
The evolution of biomedical EPR (ESR)
Abstract
Since the first EPR/ESR spectrum of a paramagnetic substance was published over 70 years ago, the technical improvements did not occur until after/during World War II with the advent of radar technology. The approaches to biomedical problems started somewhat later with the real burst of activity starting after the birth of the spin label technique about 50 years ago. The applications to proteins, then membranes and nucleic acids, and later applications to cells and eventually in-vivo on small animals and now humans has led EPR/ESR to finally being recognized as a uniquely powerful technique in the toolbox of techniques probing macromolecules and their interactions, free radical biology and its eventual value as a diagnostic technique. This article gives an overview of EPR/ESR studies of biomedically related systems, including proteins and enzymes. It presents a very personal historical perspective, briefly reviews the origins of the technique and reflects on possible future directions. As with NMR, advances in molecular biology and technology drastically changed the nature and focus of the technique, particularly the site directed spin labeling method that has been invaluable in determining protein and macromolecular structure by both EPR and NMR.
Abbreviations
EPR | electron paramagnetic resonance |
ESR | electron spin resonance |
CW | continuous wave |
DEER | Double electron-electron resonance spectroscopy |
ENDOR | electron nuclear double resonance spectroscopy |
ESEEM | electron-spin-echo-envelope-modulation |
G | Gauss |
dimensionless electron spin g-factor (related to | |
the electron gyromagnetic ratio | |
PELDOR | pulsed electron double resonance spectroscopy |
RF | radiofrequency |
SDSL | site-directed spin labeling |
T | Tesla (=10,000 G) |
1.Introduction and early history
Given the two, albeit equivalent, names for this branch of magnetic resonance spectroscopy (EPR, ESR) one might guess that it evolved from two or more areas of the physical and natural sciences. In fact, EPR (electron paramagnetic resonance) is usually attributed to the physicists while ESR is attributed to the chemists and biologists. In fact one of the professional societies representing this field is called the I.E.S. or the International EPR/ESR Society. Some of this mystique may become more apparent as the evolving history in this chapter develops.
Yevgeny Zavoisky discovered the magnetic resonance process in 1944, conducting his experiments at Kazan State University in the Russian Republic at the height of the 2nd World War and published his seminal works in English in the leading Russian physics journal of the time [106–108]. However, he failed to be credited with the full impact of his discovery. Today he is often regarded as the father of ESR (electron spin resonance). It was Zavoisky who first discovered that the RF field must be applied perpendicular to the static field in order to see the resonance phenomenon. This is important since he put together the spin with the lattice and, for this reason, he is even viewed by some as the father of all magnetic resonance. Yet Zavoisky’s experiments were doubted by the Russian Academy of Sciences, requiring him to travel to Moscow to repeat the work. It should also be noted that this was during the height of WW2 and many Soviet scientists were moved to Kazan in order to protect Russian high level science from an anticipated invasion of Moscow by the Nazis.
Nonetheless, the war ended successfully and Zavoisky was later awarded the Stalin Prize, the Lenin Prize and Hero of Socialist Labour as well as two Orders of Lenin. He became an Associate Member of the Russian Academy of Sciences and was later elected an Academician. Most of Zavoisky’s work was published in Russian journals and, since there was an element of both distrust and disdain for Russian science by the rest of the world, his work was largely ignored. From what we know Zavoisky might also have observed the first NMR signal, but he did not follow it up (at least not published). As noted above, had the circumstances of the times been less tense and scientific dissemination been easier, he might have been better known in the West. As it was, the 1952 Nobel Prize in Physics went to Bloch and Purcell [102]. Yet there are several people who feel that he deserved the Nobel Prize in magnetic resonance, not those recognized solely for NMR, and some have said that perhaps these Nobel laureates read his papers, which ultimately led to the basis for their famous NMR experiments?11
Needless to say, Zavoisky’s work is very much acknowledged these days. There is an annual Zavoisky Prize ceremony in Kazan, the capital of Tatarstan which sponsors this event. Kazan University houses a museum with his original equipment and notebooks that his daughter and past students maintain.22 The Zavoisky Prize is considered the highest award in EPR/ESR and is given to esteemed scientists in the magnetic resonance field whose contributions encompass paramagnetic substances [82].
Since the initial observation of EPR (electron paramagnetic resonance) signals, the technique and its applications have undergone phenomenal evolutionary changes, just as NMR continues to evolve. While the field initially flourished in physics departments in the U.K., the early work did not focus on biomedical applications, but rather paramagnetic metals and their complexes. This eventually led to studies of metalloproteins, which were a challenge in the early years with conventional CW instrumentation. Some early workers attempted to visualize biological (organic free radicals) despite their relatively low concentrations, then some very courageous scientists attempted to study tissue excised from live animals (ex vivo EPR) [89,92], then animals [9] and now humans (clinical in-vivo EPR) [93].
