
FTIR spectroelectrochemistry combined with a light-induced difference technique: Application to the iron-quinone electron acceptor in photosystem II
Abstract
Photosystem II (PSII) in plants and cyanobacteria performs light-driven water oxidation to obtain electrons necessary for CO2 fixation. In PSII, a series of electron transfer reactions take place from the Mn4CaO5 cluster, the catalytic site of water oxidation, to a plastoquinone molecule via several redox cofactors. Light-induced Fourier transform infrared (FTIR) difference spectroscopy has been extensively used to investigate the structures and reactions of the redox cofactors in PSII. Recently, FTIR spectroelectrochemistry combined with the light-induced difference technique was applied to study the mechanism of electron-transfer regulation in PSII involving the quinone electron acceptors, QA and QB, and the non-heme iron that bridges them. In this mini-review, this combined FTIR method is introduced, and obtained results about the redox reactions of the non-heme iron and QB, involving the long-range interaction of the Mn4CaO5 cluster with the electron-acceptor side, are summarized.
1.Introduction
Photosystem II (PSII), one of the major protein complexes that work in oxygenic photosynthesis, has a unique function of light-induced water oxidation [67]. Electrons from water are ultimately used for CO2 reduction and released protons generate a proton gradient across the thylakoid membrane to synthesize ATP. In addition, molecular oxygen, a byproduct of water oxidation, makes an oxygenic atmosphere, which is essential for sustenance of life on the earth. In photosystem II, light absorption triggers charge separation between the chlorophyll dimer (P680) and the pheophytin (Pheo) electron acceptor to produce P680+Pheo− [51]. On the electron-acceptor side, an electron on Pheo− is transferred to the primary quionone electron acceptor (QA) and then to the secondary quinone electron acceptor (QB) [16,38,48], while on the electron donor side, the electron hole on P680+ is transferred to the redox-active tyrosine (YZ) then to the Mn4CaO5 cluster, at which water oxidation takes place via a cycle of five intermediates called
Fig. 1.
Redox cofactors and the electron transfer pathway (blue arrows) in PSII. For the PSII structure, the X-ray crystallographic structure at 1.9 Å resolution (PDB code: 3ARC) [63] was used. The expanded view of the electron-acceptor side shows the arrangements of QA, QB, and the non-heme iron with their molecular interactions.
![Redox cofactors and the electron transfer pathway (blue arrows) in PSII. For the PSII structure, the X-ray crystallographic structure at 1.9 Å resolution (PDB code: 3ARC) [63] was used. The expanded view of the electron-acceptor side shows the arrangements of QA, QB, and the non-heme iron with their molecular interactions.](https://ip.ios.semcs.net:443/media/bsi/2016/5-3/bsi-5-3-bsi146/bsi-5-bsi146-g001.jpg)
The electron transfer reaction between QA and QB is controlled by the gap of their redox potentials (
Light-induced Fourier transform infrared (FTIR) difference spectroscopy has been extensively used to investigate the structures and reactions of the redox cofactors in PSII [5,11,13,14,39–43]. In this method, photoreactions are initiated by illumination of continuous-wave light or flashes from a pulse laser, and the structural changes of the cofactors and surrounding protein moieties are studied by detecting small infrared absorption changes. On the other hand, an FTIR spectroelectrochemical method has been used to investigate redox reactions of biomolecules and proteins [3,6,7,20,34,35,37]. In these studies, the redox reactions were triggered by changing the electrode potential and FTIR difference spectra were measured. Spectroelectrochemistry utilizing UV-Vis and fluorescence spectroscopies has been used to determine the
2.Instrumental setup of FTIR spectroelectrochemistry combined with light-induced FTIR difference spectroscopy
Figure 2A shows a schematic view of an optically transparent thin-layer electrode (OTTLE) cell used in our FTIR spectroelectrochemical measurements. This cell is similar to the ones previously reported [7,37]. Because strong infrared absorption of water interferes with the bands of proteins, a very thin-layer metal mesh, for example 6 µm in thickness, is used as a working electrode. When an Au working electrode is used, the surface of the electrode is modified with thiols to prevent irreversible adsorption and denaturation of proteins [62]. A commercially obtained Ag/AgCl/3 M KCl reference electrode (Cypress Systems Inc., 66-EE009; +208 mV vs. SHE) with a diameter of 2 mm and a Pt black wire as a counter electrode are arranged in the Teflon body (Fig. 2A).
Fig. 2.
