C , Chatzipanagiotou S , Tzivos D , Tzavellas EO , Boufidou F , Liappas IA . Serum cytokine concentrations in alcohol-dependent individuals without liver disease. Alcohol. (2004) ;32: (3):243–7. |
[137] | Felver ME , Mezey E , McGuire M , Mitchell MC , Herlong HF , Veech GA , et al. Plasma Tumor Necrosis Factor α Predicts Decreased Long-Term Survival in Severe Alcoholic Hepatitis. Alcoholism: Clinical and Experimental Research. (1990) ;14: (2):255–9. |
[138] | Hillmer AT , Sandiego CM , Hannestad J , Angarita GA , Kumar A , McGovern EM , et al. In vivo imaging of translocator protein, a marker of activated microglia, in alcohol dependence. Mol Psychiatry. (2017) ;22: (12):1759–66. |
[139] | Kalk NJ , Guo Q , Owen D , Cherian R , Erritzoe D , Gilmour A , et al. Decreased hippocampal translocator protein (18 kDa) expression in alcohol dependence: a [11 C]PBR28 PET study. Transl Psychiatry. (2017) ;7: (1):e996–e996. |
[140] | Kim SW , Wiers CE , Tyler R , Shokri-Kojori E , Jang YJ , Zehra A , et al. Influence of alcoholism and cholesterol on TSPO binding in brain: PET [11C]PBR28 studies in humans and rodents. Neuropsychopharmacology. (2018) ;43: (9):1832–9. |
[141] | Tyler RE , Kim SW , Guo M , Jang YJ , Damadzic R , Stodden T ,et al.. Detecting neuroinflammation in the brain following chronic alcohol exposure in rats: A comparison between in vivo and in vitro TSPO radioligand binding. European Journal of Neuroscience. (2019) ;50: (1):1831–42. |
[142] | He J , Crews FT . Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol. (2008) ;210: (2):349–58. |
[143] | Byun K , Bayarsaikhan D , Bayarsaikhan E , Son M , Oh S , Lee J ,et al.. Microglial AGE-Albumin Is Critical in Promoting Alcohol-Induced Neurodegeneration in Rats and Humans. PLoS One. (2014) ;9: (8). |
[144] | Dennis CV , Sheahan PJ , Graeber MB , Sheedy DL , Kril JJ , Sutherland GT . Microglial proliferation in the brain of chronic alcoholics with hepatic encephalopathy. Metab Brain Dis. (2014) ;29: (4):1027–39. |
[145] | Marshall SA , McKnight KH , Blose AK , Lysle DT , Thiele TE . Modulation of Binge-like Ethanol Consumption by IL-10 Signaling in the Basolateral Amygdala. J Neuroimmune Pharmacol. (2017) ;12: (2):249–59. |
[146] | Qin L , Crews FT . Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. Journal of Neuroinflammation. (2012) ;9: (1):130. |
[147] | Olmos G , Lladó J . Tumor Necrosis Factor Alpha: A Link between Neuroinflammation and Excitotoxicity. Mediators of Inflammation. (2014) ;2014: :1–12. |
[148] | Karlsson C , Schank JR , Rehman F , Stojakovic A , Björk K , Barbier E ,et al.. Proinflammatory signaling regulates voluntary alcohol intake and stress-induced consumption after exposure to social defeat stress in mice. Addiction Biology. (2017) ;22: (5):1279–88. |
[149] | Breese GR , Knapp DJ , Overstreet DH , Navarro M , Wills TA , Angel RA . Repeated Lipopolysaccharide (LPS) or Cytokine Treatments Sensitize Ethanol Withdrawal-Induced Anxiety-Like Behavior. Neuropsychopharmacol. (2008) ;33: (4):867–76. |
[150] | Ganguly P , Honeycutt JA , Rowe JR , Demaestri C , Brenhouse HC . Effects of early life stress on cocaine conditioning and AMPA receptor composition are sex-specific and driven by TNF. Brain, Behavior, and Immunity. (2019) ;78: :41–51. |
[151] | Crews FT , Sarkar DK , Qin L , Zou J , Boyadjieva N , Vetreno RP . Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol Res. (2015) ;37: (2):331–51. |
[152] | Amato L , Minozzi S , Vecchi S , Davoli M . Benzodiazepines for alcohol withdrawal. Cochrane Database of Systematic Reviews. 2010;(3). |