1.1.Scope of this article
The evolution of this method here will address only biomedical aspects and applications. Hence, a lot of important work on organic free radicals (except those that are biologically relevant), metals and metal complexes except for metalloproteins and metal ion: biomacromolecular complexes will not be covered. It is important to note that much of the early work in this field was instrument/sensitivity limited to stable biological radicals and those proteins and enzymes with paramagnetic metal ion centers. The general approach of utilizing EPR to study any macromolecular system in general, i.e., by incorporation of a stable, versatile paramagnetic ‘label’ (as shown with fluorescence extrinsic labeling), did not evolve until the spin labeling method was conceived in the early 1960s at Stanford University. Needless to say, the spin label approach opened up EPR applications to biomedical and in-vivo systems in a broad sense as covered in this article. Another indirect ‘labeling/visualization’ method evolved from applications with organic spin traps (e.g. nitrones) which are diamagnetic precursors to stable aminoxyl (nitroxide) radical adducts that form after reaction with a reactive, short-lived radical. Last, the discovery/development of extremely stable, inert paramagnetic compounds and substances revolutionized in-vivo applications since bioreduction/biodestruction/disproportionation reactions of radical probes and spin traps were troublesome obstacles to the technique. It is fair to say that I had the privilege of taking part in and observing a significant number of developments in the field.
1.2.Personal perspective
It is an honor to follow two friends and colleagues, the late Iain Campbell and Stanley J. Opella, who introduced the NMR perspective in earlier issues of this review series [19,74]. I first met Iain Campbell whilst both of us were postdoctorates in Oxford in 1967. During a visit to Stanford by Rex Richards my PhD advisor, the late Harden M. McConnell, introduced me and suggested that I contact Prof. Richards as soon as I arrived. There I met Iain, who was part of a group of very important budding scientists trying to tackle NMR biological/biomedical problems. While working with the late Sir David Phillips, the noteworthy protein crystallographer in the biophysics unit the Department of Biology, my assignment was to incorporate magnetic resonance methods into solving aspects of protein structure. Iain and I became close friends and he looked after me many times on subsequent visits to Oxford over the decades that followed. It was very sad to learn of his passing not long after his review was published in this series. It is sad to realize that all of the people mentioned above, save Stan Opella (who was also a McConnell PhD) are gone.
My work in the laboratory of Harden M. McConnell began just after his move from CalTech to Stanford in the early 1960s, where the technique of spin labeling was conceived and developed. As noted above in Section 1.1, this technique was somewhat revolutionary to expanding biomedical EPR applications at the time, and was quickly shown to answer many structural, dynamical and mechanistic aspects of protein and enzyme structure as well as probing more complex systems. Hence, while some historical contributions to the biomedical area preceded spin labeling and modern day in-vivo EPR, the early work at Stanford on proteins and enzymes and later membranes containing paramagnetic labels was a major area of study. The big sensitivity limitation with metal containing systems was the fact that the relaxation times of many paramagnetic metal ions were quite short. This meant that experiments could be performed only at low temperatures, sometimes in liquid helium, and the system was in the frozen solid state sometimes creating linewidth and resolution problems.
1.3.Magnets and frequency
Unlike NMR, the resolution/homogeneity problem is not major in EPR spectroscopy as the typical linewidths are rarely less than 0.5 G (although this relatively large linewidth should imply resolution problems, where encountered it is overcome at high frequencies). Although examples exist with organic radicals of somewhat smaller linewidths, particularly the trityl radicals, resolution has never really been as major a problem, except when a sample contains more than one radical species. While in NMR it’s very important to have highly stable magnets with very high homogeneity and no drift, this has never really plagued EPR spectroscopy as much. Nonetheless, sometime later (around the 1980–1990s), superconducting magnets became available for frequencies of the order of 10 times that of the typical X-band spectrometer. Certainly, the advent of e.g. W-band (95 GHz) and higher frequencies/magnetic fields offered higher sensitivity and resolution [40].
The most common EPR spectrometer is in the X-band frequency region, i.e. 9.5 GHz, with at field strength of 3500 G (0.35 T). Depending on the gyromagnetic ratio,
Fig. 1.
Schematic of an EPR spectrometer. The microwave source on the more recent instruments are Gunn oscillators instead of klystrons. The recorder is substituted by an analog to digital converter and a PC nowadays.