(A) An optically transparent thin-layer electrode (OTTLE) cell designed for light-induced difference FTIR spectroscopy. (B) Instrumental setup for measurements of flash-induced FTIR difference spectra of a sample in an OTTLE cell, in which the potential of the working electrode is controlled by a potentiostat.
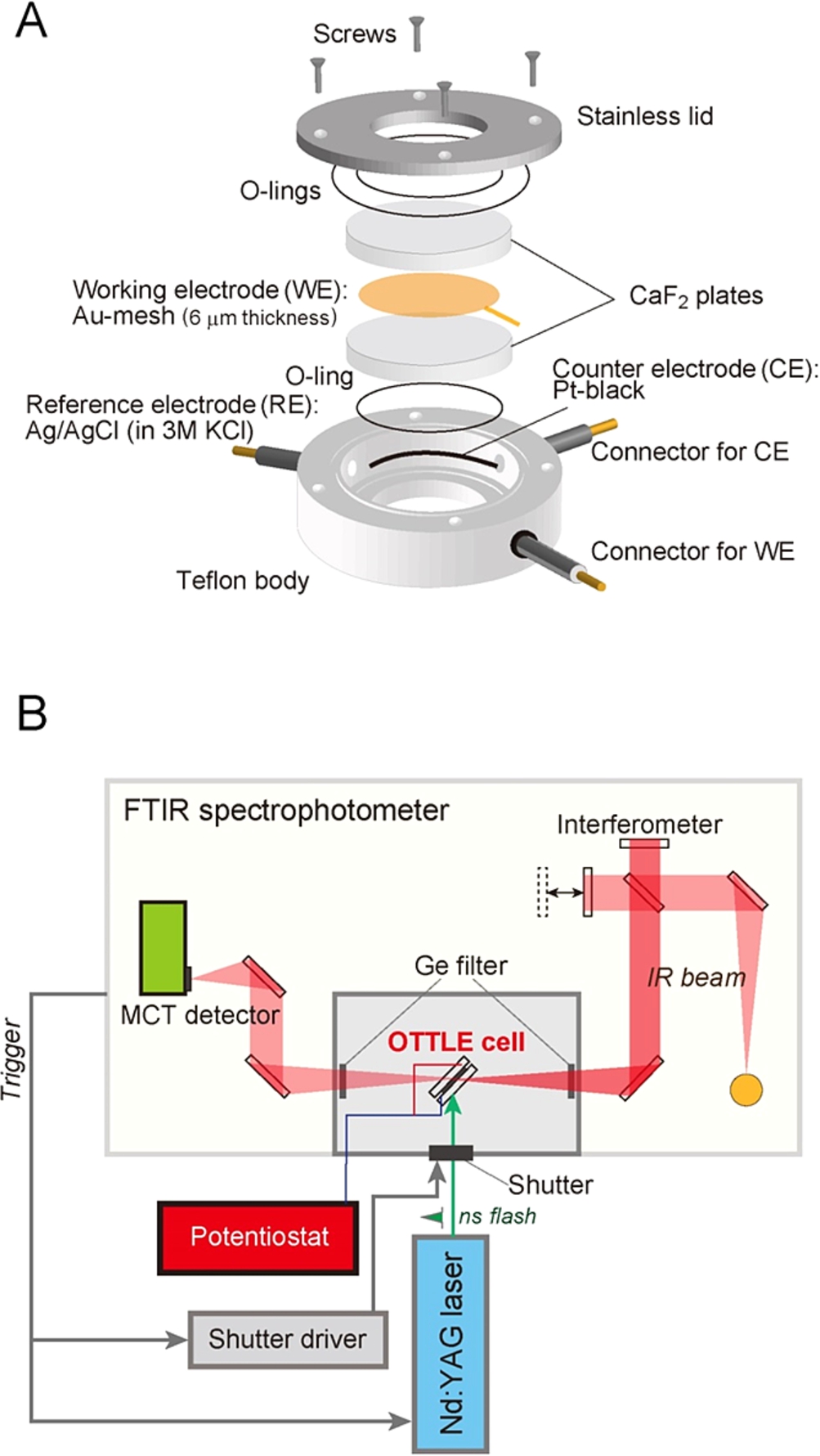
Because redox cofactors located inside a protein are difficult to interact with an electrode directly, redox mediators are necessary for electrochemical measurements. Several mediators are used to cover the potential regions of target cofactors. For example, for the measurement of the non-heme iron in PSII, Ru(NH3)6Cl2 (
For photosensitive proteins like PSII, there is a merit to combine the spectroelectrochemistry and a light-induced difference technique for studies of redox cofactors. The
For light-induced FTIR difference measurements of electrochemically controlled samples, the OTTLE cell wired with a potentiostat is set in the sample room of FTIR spectrophotometer (Fig. 2B), and light illumination is performed, for example by flashes from a Q-switched Nd:YAG laser (532 nm; ∼7 ns width), which are synchronized to FTIR scans by triggers from the spectrophotometer [39]. The temperature of the OTTLE cell is controlled by circulating cold water in a copper holder. Details of the methodology of light-induced FTIR difference spectroscopy and application to PSII have been described in previous review articles [5,11,13,14,39–43].