[153] | Collins MA , Neafsey EJ . Excitotoxicity and Adult Brain Damage: An Experimentally Unproven Chain-of-Events. Front Mol Neurosci. 2016;9. |
[154] | Breese CR , Freedman R , Leonard SS . Glutamate receptor subtype expression in human postmortem brain tissue from schizophrenics and alcohol abusers. Brain Research. (1995) ;674: (1):82–90. |
[155] | Kebir O , Gorsane M-A , Blecha L , Krebs M-O , Reynaud M , Benyamina A . Association of inflammation genes with alcohol dependence/abuse: a systematic review and a meta-analysis. Eur Addict Res. (2011) ;17: (3):146–53. |
[156] | Marcos M , Gómez-Munuera M , Pastor I , González-Sarmiento R , Laso F-J . Tumor necrosis factor polymorphisms and alcoholic liver disease: a HuGE review and meta-analysis. Am J Epidemiol. (2009) ;170: (8):948–56. |
[157] | Nguyen-Khac E , Houchi H , Daoust M , Dupas JL , Naassila M . Lack of association between tumour necrosis factor receptor types 1 and 2 gene polymorphism and severe acute alcoholic hepatitis. Eur J Gastroenterol Hepatol. (2010) ;22: (7):794–800. |
[158] | Yamauchi M , Takamatsu M , Maezawa Y , Takagi M , Araki T , Satoh S ,et al.. Polymorphism of Tumor Necrosis Factor-β and Alcohol Dehydrogenase Genes and Alcoholic Brain Atrophy in Japanese Patients. Alcoholism: Clinical and Experimental Research.7S-10S. (2001) ;25: (s2). |
[159] | Yogeetha BS , Haupt LM , McKenzie K , Sutherland HG , Okolicsyani RK , Lea RA ,et al.. BDNF and TNF-α polymorphisms in memory. Mol Biol Rep. (2013) ;40: (9):5483–90. |
[160] | Zhao P , Leonoudakis D , Abood ME , Beattie EC . Cannabinoid receptor activation reduces TNFalpha-induced surface localization of AMPAR-type glutamate receptors and excitotoxicity. Neuropharmacology. (2010) ;58: (2):551–8. |
[161] | Ceccarini J , Hompes T , Verhaeghen A , Casteels C , Peuskens H , Bormans G ,et al.. Changes in Cerebral CB1 Receptor Availability after Acute and Chronic Alcohol Abuse and Monitored Abstinence. J Neurosci. (2014) ;34: (8):2822–31. |
[162] | Basavarajappa BS , Cooper TB , Hungund BL . Chronic ethanol administration down-regulates cannabinoid receptors in mouse brain synaptic plasma membrane. Brain Res. (1998) ;793: (1-2):212–8. |
[163] | Hungund BL , Basavarajappa BS . Role of Endocannabinoids and Cannabinoid CB1 Receptors in Alcohol-Related Behaviors. Annals of the New York Academy of Sciences. (2004) ;1025: (1):515–27. |
[164] | Gahring LC , Carlson NG , Wieggel WA , Howard J , Rogers SW . Alcohol Blocks TNFα but Not Other Cytokine-Mediated Neuroprotection to NMDA. Alcoholism: Clinical and Experimental Research. (1999) ;23: (10):1571–9. |
[165] | Marchetti L , Klein M , Schlett K , Pfizenmaier K , Eisel ULM . Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. (2004) ;279: (31):32869–81. |
[166] | Mattson MP , Goodman Y , Luo H , Fu W , Furukawa K . Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. (1997) ;49: (6):681–97. |
[167] | Zhao C , Deng W , Gage FH . Mechanisms and Functional Implications of Adult Neurogenesis. Cell. (2008) ;132: (4):645–60. |
[168] | Ben-Hur T , Ben-Menachem O , Furer V , Einstein O , Mizrachi-Kol R , Grigoriadis N . Effects of proinflammatory cytokines on the growth, fate, and motility of multipotential neural precursor cells. Molecular and Cellular Neuroscience. (2003) ;24: (3):623–31. |
[169] | Iosif RE , Ekdahl CT , Ahlenius H , Pronk CJH , Bonde S , Kokaia Z ,et al.. Tumor Necrosis Factor Receptor 1 Is a Negative Regulator of Progenitor Proliferation in Adult Hippocampal Neurogenesis. J Neurosci. (2006) ;26: (38):9703–12. |
