
Clinical Development of FGFR3 Inhibitors for the Treatment of Urothelial Cancer
Abstract
The fibroblast growth factor receptor 3 (FGFR3) plays critical roles in driving oncogenesis of a subset of patients with urothelial carcinomas (UC). Growing evidence from preclinical studies suggests that FGFR3 inhibition can reduce proliferation and survival in vitro and in vivo models of FGFR3-altered UC. Early clinical trials investigating selective FGFR3 inhibitor have reported preliminary signs of antitumor activity in advanced UC patients with selected FGFR3 mutations or fusions. Currently, phase 3 trials with erdafitinib and rogaratinib are enrolling patients with known FGFR3 alterations. Future combinations with targeted therapies or immune checkpoint inhibitors may increase the efficacy of selective FGFR3 inhibitors. Herein, we discuss current clinical development of FGFR3 inhibitors as well as unsolved questions with regards to patient selection, management of toxicities and mechanisms of resistance to selective FGFR3 inhibitors.
INTRODUCTION
Metastatic urothelial carcinoma (UC) is frequent and has a poor prognosis. The clinical management of advanced UC has improved over the last decade mainly owing to novel immunotherapies targeting immune checkpoint receptors. However, anti-programmed cell death 1 (PD1) and anti-programmed death-ligand 1 (PDL1) monoclonal antibodies yield a tumor response in only one-fifth of the treated patients [1–11]. Innovative strategies aiming to improve metastatic UC treatment efficacy have learned from targeted therapies in solid tumors such as lung and breast cancers. Given the need for alternative treatments in advanced UC, there is a growing interest in targeting oncogenic pathways in UC. Over the last few years, genome sequencing techniques have led to a better understanding of the molecular biology of UC. Recently, the Cancer Genome Atlas (TCGA) project has elucidated the high degree of heterogeneity underlying cancer cell development [12, 13] and has led to the classification of muscle-invasive bladder cancer (MIBC) into molecular subtypes. These subtypes exhibit distinct sensitivity to therapies owing to their distinct genomic and transcriptomic features. DNA and epigenetic alterations have been identified in up to 60% of bladder cancers. Importantly, potential targetable alterations involving the fibroblast growth factor receptor 3 (FGFR3) have been found in up to 20% of MIBC making these molecular aberrations highly attractive for pharmacological inhibition. Genomic alterations of FGFR3 are among the best described oncogenic pathway in UC [14] and have led to extensive and ongoing investigations of FGFR3-targeted therapies in this disease.
In this review, we highlight the diverse oncogenic FGFR3 signaling mechanisms in UC, current clinical development of FGFR3 inhibitors and finally perspective and challenges of anti-FGFR3 therapy in UC.
MATERIAL AND METHODS
A review of the literature has been conducted in February 2018 using the pubmed Medline database, Cochrane database, ascopubs.org, esmo.org, clinicaltrials.org databases and scholar.google.com following PRISMA (Preferred Reporting Items for Systemic Reviews and Meta-analysis) guidelines. We searched for clinical trials articles with the following keywords: “Receptors, Fibroblast Growth Factor”, “Urinary Bladder Neoplasms”, “Carcinoma, Transitional Cell” in pubmed database; “Fibroblast Growth Factor” and “urothelial carcinoma” or “Fibroblast Growth Factor” and “bladder cancer” in Cochrane database; “fibroblast growth factor receptor inhibitor” in ascopubs.org; “fibroblast growth factor receptor inhibitor” in esmo.org database; and “fibroblast growth factor receptor 3” AND “bladder cancer”, “fibroblast growth factor receptor 3” AND “urothelial carcinoma”, “fibroblast growth factor receptor 3” AND “urothelial cancer” in scholar.google.com (Supplementary Figure 1). Search results were restricted to English language only. Studies were selected based on title and abstract reading by two authors (TI, YL). Then, for relevant abstracts, the full text was reviewed; discrepancies were resolved via consensus after discussion between the authors. Duplicates, clinical studies which did not include clinical outcome measures were excluded. The search was complemented by additional sources, mainly the reference lists of evaluated studies and meeting abstracts. From clinical.gov database, the search was performed using the following terms: “Cancer” and “Fibroblast Growth Factor”, “carcinoma” and “Fibroblast Growth Factor Receptor”. A total of 117 trials were identified. Eighty-one studies were excluded due to other types of cancer or the absence of FGFR3 inhibitors. A total of 36 trials were selected.
THE FGFR3 PATHWAY AND FGFR3 ALTERATIONS
The FGFR family contains four highly conserved transmembrane tyrosine kinase receptors (FGFR1-4) with 22 identified ligands to date [15, 16]. Each ligand needs to interact with extracellular matrix proteins to bind to its receptor, mainly with the heparan sulphate proteoglycans (HSPG) [15–17]. The dimerization of FGFR leads to the phosphorylation of the intracellular tyrosine kinase domains which results in the activation of a cascade of downstream events including the mitogen-activated protein kinase (MAPK), the signal transducer and activator of transcription (STAT), the phosphoinositide-3-kinase (PI3K)/Akt, the nuclear factor-kappa B, and the PLC-gamma DAG/PKC/IP3-Ca2 + pathways resulting in DNA transcription (Fig. 1). These pathways have critical roles in cell proliferation, metabolism and survival [18, 19].
Fig. 1.
FGFR signaling pathways.
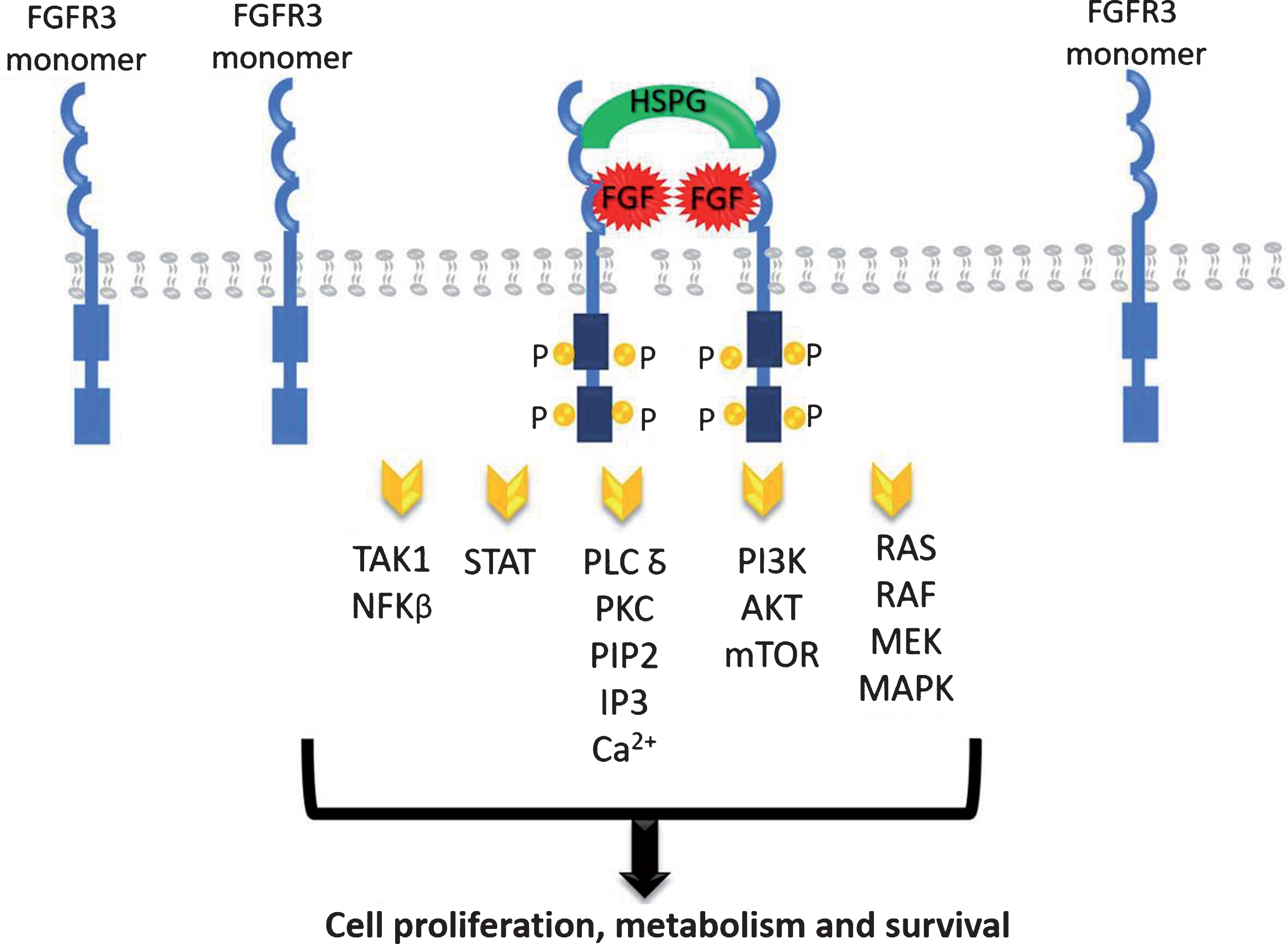
FGFRs are present in many types of normal and tumor cells and have been shown to play an important role in tumor cell growth, survival, and migration as well as in maintaining tumor angiogenesis. FGFR activating mutation, gene amplification, and translocation have been associated with neoplastic progression and tumor vascularization in multiple cancer types, including breast, lung, prostate, endometrial, gastric, and UC [20].
