
CD274 (PD-L1) Copy Number Changes (Gain) & Response to Immune Checkpoint Blockade Therapy in Carcinomas of the Urinary Tract
Abstract
BACKGROUND:
Immune checkpoint inhibitors are an important therapeutic option for urothelial carcinoma, but durable responses are achieved in a minority of patients. Identifying pre-treatment biomarkers that may predict response to these therapies or who exhibit intrinsic resistance, is of paramount importance.
OBJECTIVE:
To explore the prevalence of PD-L1 copy number alteration in urothelial carcinoma and correlate with response to immune checkpoint inhibitors.
METHODS:
We analyzed a cohort of 1050 carcinomas of the bladder and upper urinary tract that underwent targeted next generation sequencing, prospectively. We assessed PD-L1 protein expression, copy number status (next generation sequencing/FISH), and detailed treatment response.
RESULTS:
We identified 9 tumors with PD-L1 amplification and 9 tumors with PD-L1 deletion. PD-L1 protein expression was the highest in PD-L1 amplified tumors. Of the 9 patients whose tumors harbored PD-L1 amplification, 6 received immunotherapy with 4 deriving clinical benefit, and 2 achieving durable response. Of the 9 patients whose tumors had PD-L1 copy number losses, 4 received immunotherapy with 3 experiencing disease progression.
CONCLUSIONS:
PD-L1 copy number alterations may serve as potential biomarkers of response to immunotherapy in urothelial carcinoma patients, if validated in larger cohorts.
INTRODUCTION
Immune checkpoint inhibitors (ICIs) induce durable responses in a minority of patients with locally advanced and metastatic urothelial carcinoma (UC), but the degree and durability of response varies in those that do benefit clinically [1]. There is thus an urgent need to identify pre-treatment biomarkers to identify patients likely to respond to ICI or who exhibit intrinsic resistance. One potential biomarker of ICI response is copy number change at 9p24.1, which includes the loci for both PD-L1/CD274 and PD-L2/PDCD1LG2 [2–6]. In tumors with PD-L1 amplification, increased production of PD-L1 is thought to result in its greater binding to the PD-1 receptor on cytotoxic T-cells, leading to immune evasion [7]. While prior studies have indicated that amplification of PD-L1 may be associated with response to antibodies targeting PD-L1/PD-1 in a variety of cancer types, the association between PD-L1 copy number alterations and PD-L1 expression and the clinical implications of PD-L1 amplification in UC remain poorly characterized [2–4, 6].
MATERIALS AND METHODS
This study was approved by the institutional review board (12–245, 06–107) and informed consent was obtained from all participants. To define the prevalence of PD-L1 copy number alterations in tumors of the urinary tract, we analyzed a cohort of 1050 carcinomas of the bladder and upper urinary tract that were prospectively sequenced using the MSK-IMPACT next generation sequencing (NGS) assay [8]. Amplifications were defined as a fold change≥2.0 and deletions were defined as fold change≤–2.0 (ref 5). Fluorescence in situ hybridization (FISH) for JAK2/INSL6 and PD-L1/PD-L2 genes was performed, as previously described [3], on 3 of the tumors with PD-L1 amplification by MSK-IMPACT that had tissue available and on 2 additional cases, due to high expression of PD-L1 by immunohistochemistry (IHC). An amplified signal was defined as > 10:1 ratio of PD-L1 signal to the reference centromeric probe. IHC for PD-L1 (clone E1L3N) was performed on 8 and 4 tumors with PD-L1 amplification and deletion, respectively, and scored using the combined positivity score (CPS), which is calculated by the sum of PD-L1-expressing tumor and immune cells, divided by the total number of tumor cells and multiplied by 100. A cutoff of 10 is considered clinically actionable [1]. We also performed PD-L1 IHC staining on 74 UC tumors that did not harbor CD274/PD-L1 amplification by MSK-IMPACT. The cBioPortal.32e34 platform was used to analyze data from The Cancer Genome Atlas [9].