[170] | Wu JP , Kuo JS , Liu YL , Tzeng SF . Tumor necrosis factor-alpha modulates the proliferation of neural progenitors in the subventricular/ventricular zone of adult rat brain. Neurosci Lett. (2000) ;292: (3):203–6. |
[171] | Liu Y-P , Lin H-I , Tzeng S-F . Tumor necrosis factor-α and interleukin-18 modulate neuronal cell fate in embryonic neural progenitor culture. Brain Research. (2005) ;1054: (2):152–8. |
[172] | Peng H , Whitney N , Wu Y , Tian C , Dou H , Zhou Y ,et al.. HIV-1-infected and/or immune-activated macrophage-secreted TNF-α affects human fetal cortical neural progenitor cell proliferation and differentiation. Glia. (2008) ;56: (8):903–16. |
[173] | Widera D , Mikenberg I , Elvers M , Kaltschmidt C , Kaltschmidt B . Tumor necrosis factor α triggers proliferation of adult neural stem cells via IKK/NF-κB signaling. BMC Neuroscience. (2006) ;7: (1):64. |
[174] | Depuydt B , Loo GV , Vandenabeele P , Declercq W . Induction of apoptosis by TNF receptor 2 in a T-cell hybridoma is FADD dependent and blocked by caspase-8 inhibitors. Journal of Cell Science. (2005) ;118: (3):497–504. |
[175] | Marchetti L , Klein M , Schlett K , Pfizenmaier K , Eisel ULM . Tumor Necrosis Factor (TNF)-mediated Neuroprotection against Glutamate-induced Excitotoxicity Is Enhanced by N-Methyl-D-aspartate Receptor Activation Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-κb pathway. J Biol Chem. (2004) ;279: (31):32869–81. |
[176] | Yang L , Lindholm K , Konishi Y , Li R , Shen Y . Target Depletion of Distinct Tumor Necrosis Factor Receptor Subtypes Reveals Hippocampal Neuron Death and Survival through Different Signal Transduction Pathways. J Neurosci. (2002) ;22: (8):3025–32. |
[177] | Chen Z , Palmer TD . Differential roles of TNFR1 and TNFR2 signaling in adult hippocampal neurogenesis. Brain Behav Immun. (2013) ;30: :45–53. |
[178] | Golan H , Levav T , Mendelsohn A , Huleihel M . Involvement of Tumor Necrosis Factor Alpha in Hippocampal Development and Function. Cereb Cortex. (2004) ;14: (1):97–105. |
[179] | Neumann H , Schweigreiter R , Yamashita T , Rosenkranz K , Wekerle H , Barde Y-A . Tumor Necrosis Factor Inhibits Neurite Outgrowth and Branching of Hippocampal Neurons by a Rho-Dependent Mechanism. J Neurosci. (2002) ;22: (3):854–62. |
[180] | Belmer A , Patkar OL , Lanoue V , Bartlett SE . 5-HT1A receptor-dependent modulation of emotional and neurogenic deficits elicited by prolonged consumption of alcohol. Sci Rep. (2018) ;8: (1):2099. |
[181] | Crews FT , Nixon K . Mechanisms of Neurodegeneration and Regeneration in Alcoholism. Alcohol Alcohol. (2009) ;44: (2):115–27. |
[182] | Geil CR , Hayes DM , McClain JA , Liput DJ , Marshall SA , Chen KY ,et al.. Alcohol and adult hippocampal neurogenesis: promiscuous drug, wanton effects. Prog Neuropsychopharmacol Biol Psychiatry. (2014) ;54: :103–13. |
[183] | Herrera DG , Yague AG , Johnsen-Soriano S , Bosch-Morell F , Collado-Morente L , Muriach M ,et al.. Selective impairment of hippocampal neurogenesis by chronic alcoholism: protective effects of an antioxidant. Proc Natl Acad Sci USA. (2003) ;100: (13):7919–24. |
[184] | Nixon K , Crews FT . Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. J Neurochem. (2002) ;83: (5):1087–93. |
[185] | Richardson HN , Chan SH , Crawford EF , Lee YK , Funk CK , Koob GF ,et al.. Permanent impairment of birth and survival of cortical and hippocampal proliferating cells following excessive drinking during alcohol dependence. Neurobiol Dis. (2009) ;36: (1):1–10. |