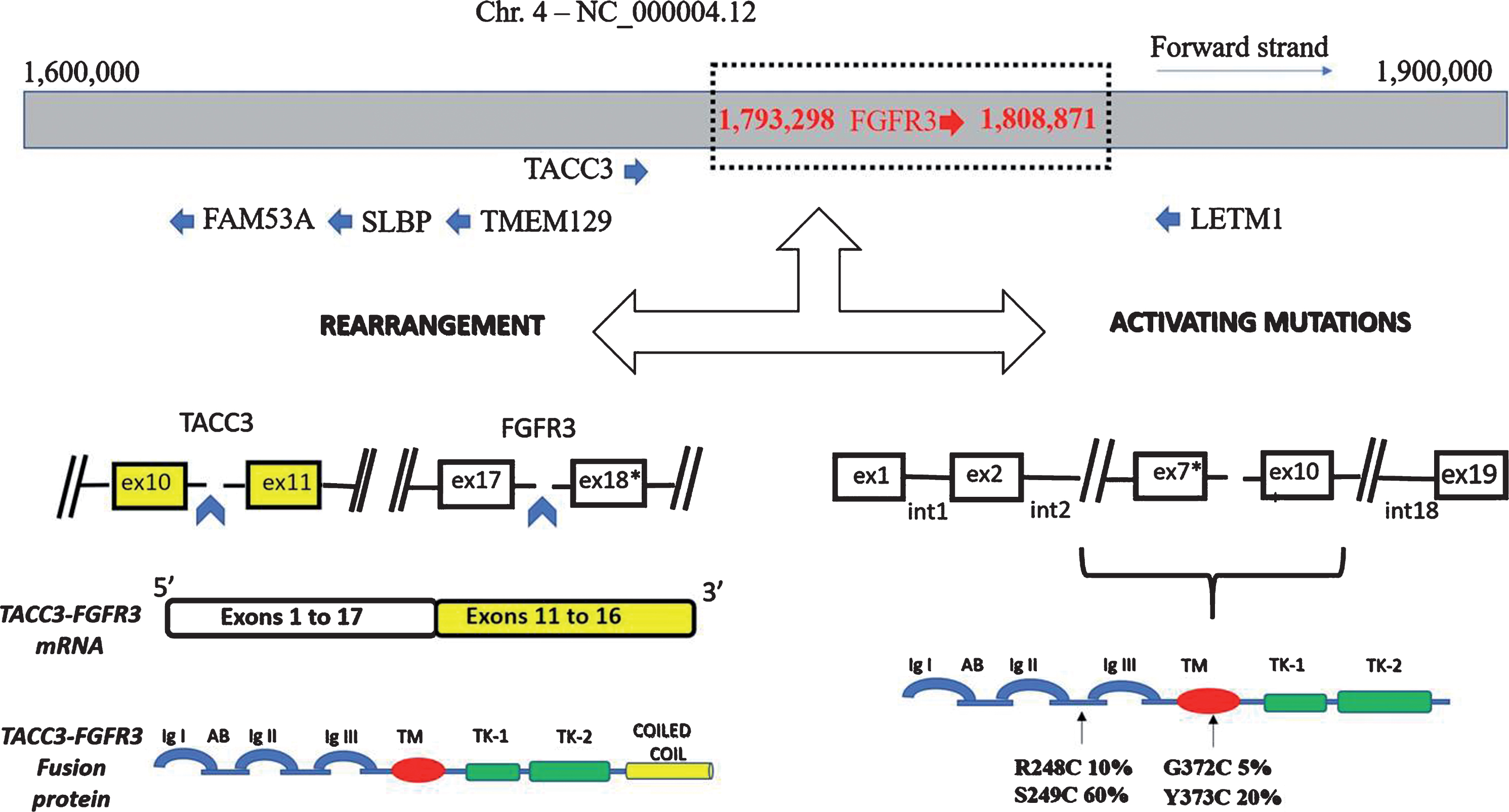
FGFR3 gene is located on the short arm of the chromosome 4 (location p16.3) and contains 19 exons and 18 introns spanning 16.5 kb (Fig. 2). In an analysis of 4,853 tumors by next-generation sequencing (NGS), aberrant FGFR3 have been identified in 22% of UC, 4% of glioma, 3% of carcinoma of unknown primary and endometrial carcinoma, 2% of pancreatic, ovarian, and gastric carcinoma [21]. In addition, nearly 5% of glioblastomas harbour the FGFR3-TACC3 rearrangement which is mutually exclusive with MET and EGFR alterations. FGFR3 overexpression may be detected by immunohistochemistry (IHC) while fusions and mutations could be detected by in situ hybridization methods (fluorescence FISH or chromogenic-CISH) as well as quantitative real-time PCR, targeted sequencing, and NGS [22, 23]. However, these techniques may be limited by intratumor heterogeneity and results may vary according to the segment and the type of tissue obtained at biopsy [23]. Detecting genomic alterations in circulating tumor DNA (ctDNA) may be an alternative solution to overcome this barrier [23].
Fig. 2.
FGFR3 gene alterations.
DYSREGULATION OF FGFR SIGNALING IN UC
Aberrant FGFR3 alterations have been described in 15-20% and up to 60% of MIBC and non-muscle invasive bladder cancer (NMIBC), respectively [13, 24]. Mutations represent nearly 70% of the alterations, rearrangements 20%, and overexpression 10% (Fig. 2 & 3). The most common mutations involve exon 7 and 10 and are: S249C (60%), Y375C (20%), R248C (10%), and G372C (5%) mutations. As a result, FGFR3 becomes constitutively activated, even in the absence of its ligands, through ligand-independent dimerization [25, 26]. Mutations in the extracellular or transmembrane domains stabilize receptor dimerization through generation of novel disulphide or hydrogen bonds, thus promoting constitutive activity. Activating mutations within the kinase domain stimulate ligand-independent function, although they are relatively rare. In the context of fusion aberrations, several partners have been described including the transforming acid coiled-coil 3 (TACC3), the TNFAIP3 Interacting Protein 2 (TNIP2), and the BAI1-associated protein 2-like 1(BAIAP2L1) leading to constitutively active fusion proteins [27] (Fig. 2). The FGFR3-TACC3 and FGFR3-BAIAP2L1 fusion products exhibit constitutive FGFR3 kinase activity and promote cell proliferation and transformation while constitutive dimerization of FGFR3-BAIAP2L1 mediated by a protein – protein interaction domain in BAIAP2L1 is required for ligand-independent activity.
Table 1
Ongoing trials of FGFR3 inhibitors in UC as of September 2018
| Trial | Type | Drug | Status |
| NCT 03473756 | Phase 1b/2 | Rogaratinib + atezolizumab | Recruiting |
| NCT 03410693 (FORT-1) | Phase 2-3 | Rogaratinib vs chemotherapy | Recruiting |
| NCT02872714 (Fight-01) | Phase 2 | INCB054828 | Recruiting |
| NCT03473743 | Phase 1-2 | Erdafitinib + JNJ-63723283 | Recruiting |
| NCT00790426 | Phase 2 | Dovitinib | Recruiting |
| NCT02925533 | Phase 1b | B-701 + Pembrolizumab | Recruiting |
| NCT03390504 (THOR) | Phase 3 | JNJ-42756493 | Recruiting |
Fig. 3.
Pattern of FGFR3 DNA alterations (from cBioportal website).
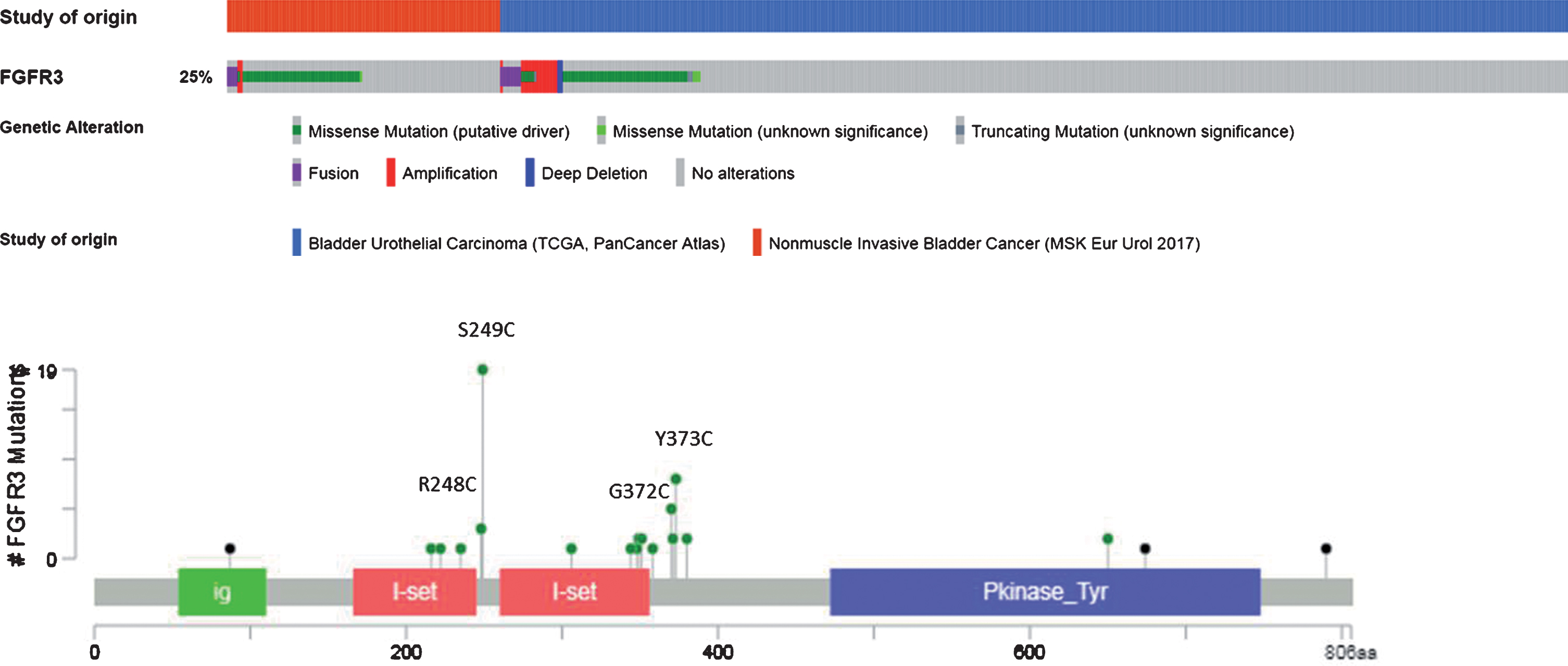
Finally, the molecular mechanisms driving protein overexpression in the presence of a wild-type FGFR3 gene are still under investigation. Some studies showed that FGFR3 overexpression could be the result of gene amplification [28] or regulation by microRNAs mainly miRNA-99a and miRNA-100 [12]. Recently, human tumors with FGFR3-TACC3 fusions were shown to cluster within transcriptional subgroups that are characterized by the activation of mitochondrial function that initiate the chain of metabolic responses that drive mitochondrial metabolism [29].