RESULTS
We identified 7 tumors (0.66%) with PD-L1 amplification and 9 (0.86%) with PD-L1 deletion (Fig. 1, S-Tables 1 and 2, S-Molecular Data). Two additional tumors that did not undergo MSK-IMPACT were found to harbor PD-L1 amplification by FISH after diffuse PD-L1 expression was identified by IHC. The histopathologic tumor types with PD-L1 amplification included UC, not otherwise specified (n = 5), small cell/neuroendocrine carcinoma (n = 3), and a poorly differentiated carcinoma with clear cell adenocarcinoma features (n = 1). Within the same cohort of 1050 tumors, 25 additional tumors were of small cell/neuroendocrine carcinoma histology that did not harbor CD274/PD-L1 amplification.
Fig. 1
Molecular profiling and immunohistochemistry. This oncoprint depicts the results of molecular profiling and IHC for PD-L1, as well as other relevant alterations. Tumors with PD-L1 amplification were associated with better response to ICI treatment. The one tumor with PD-L1 deletion with durable response is an HPV-associated carcinoma.
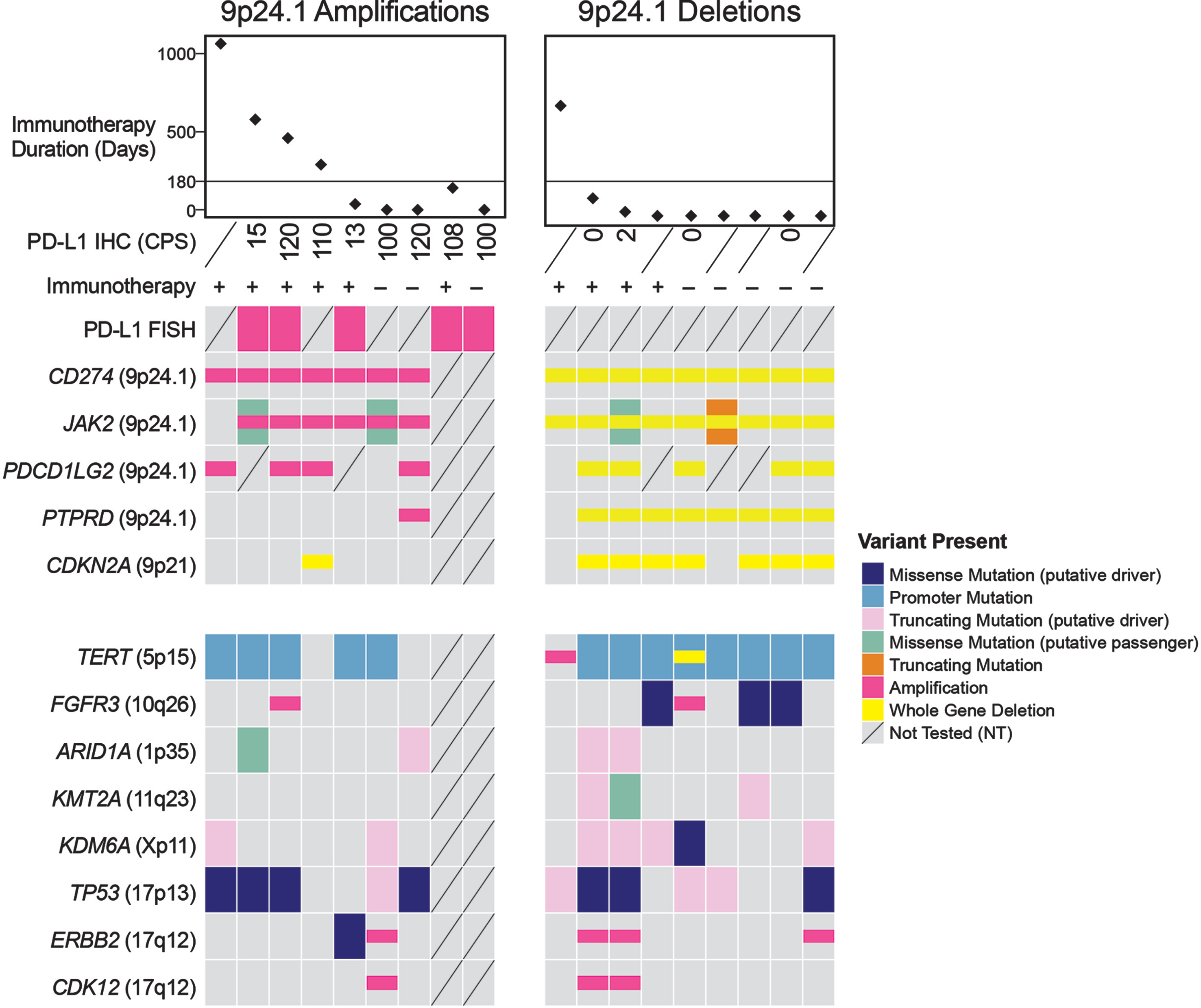
The CPS range in PD-L1 amplified tumors was 13 to 120 (median: 104, n = 8) versus UC tumors without PD-L1 amplification (range 0–70, median: 2, n = 74; p < 0.0001, S-Figure 1). Notably, high CPS in CD274/PD-L1-amplified tumors was primarily driven by PD-L1 expression on tumor cells, not immune cells, as six of the eight tumors tested using IHC expressed PD-L1 on nearly 100%of tumor cells. For comparison, from the 74 tumors without CD274/PD-L1 amplification that were stained with PD-L1, only 14 (19%) had a CPS≥10. In six of these 14 tumors, PD-L1 expression was identified nearly exclusively on immune cells, not on tumor cells (CPS range: 10–20). Moreover, three tumors with the highest levels of PD-L1 expression (CPS of 40, 65 and 70) and no CD274/PD-L1 amplification exhibited sarcomatoid (n = 2) and squamous differentiation (n = 1), which are known to be associated with high PD-L1 expression levels [10, 11]. The remaining five tumors expressed PD-L1 on 45%, 15%, 15%, 10%and 8%of tumor cells, corresponding to a CPS of 50, 20, 18, 15 and 11, respectively. Our results suggest that tumors with very high levels of PD-L1 expression on tumor cells may be enriched for CD274/PD-L1 gene amplification, particularly those that don’t have squamous or sarcomatoid morphology. Furthermore, our findings indicate that there are other mechanisms of PD-L1 overexpression beyond CD274/PD-L1 gene amplification that may be secondary to tumor-intrinsic factors or related to tumor microenvironment, which require further investigation.