3.FTIR spectroelectrochemical study on the non-heme iron
The non-heme iron connects QA and QB by a molecular bridge, QA-His214(D2)-Fe-His215(D1)-QB (Fig. 1) [18,21,63]. In addition to these His ligands, other two His residues, D1-His272 and D2-His268, and a bicarbonate ion function as ligands to the non-heme iron [18,21,63]. Under physiological conditions, the non-heme iron is not involved in the electron transfer reaction from QA to QB [16,38,48], because of its high
Figure 3 shows flash-induced FTIR difference spectra of the O2-evolving (a, black line) and Mn-depleted (c, red line) PSII membranes of spinach at pH 6.5 measured in an electrolytic solution at +600 mV (vs. SHE) [28]. Bands at 1339, 1258, 1229, 1109, and 1101 cm−1, which were observed in both spectra, are typical of the Fe2+/Fe3+ difference signals [4,22,44,60,61]. The 1339(+)/1229(−) cm−1 bands were attributed to the symmetric CO stretching vibrations of the bicarbonate ligand [22], while the positive band at 1258 cm−1 and a part of the 1229 cm−1 negative band were assigned to the CO stretching vibration of a Tyr side chain (either D1-Tyr246 or D2-Tyr244) structurally coupled to the non-heme iron using [4-13C]Tyr labeling [60]. In addition, the 1109(+)/1101(−) peaks were assigned to the CN stretching vibrations of the imidazole ring of the His ligands [4]. In the difference spectrum of intact PSII, signals were also observed at 1439, 1419, 1404 and 1365 cm−1, which arise from the transition from the dark-stable
Fig. 3.
(a)
![(a) S2Fe2+/S1Fe3+ (black line) and S2/S1 (green line) FTIR difference spectra of the intact PSII membranes isolated from spinach. (b) An Fe2+/Fe3+ difference spectrum of the intact PSII membranes obtained by subtraction of the S2/S1 spectrum from the S2Fe2+/S1Fe3+ spectrum in (a). (c) An Fe2+/Fe3+ difference spectrum of the Mn-depleted PSII membranes. The S2Fe2+/S1Fe3+ difference spectrum of the intact PSII (a, black line) and the Fe2+/Fe3+ difference spectrum of the Mn-depleted PSII (c, red line) were measured at +600 mV at pH 6.5. The S2/S1 difference spectrum of the intact PSII (a, green line) was measured at +430 mV at pH 5.5. Reproduced from the data in Ref. [28].](https://ip.ios.semcs.net:443/media/bsi/2016/5-3/bsi-5-3-bsi146/bsi-5-bsi146-g003.jpg)
Fig. 4.
(A) Major signals around 1240 cm−1 in the Fe2+/Fe3+ difference spectra of the intact (left panel) and Mn-depleted (right panel) PSII membranes at a series of electrode potentials. (B) Semilogarithmic Nernst plots of the redox reaction of the non-heme iron in the intact (blue circles) and Mn-depleted (red circles) PSII membranes. The regression lines (dashed line) with slopes and intercepts are also shown. (C) Plots with theoretical Nernstian curves assuming one-electron redox processes with
![(A) Major signals around 1240 cm−1 in the Fe2+/Fe3+ difference spectra of the intact (left panel) and Mn-depleted (right panel) PSII membranes at a series of electrode potentials. (B) Semilogarithmic Nernst plots of the redox reaction of the non-heme iron in the intact (blue circles) and Mn-depleted (red circles) PSII membranes. The regression lines (dashed line) with slopes and intercepts are also shown. (C) Plots with theoretical Nernstian curves assuming one-electron redox processes with Em=+468 mV (intact) and +486 mV (Mn-depleted). Reproduced from the data in Ref. [28].](https://ip.ios.semcs.net:443/media/bsi/2016/5-3/bsi-5-3-bsi146/bsi-5-bsi146-g004.jpg)
From the intensities of the major signals around 1240 cm−1 in the thus-obtained Fe2+/Fe3+ spectra at a series of electrode potentials between +350 and +600 mV (Fig. 4A), the molar ratios of Fe3+ and Fe2+ were estimated and analyzed in a semilogarithmic Nernst plot (Fig. 4B) [28]. A linear relationship with slopes of 65 and 62 mV was obtained for the intact and Mn-depleted PSII samples, respectively; These slopes are similar to the theoretical value of 56 mV at 10°C (measuring temperature). The data from both PSII samples well followed the theoretical one-electron Nernstian curves (Fig. 4C), and the
Fig. 5.