CLINICAL FEATURES ASSOCIATED WITH FGFR3 GENE ALTERATION
The prognostic value of FGFR3 alterations in UC is still unclear, as some studies have shown no association between FGFR3 gene alterations and tumor recurrence or patient survival [30, 31], while others retrospective studies showed that FGFR3 mutations are associated with lower risk of progression [32]. In very early disease, the frequency of FGFR3 mutations is highest [24]. In stage Ta tumors, several studies indicate that those with mutations appear at lower risk of recurrence and progression [30, 31]. Similarly, in stage T1 tumors, favorable outcome is associated with the presence of mutations [32]. In MIBC, the mutational landscape of FGFR3 altered UC is different and the presence of FGFR3 mutations was found to be associated with a higher frequency of CDKN2A deletion than in NMIBC [13, 33]. FGFR3-mutated NMIBC but not wild-type tumors, hemizygous or homozygous deletion of CDKN2A was a predictor of disease progression that was independent of tumor grade and stage. MIBC with FGFR3 mutation and CDKN2A deletion represent tumors that have progressed from NMIBC. To date, it is still not clear whether such patients have distinct disease outcome. Longitudinal genomic studies will be helpful to decipher molecular disease history of FGFR3 altered UC. These data suggest that the effects of FGFR activation, and thus response to FGFR3 inhibitors, may be highly context dependent.
In the context of metastatic disease, FGFR3 gene alterations are not associated with progression-free survival or overall survival after first-line chemotherapy [34].
Importantly, FGFR3 altered UC may exhibit different immune landscape. TCGA has identified multiple transcriptomic subtypes in MIBC, and thus provide potential insights into response to existing chemotherapies and novel targeted agents [13]. These data show that elevated expression of FGFR3 and the presence of FGFR3 mutations, amplification, and FGFR3-TACC3 fusions are enriched in the subtype of MIBC referred to as papillary-like/Cluster I. Multiple reports showed that Cluster 1 subtype is characterized by a low immune infiltration referred as to immune desert. In this context, several small retrospectives analyses suggest that papillary-like cluster I subtype is more resistant to immune checkpoint inhibitors (ICI) and warrant prospective validation [35, 36] while another study suggested that FGFR3 alterations did not preclude response to nivolumab [37].
Clinical activity of FGFR3 inhibitors
The high frequency of FGFR3 alterations observed in UC has led to the interest of exploring this pathway as a potential therapeutic target. A number of preclinical studies involving bladder cancer cell lines and xenograft models illustrated the anti-tumor activity of drugs inhibiting FGFR mediated signaling pathways [38–41]. In view of the pre-clinical evidence indicating oncogenic addiction in FGFR dysregulated xenografts, clinical development of FGFR inhibitors focus now on FGFR aberrant tumors after early drug development trials reported encouraging results, mainly in patients with UC harboring genomic alterations. FGFR targeting is investigated using different strategies such as selective and non-selective tyrosine kinase inhibitors, monoclonal antibodies and antibody drug conjugates (Table 1).
Monoclonal antibody
B701 is a human immunoglobulin G1 monoclonal antibody directed against FGFR3, with potential antineoplastic activity. Upon intravenous administration, the anti-FGFR3 monoclonal antibody B-701 specifically binds to and inhibits both wild-type and mutated forms of FGFR3. This may result in the inhibition of FGFR3 phosphorylation, thereby preventing its activation and FGFR3-mediated signal transduction pathways. This results in the inhibition of cell proliferation and the induction of cell death in FGFR3-expressing tumor cells in multiple preclinical models. B-701 was investigated in combination with docetaxel in subjects with advanced or metastatic UC (NCT02401542). Preliminary results of the phase 1 show that B-701 combined with docetaxel is safe and effective in some patients who failed platinum-based chemotherapy in UC [42]. Some activity was seen in the FGFR3 Mutation/Fusion patients compared to wild-type patients. A Phase 2 expansion is currently enrolling FGFR3 M/F patients (B-701 monotherapy vs. combination B-701 + D).
MFGR1877S (R3Mab), is a FGFR3-specific human monoclonal antibody which shows excellent activity in preclinical UC models. Upon administration, the anti-FGFR3 antibody MFGR1877S binds to and inhibits FGFR3, which may result in the inhibition of both FGFR3 phosphorylation and FGFR3-mediated signal transduction pathways. Long-term stable disease was reported in five of ten MIBC patients in a phase 1 study [43].
Antibody drug conjugate
LY3076226 is an antibody-drug conjugate, comprised of anti-FGFR-3 antibody conjugated to a microtubule inhibitor, DM4, for the treatment of advanced or metastatic cancer. A phase I clinical trial (NCT02529553) is investigating LY3076226 in advanced or metastatic cancer (including multiple myeloma and lymphoma), locally advanced, unresectable, or metastatic urothelial carcinoma with overexpression or alterations in FGFR3.
Non-selective FGFR tyrosine kinase inhibitors
Tyrosine kinase inhibitors (TKIs) inhibit the kinase activity of the receptors by preventing binding of ATP. Initial development focused on nonselective TKIs that had most potent activity against PDGF and VEGF receptors but also were shown to have some level of activity against other related receptors including FGFRs. These TKIs include dovitinib, ponatinib, pazopanib, nintedanib, lucitanib, brivanib and lenvatinib, which tend to have higher activity against FGFR1 than FGFR3. These nonselective FGFR TKIs are compounds that bind to the relatively conserved ATP-binding domain in receptor tyrosine kinases and lack kinase selectivity. Overall, multitargeted TKIs have not shown significant efficacy in UC. The main issues with non-selective FGFR TKIs are the lack of specificity against FGFR3 and their toxicities.
Dovitinib was investigated in a phase 2 trial in patients with progressive FGFR3-mutated or FGFR3 wild-type advanced UC [44]. Forty-four patients with advanced UC who had progressed after one to three platinum-based and/or combination chemotherapy regimens were given dovitinib at a fixed dose of 500 mg once daily on a 5-days-on/2-days-off schedule. Dovitinib was well tolerated, but had very limited single-agent activity in previously-treated patients with advanced UC, regardless of FGFR3 mutation status (3% in FGFR3 WT and 0% in FGFR3 mut). Brivanib, a VEGFR2 and FGFR1 inhibitor similarly showed disappointing results in patients with advanced UC. A Phase 2 trial of pazopanib as single agent in highly pretreated patients with advanced UC reported partial response in seven patients and stable disease in 14 out of 41 patients [45]. FGFR3 S249C mutation was found in a resistant patient and no FGFR-related alterations in the two responders, suggesting that response was related to other targets of this agent [46]. A recent clinical report described a durable (>6 months) response to pazopanib in a patient whose tumor contained amplified FGF19 and a point mutation in FGFR3 (S249C) [47].
Selective FGFR tyrosine kinase inhibitors
More encouraging data have been reported with selective FGFR tyrosine kinase inhibitors. These agents include erdafitinib (JNJ 42756493), infigratinib (BGJ398), Rogaritinib (BAY 1163877), AZD4547, Pemigatinib (INCB54828), TAS-120, LY2874455, DEBIO 1347, PD173074 and BLU9931.
Erdafitinib
Erdafinitib is a pan-FGFR tyrosine kinase inhibitor with IC-50 values in the low nanomolar range for all members of the FGFR family. It has demonstrated potent inhibition of cell proliferation with IC50 values ranging from 1 to 1000 nM in the FGFR pathway-activated cancer cell lines. Also, erdafitinib has been shown to have in vivo antitumor activity in several murine models of FGFR-driven bladder cancer models [38]. A phase 1 trial has assessed different doses and regimens of erdafitinib from 0.5 mg to 12 mg daily or 10 mg or 12 mg administered intermittently (1 week on/1 week off) [48]. Sixty-five patients across different advanced solid tumors with or without FGFR1-4 alterations (amplification, translocations and mutations) were treated in the dose escalation phase, 23 patients had FGFR genetic aberration. Importantly, maximum-tolerated dose was not defined. Nine milligrams daily was considered as the initial RP2D; however, tolerability was improved with intermittent schedules, and 10 mg administered on a 7-days-on/7-days-off schedule was considered the final RP2D. No response was seen in patients with unknown or no FGFR aberration. Among 23 response-evaluable patients with tumor FGFR pathway alterations, four confirmed responses and one unconfirmed partial response were observed in patients with glioblastoma, UC and endometrial cancer (all with FGFR2 or FGFR3 translocations); 16 patients had stable disease. For all subjects with UC, overall response rate across dose levels was 40%. At the 9 mg dose level, ORR was 55% for response-evaluable subjects with UC who harbored selected FGFR aberrations [49]. The most common adverse events were hyperphosphatemia, dry mouth, asthenia, stomatitis and decrease appetite.
In a global phase 2 study BLC2001 (NCT02365597), patients with mUC and specific FGFR2/FGFR3 mutations or translocations were randomized 1:1 to 28-day cycles of oral 6 mg/d continuous dosing or 10 mg/day intermittent 7 days on/7 days off dosing. Both regimens have shown promising efficacy and tolerability [50]. Based on these results and erdafitinib pharmacometric modeling, dosing was optimized at 8 mg/day continuously and further uptitrated to 9 mg/day if no significant treatment-related adverse events were observed during the first cycle. In the latter cohort, 99 patients were enrolled and treated with 8 mg daily for 28 days, with escalation to 9 mg allowed in the absence of significant adverse events [51]. All patients had metastatic or surgically unresectable UC with FGFR3 mutation or FGFR2 or FGFR3 fusion. Prior treatment with chemotherapy and/or immune checkpoint inhibitors was allowed. Treatment-related adverse events were manageable, with 10 percent of patients discontinuing treatment due to symptoms. There were no treatment-related deaths and no Grade 4 events. The most common adverse events were Grade 1 and 2, including hyperphosphatemia (72 patients, Grade≥3 in 2 patients), stomatitis (54 patients, Grade≥3 in 9 patients), and diarrhea (37 patients, Grade≥3 in 4 patients). Treatment with erdafitinib met its primary objective with a 40% overall response rate, including complete response, in three percent of patients and partial response, or tumor shrinkage, in 37 percent. An additional 39% of patients had stable disease without progression. Preliminary data from the trial indicate a median overall survival of 13.8 months. Based on the Phase 2 study, the U.S. Food and Drug Administration granted a breakthrough therapy designation to erdafitinib in 2018.