Few studies have explored the correlation between CD274/PD-L1 copy number gain and response to ICIs [2–4, 6]. Some reports suggest that CD274/PD-L1 amplification may be a predictor of favorable response to ICI therapy independent of tumor mutational burden [2–4]. In our cohort of the 9 patients whose tumors harbored CD274/PD-L1 amplification, 6 received ICIs (Fig. 2A-D, S-Figures 2, 3, S-Table 1), five of whom had recurrent or metastatic disease at the time of initiation of ICI therapy. Clinical benefit, defined as time on ICI treatment of 180 days or greater was achieved in 4 of the patients, including 2 patients with durable responses of 68 (Case 1, S-Figure 2) and 29 months (Cases 2) [12]. More details about the cohort are available in S-Table 1. While our findings indicate that patients whose tumors harbor CD274/PD-L1 amplification may have higher response rate to ICI, our cohort of CD274/PD-L1-amplified tumors is very small and cannot explain the response to ICI in patients whose tumors don’t harbor such genetic alterations. We further explored whether tumor mutation burden (TMB) was associated with response to ICI therapy in this cohort beyond PD-L1 copy number alterations. TMB data were available for of 7 of 9 PD-L1-amplified and all 9 PD-L1-deleted tumors (see supplementary molecular data table). For the 4 patients who derived clinical benefit, TMB was 7.9, 9.7, 11.2 and 2.6 mutations/megabase, respectively. Using the current cutoff of 10 mutations/ megabase based on recent FDA approval of pembrolizumab in TMB-high tumors, two of these four cases would not be considered TMB-high, while a third case has a borderline TMB value (9.7 mutations/ megabase). Conversely, at least 2 patients with TMB high tumors and PD-L1 gene deletion (15.8 and 25.5 mutations/ megabase, respectively) exhibited progressive disease while on immunotherapy. These findings suggest that TMB by itself does not explain the response to immunotherapy in the 4 patients whose tumors harbored PD-L1 gene amplification.
Fig. 2
Copy Number Assessment, Histopathology & Imaging. A copy number plot (A, Case 4) with relative (Log2) tumor/normal ratios (y-axis) and corresponding chromosomes (x-axis) is displayed, with each blue dot representing an individual probe region. Amplified regions are shown in red and the inset shows a 20-fold gain based on the FACETS algorithm (inset; chromosome 9, arrow indicates 9p24.1 amplicon). Representative histopathology (B), PD-L1 IHC (C) and FISH using probes for PD-L1/PD-L2 (D) is shown. A copy number plot for a second patient is depicted (E, Case 10) and genomic deletions are represented with red dots, confirmed using the FACETS algorithm (inset; chromosome 9, arrow indicates 9p24.1 amplicon). Baseline CT imaging for this patient (Case 10) prior to initiation of immunotherapy (F) and at the time of disease progression, at 735 days of follow up (G, H), is shown.