Fe2+/Fe3+ difference spectra of the intact (blue line) and Mn-depleted (red line) PSII membranes in the regions of the C=O stretch of COOH (A), and the CN stretch of His (B). Reproduced from Ref. [28].
![Fe2+/Fe3+ difference spectra of the intact (blue line) and Mn-depleted (red line) PSII membranes in the regions of the C=O stretch of COOH (A), and the CN stretch of His (B). Reproduced from Ref. [28].](https://ip.ios.semcs.net:443/media/bsi/2016/5-3/bsi-5-3-bsi146/bsi-5-bsi146-g005.jpg)
The Fe2+/Fe3+ difference spectra also showed some changes in the COOH and His CN regions upon Mn depletion (Fig. 5). A negative peak at 1750 cm−1 was observed in the region of the C=O stretching vibration of COOH in intact PSII, whereas this band was not detected in Mn-depleted PSII (Fig. 5A). It was suggested that the
4.FTIR spectroelectrochemical study on QB
In contrast to many reports of the measurement of
Fig. 6.
(A) Light-induced FTIR difference spectra upon single reduction of QB (a) and QA (b) of Mn-depleted PSII core complexes isolated from Thermosynechococcus elongatus. (B) Expanded spectra of the ester C=O region of the
![(A) Light-induced FTIR difference spectra upon single reduction of QB (a) and QA (b) of Mn-depleted PSII core complexes isolated from Thermosynechococcus elongatus. (B) Expanded spectra of the ester C=O region of the QB−/QB (a) and QA−/QA (b) difference spectra. (C) Hydrogen-bond interactions of pheophytin electron acceptors located near QA and QB in PSII (PDB code: 3ARC [63]). Spectra in panels A and B are reproduced from Ref. [59].](https://ip.ios.semcs.net:443/media/bsi/2016/5-3/bsi-5-3-bsi146/bsi-5-bsi146-g006.jpg)
Fig. 7.
(A) The 1745 cm−1 band specific to
![(A) The 1745 cm−1 band specific to QB−/QB at a series of the electrode potentials for the intact (left panel) and Mn-depleted (right panel) PSII core complexes from T. elongatus. The contribution of the QA−/QA signal (typically at 1721 cm−1) was eliminated by subtraction of the QA−/QA difference spectrum measured at +50 mV from each spectrum. (B) Semi-logarithmic Nernst plots of the redox reactions of QB in the intact (blue circles) and Mn-depleted (red circles) PSII core complexes. α indicates the intensity ratio of the 1745 cm−1 peak at each electrode potential relative to the intensity at +250 mV, at which QB is fully oxidized. The regression lines (dashed lines) with slopes and intercepts are also shown. (C) Fitting of the experimental intensity ratios (α) with theoretical Nernst curves for the intact (blue lines) and Mn-depleted (red lines) PSII samples. Dashed lines reveal the theoretical curves of two-electron reactions with Em of +155 (intact) and +132 (Mn-depleted) mV, while solid lines reveal the simulated curves with Em values of the first and second reduction of QB as fitting parameters: Em(QB−/QB)=+93 mV and Em(PQH2/QB−)=+213 mV for intact PSII, and Em(QB−/QB)=+87 mV and Em(PQH2/QB−)=+157 mV for Mn-depleted PSII. Reprinted from Ref. [27].](https://ip.ios.semcs.net:443/media/bsi/2016/5-3/bsi-5-3-bsi146/bsi-5-bsi146-g007.jpg)
This marker peak of QB at 1745 cm−1 was used to examine the redox state of QB at a series of the electrode potentials (Fig. 7A) [27]. Note that in this method, the QB signal is obtained as a light-induced change of QB, typically one electron reduction of QB, after reaching equilibrium of electrochemical reactions, and hence electrochemically-induced changes are not directly detected in FTIR spectra. The semilogarithmic Nernst plots of the relative intensities of this peak against the electrode potential (Fig. 7B) showed virtually linear relationships with slopes of 30 ± 2 and 39 ± 1 mV for intact and Mn-depleted PSII, respectively. These slopes are closer to 28 mV, the theoretical value of a two-electron reaction at 10°C (measurement temperature), than 56 mV, that of a one-electron reaction. The apparent redox potentials (
Fig. 8.