Infigratinib
Infigratinib (BGJ398), is an orally bio-available, selective and ATP competitive pan-FGFR TKI. At the cellular level, infigratinib selectively inhibits the kinase activity of FGFR1, FGFR2, FGFR3 and FGFR4 with IC50 values of 2-8 nM for FGFR1-3. Consistent with inhibition of FGFR autophosphorylation, infigratinib inhibits FGFR downstream signalling and proliferation of human cancer cell lines harbouring genetic alterations of the FGFRs including lung, breast, gastric and urothelial cancers. In mice, infigratinib showed a significant, dose-dependent anti-tumor activity against RT112 xenografts tumors [40].
A phase 1 study investigated infigratinib in patients with solid tumors carrying FGFR genetic alterations (mutation or fusion) [52]. Ninety-four patients received infigratinib once or twice daily in 28-day cycles in escalating cohorts. Maximal tolerated dose was 125 mg qd 3 weeks on and 1 week off. In terms of efficacy, four out of five patients with FGFR3 mutated UC had tumor regression. Antitumor activity observed in this phase 1 trial (NCT01004224) led to initiation of an extended cohort of genetically-selected patients to further characterize infigratinib activity in UC. In this extended cohort, genetically selected and previously platinum-treated UC patients were treated with infigratinib administered orally at 125 mg/day on a 3 weeks on, 1 week off schedule until unacceptable toxicity or progression [53]. Among 67 patients treated, an overall response rate of 25.4% was observed and an additional 38.8% of patients had disease stabilization, translating to a disease control rate of 64.2%. The most common toxicities were those expected with the pharmacodynamic inhibition of FGFR, e.g, hyperphosphatemia and decreased appetite.
Rogaratinib
Rogaratinib (BAY 1163877) is an oral inhibitor of all FGFR subtypes with low nanomolar binding pan-FGFR kinase inhibitor [54]. Rogaratinib has been assessed in a phase 1 trial in which patients with advanced UC were selected based on high FGFR1-3 mRNA expression in biopsy specimens [55]. The test used in this study was based on RNAscope technology which is an in situ hybridization (ISH) assay for detection of FGFR1-3 RNA within intact cells. This test enables simultaneous signal amplification and background noise suppression and is compatible with routine formalin-fixed, paraffin-embedded tissue specimens. Patients were treated with rogaratinib 800 mg twice daily until tumor progression, untolerable toxicity or withdrawal. Of 219 prescreened patients, 45% were found to be FGFR positive. Of those, 87% of samples were positive for FGFR3 mRNA, 5% for FGFR1 mRNA and 8% were double FGFR mRNA-positive (FGFR1/2, 1/3 or 2/3). Frequency of FGFR3 activating mutations was only 7%, all of which also had high FGFR3 mRNA. Among the 51 patients evaluable for response, ORR was 24% and disease control rate was 73%.
AZD4547
AZD4547 is a potent and selective inhibitor if FGFR1,2 and 3 receptor tyrosine kinases (IC50 < 5nM). AZD4547 was shown to inhibit proliferation in a dose-dependent manner in cancer cell lines known to overexpress FGFRs [56]. Oral treatment of mice bearing FGFR-amplified cancer cell lines resulted on dose-dependent tumor growth inhibition. The preliminary results of two phase I studies (NCT00979134 and NCT01213160) assessing the safety and tolerability of AZD4547 in advanced solid tumors have been reported [57]. In the first study which has been performed in a western population, one bladder cancer patient harboring FGFR-amplification had SD [58]. The specificity of AZD4547 is the activity in tumor selected for FGFR1 and FGFR2 amplification. However, neither FGFR1 amplification nor FGFR2 amplification are common in UC. AZD4547 is currently investigated in association with durvalumab in BISCAY (NCT02546661) study for chemo-treated metastatic patients with tumors that have FGFR3 mutations or fusions.
Pemigatinib
Pemigatinib (INCB054828) is a potent inhibitor of the kinase activity of FGFR1, FGFR2 and FGFR3 and has been shown to inhibit growth in several tumor models. In a phase 1/2 study, patients with refractory advanced solid tumors received pemigatinib qd, 2 weeks on/ 1 week off [59]. Recommended phase 2 dose was 13.5 mg qd and was well-tolerated. As for other specific FGFR inhibitors, hyperphosphatemia was the most common adverse event and preliminary efficacy in patients with FGFR aberration was observed. Fight-01 is an open-label phase 2 trial investigating pemigatinib in UC [60]. Preliminary results indicate that pemigatinib is well-tolerated and showed activity in previously-treated UC patients. Overall response rate including unconfirmed partial responses was 25% among 64 patients with FGFR3 mutations/fusions. Only one patient achieved partial response among 40 patients with other FGFR/FGFR gene alterations (e.g, FGFR10 amplification, FGFR1 amplification).
FURTHER DEVELOPMENT OF FGFR TARGETED THERAPIES IN UC
Demonstrating the efficacy in metastatic setting
Proof-of-concept of aforementioned phase 1/2 studies demonstrated that pan-FGFR inhibitors are active as single agents in patients with metastatic UC with selected FGFR mutations and translocations and have opened a promising therapeutic avenue in UC. Two phase 3 trials are now ongoing to demonstrate superiority of single agent pan-FGFR inhibitor over standard of care options in refractory subjects with selected FGFR3 gene aberrations. Erdafitinib, the most active FGFR inhibitor based on the data from the phase 1 and phase 2 studies is currently investigated in THOR study. THOR (NCT03390504) is a randomized open-label multicenter, global phase 3 study of erdafitinib versus standard of care in patients with advanced UC and selected FGFR aberrations who have progressed on or after one prior line of therapy. The primary objective of THOR is to evaluate efficacy of erdafitinib versus chemotherapy or pembrolizumab in patients with advanced UC harboring selected FGFR aberrations (mutations or fusions) The primary endpoint is overall survival and will be evaluated in two cohorts: (i) erdafitinib versus chemotherapy (docetaxel or vinflunine) in patients who have received prior anti-PD(L)1 agent (ii) erdafitinib versus pembrolizumab in patients who have not received prior anti-PD(L)1 agent. Secondary endpoints include progression-free survival, response rate, quality of life and safety profile. Exploratory objectives will evaluate DNA, RNA and protein biomarkers in tissue and blood samples to predict tumor response or resistance. The two cohorts will be assessed independently and 631 patients are needed.
FORT-1 study (NCT03410693) is a randomized open-label, multicenter phase 2/3 study to evaluate the efficacy and safety of rogaratinib compared to chemotherapy in patients with FGFR positive advanced UC who have received prior platinum containing chemotherapy. The primary endpoint will be overall survival and usual secondary endpoints (PFS, response rate, safety) will be assessed. Overall, 400 patients will be enrolled into the arms: rogaratinib or chemotherapy arm. In this study, molecular tests encompass mutations, fusions but also FGFR1 or FGFR3 overexpression using an RNA in situ hybridization (RNA-ISH) test as described above. Given these criteria, approximately, 40% of UC patients with advanced UC may be identified as FGFR-positive.
Investigating the activity in early stage of UC
The clinical course of UC is dominated by frequent recurrence and/or progression of NMIBC to MIBC. Most new UC cases are non-muscle invasive at diagnosis. Therefore, there is a need to explore the clinical efficacy of novel therapies in non-metastatic UC including NMIBC. As described above, FGFR3 has been involved as a critical driver of NMIBC carcinogenesis. FGFR3 mutations and/or overexpression have been found in up to 70% of low-grade NMIBC. These mutations result in clinical phenotype dominated by frequent NMIBC recurrences with infrequent progression to MIBC. But, in the context of CDKN2A deletions, FGFR3 mutations drive the progression to MIBC. Dovitinib was investigated in BCG-refractory NMIBC patients harboring FGFR3 gene alterations. A multicenter phase 2 trial was conducted to assess the 6-month TURBT-confirmed complete response in 13 BCG-unresponsive NMIBC patients with increased phosphorylated FGFR3 expression analyzed by immunohistochemistry or FGFR3 mutations [61]. The primary endpoint was not met. The 6-month CR rate was only 8% but 33% in patients (1 out of 3) with FGFR3 mutations. However, long-term dovitinib administration was not feasible due to frequent toxicity as all patients developed grade 3/4 events. These data suggest that one key issue in this setting is to develop tolerable regimen in these asymptomatic and curable patients.
Non-metastatic MIBC is another area of intensive investigation of FGFR inhibitors. Several trials are investigating FGFR inhibitors in adjuvant setting. An open-label phase 2 study will assess the safety and efficacy of adjuvant INCB054828 in patients with pT3-4 and/or pN1-N3 UC harboring FGFR3 mutations/fusions. The most attractive setting remains neo-adjuvant setting by collecting pre and post tumor samples for pharmacodynamic analysis of FGFR inhibition. Furthermore, bladder sparing strategies may be investigated if pilot studies in neo-adjuvant studies would be able to demonstrate high complete and durable response rate in MIBC.
Combining FGFR inhibitors
Not all patients with FGFR3 aberration respond to FGFR inhibition which raises the question of which strategy may be envisioned to improve the proportion of patients benefiting from FGFR inhibitors. The rationale of various combinations may be investigated based on the off-target mechanisms of resistance discussed below, e.g PI3K and FGFR3 inhibitors; EGFR and FGFR3 inhibitors.