[186] | Nixon K , Crews FT . Temporally Specific Burst in Cell Proliferation Increases Hippocampal Neurogenesis in Protracted Abstinence from Alcohol. J Neurosci. (2004) ;24: (43):9714–22. |
[187] | Rice AC , Bullock MR , Shelton KL . Chronic ethanol consumption transiently reduces adult neural progenitor cell proliferation. Brain Research. (2004) ;1011: (1):94–8. |
[188] | Åberg E , Hofstetter CP , Olson L , Brené S . Moderate ethanol consumption increases hippocampal cell proliferation and neurogenesis in the adult mouse. Int J Neuropsychopharmacol. (2005) ;8: (4):557–67. |
[189] | Vengeliene V , Bilbao A , Molander A , Spanagel R . Neuropharmacology of alcohol addiction. Br J Pharmacol. (2008) ;154: (2):299–315. |
[190] | Lee J , Duan W , Mattson MP . Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. Journal of Neurochemistry. (2002) ;82: (6):1367–75. |
[191] | Costa M-A , Girard M , Dalmay F , Malauzat D . Brain-derived neurotrophic factor serum levels in alcohol-dependent subjects 6 months after alcohol withdrawal. Alcohol Clin Exp Res. (2011) ;35: (11):1966–73. |
[192] | Köhler S , Klimke S , Hellweg R , Lang UE . Serum brain-derived neurotrophic factor and nerve growth factor concentrations change after alcohol withdrawal: preliminary data of a case-control comparison. Eur Addict Res. (2013) ;19: (2):98–104. |
[193] | Costa M-A , Girard M , Dalmay F , Malauzat D . Brain-Derived Neurotrophic Factor Serum Levels in Alcohol-Dependent Subjects 6 Months After Alcohol Withdrawal. Alcoholism: Clinical and Experimental Research. (2011) ;35: (11):1966–73. |
[194] | MacLennan AJ , Lee N , Walker DW . Chronic ethanol administration decreases brain-derived neurotrophic factor gene expression in the rat hippocampus. Neurosci Lett. (1995) ;197: (2):105–8. |
[195] | Pandey SC , Zhang D , Mittal N , Nayyar D . Potential role of the gene transcription factor cyclic AMP-responsive element binding protein in ethanol withdrawal-related anxiety. J Pharmacol Exp Ther. (1999) ;288: (2):866–78. |
[196] | Tapia-Arancibia L , Rage F , Givalois L , Dingeon P , Arancibia S , Beaugé F . Effects of alcohol on brain-derived neurotrophic factor mRNA expression in discrete regions of the rat hippocampus and hypothalamus. J Neurosci Res. (2001) ;63: (2):200–8. |
[197] | Dalçik H , Yardimoglu M , Filiz S , Gonca S , Dalçik C , Erden BF . Chronic ethanol-induced glial fibrillary acidic protein (GFAP) immunoreactivity: an immunocytochemical observation in various regions of adult rat brain. Int J Neurosci. (2009) ;119: (9):1303–18. |
[198] | Franke H . Influence of chronic alcohol treatment on the GFAP-immunoreactivity in astrocytes of the hippocampus in rats. Acta Histochem. (1995) ;97: (3):263–71. |
[199] | Korbo L . Glial cell loss in the hippocampus of alcoholics. Alcohol Clin Exp Res. (1999) ;23: (1):164–8. |
[200] | Miguel-Hidalgo JJ , Rajkowska G . Comparison of prefrontal cell pathology between depression and alcohol dependence. J Psychiatr Res. (2003) ;37: (5):411–20. |
[201] | Miguel-Hidalgo JJ . Lower packing density of glial fibrillary acidic protein-immunoreactive astrocytes in the prelimbic cortex of alcohol-naive and alcohol-drinking alcohol-preferring rats as compared with alcohol-nonpreferring and Wistar rats. Alcohol Clin Exp Res. (2005) ;29: (5):766–72. |
[202] | Rintala J , Jaatinen P , Kiianmaa K , Riikonen J , Kemppainen O , Sarviharju M ,et al.. Dose-dependent decrease in glial fibrillary acidic protein-immunoreactivity in rat cerebellum after lifelong ethanol consumption. Alcohol. (2001) ;23: (1):1–8. |
[203] | Kelso ML , Liput DJ , Eaves DW , Nixon K . Upregulated vimentin suggests new areas of neurodegeneration in a model of an alcohol use disorder. Neuroscience. (2011) ;197: :381–93. |