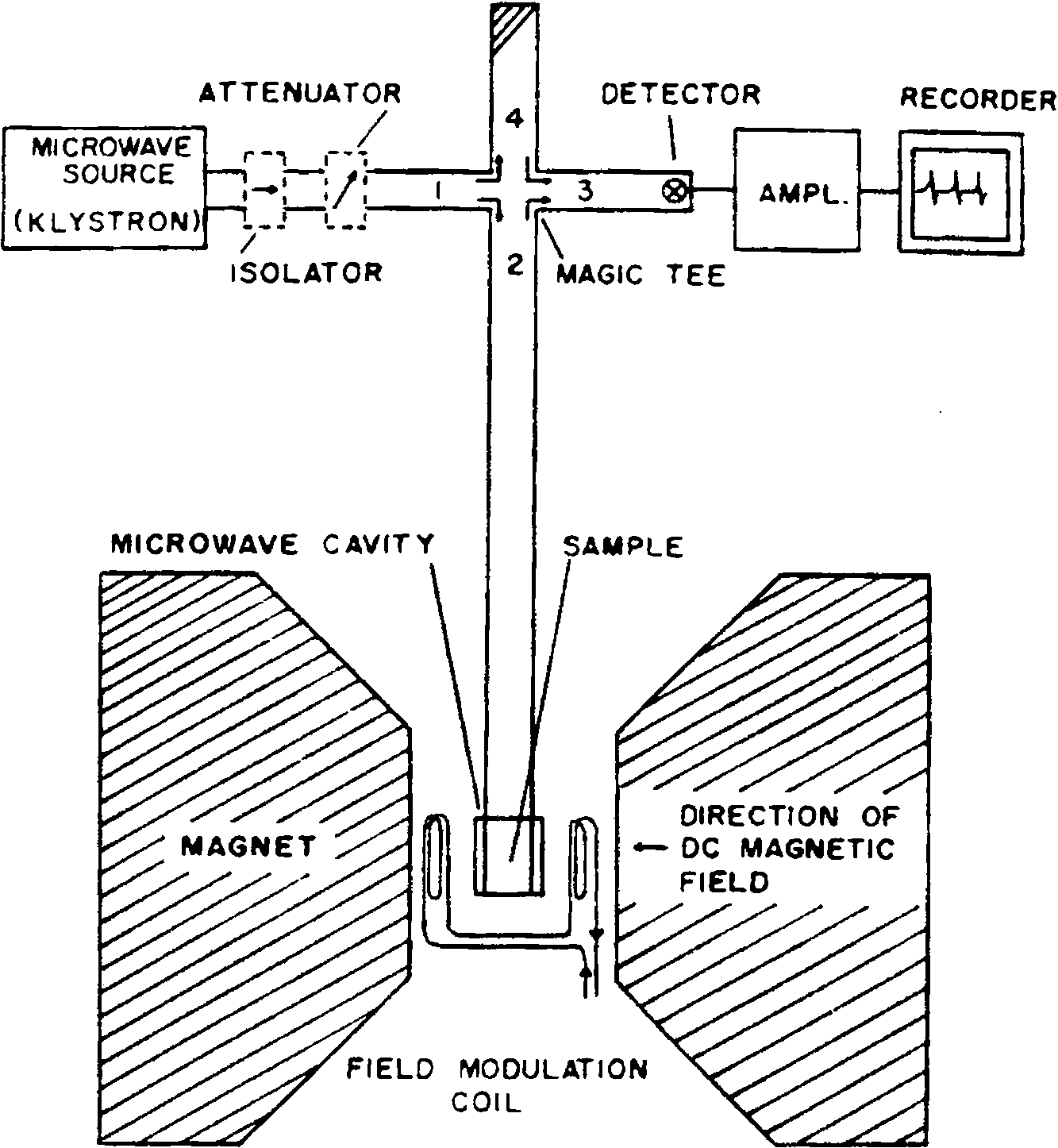
The EPR experiment was for many years a continuous wave (CW) experiment where one fixed the frequency/power source and swept the magnetic field over ranges of ca. 100–1000 or 2000 Gauss. It was really only in the period starting about 1980 or so where pulsed EPR methods became more generally available and later pulsed Fourier transform methods, developed from that used in NMR. In fact the major limitations still relate back to short electron spin relaxation times, meaning that large powerful pulses were necessary. This involved travelling wave tubes (TWTs) that required kilowatts of power and this took many years of refinement and development, accomplished commercially by Bruker BioSpin. And, of course, as the frequency increased the magnetic field increased proportionately.
It should also be noted that the sensitivity in EPR is less problematic than as in NMR, where e.g. at room temperature, the population differences between the ground and excited spin states are very small. Since EPR operates at energies that are 1000 times or larger than in NMR the Boltzmann differences are significantly larger meaning one can excite a much larger percentage of the spins in the sample for an EPR experiment compared to that of an NMR. Yet the incentive for commercial development of EPR instrumentation has always been somewhat less and the pricing relatively more expensive, since the technique requires paramagnetic substances vs. stable nuclei which one can observe in natural abundance (e.g., protons, carbon-13, P-31 isotopes, etc.). Perhaps more important was the advent of MRI which resulted in a tremendous increase in the sale of NMR instrumentation in general. The EPR community was essentially dependent on Bruker, which was always involved in developing research EPR instruments; however, the priorities meant that developments were also somewhat slower at times than the burgeoning MRI field.