Emerging data from retrospective analyses indicate that patients with FGFR3-altered UC have poorer response to immune checkpoint inhibitors (ICI). The response to ICI is largely dependent, among others, on pre-existing CD8 T cell infiltration. TCGA study showed that UC can be classified via gene expression signature into several subtypes. Luminal 1, papillary-like UC, are characterized by immune desert lacking immune cell infiltrate [13, 62]. Luminal 1 tumors are also enriched for FGFR3 mutations and gene fusions. Taken together, these data suggest that luminal 1 UC have evidence of FGFR3 aberrations and also exhibit a cold immune microenvironment, explaining the lower response of this subtype to ICI. Recent data from BLC2001 study show that across all treatment regimens, 33 patients had received PD(L)1 inhibitors prior to BLC2001 enrollment [35]. Only two out of 33 (6%) patients had responded to prior ICI. In the phase 1 study investigating rogaratinib, 10 patients with FGFR3 overexpression had prior ICI treatment. Of those, 9 patients (90%) experienced progressive disease as best response to ICI and one patient achieved stable disease for 9 months. All 10 patients expressed low PDL1 mRNA levels [36]. However, in another retrospective study, FGFR3 alterations did not preclude response to nivolumab and were not associated with decrease of CD8 T cell infiltration [37]. These results stress the need of prospective data to properly address the question. One strategy aims to switch cold tumor into hot tumor by inhibiting molecular oncogene. Several data from lung cancer and melanomas suggest that oncogene-addicted tumors exhibit poor T-cell infiltration [63]. Inhibiting these oncogenes may induce immune cell infiltration. Based on this rationale, several phase 1 studies are currently investigating the combination of FGFR3 inhibitors and ICI (NCT03473743, NCT03123055, NCT03473756).
CLINICAL CHALLENGES WITH FGFR INHIBITORS
Dosing regimens
Optimal dosing regimen remains critical for targeted agents. Three criteria should be taken into account for determining the best dosing regimen: optimal pharmacodynamic effect, optimal dose-intensity and clonal competitive release. Current paradigms indicate that some level of competition between genetically distinct tumor clones exist and therefore should be exploited for therapeutic purposes. An alternative strategy to high dose (meaning at near-maximum tolerated dose) regimens implies the administration of reduced dose of therapy on a continuous schedule. This type of regimen facilitates better dose-intensity and better control of tumor growth than full-dose therapy while intermittent dose with drug holidays may fail to control tumor growth for a long time in preclinical models. The underlying hypothesis is that elimination of a dominant drug-sensitive clone might allow the competitive release of resistant subclones to undergo accelerated growth resulting in rapid disease progression. BLC2001 results support this hypothesis in intermittent dosing as response rate was better in the 6 mg continuous dosing vs 10 mg intermittent dosing [50]. Ideally, the optimal dose of targeted therapies should be determinated on pharmacodynamics data. On-target toxicities from pan-FGFR inhibitors include hyperphosphatemia as well as skin and eye dryness. As a result, hyperphosphatemia should be observed in case of optimal FGFR inhibition. In BLC2001, hyperphosphatemia was more frequent in the continuous dosing (62% and 69% at 6 mg and 8 mg continuous dose versus 46% in the intermittent dose regimen). Based on the PK/PD data modeling, hyperphosphatemia was associated with better outcome in BLC2001. As a result, the dose was increased further to 8 mg continuous daily dosing with up-titration to 9 mg continuous daily in those whose serum phosphate levels remained normal (<5.5 mg/dl). The latter dosing regimen results in improved clinical activity with 40% of patients achieving response versus 24% in the intermittent dosing regimen and 35% in the 6 mg continuous regimen. Infigratinib is given at 125 mg daily using an intermittent schedule (3-weeks-on/1-week-off schedule). Rogaritinib is given at 800 mg continuously but only 45% of patients experience any grade hyperphosphatemia. For both infigratinib and rogaritinib, response rate is around 25%. These data suggest that dosing regimen of FGFR inhibitor may be critical for optimal antitumor activity.
Patient selection
Patient selection is one of the key issues in the field of targeted therapies. Distinct alterations in FGFR3 may sensitize to FGFR inhibitors differently. The majority of clinical trials select patients on the presence of FGFR3 mutations or translocations. But, as discussed above, FORT-1 allowed patients with FGFR3 overexpression as assessed by ISH to be eligible. To date, there is no data indicating whether FGFR3 overexpression may add useful information for selecting the best target population for FGFR3 inhibitor. In the phase 1 study of rogaratinib, 50% of patients were found to be FGFR1-3 positive on archival biopsies. Of those, nearly 94% were positive for FGFR3 mRNA. Among patients with FGFR overexpression, 17% harbor FGFR3 mutation suggesting that testing for FGFR3 expression may broaden the target population. Seven out of eleven patients with a complete or partial response showed FGFR3 mRNA overexpression without FGFR3 gene mutation. However, only 8% of all screened patients had FGFR3 activating mutations suggesting that molecular testing for FGFR3 mutations was less sensitive than that of used in BLC2001 or BGJ398 study. FGFR3 or its ligands (e.g, FGF3, FGF4) amplifications are not molecular criteria for enrolling patients in clinical trial with FGFR3 inhibitors. However, robust data are needed to address UC addiction to amplification of FGFR members. Fight-01 study with pemigatinib includes one cohort dedicated to molecular aberrations beyond FGFR3 mutations and fusions. The data may be reported early 2019.
In terms of DNA genetic alterations, not all FGFR3 aberrations confer sensitivity to FGFR3 inhibitors. The K650E mutation located in the kinase domain is a FGFR3-activating mutation that leads to destabilization of the inactive conformation of the kinase domain and stabilization of the active conformation of the activation loop of the kinase domain of the receptor. However, this mutation should not be associated with sensitivity to most FGFR3 inhibitors currently investigated. Even though this residue is not in direct contact with infigratinib, this mutation destabilizes the inactive conformation to which infigratinib binds and may explain the lower activity of infigratinib against K650E-mutant FGFR3. A thorough analysis of activity of FGFR inhibitors according to genetic alterations is needed to move forward to better selection of patients eligible to anti-FGFR therapy. This issue raises the question as to which molecular testing should be used. Selection of patient eligible to erdafitinib refers to PCR-based strategy while others study with pemigatinib or infigratinib use NGS to select the patients. All these tests required recent archival tumor samples and tumor biopsy material is often limited. Increasing evidence is emerging to support screening for FGFR aberrations in circulating tumor DNA extracted from plasma or urine [53]. This issue is critical to conduct clinical trials in non-metastatic setting whereby turn-around for molecular screening should be fast enough to start off therapy as soon as possible.
Management of toxicities
Non-selective inhibitors, such as dovitinib or pazopanib, are not well tolerated with serious adverse events occurring in one-fourth of the patients and grade≥3 toxicity rates between 60 and 100% [44]. These adverse events are related in part to the inhibition of angiogenesis and include hypertension, cardiovascular events, and proteinuria [64]. Other commonly reported toxicities are shared with other TKIs, including vomiting, diarrhea and skin reactions [64]. Selective inhibitors have much more acceptable safety profiles with nearly 10% of the patients discontinuing the drug owing to serious adverse events, and grade≥3 toxicity rates between 10 and 15% [48]. As discussed above, the main toxicities are hyperphosphatemia which could be easily managed by phosphate binders. Other toxicities include skin and eye dryness, keratopathy and retinal pigment epithelial detachment which are rare and reversible. Obviously, clinical experience with these drugs remains still limited and long-term consequences of FGFR inhibition have to be properly investigated.
Primary and acquired resistance
As seen in most cancer therapies, resistance to FGFR inhibitors is ineluctable. Patients with initial response to FGFR inhibitors ultimately relapse on therapy, generally within 12 to 18 months. Analysis of post-progression biopsy specimens remains extremely rare in the context of FGFR inhibitors. Therefore, mechanisms of resistance to FGFR inhibitors are largely unknown. Theoretically, mechanisms of resistance to TKIs should be heterogeneous and encompass on-target genetic alterations (e.g, FGFR resistance mutations, FGFR gene amplification) and off-target mechanisms of resistance (e.g, upregulation of by-pass signaling pathways). Exploratory studies are ongoing to figure out the mechanisms of both primary and acquired resistance. On-target mechanisms of resistance could result from mutations in the ATP binding site of the receptor tyrosine kinase which confer resistance to inhibitors as seen in lung cancer. Recurrent point mutations in the FGFR2 kinase domain were found in patients with FGFR2-fusion positive cholangiocarcinoma developing acquired resistance to infigratinib [65]. All these mutations (p549H, p549K, pV564F, pE565A, pL617V, pK659M, pK641R) compromise inhibition by infigratinib. Especially, p.V564F gatekeeper mutation confers resistance by inducing a steric clash with infigratinib in its FGFR2 binding pocket. The other mutation destabilizes the inactive conformation of the kinase. As FGFR2 and FGFR3 share nearly 90% amino acid homology in their kinase domains, all six corresponding residues in FGFR3 would be expected to play a similar role in resistance to at least infigratinib in UC. In preclinical model, exposure TEL-FGFR3-expressing BaF3 cells to various dose of infigratinib resulted in resistant colonies harboring distinct FGFR3 mutations. All these mutations (pN540K, pV555L, pV555M, pL608V and pK650E mutations) result in amino acids changes that corresponded to those seen in FGFR2 in patients with cholangiocarcinoma. However, such mutations seem to be very rare in UC. In the infigratinib study in UC, samples from 22 patients who progressed while on treatment were analyzed for novel resistance mutations [53]. Recurrent mutations were not detected. FGFR3 gatekeeper mutations (V443L, V443M and L496V) were detected in the cell free DNA of only 4 patients during treatment. Taken together, these preliminary data stress the need of systematic analyses of cfDNA and tissue samples collected at time of acquired resistance in ongoing prospective studies of FGFR inhibitors.