(A) Diagram of the redox potentials of the electron transfer components in PSII. (B) The effect of Mn depletion on the redox potentials of single reduction of QA and QB. Solid and dashed black arrows indicate forward and backward electron transfer, respectively. The redox potential levels of intact and Mn-depleted PSII are expressed by blue and red bars, respectively, in (B). aValues from Refs [1,24,26,30,31,56,57]. bValues from Refs [29,51]. Reprinted from Ref. [27].
![(A) Diagram of the redox potentials of the electron transfer components in PSII. (B) The effect of Mn depletion on the redox potentials of single reduction of QA and QB. Solid and dashed black arrows indicate forward and backward electron transfer, respectively. The redox potential levels of intact and Mn-depleted PSII are expressed by blue and red bars, respectively, in (B). aValues from Refs [1,24,26,30,31,56,57]. bValues from Refs [29,51]. Reprinted from Ref. [27].](https://ip.ios.semcs.net:443/media/bsi/2016/5-3/bsi-5-3-bsi146/bsi-5-bsi146-g008.jpg)
By determining the
5.Concluding remarks
The combined method of thin-layer electrochemistry and light-induced FTIR difference spectroscopy was applied to study the redox properties of the non-heme iron and the terminal quinone QB on the electron-acceptor side of PSII. In particular, the FTIR signal specific to QB reduction enabled the first direct measurement of the
Although the mechanism of electron transfer regulation in PSII was clarified by determination of the
Acknowledgements
This study was supported by JSPS KAKENHI (25410009 to Y.K., 24000018, 24107003, and 25291033 to T.N.).
References
[1] | S.I. Allakhverdiev, T. Tsuchiya, K. Watabe, A. Kojima, D.A. Los, T. Tomo, V.V. Klimov and M. Mimuro, Redox potentials of primary electron acceptor quinone molecule (QA)− and conserved energetics of photosystem II in cyanobacteria with chlorophyll a and chlorophyll d, Proc. Natl. Acad. Sci. USA 108: ((2011) ), 8054–8058. |
[2] | A.J. Bard and L.R. Faulkner, Electrochemical Methods, 2nd edn, Wiley, New York, (2001) . |
[3] | M. Bauscher, E. Nabedryk, K. Bagley, J. Breton and W. Mäntele, Investigation of models for photosynthetic electron acceptors: Infrared spectroelectrochemistry of ubiquinone and its anions, FEBS Lett. 261: ((1990) ), 191–195. |
[4] | C. Berthomieu and R. Hienerwadel, Iron coordination in photosystem II: Interaction between bicarbonate and the QB pocket studied by Fourier transform infrared spectroscopy, Biochemistry 40: ((2001) ), 4044–4052. |
[5] | C. Berthomieu and R. Hienerwadel, Fourier transform infrared (FTIR) spectroscopy, Photosynth. Res. 101: ((2009) ), 157–170. |
[6] | S.P. Best, Spectroelectrochemistry of hydrogenase enzymes and related compounds, Coord. Chem. Rev. 249: ((2005) ), 1536–1554. |
[7] | S.P. Best, S.J. Borg and K.A. Vincent, Infrared spectroelectrochemistry, in: Spectroelectrochemistry, W. Klaim and A. Klein, eds, Royal Society of Chemistry, Cambridge, UK, (2008) , pp. 1–30. |
[8] | J. Bowes, A.R. Crofts and S. Itoh, A high potential acceptor for photosystem II, Biochim. Biophys. Acta 547: ((1979) ), 320–335. |
[9] | J. Cao and Govindjee, Chlorophyll a fluorescence transient as an indicator of active and inactive photosystem II in thylakoid membranes, Biochim. Biophys. Acta 1015: ((1990) ), 180–188. |
[10] | T. Cardona, A. Sedoud, N. Cox and A.W. Rutherford, Charge separation in photosystem II: A comparative and evolutionary overview, Biochim. Biophys. Acta 1817: ((2012) ), 26–43. |
[11] | H.-A. Chu, Fourier transform infrared difference spectroscopy for studying the molecular mechanism of photosynthetic water oxidation, Frontiers Plant Sci. 4: ((2013) ), Article 146. |
[12] | A.R. Crofts and C.A. Wraight, The electrochemical domain of photosynthesis, Biochim. Biophys. Acta 726: ((1983) ), 149–185. |