2.In-vivo EPR: History and developments
2.1.Ex-vivo studies of living substances
The major consideration here is nonresonant absorption/water heating that occurs in much of the microwave frequency region. Considering that an X-band 9.5 GHz EPR instrument is in the same general frequency range as that of a microwave oven and specifically in the radar range, the electric field component of the electromagnetic radiation will excite water molecules in a biological sample, resulting in eventual heating and evaporation.
The initial EPR studies on isolated cells and tissues were published in the early 1950s (for extensive reviews see [89–91]). To some degree it was spurred by Michaelis’ proposition that many enzyme mechanisms were via a series of one-electron oxidation-reduction steps, consequently resulting in free radical intermediates [69]. The other major speculation was that free radicals might be linked to cancer, hence many of the early EPR studies focused on malignant cells and tissues [90]. The results were complicated by the quite low sensitivity of homemade or commercially available EPR spectrometers, the problem of nonresonant microwave absorption which led to additional sensitivity losses and the possibility of significant microwave heating.
Some of these problems were addressed in three different ways, unfortunately leading at times to different results.
(1) Removal of the water from cells and tissues by drying – usually by lyophilization. With the absence of water, non-resonant microwave absorption was essentially eliminated and one could pack more material into the sample tube. But, the drying process gave rise to changes in the system [44]. The most important changes were artifacts due to physicochemical factors, specifically the generation of free radical signals from interactions of dried cell constituents with O2. In addition, free radicals were generated by mechanical forces associated with freezing [97]. Hence samples processing was a key source of artifacts in many cases. The first studies of lyophilized cells and tissues were reported by Commoner and colleagues where they described experiments at 9 GHz on 250 mg of freeze-dried bacteria (Pseudomonas fluorescens) in which a small but reproducible singlet was detected at
(2) Freezing the sample to minimize dielectric losses (since the dielectric constant of ice is much less than that of liquid water). This also significantly slowed down most biochemical processes as well. However mechanical forces associated with freezing can generate free radicals. In addition, frozen organic radicals easily undergo microwave power saturation. In the end, freezing did not always stop all reactions and some still proceeded if not kept at very, very low temperature. There were also contributions from visible and UV light exposure even to frozen samples, which were additional sources of possible artifacts.
(3) Study ‘natural’ tissues in sample holders in resonant cavities which minimized nonresonant microwave losses. The aqueous flat cell was key to these and all future biochemical studies, which allowed the reproducible observation of aqueous samples. The sample is placed in a region of the resonant cavity with the absolute minimum electric field contribution and the maximum magnetic field contribution. The quartz tissue cell has a removable plate and (usually) a slight depression where thin slices of solid tissues can be mounted and kept from drying with a cover slide. Hence the artifactual issues mentioned earlier were significantly eliminated although temporal aspects of the sample/tissue metabolism had to be accounted for during the measurement. Aspects such as nutrient perfusion were difficult given the limited volume of the sample holder; hence maintaining steady-state metabolism was difficult [92]. Of course, sensitivity is directly related to the amount of sample, which was limited by the dimensions of the sample cell. Some tricks to overcome these problems involve the use of larger volume multimode cavities and, of course, time averaging techniques. On the other hand, power saturation is not an issue since the water in the sample actually contributes to losses of microwave power.
In the past two decades there have been many improvements in microwave resonators, sample holders, and EPR spectrometers in general such that it is now possible to use relatively large biological samples, even at 9 GHz, although lower frequencies allow even larger samples [25]. In fact Commoner later recognized the disadvantages with using dried materials and reported work with aqueous preparations. The first EPR signals in live cells were attributed to processes associated with photosynthesis [22]. They also observed EPR signals from functioning submitochondrial particles [23]. This work was probably one of the pioneering studies which eventually led to investigations using EPR to study redox enzymes. Then, with subsequent newer instrumentation, they studied free radicals associated with function in surviving mammalian tissues [24]. The signals were very broad lines at
Commoner and others looked at quickly frozen samples. The much more complex EPR spectra were quite different from the single line usually found in wet systems which was a relatively intense organic radical that was easily power saturated and not prominent in the higher microwave power frozen sample spectra. These frozen samples showed several paramagnetic trace elements, which were not observed in unfrozen samples due to their short relaxation times and consequently broad lines [95]. Actually varying the microwave power is almost diagnostic in differentiating between (organic) free radicals and paramagnetic metal ions in complex biological samples, and can be fine tuned to distinguish different classes of paramagnetic species.