Another issue consists in the intra-tumor heterogeneity which is a common hurdle to precision-medicine in solid tumors, especially for patient selection. Differential activation of FGFR transduction pathway and co-occuring oncogenic events are likely to be critical in determining whether or not FGFR3 positive UC depend on FGFR3 pathways for survival. Recent reports in other oncogene-addicted tumors suggest that clonal event and early truncal events are critical in terms of therapeutic targeting potential. In UC, the importance of clonality in response to FGFR3 mutations and fusions has to be investigated. As of result, off-target mechanisms of resistance may be predominant in UC developing acquired resistance to FGFR inhibitors [66, 67]. By performing a synthetic lethality screen for AZD4547 using a short hairpin RNA library targeting the human kinome in FGFR3-TACC3 fusion positive RT112 cell line, multiple members of the phosphoinositide 3-kinase pathway have been associated with resistance to FGFR inhibition [66]. Inhibition of PI3KCA enhances the efficacy of FGFR inhibitors. These data suggest that resistance pathway to FGFR inhibitors often converge on the PI3K pathway and thus provide a rationale to treat FGFR3-altered UC with a combination of FGFR and PI3K inhibitors. In other study using similar strategy with RNA interference genetic screens, EGFR receptor has been shown to limit sensitivity to FGFR inhibition in FGFR3-mutant and FGFR3–translocated cell lines. Combination of FGFR and EGFR inhibitors overcome the resistance mechanisms in vitro and in vivo [67].
CONCLUSION
Early clinical data from phase 1 and 2 studies demonstrate that targeting FGFR3 aberrations is a promising strategy to improve the outcome of a subset of metastatic UC. Current phase 3 trials with erdafitinib (THOR study) and rogaratinib (FORT-1 study) are ongoing to demonstrate whether FGFR inhibitors improve outcome over standard of care in FGFR3-altered metastatic UC. A better understanding of biology of UC harboring FGFR3 mutations or fusions as well as mechanisms of resistance to FGFR inhibitors will provide useful insights into how combining FGFR3 inhibitors in the future. Taken together, these data show that FGFR targeting may change the clinical practice in UC harboring FGFR3 aberrations.
FUNDING
The authors report no funding.
CONFLICTS OF INTEREST
Dr. LORIOT reports grants, personal fees, non-financial support and other from Janssen, personal fees and other from Incyte, during the conduct of the study; personal fees, non-financial support and other from Astra Zeneca, grants, personal fees and non-financial support from MSD, personal fees and non-financial support from BMS, personal fees from clovis, personal fees and non-financial support from Seattle Genetics, personal fees from Pfizer, outside the submitted work; In addition, Dr. LORIOT has a patent Cancer Treatment (US: 62/455211ÞUROPE: 17209098.7) pending, and a patent Cancer Treatment (WO2018/141921) pending.
The other authors have no conflicts of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/BLC-180205.
ACKNOWLEDGMENTS
The authors have no acknowledgments.
REFERENCES
[1] | Powles T , Eder JP , Fine GD , Braiteh FS , Loriot Y , Cruz C , Bellmunt J , Burris HA , Petrylak DP , Teng SL , Shen X , Boyd Z , Hegde PS , Chen DS , Vogelzang NJ . MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. (2014) ;515: (7528):558–62. |
[2] | Powles T , Duran I , van der Heijden MS , Loriot Y , Vogelzang NJ , De Giorgi U , Oudard S , Retz MM , Castellano D , Bamias A , Flechon A , Gravis G , Hussain S , Takano T , Leng N , Kadel EE>3rd , Banchereau R , Hegde PS , Mariathasan S , Cui N , Shen X , Derleth CL , Green MC , Ravaud A . Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): A multicentre, open-label, phase 3 randomised controlled trial. Lancet. (2018) 24;391: (10122):748–57. |
[3] | Rosenberg JE , Hoffman-Censits J , Powles T , van der Hei-jden MS , Balar AV , Necchi A , Dawson N , O’Donnell PH , Balmanoukian A , Loriot Y , Srinivas S , Retz MM , Grivas P , Joseph RW , Galsky MD , Fleming MT , Petrylak DP , Perez-Gracia JL , Burris HA , Castellano D , Canil C , Bellmunt J , Bajorin D , Nickles D , Bourgon R , Frampton GM , Cui N , Mariathasan S , Abidoye O , Fine GD , Dreicer R . Ate-zolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet. (2016) ;387: (10031):1909–20. |
[4] | Petrylak DP , Powles T , Bellmunt J , Braiteh F , Loriot Y , Morales-Barrera R , Burris HA , Kim JW , Ding B , Kaiser C , Fasso M , O’Hear C , Vogelzang NJ . Atezolizumab (MPDL3280A) Monotherapy for Patients With Metastatic Urothelial Cancer: Long-term Outcomes From a Phase 1 Study. JAMA Oncol. (2018) ;4: (4):537–44. |
[5] | Balar AV , Galsky MD , Rosenberg JE , Powles T , Petrylak DP , Bellmunt J , Loriot Y , Necchi A , Hoffman-Censits J , Perez-Gracia JL , Dawson NA , van der Heijden MS , Dreicer R , Srinivas S , Retz MM , Joseph RW , Drakaki A , Vaisham-payan UN , Sridhar SS , Quinn DI , Duran I , Shaffer DR , Eigl BJ , Grivas PD , Yu EY , Li S , Kadel EE 3rd , Boyd Z , Bourgon R , Hegde PS , Mariathasan S , Thåström A , Abidoye OO , Fine GD , Bajorin DF ; IMvigor210 Study Group. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet. (2017) ;389: (10064):67–76. |
[6] | Balar AV , Castellano D , O’Donnell PH , Grivas P , Vuky J , Powles T , Plimack ER , Hahn NM , de Wit R , Pang L , Savage MJ , Perini RF , Keefe SM , Bajorin D , Bellmunt J . First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. (2017) ;18: (11):1483–92. |
[7] | Sharma P , Callahan MK , Bono P , Kim J , Spiliopoulou P , Calvo E , Pillai RN , Ott PA , de Braud F , Morse M , Le DT , Jaeger D , Chan E , Harbison C , Lin CS , Tschaika M , Azrile-vich A , Rosenberg JE . Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): A multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol. (2016) ;17: (11):1590–8. |
[8] | Sharma P , Retz M , Siefker-Radtke A , Baron A , Necchi A , Bedke J , Plimack ER , Vaena D , Grimm MO , Bracarda S , Arranz JA , Pal S , Ohyama C , Saci A , Qu X , Lambert A , Krishnan S , Azrilevich A , Galsky MDplimack. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. (2017) ;18: (3):312–22. |
[9] | Massard C , Gordon MS , Sharma S , Rafii S , Wainberg ZA , Luke J , Curiel TJ , Colon-Otero G , Hamid O , Sanborn RE , O’Donnell PH , Drakaki A , Tan W , Kurland JF , Rebe-latto MC , Jin X , Blake-Haskins JA , Gupta A , Segal NH . Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J Clin Oncol. (2016) ;34: (26):3119–25. |
[10] | Plimack ER , Bellmunt J , Gupta S , Berger R , Chow LQ , Juco J , Lunceford J , Saraf S , Perini RF , O’Donnell PH . Safety and activity of pembrolizumab in patients with locally advanced or metastatic urothelial cancer (KEYNOTE-O12): A non-randomised, open-label, phase 1b study. Lancet Oncol. (2017) ;18: (2):212–20. |
[11] | Bellmunt J , de Wit R , Vaughn DJ , Fradet Y , Lee JL , Fong L , Vogelzang NJ , Climent MA , Petrylak DP , Choueiri TK , Necchi A , Gerritsen W , Gurney H , Quinn DI , Culine S , Sternberg CN , Mai Y , Poehlein CH , Perini RF , Bajorin DF ; KEYNOTE-045 Investigators. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med. (2017) ;376: (11):1015–26. |
[12] | Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. (2014) ;507: (7492):315–22. |
[13] | Robertson AG , Kim J , Al-Ahmadie H , Bellmunt J , Guo G , Cherniack AD , Hinoue T , Laird PW , Hoadley KA , Akbani R , Castro MAA , Gibb EA , Kanchi RS , Gordenin DA , Shukla SA , Sanchez-Vega F , Hansel DE , Czerniak BA , Reuter VE , Su X , de Sa Carvalho B , Chagas VS , Mungall KL , Sadeghi S , Pedamallu CS , Lu Y , Klimczak LJ , Zhang J , Choo C , Ojesina AI , Bullman S , Leraas KM , Lichtenberg TM , Wu CJ , Schultz N , Getz G , Meyerson M , Mills GB , McConkey DJ ; TCGA Research Network, Weinstein JN, Kwiatkowski DJ, Lerner SP. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. (2017) ;171: (3):540–56. |
[14] | Cappellen D , De Oliveira C , Ricol D , de Medina S , Bourdin J , Sastre-Garau X , Chopin D , Thiery JP , Rad-vanyi F . Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. (1999) ;23: (1): 18–20. |
[15] | Dieci MV , Arnedos M , Andre F , Soria JC . Fibroblast growth factor receptor inhibitors as a cancer treatment: From a biologic rationale to medical perspectives. Cancer Discov. (2013) ;3: (3):264–79. |
[16] | Thompson LM , Plummer S , Schalling M , Altherr MR , Gusella JF , Housman DE , Wasmuth JJ . A gene encoding a fibroblast growth factor receptor isolated from the Huntington disease gene region of human chromosome 4. Genomics. (1991) ;11: (4):1133–42. |