[13] | R.J. Debus, Protein ligation of the photosynthetic oxygen-evolving center, Coord. Chem. Rev. 252: ((2008) ), 244–258. |
[14] | R.J. Debus, FTIR studies of metal ligands, networks of hydrogen bonds, and water molecules near the active site Mn4CaO5 cluster in photosystem II, Biochim. Biophys. Acta 1847: ((2015) ), 19–34. |
[15] | B.A. Diner, Dependence of the deactivation reactions of photosystem II on the redox state of plastoquinone pool a varied under anaerobic conditions: Equilibria on the acceptor side of photosystem II, Biochem. Biophys. Acta 460: ((1977) ), 247–258. |
[16] | B.A. Diner, V. Petrouleas and J.J. Wendoloski, The iron-quinone electron-acceptor complex of photosystem II, Physiol. Plant. 81: ((1991) ), 423–436. |
[17] | P.L. Dutton, Redox potentiometry: Determination of midpoint potentials of oxidation-reduction components of biological electron-transfer systems, Methods Enzymol. 54: ((1978) ), 411–435. |
[18] | K.N. Ferreira, T.M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Architecture of the photosynthetic oxygen-evolving center, Science 303: ((2004) ), 1831–1838. |
[19] | C. Fufezan, C. Zhang, A. Krieger-Liszkay and A.W. Rutherford, Secondary quinone in photosystem II of Thermosynechococcus elongatus: Semiquinone-iron EPR signals and temperature dependence of electron transfer, Biochemistry 44: ((2005) ), 12780–12789. |
[20] | R.S.K.A. Gamage, S. Umapathy and A.J. McQuillan, OTTLE cell study of the UV-visible and FTIR spectroelectrochemistry of the radical anion and dianion of 1,4-benzoquinone in DMSO solutions, J. Electroanal. Chem. 284: ((1990) ), 229–235. |
[21] | A. Guskov, J. Kern, A. Gabdulkhakov, M. Broser, A. Zouni and W. Saenger, Cyanobacterial photosystem II at 2.9-Å resolution and the role of quinones, lipids, channels and chloride, Nat. Struct. Mol. Biol. 16: ((2009) ), 334–342. |
[22] | R. Hienerwadel and C. Berthomieu, Bicarbonate binding to the non-heme iron of photosystem II investigated by Fourier transform infrared difference spectroscopy and 13C-labeled bicarbonate, Biochemistry 34: ((1995) ), 16288–16297. |
[23] | W. Hillier and J. Messinger, Mechanism of photosynthetic oxygen production, in: Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase, T. Wydrzynski and K. Satoh, eds, Springer, Dordrecht, The Netherlands, (2005) , pp. 567–608. |
[24] | K. Ido, C.M. Gross, F. Guerrero, A. Sedoud, T.-L. Lai, K. Ifuku, A.W. Rutherford and A. Krieger-Liszkay, High and low potential forms of the QA quinone electron acceptor in photosystem II of Thermosynechococcus elongatus and spinach, J. Photochem. Photobiol. B 104: ((2011) ), 154–157. |
[25] | I. Ikegami and S. Katoh, Studies on chlorophyll fluorescence in chloroplasts II. Effect of ferricyanide on the induction of fluorescence in the presence of 3-(3,4-dichlorophynyl)-1,1-dimethylurea, Plant Cell Phys. 14: ((1973) ), 829–836. |
[26] | G.N. Johnson, A.W. Rutherford and A. Krieger, A change in the midpoint potential of the quinone QA in photosystem II associated with photoactivation of oxygen evolution, Biochim. Biophys. Acta 1229: ((1995) ), 202–207. |
[27] | Y. Kato, R. Nagao and T. Noguchi, Redox potential of the terminal quinone electron acceptor QB in photosystem II reveals the mechanism of electron transfer regulation, Proc. Natl. Acad. Sci. USA 113: ((2016) ), 620–625. |
[28] | Y. Kato and T. Noguchi, Long-range interaction between the Mn4CaO5 cluster and the non-heme iron center in photosystem II as revealed by FTIR spectroelectrochemistry, Biochemistry 53: ((2014) ), 4914–4923. |
[29] | Y. Kato, M. Sugiura, A. Oda and T. Watanabe, Spectroelectrochemical determination of the redox potential of pheophytin a, the primary electron acceptor in photosystem II, Proc. Natl. Acad. Sci. USA 106: ((2009) ), 17365–17370. |