2.2.In-vivo studies of small animals: Early attempts
Overall, studying wet, functioning isolated cells or tissues is informative. For frozen samples frozen punch biopsies are useful. Yet carrying out real time in-vivo studies would be the most valuable; however, whole animals can’t be subjected to liquid nitrogen temperatures, can’t be placed in a typical 5 mm sample tube or 1 mm flat cell and can’t be subjected to nonresonant microwave heating [52]. There were some unpublished studies by Rowlands and colleagues at the Southwest Research Institute, San Antonio, TX on whole animals using a very low frequency EPR spectrometer (about 250 MHz) and the injection of nitroxides (aminoxyl) into mice placed inside a rectangular cavity [78,79]. The signal to noise was very poor and limited, and was probably never published in a refereed journal. In 1971, J. Hutchison at the University of Aberdeen published a paper describing the development of an in-vivo EPR instrument where they obtained spectra from excised guinea pig liver, although they were unable to obtain data from an intact mouse. They also provided an analysis of the potential optimum frequency for in-vivo EPR [52]. While Hutchison suggested that frequencies around 1000 MHz (L-band) would be ideally suited for in-vivo work, the development of the prototype L-band spectrometer that has been the basis for much of the initial in-vivo EPR studies. Unpublished accounts from Washington University, St. Louis, attested to early L-band studies by Commoner and his EPR colleagues in the Department of Chemistry. We have to give a tremendous amount of credit to these true pioneers who had the ‘audacity’ to put living animals in homemade EPR machines during a period where sophisticated components and sensitive paramagnetic probes were not readily available.
The contemporary pioneering experiment was reported by Piette and colleagues in 1975 at the University of Hawaii [30]. They implanted a helix resonator in the liver of a rat and administered high doses of the piperidine nitroxide TEMPOL (or 1-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl). They followed the pharmacokinetics of the rapid disappearance of the nitroxide and also reported on the spectrum of a spin labeled drug obtained in-vivo. This work suffered from severe microwave heating of this lossy aqueous medium at 9.5 GHz radiation, causing some discomfort to the animal. The second problem was the rapid bioreduction of the six-membered ring (piperidine) nitroxide that was studied. Nonetheless, this work was a major inspiration to all of the people who entered this field in the early to mid-1980s. This study by the Piette group above is usually considered the first in-vivo EPR study even though the earlier attempts noted above cannot be overlooked.
3.In-vivo EPR: What can one see? What physiological information can EPR provide?
When thinking about experiments to observe paramagnetic centers or free radical phenomena in living systems, we first have to ask ourselves: “what can one observe?” They are basically three or four possible avenues (or combinations thereof) that have turned out to be feasible.
3.1.Direct observation of organic free radicals involved in physiological function
Ideally, one wants to observe real biological radicals, e.g. reactive oxygen species (ROS) and other ‘natural’ radicals as they occur during normal metabolism. This would be radical co-factors such as the flavin radical, the ascorbyl radical, and the various radical intermediates in metabolic oxidation-reduction reactions. However all of these exist in fairly low concentrations that are well beyond the limits of detection in many cases. In the case of the flavin radical, this might be typically observable in a few flavoenzymes in-situ; however, when looking at the whole animal or whole tissue in an in-vivo/ex-vivo experiment, one must be able to discern distinct paramagnetic/radical species and look at their temporal behavior with fairly high sensitivity and characterizability. Overall, this is a limited to a few distinct systems with tissue, cells or organs where a particular radical enzyme system exists in very high concentration, or where there are systems producing high levels of radical intermediates. Of course, there are the reactive oxygen species (ROS), such as superoxide radical anion, hydroxyl radical, and some carbon based lipoxy and peroxy radicals. These usually result from some type of oxidative stress and are usually quite short-lived and in very low (subdetectable) concentrations of the order of nanomolar or much less. Suffice it to say, commercial EPR spectrometers are really only sensitive to organic radicals in the micromolar range, perhaps
3.2.Direct observation of extrinsic radicals (spin probes/spin labels)
This approach must be to introduce stable, extrinsic radicals such as spin label aminoxyl radicals (nitroxides) or other compounds. Aminoxyl radicals were employed extensively, particularly in the early phases of in-vivo EPR spectroscopy in the 1980s; however, they suffered from several practical and instrumental limitations which restricted sensitivity. One feature is that the typical N-14 nitroxide (aminoxyl radical) yields a three line spectrum with a linewidth of 0.5 G or higher, depending on the tumbling motions of the probe, effectively decreasing the sensitivity by at least 1/3. There was some success with isotopically enriching these radicals with N-15, yielding a two – vs. three – line spectrum or 1.5 fold sensitivity increase. Further incorporation of deuterium allowed for a slightly narrower linewidth. The real limitation is that this is a very expensive reagent where the costs become almost prohibitive when injecting large animals with large doses of enriched radical. A more recent development, which is also noted elsewhere in this article, was the resurrection of Gomberg’s 100 year old triphenylmethyl radical (abbreviated as the trityl) [39]. This radical has the free electron essentially localized on the central carbon, with little or no hyperfine interaction from neighboring nuclei, allowing one to obtain EPR spectra with extremely narrow (mG) linewidths, thus resulting in very high sensitivity. On the other hand, the dynamical information provided is fairly limited since motional lineshape effects are minimal. However, this problem has been exploited as a unique, sensitive probe for dissolved oxygen by virtue of paramagnetic O2: radical Heisenberg exchange phenomenon (i.e., Heisenberg broadening). This effect has been taken advantage of by a significant number of researchers since the relationship between the Heisenberg exchange broadening and the local oxygen partial pressure can be accurately measured and calibrated at fairly low pressures, thus catapulting the technique of ‘in-vivo EPR oximetry’ into a broad range of clinical and other applications [86].