[17] | Harmer NJ , Ilag LL , Mulloy B , Pellegrini L , Robinson CV , Blundell TL . Towards a resolution of the stoichiometry of the fibroblast growth factor (FGF)-FGF receptor-heparin complex. J Mol Biol. (2004) ;339: (4):821–34. |
[18] | Chen Y , Li X , Eswarakumar VP , Seger R , Lonai P . Fibrob-last growth factor (FGF) signaling through PI 3-kinase and Akt/PKB is required for embryoid body differentiation. Oncogene. (2000) ;19: (33):3750–6. |
[19] | Itoh N , Ornitz DM . Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. (2011) ;149: (2):121–30. |
[20] | Babina IS , Turner NC . Advances and Challenges in Targeting FGFR Signalling in Cancer. Nat Rev Cancer. (2017) ;17: (5):318–32. |
[21] | Helsten T , Elkin S , Arthur E , Tomson BN , Carter J , Kurzrock R . The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin Cancer Res. (2016) ;22: (1):259–67. |
[22] | Wagle N , Berger MF , Davis MJ , Blumenstiel B , Defelice M , Pochanard P , Ducar M , Van Hummelen P , Macconaill LE , Hahn WC , Meyerson M , Gabriel SB , Garraway LA . High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. (2012) ;2: (1):82–93. |
[23] | Chae YK Ranganath K , Hammerman PS , Vaklavas C , Mohindra N , Kalyan A , Matsangou M , Costa R1 , Carneiro B , Villaflor VM , Cristofanilli M , Giles FJ . Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Onco-target. (2017) ;8: (9):16052–74. |
[24] | Hedegaard J , Lamy P , Nordentoft I , Algaba F , Høyer S , Ulhøi BP , Vang S , Reinert T , Hermann GG , Mogensen K , Thomsen MBH , Nielsen MM , Marquez M , Segersten U , Aine M , Hoglund M , Birkenkamp-Demtroder K , Fristrup N , Borre M , Hartmann A , Stöhr R , Wach S , Keck B , Seitz AK , Nawroth R , Maurer T , Tulic C , Simic T , Junker K , Horstmann M , Harving N , Petersen AC , Calle ML , Steyerberg EW , Beukers W , van Kessel KEM , Jensen JB , Pedersen JS , Malmstrom PU , Malats N , Real FX , Zwarthoff EC , ørntoft TF , Dyrskjøt L . Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell. (2016) ;30: (1):27–42. |
[25] | d’Avis PY , Robertson SC , Meyer AN , Bardwell WM , Webster MK , Donoghue DJ . Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth Differ. (1998) ;9: (1):71–8. |
[26] | di Martino E1 , L’Hote CG , Kennedy W , Tomlinson DC , Knowles MA . Mutant fibroblast growth factor receptor 3 induces intracellular signaling and cellular transformation in a cell type-and mutation-specific manner. Oncogene. (2009) ;28: (48):4306–16. |
[27] | Williams SV , Hurst CD , Knowles MA . Oncogenic FGFR3 gene fusions in bladder cancer. Hum Mol Genet. (2013) ;22: (4):795–803. |
[28] | Nord H , Segersten U , Sandgren J , Wester K , Busch C , Menzel U , Komorowski J , Dumanski JP , Malmström PU , Diaz de Stähl T . Focal amplifications are associated with high grade and recurrences in stage Ta bladder carcinoma. Int J Cancer. (2010) ;126: (6):1390–402. |
[29] | Frattini V , Pagnotta SM , Tala , Fan JJ , Russo MV , Lee SB , Garofano L , Zhang J , Shi P , Lewis G , Sanson H , Frederick V , Castano AM , Cerulo L , Rolland DCM , Mall R , Mokhtari K , Elenitoba-Johnson KSJ , Sanson M , Huang X , Ceccarelli M , Lasorella A , Iavarone A. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature. (2018) ;553: (7687):222–7. |
[30] | Hernandez S , Lopez-Knowles E , Lloreta J , Kogevinas M , Amoros A , Tardón A , Carrato A , Serra C , Malats N , Real FX . Prospective study of FGFR3 mutations as a prognostic factor in nonmuscle invasive urothelial bladder carcinomas. J Clin Oncol. (2006) ;24: (22):3664–71. |
[31] | Hernández S , López-Knowles E , Lloreta J , Kogevinas M , Jaramillo R , Amoros A , Tardón A , García-Closas R , Serra C , Carrato A , Malats N , Real FX . FGFR3 and Tp53 mutations in T1G3 transitional bladder carcinomas: Independent distribution and lack of association with prognosis. Clin Cancer Res. (2005) ;11: (15):5444–50. |
[32] | van Rhijn BW , van der Kwast TH , Liu L , Fleshner NE , Bostrom PJ , Vis AN , Alkhateeb SS , Bangma CH , Jew-ett MA , Zwarthoff EC , Zlotta AR , Bapat B . The FGFR3 mutation is related to favorable pT1 bladder cancer. J Urol. (2012) ;187: (1):310–4. |
[33] | Rebouissou S , Herault A , Letouze E , Neuzillet Y , Laplanche A , Ofualuka K , Maille P , Leroy K , Riou A , Lepage ML , Vordos D , de la Taille A , Denoux Y , Sibony M , Guyon F , Lebret T , Benhamou S , Allory Y , Radvanyi F . CDKN2A homozygous deletion is associated with muscle invasion in FGFR3-mutated urothelial bladder carcinoma. J Pathol. (2012) ;227: (3):315–24. |
[34] | Necchi A , Lo Vullo S , Raggi D , Gloghini A , Gian-natempo P , Colecchia M , Mariani L . Prognostic Effect of FGFR Mutations or Gene Fusions in Patients with Metastatic Urothelial Carcinoma Receiving First-line Platinum-based Chemotherapy: Results from a Large, Single-institution Cohort. Eur Urol Focus. (2018) ; pii: S2405-4569(18)30075-0.. |
[35] | Siefker-Radtke AO , Necchi A , Rosenbaum E , Culine S , Burgess EF , O’Donnell PH , Tagawa ST , Zakharia Y , OHagan A , Avadhani AN , Zhong B , Santiago-Walker AE , Roccia T , and Loriot Y . Efficacy of programmed death 1 (PD-1) and programmed death 1 ligand (PD-L1) inhibitors in patients with FGFR mutations and gene fusions: Results from a data analysis of an ongoing phase 2 study of erdafitinib (JNJ-42756493) in patients (pts) with advanced urothelial cancer (UC). J Clin Oncol. (2018) ;36: :(6_suppl):450. |
[36] | Joerger M , Cassier PA , Penel N , Cathomas R , Richly H , Schostak M , Janitzky A , Wermke M , Nogova L , Tai DWM , Sayehli C , Griillich C , Grande E , Navarro A , Park SH , Nogai H , Bender S , Ellinghaus P , and Schuler MH . Rog-aratinib in patients with advanced urothelial carcinomas prescreened for tumor FGFR mRNA expression and effects of mutations in the FGFR signaling pathway. J Clin Oncol. (2018) ;36: :15_suppl:4513. |
[37] | Galsky MT , Saci A , Szabo P , Wang L , Zhu J , Azrilevich A , Fischer BS , Necchi A , Siefker-Radtke AO , and Sharma P . Fibroblast growth factor receptor 3 (FGFR3), peroxisome proliferator-activated receptor gamma (PPARg), and outcomes with nivolumab (nivo) in metastatic urothelial cancer (UC). J Clin Oncol. (2018) ;36: :6_suppl, 511–511. |
[38] | Karkera JD , Cardona GM , Bell K , Gaffney D , Portale JC , Santiago-Walker A , Moy CH , King P , Sharp M , Bahleda R , Luo FR , Alvarez JD , Lorenzi MV , Platero SJ . Oncogenic Characterization and Pharmacologic Sensitivity of Activating Fibroblast Growth Factor Receptor (FGFR) Genetic Alterations to the Selective FGFR Inhibitor Erdafitinib. Mol Cancer Ther. (2017) ;16: (8):1717–26. |
[39] | Wu YM , Su F , Kalyana-Sundaram S , Khazanov N , Ateeq B , Cao X , Lonigro RJ , Vats P , Wang R , Lin SF , Cheng AJ , Kunju LP , Siddiqui J , Tomlins SA , Wyngaard P , Sadis S , Roychowdhury S , Hussain MH , Feng FY , Zalupski MM , Talpaz M , Pienta KJ , Rhodes DR , Robinson DR , Chin-naiyan AM . Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. (2013) ;3: (6):636–47. |
[40] | Guagnano V , Furet P , Spanka C , Bordas V , Le Douget M , Stamm C , Brueggen J , Jensen MR , Schnell C , Schmid H , Wartmann M , Berghausen J , Drueckes P , Zimmerlin A , Bussiere D , Murray J , Graus Porta D . Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem. (2011) ;54: (20):7066–83. |
[41] | Lamont FR , Tomlinson DC , Cooper PA , Shnyder SD , Chester JD , Knowles MA . Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo. Br J Cancer. (2011) ;104: (1):75–82. |
[42] | Bellmunt J , Picus J , Kohli M , Arriaga YE , Milowsky MI Currie G , Abella S , Pal SK . FIERCE-21: Phase 1b/2 study of docetaxel + b-701, a selective inhibitor of FGFR3, in relapsed or refractory (R/R) metastatic urothelial carcinoma (mUCC). J Clin Oncol. (2018) ;36: :(suppl; abstr 4534). |
[43] | Donnell PO , Goldman JW , Gordon MS , Shih K , Choi YJ , Lu D , Kabbarah O , Ho W , Rooney I , Lam ET . A Phase I Dose-escalation Study of MFGR1877S, a Human Monoclonal Anti-fibroblast Growth Factor Receptor 3 (FGFR3) Antibody, in Patients (pts) with Advanced Solid Tumors. Eur J Cancer. (2012) ;48: :191–2. |
[44] | Milowsky MI , Dittrich C , Duran I , Jagdev S , Millard FE , Sweeney CJ , Bajorin D , Cerbone L , Quinn DI , Stadler WM , Rosenberg JE , Lochheed M , Sen P , Squires M , Shi M , Sternberg CN . Phase 2 trial of dovitinib in patients with progressive FGFR3-mutated or FGFR3 wild-type advanced urothelial carcinoma. Eur J Cancer. (2014) ;50: (18):3145–52. |
[45] | Necchi A , Mariani L , Zaffaroni N , Schwartz LH , Gian-natempo P , Crippa F , Morosi C , Lanocita R , Sava T , Ortega C , Messina C , Sacco C , Pennati M , Daidone MG , Nicolai N , De Braud F , Gianni AM , Salvioni R . Pazopanib in advanced and platinum-resistant urothelial cancer: An open-label, single group, phase 2 trial. Lancet Oncol. (2012) ;13: (8):810–6. |