[30] | A. Krieger, A.W. Rutherford and G.N. Johnson, On the determination of redox midpoint potential of the primary quinone electron acceptor, QA, in photosystem II, Biochim. Biophys. Acta 1229: ((1995) ), 193–201. |
[31] | A. Krieger and E. Weis, Energy-dependent of chlorophyll-a-fluorescence: The involvement of proton-calcium exchange at photosystem 2, Photosynthetica 27: ((1992) ), 89–98. |
[32] | A. Krieger-Liszkay, C. Fufezan and A. Trebst, Singlet oxygen production in photosystem II and related protection mechanism, Photosynth. Res. 98: ((2008) ), 551–564. |
[33] | D. Lazár, Chlorophyll a fluorescence induction, Biochim. Biophys. Acta 1412: ((1999) ), 1–28. |
[34] | W.G. Mäntele, A.M. Wollenweber, E. Nabedryk and J. Breton, Infrared spectroelectrochemistry of bacteriochlorophylls and bacteriopheophytins: Implications for the binding of the pigments in the reaction center from photosynthetic bacteria, Proc. Natl. Acad. Sci. USA 85: ((1988) ), 8468–8472. |
[35] | F. Melin and P. Hellwig, Recent advances in the electrochemistry and spectroelectrochemistry of membrane proteins, Biol. Chem. 394: ((2013) ), 593–609. |
[36] | J. Messinger, T. Noguchi and J. Yano, Photosynthetic O2 evolution, in: Molecular Solar Fuels, T. Wydrzynski and W. Hillier, eds, Royal Society of Chemistry, Cambridge, UK, (2011) , Chapter 7. |
[37] | D. Moss, E. Nabedryk, J. Breton and W. Mäntele, Redox-linked confromational changes in proteins detected by a combination of infrared spectroscopy and protein electrochemistry, Eur. J. Biochem. 187: ((1990) ), 565–572. |
[38] | F. Müh, C. Glöckner, J. Hellomich and A. Zouni, Light-induced quinone reduction in photosystem II, Biochim. Biophys. Acta 1817: ((2012) ), 44–65. |
[39] | T. Noguchi, Light-induced FTIR difference spectroscopy as a powerful tool toward understanding the molecular mechanism of photosynthetic oxygen evolution, Photosynth. Res. 91: ((2007) ), 59–69. |
[40] | T. Noguchi, Fourier transform infrared analysis of the photosynthetic oxygen-evolving center, Coord. Chem. Rev. 252: ((2008) ), 336–346. |
[41] | T. Noguchi, Monitoring the reactions of photosynthetic water oxidation using infrared spectroscopy, Biomed. Spectrosc. Imaging 2: ((2013) ), 115–128. |
[42] | T. Noguchi, Fourier transform infrared difference and time-resolved infrared detection of the electron and proton transfer dynamics in photosynthetic water oxidation, Biochim. Biophys. Acta 1847: ((2015) ), 35–45. |
[43] | T. Noguchi and C. Berthomieu, Molecular analysis by vibrational spectroscopy, in: Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase, T. Wydrzynski and K. Satoh, eds, Springer, Dordrecht, The Netherlands, (2005) , pp. 367–387. |
[44] | T. Noguchi and Y. Inoue, Identification of Fourier transform infrared signals from the non-heme iron in photosystem II, J. Biochem. 118: ((1995) ), 9–12. |
[45] | T. Noguchi, T. Ono and Y. Inoue, Direct detection of a carboxylate bridge between Mn and Ca2+ in the photosynthetic oxygen-evolving center by means of Fourier transform infrared spectroscopy, Biochim. Biophys. Acta 1228: ((1995) ), 189–200. |
[46] | T. Noguchi and M. Sugiura, Analysis of flash-induced FTIR difference spectra of the S-state cycle in the photosynthetic water-oxidizing complex by uniform 15N and 13C isotope labeling, Biochemistry 42: ((2003) ), 6035–6042. |
[47] | G.C. Papageorgiou and Govindjee, Photosystem II fluorescence: Slow changes – Scaling from the past, J. Photochem. Photobiol. B Biol. 104: ((2011) ), 258–270. |
[48] | V. Petrouleas and A.R. Crofts, The quinone iron acceptor complex, in: Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase, T. Wydrzynski and K. Satoh, eds, Springer, Dordrecht, The Netherlands, (2005) , pp. 177–206. |