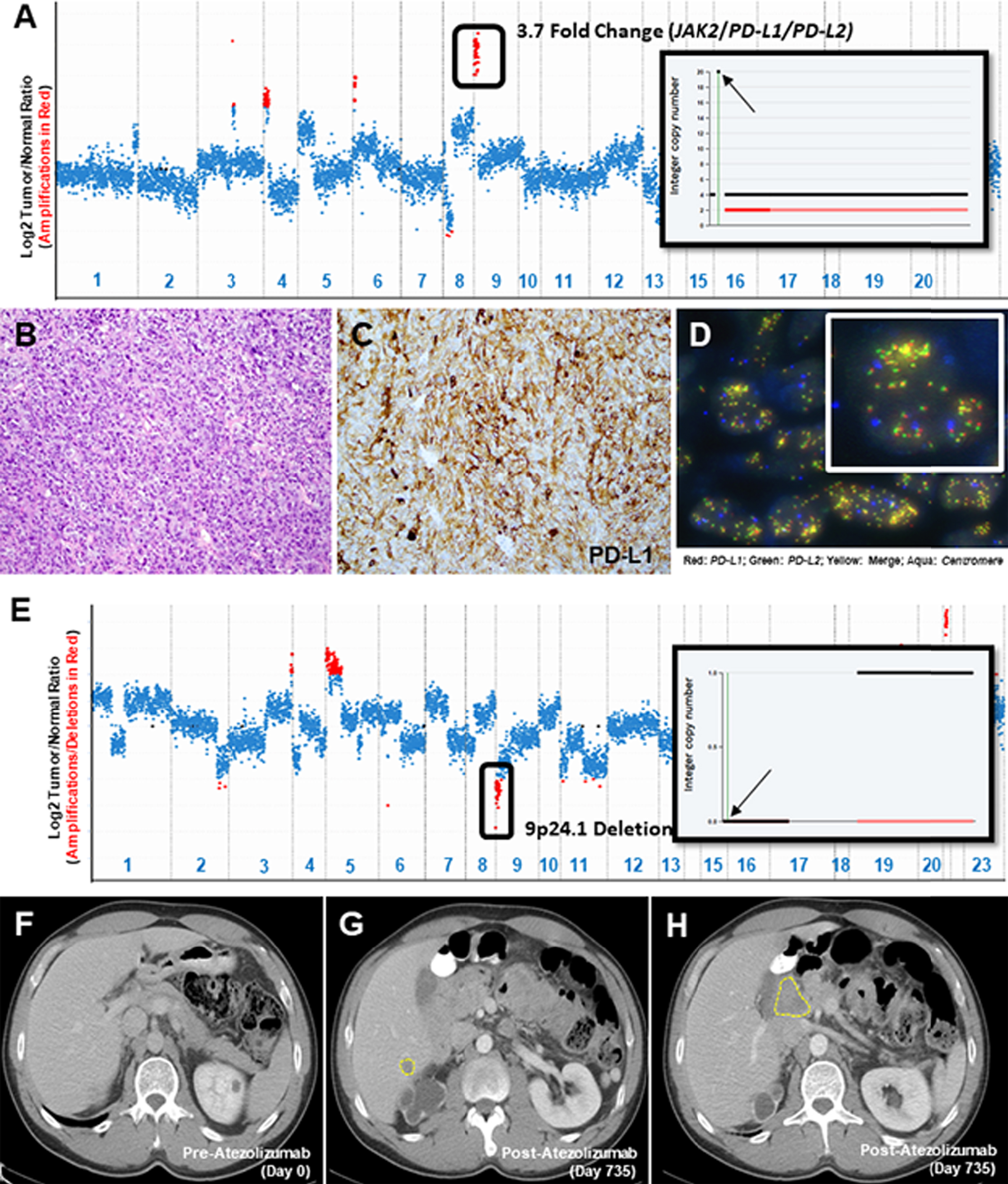
Studies of acquired resistance to ICIs in melanoma have implicated genes that regulate tumor cell interaction with T-cells (B2M) and genes involved in upregulating PD-L1 expression (JAK1, JAK2) [13]. It has thus been hypothesized that deletions encompassing the 9p24.1 locus (PD-L1, PD-L2 and JAK2) may also lead to ICI resistance. In our cohort, we identified 9 UC tumors with PD-L1 copy number losses (Fig. 1, Fig. 2E-H, S-Tables 1 and 2, S-Molecular Data). IHC was performed on five such tumors and no PD-L1 expression was detected on the tumor cells in any of them. Of the 4 patients who were treated with ICIs, 3 developed progressive disease (Case 11, 12 and 16; Fig. 2 E-H). The one patient with evidence of CD274/PD-L1 copy number loss who derived durable clinical benefit to ICI had an HPV-associated bladder carcinoma (Case 10). Whether this HPV association contributed to the durable response in this case is not clear as studies on HPV-associated carcinomas in other sites and their response to immunotherapy have yielded mixed results[14, 15]. Other co-alterations of note included a loss of function alteration in JAK2 in one patient who did not receive ICIs (Case13).