[46] | Pinciroli P , Won H , Iyer G , Canevari S , Colecchia M , Gian-natempo P , Raggi D , Pierotti MA , De Braud FG , Solit DB , Rosenberg JE , Berger MF , Necchi A . Molecular Signature of Response to Pazopanib Salvage Therapy for Urothelial Carcinoma. Clin Genitourin Cancer. (2016) ;14: (1):e81–90. |
[47] | Palma N , Morris JC , Ali SM , Ross JS , Pal SK . Exceptional Response to Pazopanib in a Patient with Urothelial Carcinoma Harboring FGFR3 Activating Mutation and Amplification. Eur Urol. (2015) ;68: (1):168–70. |
[48] | Tabernero J , Bahleda R , Dienstmann R , Infante JR , Mita A , Italiano A , Calvo E , Moreno V , Adamo B , Gazzah A , Zhong B , Platero SJ , Smit JW , Stuyckens K , Chatterjee-Kishore M , Rodon J , Peddareddigari V , Luo FR , Soria JC . Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients With Advanced Solid Tumors. J Clin Oncol. (2015) ;33: (30):3401–8. |
[49] | Soria JC , Italiano A , Cervantes A , Tabernero J , Infante J , Lara PN , Spira A , Calvo E , Moreno V , Blay JY , Lauer R , Chan N , Zhong B , Ademi Santiago-Walker A , Busso-lari , Luo FR , Xie H , Hammerman P . Safety and activity of the pan-fibroblast growth factor receptor (FGFR) inhibitor erdafitinib in phase 1 study patients with advanced urothelial carcinoma. Ann Oncol. (2016) ;27: (6):266–95. |
[50] | Loriot Y , Necchi A , Park SH , Garcia-Donas J , Huddart RA , Burgess EF , Fleming MT , Rezazadeh A , Mellado B , Var-lamov S , Joshi M , Duran I , OHagan A , Avadhani AN , Zhong B , Stuyckens K , Dosne AG , Siefker-Radtke AO, and On Behalf of the BLC2001 Study Group. Erdafitinib (ERDA; JNJ-42756493), a pan-fibroblast growth factor receptor (FGFR) inhibitor, in patients (pts) with metastatic or unre-sectable urothelial carcinoma (mUC) and FGFR alterations (FGFRa): Phase 2 continuous versus intermittent dosing. J Clin Oncol. (2018) ;36: (6_suppl):411. |
[51] | Siefker-Radtke AO , Necchi A Park SH , García-Donas J , Huddart RA , Burgess EF , Fleming MT , Rezazadeh A , Mellado B , Varlamov S , Joshi M , Duran I , Tagawa ST , OHagan A , Avadhani AN , Zhong B , De Porre P , Loriot Y , on behalf of the BLC2001 Study Group. First results from the primary analysis population of the phase 2 study of erdafitinib (ERDA; JNJ-42756493) in patients (pts) with metastatic or unresectable urothelial carcinoma (mUC) and FGFR alterations (FGFRalt). J Clin Oncol. (2018) ;36: (15_suppl):4503–4503. |
[52] | Nogova L , Sequist LV , Perez Garcia JM , Andre F , Delord JP , Hidalgo M , Schellens JH , Cassier PA , Camidge DR , Schuler M , Vaishampayan U , Burris H , Tian GG , Campone M , Wainberg ZA , Lim WT , LoRusso P , Shapiro GI , Parker K , Chen X , Choudhury S , Ringeisen F , Graus-Porta D , Porter D , Isaacs R , Buettner R , Wolf J . Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J Clin Oncol. (2017) ;35: (2):157–65. |
[53] | Pal SK , Rosenberg JE , Hoffman-Censits JH , Berger R , Quinn DI , Galsky MD , Wolf J , Dittrich C , Keam B , Delord JP , Schellens JHM , Gravis G , Medioni J , Maroto P , Sriuran-pong V , Charoentum C , Burris HA , Grünwald V , Petrylak D , Vaishampayan U , Gez E , De Giorgi U , Lee JL , Voort-man J , Gupta S , Sharma S , Mortazavi A , Vaughn DJ , Isaacs R , Parker K , Chen X , Yu K , Porter D , Graus Porta D , Bajorin DF . Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov. (2018) ;8: (7):812–21. |
[54] | Collin MP , Lobell M , Hübsch W , Brohm D , Schirok H , Jautelat R , Lustig K , Bomer U , Vohringer V , Heroult M , Grünewald S , Hess-Stumpp H . Discovery of Rogaratinib (BAY 1163877): A pan-FGFR Inhibitor. ChemMedChem. (2018) ;13: (5):437–45. doi: 10.1002/cmdc.201700718. |
[55] | Joerger M , Cassier P , Penel N , Cathomas R , Richly H , Schostak M , Janitzky A , Wermke M , Nogova L , Wai-Meng Tai D , Sayehli C , Grüllich C , Grande E , Navarro A , Park SH , Gillessen S , Noga Hi , Bender S , Elling-haus P , and Schuler MH . Rogaratinib treatment of patients with advanced urothelial carcinomas prescreened for tumor FGFR mRNA expression. Journal of Clinical Oncology. (2018) 36: (6_suppl):494. |
[56] | Gavine PR , Mooney L , Kilgour E , Thomas AP , Al-Kadhimi K , Beck S , Rooney C , Coleman T , Baker D , Mellor MJ , Brooks AN , Klinowska T . AZD4547: An orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. (2012) ;72: (8):2045–56. |
[57] | Paik PK , Shen R , Berger MF , Ferry D , Soria JC , Math-ewson A , Rooney C , Smith NR , Cullberg M , Kilgour E , Landers D , Frewer P , Brooks N , Andrei F . A Phase Ib Open-Label Multicenter Study of AZD4547 in Patients with Advanced Squamous Cell Lung Cancers. Clin Cancer Res. (2017) ;23: (18):5366–73. |
[58] | Andre F , Ranson m , Dean E , Varga A , van der Noll R , Stockman PK , Ghiorghiu D , Kilgour E , Smith PD , Macpherson M , Lawrence P , Hastie A , Schellens JHM . Results of aphase I study of AZD4547, an inhibitor of fibroblast growth factor receptor (FGFR), in patients with advanced solid tumors. Cancer Res. (2013) ;73: (8 Suppl):Abstract nr LB-145. |
[59] | Saleh M , Gutierrez M , Subbiah V , Smith DC , Feliz L , Zhen H , Ji T , Nemunaitis J . Preliminary results from a phase 1/2 study of INCB054828, a highly selective fibroblast growth factor receptor (FGFR) inhibitor, in patients (pts) with advanced malignancies. Mol Cancer Ther. (2018) ;17: (1 Suppl):Abstract nr A098. |
[60] | Necchi A , Pouessel D , Leibowitz-Amit R , Flechon A , Gupta S , Barthelemy P , Maio M , Zhu X , Asatiani E , Serbest G , Zhen H , Loriot Y . Interim results of fight-201, a phase II, open-label, multicenter study of INCB054828 in patients (pts) with metastatic or surgically unresectable urothe-lial carcinoma (UC) harboring fibroblast growth factor (FGF)/FGF receptor (FGFR) genetic alterations (GA). Ann Oncol. (2018) ;29: (Suppl 8): Abstract 900P. |
[61] | Hahn NM , Bivalacqua TJ , Ross AE , Netto GJ , Baras A , Park JC , Chapman C , Masterson TA , Koch MO , Bihrle R , Foster RS , Gardner TA , Cheng L , Jones DR , McElyea K , Sandusky GE , Breen T , Liu Z , Albany C , Moore ML , Loman RL , Reed A , Turner SA , De Abreu FB , Gallagher T , Tsongalis GJ , Pli-mack ER , Greenberg RE , Geynisman DM . A Phase II Trial of Dovitinib in BCG-Unresponsive Urothelial Carcinoma with FGFR3 Mutations or Overexpression: Hoosier Cancer Research Network Trial HCRN 12-157. Clin Cancer Res. (2017) ;23: (12):3003–11. |
[62] | Mariathasan S , Turley SJ , Nickles D , Castiglioni A , Yuen K , Wang Y , Kadel EE III , Koeppen H , Astarita JL , Cubas R , Jhunjhunwala S , Banchereau R , Yang Y , Guan Y , Chalouni C , Ziai J , Şenbabaoğlu Y , Santoro S , Sheinson D , Hung J , Giltnane JM , Pierce AA , Mesh K , Lianoglou S , Riegler J , Carano RAD , Eriksson P , Hoglund M , Somarriba L , Halli-gan DL , van der Heijden MS , Loriot Y , Rosenberg JE , Fong L , Mellman I , Chen DS , Green M , Derleth C , Fine GD , Hegde PS , Bourgon R , Powles T . TGFß attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) ;554: (7693):544–8. |
[63] | Spranger S , Gajewski TF . Impact of oncogenic pathway on evasion of antitumour immune responses. Nat rev Cancer. (2018) ;18: (3):139–47. |
[64] | Hierro C , Rodon J and Tabernero J . Fibroblast growth factor (FGF) Receptor/FGF inhibitors: Novel targets and strategies for optimization of response of solid tumors. Semin Oncol. (2015) ;42: (6):801–19. |
[65] | Goyal L , Saha SK , Liu LY , Siravegna G , Leshchiner I , Ahro-nian LG , Lennerz JK , Vu P , Deshpande V , Kambadakone A , Mussolin B , Reyes S , Henderson L , Sun JE , Van Seven-ter EE , Gurski JM Jr , Baltschukat S , Schacher-Engstler B , Barys L , Stamm C , Furet P , Ryan DP , Stone JR , Iafrate AJ , Getz G , Porta DG , Tiedt R , Bardelli A , Juric D , Corcoran RB , Bardeesy N , Zhu AX . Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov. (2017) ;7: (3):252–63. |
[66] | Wang L , Sustic T , Leite de Oliveira R , Lieftink C , Halo-nen P , van de Ven M , Beijersbergen RL , van den Heuvel MM , Bernards R , van der Heijden MS . A Functional Genetic Screen Identifies the Phosphoinositide 3-kinase Pathway as a Determinant of Resistance to Fibroblast Growth Factor Receptor Inhibitors in FGFR Mutant Urothelial Cell Carcinoma. Eur Urol. (2017) ;71: (6):858–62. |
[67] | Datta J , Damodaran S , Parks H , Ocrainiciuc C , Miya J , Yu L , Gardner EP , Samorodnitsky E , Wing MR , Bhatt D , Hays J , Reeser JW , Roychowdhury S . Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol Cancer Ther. (2017) ;16: (4):614–24. |