A very clever application, particularly for long-term localized studies of oxygen tension in humans and animals involves the incorporation of inert, solid free radical compounds, frequently based on charcoal or charred wood. This was exploited substantially over the decades from the 1980s onward at the University of Illinois EPR Center. The best probe for humans, India ink, provides very sensitive stable radical signals that can be imaged and/or utilized for measuring oxygen pressure. It also had the distinct advantage of prior FDA approval as a marker on the skin of patients awaiting surgery. India ink has proven to be a very powerful probe for measuring cutaneous and subcutaneous oxygen tension [56,94]. One of the most successful class of oxygen probes were based on lithium phthalocyanine compounds, which were intensely paramagnetic in the (insoluble) solid state [56,64].
3.3.Nitric oxide (NO): A naturally occurring free radical
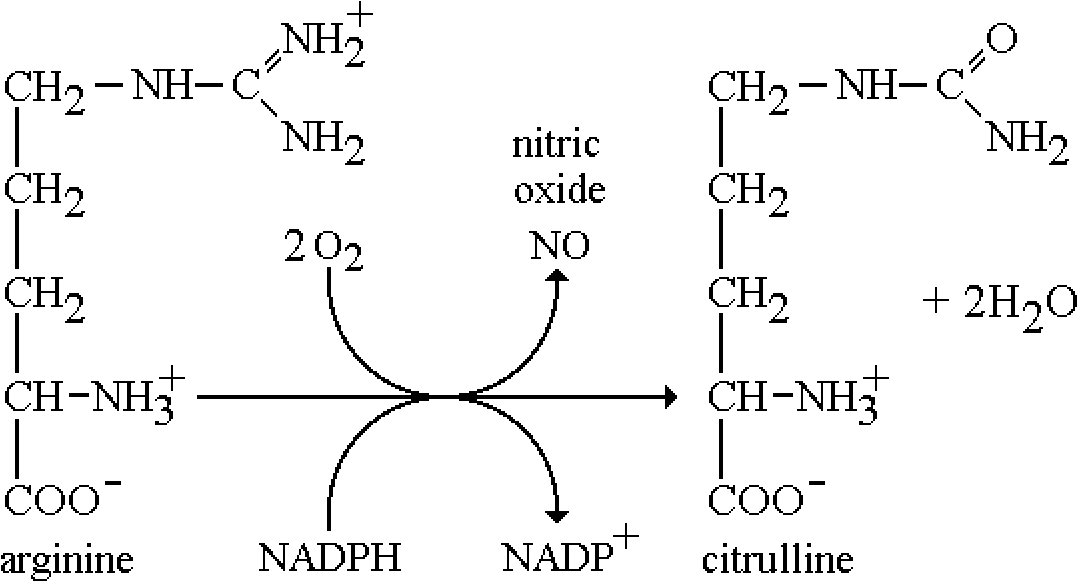
Nitric oxide is involved in muscle relaxation and is a product of the enzyme catalyzed conversion of arginine by nitric oxide synthase. The basal levels of NO, however, are sub-micromolar, hence too small to observe in-vivo. In addition the paramagnetic gas NO is effectively EPR silent. However, it was shown by Vanin’s group [62] and Lai and Komarov [63] that one can ‘trap’ NO as the ferrous iron complex in a coordination complex involving dithiocarbamates. The two most useful compounds are the water soluble, and the lipid soluble (blood brain barrier permeable). A schematic of the chemistry is shown in Fig. 2. While the levels in-vivo are too low to observe, even after long-term accumulation, there are pathological states where NO levels are exceptionally high, approaching sub-millimolar concentrations. Specifically, these are during septic shock, when the inducible nitric oxide synthase is expressed at high, almost uncontrollable levels, as a defense mechanism to invading infectious substances such as lipopolysaccharide, along with generating high concentrations of superoxide as well. The pathological result is a severe lowering of blood pressure, which leads to anaphylactic shock and sometimes death. The other region where the levels are high is in particular regions of the brain during seizures, particularly epilepsy. Several groups were successful in monitoring these levels ex-vivo and Fujii and Berliner and colleagues were able to directly measure ‘trapped’ NO in living mice and rats [7,35,36]. Yoshimura and coworkers were able to image NO in the abdominal section of a septic rat, although the large linewidth of the radical signal, ca. 3 Gauss, presents a limitation to the image resolution since the field gradients needed for such a study exceed those necessary for aminoxyl radicals [105]. Nonetheless, this is a unique demonstration of the power of EPR in ‘directly’ measuring a biological free radical in living animals. We later demonstrated that this trapped NO complex was an excellent MRI contrast agent, allowing much higher resolution imaging by virtue of the efficient paramagnetic relaxation of water protons in the tissue near the NO [37]. Hence a temporal measurement of NO generation was imaged ‘indirectly’ but locally in the MRI image. They were able to demonstrate the effects of nitric oxide synthase inhibitors where the MRI contrast was diminished in correlation with the reduced levels of generated NO. Later in-vivo studies on mice and rats demonstrated that elevated NO levels in the brain occurred during seizures in epileptic model systems [54].