Notable recurring molecular co-alterations in tumors with PD-L1 loss included activating FGFR3 alterations in 4 cases (Case12, 14, 15, 17). FGFR pathway alteration has been associated with a poor prognosis and resistance to immunotherapy [16]. Thus PD-L1 deletions should be further investigated for their co-occurrence with FGFR3 alterations as a possible contributory mechanism of ICI resistance.
CONCLUSION
CD274/PD-L1 amplification was associated with high PD-L1 expression as compared to non-amplified UC tumors. Combining our single-institution experience with the TCGA dataset (10 amplified and 5 deleted cases), the combined incidence of amplification events at the 9p24.1 locus was 1.16%(17 of 1456) and that of corresponding deletion events was 0.96%(14 of 1456; see S-Table 3 for a list of the TCGA cases with amplification and deletion) [17]. As four of six patients with CD274/PD-L1 amplification treated with ICI derived clinical benefit, our results suggest that CD274/PD-L1 amplification may contribute to ICI sensitivity and warrants further investigation as a potentially predictive biomarker of response to ICI.
Limitations of the current study include its retrospective design and the fact that all patients studied were treated at a single tertiary institution, factors that may have led to selection bias.
As the incremental cost of profiling tumors for PD-L1 amplification and deletions is minimal for patients undergoing comprehensive NGS-based molecular profiling, PD-L1 copy number analysis may be an easily measured and cost effective biomarker that, in combination with other tumor and host factors, may provide better prediction for response to ICI therapy. Given the rarity of CD274/PD-L1 amplification in UC, future studies on the influence of CD274/PD-L1 copy number alterations on ICI response will need to combine prospective genomic datasets with detailed demographic and treatment data generated at multiple institutions.
ACKNOWLEDGMENTS
The authors have no acknowledgments.
FUNDING
This work was supported by Cycle for Survival (HA, DS), R01 CA233899 (HA), P01CA221757 (HA, DS), SPORE in Bladder Cancer P50CA221745, a Sloan Kettering Institute for Cancer Research Cancer Center Support Grant (P30CA008748), and the Marie-Josée and Henry R Kravis Center for Molecular Oncology.
AUTHOR CONTRIBUTIONS
SG, CMV, YZ, SKT, SWF, AG, YBC, SJS, MYT, SAF, GI, JER, DFB, BHB, EJP, DSR, ML, JCC, DBS, VER and HAA performed the research. SG, CMV and HAA designed the research study, analyzed the data and wrote the manuscript.