[204] | Monje ML , Toda H , Palmer TD . Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science. (2003) ;302: (5651):1760–5. |
[205] | Cacci E , Claasen J-H , Kokaia Z . Microglia-derived tumor necrosis factor-α exaggerates death of newborn hippocampal progenitor cells in vitro. Journal of Neuroscience Research. (2005) ;80: (6):789–97. |
[206] | Henriques JF , Portugal CC , Canedo T , Relvas JB , Summavielle T , Socodato R . Microglia and alcohol meet at the crossroads: Microglia as critical modulators of alcohol neurotoxicity. Toxicology Letters. (2018) ;283: :21–31. |
[207] | McCarthy GM , Farris SP , Blednov YA , Harris RA , Mayfield RD . Microglial-specific transcriptome changes following chronic alcohol consumption. Neuropharmacology. (2018) ;128: :416–24. |
[208] | Yang Q , Zhang Z , Gregg EW , Flanders WD , Merritt R , Hu FB . Added Sugar Intake and Cardiovascular Diseases Mortality Among US Adults. JAMA Intern Med. (2014) ;174: (4):516–24. |
[209] | Rock RB , Gekker G , Hu S , Sheng WS , Cheeran M , Lokensgard JR ,et al.. Role of Microglia in Central Nervous System Infections. Clin Microbiol Rev. (2004) ;17: (4):942–64. |
[210] | Vezzani A , Moneta D , Richichi C , Aliprandi M , Burrows SJ , Ravizza T ,et al.. Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia. (2002) ;43: (Suppl 5):30–5. |
[211] | Koo JW , Duman RS . IL-1β is an essential mediator of the antineurogenic and anhedonic effects of stress. PNAS. (2008) ;105: (2):751–6. |
[212] | Qin L , He J , Hanes RN , Pluzarev O , Hong J-S , Crews FT . Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. Journal of Neuroinflammation. (2008) ;5: (1):10. |
[213] | Zunszain PA , Anacker C , Cattaneo A , Choudhury S , Musaelyan K , Myint AM ,et al.. Interleukin-1 β: A New Regulator of the Kynurenine Pathway Affecting Human Hippocampal Neurogenesis. Neuropsychopharmacology. (2012) ;37: (4):939–49. |
[214] | Zou J , Crews FT . Inflammasome-IL-1β Signaling Mediates Ethanol Inhibition of Hippocampal Neurogenesis. Front Neurosci. (2012) ;6: (77). |
[215] | Crews F , Nixon K , Kim D , Joseph J , Shukitt-Hale B , Qin L ,et al.. BHT Blocks NF-κB activation and Ethanol-Induced Brain Damage. Alcoholism: Clinical and Experimental Research. (2006) ;30: (11):1938–49. |
[216] | Zou J , Crews F . Induction of Innate Immune Gene Expression Cascades in Brain Slice Cultures by Ethanol: Key Role of NF-κB and Proinflammatory Cytokines. Alcoholism: Clinical and Experimental Research. (2010) ;34: (5):777–89. |
[217] | Leclercq S , Cani PD , Neyrinck AM , Stärkel P , Jamar F , Mikolajczak M ,et al.. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain, Behavior, and Immunity. (2012) ;26: (6):911–8. |
[218] | Iimuro Y , Gallucci RM , Luster MI , Kono H , Thurman RG . Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology. (1997) ;26: (6):1530–7. |
[219] | Ponnappa BC , Israel Y , Aini M , Zhou F , Russ R , Cao Q ,et al.. Inhibition of tumor necrosis factor alpha secretion and prevention of liver injury in ethanol-fed rats by antisense oligonucleotides. Biochemical Pharmacology. (2005) ;69: (4):569–77. |
[220] | Rajayer SR , Jacob A , Yang W-L , Zhou M , Chaung W , Wang P . Cold-Inducible RNA-Binding Protein Is an Important Mediator of Alcohol-Induced Brain Inflammation. PLOS ONE. (2013) ;8: (11):e79430. |
[221] | Qin L , Crews FT . Focal Thalamic Degeneration from Ethanol and Thiamine Deficiency is Associated with Neuroimmune Gene Induction, Microglial Activation, and Lack of Monocarboxylic Acid Transporters. Alcoholism: Clinical and Experimental Research. (2014) ;38: (3):657–71. |