Fig. 2.
Biosynthetic pathway for the generation of NO. The reaction is catalyzed by nitric oxide synthase (NOS).
3.4.Incorporation of aminoxyl (aka nitroxide) radicals
This was the first application of extrinsic radicals to in-vivo systems. This is basically incorporating the compounds, aminoxyl (aka nitroxide) radicals that are used in spin labeling, but targeting them (if possible) to specific regions of the body, organs, etc. In fact, the work from the Piette group was one of the earliest examples [30]. We initially worked on the same X-band system after Piette trying to measure EPR at the carotid artery in a rat, but realized quickly that the microwave heating at 9.5 GHz was a real issue [unpublished results]. L-band was obviously the frequency region of choice, where we started with EPR imaging of plants, such as celery [10]. The obstacles/limitations of this approach are at least two-fold: the large linewidths and hyperfine splitting reducing total sensitivity, i.e., >0.5 Gauss (unless one uses isotopically enriched labels/probes); and bioreduction/biodestruction, distribution and elimination by various excretion mechanisms. While the latter processes are reasonably well understood from pharmacokinetic studies, the real problem lies in the antioxidant processes that go on in living cells via NADH, vitamin C, etc. The kinetics of this process are extremely fast with the six-membered piperidine nitroxides (intracellular
The other advantages of spin probes, in general, is that the functionality can be modified to fit the target, including the possibility to block the ability to cross cell membranes using charged labels, and conversely employing ‘caged’ compounds that cross the cell membrane and be then are hydrolyzed inside the cell to a charged compound that could not transported back out again. Surprisingly, there was very little success with very specific/affinity based spin probes, even conjugated to an antibody, probably because the final local concentrations that could be achieved were not high enough for sensitive detection, particularly due to bioreduction of the paramagnetic N–O∙ moiety to the diamagnetic N–OH (hydroxylamine).
3.5.In-vivo EPR oximetry
The last problem concerning sensitivity of detecting aminoxyl/nitroxide radicals is the oxygen broadening effect on the EPR linewidth. But this is not an obstacle if taken advantage of properly and, in fact, the field of EPR oximetry as described above has flourished based on the Heisenberg exchange broadening effect. In fact, aminoxyl/nitroxide radicals compounds were actually first used in the development of EPR oximetry in-vitro [32,86]. However, the price paid with line broadening is of course sensitivity, requiring large concentrations of label to compensate. Nonetheless, as mentioned above, this has been a popular, useful method which benefitted tremendously with the discovery and use of other types of materials mentioned earlier that had strong signals, narrower linewidths, inertness to bioreduction and the feasibility of safely implanting them in an animal or human. Lithium phthalocyanine could be implanted into tissue encapsulated in a gas permeable inert polymer. This probe was later patented as Oxylite© and Oxyspin©, a biomaterial that could be implanted in tissue, stem cells, etc. that reported oxygen tension via the same type of Heisenberg exchange line broadening of its single narrow linewidth line [20,68,75,76,98]. Some other materials were also encapsulated, particularly powdered carbon chars, that were used for the same purpose [43]. Amazingly, with most materials the oxygen linewidth calibration curve was quite linear, reversible and very reproducible. The latter approach has been used for long-term temporal studies of the vascularity in the feet of diabetic patients, where the oxygen tension is remotely monitored over long period of times with surface coil probes with no adverse effects on the patient and the is implanted material removable later [55,56,103].