[49] | V. Petrouleas and B.A. Diner, Identification of Q400, a high-potential electron acceptor of photosystem II, with the iron of the quinone-iron acceptor complex, Biochim. Biophys. Acta 849: ((1986) ), 264–275. |
[50] | M.P.J. Pulles, H.J. van Gorkom and J.G. Willemsen, Absorbance changes due to charge-accumulating species in system 2 of photosynthesis, Biochim. Biophys. Acta 449: ((1976) ), 536–540. |
[51] | F. Rappaport and B.A. Diner, Primary photochemistry and energetics leading to the oxidation of the (Mn)4Ca cluster and to the evolution of molecular oxygen in photosystem II, Coord. Chem. Rev. 252: ((2008) ), 259–272. |
[52] | G. Renger, Photosynthetic water splitting: Apparatus and mechanism, in: Photosynthesis: Plastid Biology, Energy Conversion and Carbon Assimilation, J.J. Eaton-Rye, B.C. Tripathy and T.D. Sharkey, eds, Springer, Dordrecht, The Netherlands, (2012) , Chapter 17. |
[53] | H.H. Robinson and A.R. Crofts, Kinetics of the oxidation-reduction reactions of the photosystem II quinone acceptor complex, and the pathway for deactivation, FEBS Lett. 153: ((1983) ), 221–226. |
[54] | A.W. Rutherford, A. Osyczka and F. Rappaport, Back-reactions, short-circuits, leaks and other energy wasteful reactions in biological electron transfer: Redox tuning to survive life in O2, FEBS Lett. 586: ((2012) ), 603–616. |
[55] | A. Sedoud, N. Cox, M. Sugiura, W. Lubitz, A. Boussac and A.W. Rutherford, Semiquinone-iron complex of photosystem II: EPR signals assigned to the low-field edge of the ground state doublet of QA∙−Fe2+ and QB∙−Fe2+, Biochemistry 50: ((2011) ), 6012–6021. |
[56] | T. Shibamoto, Y. Kato, R. Nagao, T. Yamazaki, T. Tomo and T. Watanabe, Species-dependence of the redox potential of the primary quinone electron acceptor QA in photosystem II verified by spectroelectrochemistry, FEBS Lett. 584: ((2010) ), 1526–1530. |
[57] | T. Shibamoto, Y. Kato, M. Sugiura and T. Watanabe, Redox potential of the primary plastoquinone electron acceptor QA in photosystem II from Thermosynechococcus elongatus determined by spectroelectrochemistry, Biochemistry 48: ((2009) ), 10682–10684. |
[58] | T. Shibamoto, Y. Kato and T. Watanabe, Spectroelectrochemistry of cytochrome b559 in the D1-D2-Cyt b559 complex from spinach, FEBS Lett. 582: ((2008) ), 1490–1494. |
[59] | H. Suzuki, M. Nagasaka, M. Sugiura and T. Noguchi, Fourier transform infrared spectrum of the secondary quinone electron acceptor QB in photosystem II, Biochemistry 44: ((2005) ), 11323–11328. |
[60] | R. Takahashi, A. Boussac, M. Sugiura and T. Noguchi, Structural coupling of a tyrosine side chain with the non-heme iron center in photosystem II as revealed by light-induced Fourier transform infrared difference spectroscopy, Biochemistry 48: ((2009) ), 8994–9001. |
[61] | R. Takahashi, K. Hasegawa, A. Takano and T. Noguchi, Structures and binding sites of phenolic herbicides in the QB pocket of photosystem II, Biochemistry 49: ((2010) ), 5445–5454. |
[62] | I. Taniguchi, K. Toyosawa, H. Yamaguchi and K. Yasukouchi, Voltammetric response of horse heat cytochrome c at a gold electrode in the presence of sulfur bridged bipyridines, J. Electroanal. Chem. 140: ((1982) ), 187–193. |
[63] | Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å, Nature 473: ((2011) ), 55–60. |
[64] | H.J. van Gorkom, Identification of reduced primary electron-acceptor of photosystem II as a bound semiquinone anion, Biochim. Biophys. Acta 347: ((1974) ), 439–442. |
[65] | I. Vass, Role of charge recombination processes in photodamage and photoprotection of the photosystem II complex, Physiol. Plant 142: ((2011) ), 6–16. |
[66] | I. Vass and K. Cser, Janus-faced charge recombinations in photosystem II photoinhibition, Trends Plant Sci. 14: ((2009) ), 200–205. |
[67] | T. Wydrzynski and K. Satoh (eds), Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase, Springer, Dordrecht, The Netherlands, (2005) . |