[222] | Kane CJM , Phelan KD , Douglas JC , Wagoner G , Johnson JW , Xu J ,et al. Effects of Ethanol on Immune Response in the Brain: Region-Specific Changes in Adolescent Versus Adult Mice. Alcoholism: Clinical and Experimental Research. (2014) ;38: (2):384–91. |
[223] | Qin L , Crews FT . Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. Journal of Neuroinflammation. (2012) ;9: (1):130. |
[224] | Pascual M , Baliño P , Aragón CMG , Guerri C . Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: Role of TLR4 and TLR2. Neuropharmacology. (2015) ;89: :352–9. |
[225] | Whitman BA , Knapp DJ , Werner DF , Crews FT , Breese GR . The Cytokine mRNA Increase Induced by Withdrawal from Chronic Ethanol in the Sterile Environment of Brain is Mediated by CRF and HMGB1 Release. Alcoholism: Clinical and Experimental Research. (2013) ;37: (12):2086–97. |
[226] | Doremus-Fitzwater TL , Buck HM , Bordner K , Richey L , Jones ME , Deak T . Intoxication- and Withdrawal-Dependent Expression of Central and Peripheral Cytokines Following Initial Ethanol Exposure. Alcoholism: Clinical and Experimental Research. (2014) ;38: (8):2186–98. |
[227] | Baxter-Potter LN , Henricks AM , Berger AL , Bieniasz KV , Lugo JM , McLaughlin RJ . Alcohol vapor exposure differentially impacts mesocorticolimbic cytokine expression in a sex-, region-, and duration-specific manner. Neuroscience. (2017) ;346: :238–46. |
[228] | Vetreno RP , Qin L , Crews FT . Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiology of Disease. (2013) ;59: :52–62. |
[229] | Umhau JC , Schwandt M , Solomon MG , Yuan P , Nugent A , Zarate CA ,et al.. Cerebrospinal Fluid Monocyte Chemoattractant Protein-1 in Alcoholics: Support for a Neuroinflammatory Model of Chronic Alcoholism. Alcoholism: Clinical and Experimental Research. (2014) ;38: (5):1301–6. |
[230] | Zahr NM . Peripheral TNFα elevations in abstinent alcoholics are associated with hepatitis C infection. PLoS One. (2018) ;13: (2). |
[231] | Heberlein A , Käser M , Lichtinghagen R , Rhein M , Lenz B , Kornhuber J ,et al.. TNF-α and IL-6 serum levels: Neurobiological markers of alcohol consumption in alcohol-dependent patients? Alcohol. (2014) ;48: (7):671–6. |
[232] | Kiefer F , Jahn H , Schick M , Wiedemann K . Alcohol intake, tumour necrosis factor-alpha, leptin and craving: factors of a possibly vicious circle? Alcohol Alcohol. (2002) ;37: (4):401–4. |
[233] | Nicolaou C , Chatzipanagiotou S , Tzivos D , Tzavellas EO , Boufidou F , Liappas IA . Serum cytokine concentrations in alcohol-dependent individuals without liver disease. Alcohol. (2004) ;32: (3):243–7. |
[234] | Sierksma A , Patel H , Ouchi N , Kihara S , Funahashi T , Heine RJ ,et al.. Effect of Moderate Alcohol Consumption on Adiponectin, Tumor Necrosis Factor-α, and Insulin Sensitivity. Diabetes Care. (2004) ;27: (1):184–9. |
[235] | Felver ME , Mezey E , McGuire M , Mitchell MC , Herlong HF , Veech GA ,et al.. Plasma Tumor Necrosis Factor α Predicts Decreased Long-Term Survival in Severe Alcoholic Hepatitis. Alcoholism: Clinical and Experimental Research. (1990) ;14: (2):255–9. |
[236] | Gonzalez-Quintela A , Campos J , Loidi L , Quinteiro C , Perez L-F , Gude F . Serum TNF-α levels in relation to alcohol consumption and common TNF gene polymorphisms. Alcohol. (2008) ;42: (6):513–8. |
[237] | Daniluk J , Szuster-Ciesielska A , Drabko J , Kandefer-Szerszeń M . Serum cytokine levels in alcohol-related liver cirrhosis. Alcohol. (2001) ;23: (1):29–34. |