CONFLICT OF INTEREST
The authors (SG, CMV, YZ, SKT, SWF, AG, YBC, SJS, MYT, SAF, GI, JER, DFB, BHB, EJP, DSR, ML, JCC, DBS, VER and HAA) have no conflicts of interest for the work presented in the current study. EJ Pietzak is on the Scientific Advisory Board of Merck. DB Solit has consulted for/received honoraria from Pfizer, Loxo Oncology, Lilly Oncology, Q.E.D. Therapeutics, Vivideon Therapeutics, and Illumina. SA Funt has received research funding from Genentech and AstraZeneca/MedImmune; travel expenses from AstraZeneca/MedImmune; is a consultant for AstraZeneca/MedImmune and has stock ownership in Urogen Pharma, Allogene Therapeutics, Neogene Therapeutics and Kite Pharma. JE Rosenberg has received research funding from Incyte and AstraZeneca, as well as institutional research funding from Genentech, Oncogenex, Agensys, Mirati Therapeutics, Novartis, Viralytics, Genentech/Roche, Seattle Genetics and Bayer and has patents/royalties/other intellectual property pertaining to predictors of platinum sensitivity. He has received honoraria from UpToDate, Bristol-Myers Squibb, AstraZeneca, Medscape, Vindico, Peerview, Chugai Pharma; travel expenses from Genentech/Roche and Bristol-Myers Squibb; consults for Lilly, Merck, Agensys, Roche/Genentech, Sanofi, AstraZeneca/MedImmune, Bristol-Myers Squibb, EMD Serono, Seattle Genetics, Bayer, Inovio Pharmaceuticals, BioClin Therapeutics, QED Therapeutics, Adicet Bio, Sensei Biotherapeutics, Fortress Biotech, Pharmacyclics and western oncolytics and has stock ownership in Merck and Illumina. H Al-Ahmadie is a Consultant for Bristol-Myers-Squibb, AstraZeneca, Janssen Biotech and Paige.AI. JER, BHB and HAA are Editorial Board Members of this journal, but were not involved in the peer-review process nor had access to any information regarding its peer-review.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/BLC-201532.
REFERENCES
[1] | Teo MY , Rosenberg JE . Refining existing knowledge and management of bladder cancer. Nat Rev Urol. 2018. |
[2] | Goodman AM , et al. Prevalence of PDL1 Amplification and Preliminary Response to Immune Checkpoint Blockade in Solid Tumors. JAMA Oncol. (2018) ;4: (9):1237–44. |
[3] | Gupta S , et al. JAK2/PD-L1/PD-L2 (9p24.1) amplifications in renal cell carcinomas with sarcomatoid transformation: implications for clinical management. Mod Pathol. (2019) ;32: (9):1344–58. |
[4] | Gupta S , et al. JAK2, PD-L1, and PD-L2 (9p24.1) amplification in metastatic mucosal and cutaneous melanomas with durable response to immunotherapy. Hum Pathol. (2019) ;88: :87–91. |
[5] | Gupta S , et al. Next-Generation Sequencing-Based Assessment of JAK2, PD-L1, and PD-L2 Copy Number Alterations at 9p24.1 in Breast Cancer: Potential Implications for Clinical Management. J Mol Diagn. (2019) ;21: (2):307–17. |
[6] | Ikeda S , et al. Metastatic basal cell carcinoma with amplification of PD-L1: exceptional response to anti-PD1 therapy. NPJ Genom Med. (2016) ;1: :1. |
[7] | Taube JM , et al. Implications of the tumor immune microenvironment for staging and therapeutics. Mod Pathol. (2018) ;31: (2):214–34. |
[8] | Cheng DT , et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. (2015) ;17: (3):251–64. |
[9] | Gao J , et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal.pl. (2013) ;6: (269):1. |
[10] | Guo CC , et al. Dysregulation of EMT Drives the Progression to Clinically Aggressive Sarcomatoid Bladder Cancer. Cell Rep. (1793) ;27: (6):1781–1793 e4. |
[11] | Reis H , et al. PD-L1 Expression in Urothelial Carcinoma With Predominant or Pure Variant Histology: Concordance Among 3 Commonly Used and Commercially Available Antibodies. Am J Surg Pathol. (2019) ;43: (7):920–7. |
[12] | Snyder A , et al. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLoS Med. (2017) ;14: (5):e1002309. |
[13] | Zaretsky JM , et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. (2016) ;375: (9):819–29. |
[14] | Zandberg DP , et al. Durvalumab for recurrent or metastatic head and neck squamous cell carcinoma: Results from a single-arm, phase II study in patients with> /=25%tumour cell PD-L1 expression who have progressed on platinum-based chemotherapy. Eur J Cancer. (2019) ;107: :142–52. |
[15] | Ferris RL , et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. (2016) ;375: (19):1856–67. |
[16] | Sweis RF , et al. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol Res. (2016) ;4: (7):563–8. |
[17] | Robertson AG , et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. (2017) ;171: (3):540–556 e25. |