4.In-vivo EPR and imaging
4.1.L-band in-vivo EPR
The first reports of an in-vivo L-band experiment (vs. minimally invasive X-band experiments from the Piette group discussed earlier [30]) was in the early 1980s by S.J. Lukiewicz and S.G. Lukiewicz at the Society of Magnetic Resonance Meeting in 1984 and an International Pigment Cell Conference [65], where the authors detected the in-vivo EPR spectrum of melanin radicals in melanoma tumor cells implanted in the tail of a mouse. The first in-vivo oximetry studies were also done at L-band [88].
Progress with in-vivo EPR and imaging development in our laboratory first started with single loop L-band surface coils evaluating the feasibility of imaging biological systems at this frequency [34]. The first biological EPR imaging experiment addressed capillary diffusion of the nitroxide TEMPOL in celery in the presence of a 20–30 Gauss/cm field gradient, where we could distinguish different capillaries in the plant structure [10]. About two years later, in a collaboration with S.J. Lukiewicz, we reported the first in-vivo image of aminoxyl/nitroxide radical flow in a melanoma tumor cell line implanted in the tail of a live mouse [11]. Given the microwave engineering and computing power at the time, the resolution was not phenomenal. The Swartz group at the University of Illinois focused instead on the development of in-vivo high resolution spectroscopy [2]. They utilized in-vivo EPR oximetry using carbon chars and India ink and also demonstrated the observation of reactive free radicals. The Illinois group also refined imaging procedures for biologically relevant studies [3,26,27]. The field started to really blossom in the late 1980s in laboratories in Yamagata (Japan) [104] and Johns Hopkins University, where Zweier and Kuppusamy [109] observed nitroxide radicals in the intact beating heart. Further developments at very low field (i.e., 250 MHz frequency) and in-vivo EPR imaging were initiated by Halpern and coworkers using a birdcage-like capacitively coupled resonator [41].
The loop-gap resonator, developed by Froncisz and Hyde, turned out to be a major breakthrough for in-vivo EPR as well as other in-situ applications [31]. While first demonstrated to show significantly improved sensitivity at X-band, constructing the lower frequency L-band analog was straightforward; in fact, the tolerances were more forgiving than for X-band. Subsequently the loop-gap resonator and a closely related reentrant cavity developed by Sotgiu and colleagues [83] became quite common to in-vivo EPR instrumentation. The L-band reentrant cavity resonator, which was quite suitable for whole body experiments on mice, was developed at the University of L’Aquila, Italy and then later at the Milwaukee EPR Center [38,83]. We invited Antonello Sotgiu from L’Aquila to our laboratory where he developed a reentrant resonator for our pharmacokinetic studies on the rates of biodestruction/elimination of several aminoxyl/nitroxide probes in mice and rats [15].
4.2.In-vivo instrumentation with human subjects
Perhaps the first demonstration of in-vivo EPR on humans was published by Herrling and Groth in Berlin at the former East German Academy of Sciences, where they were measuring the antioxidant and inflammation behavior of human skin [33,46,47]. The spin probe, TEMPOL, is very sensitive to bioreduction and can be monitored directly using surface coils. In addition, one can image several millimeters into the derma. Herrling and Groth and colleagues monitored the kinetics of bioreduction of the spin probe under normal conditions and compared them under the influence of irritants initiated by UV-A and UV-B illumination. In fact, they eventually developed an accepted measurement parameter for the antioxidant capacity of various sunscreens in the cosmetic industry by quantitating the ability of each skin cream to reverse the effects of UV irradiation and subsequent inflammation and radical generation on the bioreduction rates of the spin probe [47]. This work was followed by the Swartz group who showed that a tattoo (which is composed of India ink) could be monitored by placing an in-vivo EPR surface coil on the arm of a human. A tourniquet was tied above the tattoo of a human volunteer, restricting the blood supply and hence changing oxygen levels in the derma could be accurately monitored. This eventually led to temporal studies on blood circulation in the feet of diabetics as a real, significant clinical monitor of deep vein circulation in a patient [55].
Besides recognizing that lower frequencies (i.e., L-band 1 GHz or lower) are most suitable from both the nonresonant absorption problem, the issue of human, clinical in-vivo EPR had additional requirements, particularly that the depth penetration had to exceed a cm or so that was achieved with small animals. Hence, the novel development of implantable resonators evolved from the Swartz group, then already at Dartmouth College Medical School, with the use of wire surface type coils with ‘memory’ metals in the coils. Since the goal was to measure oxygen pressure, the coil was also fitted with an encapsulated inert paramagnetic probe, such as Oxyspin©. In fact, the Swartz group along with a consortium of workers at Dartmouth, Emory University and the Univ. of Brussels, have embarked on studies to localize areas for eventual surgical removal or correction of regions that have reduced oxygen tension due to tumors, or as a calibration of oxygen pressures suitable for subsequent radiation therapy that has also been a major thrust in the Univ. of Chicago group as well, who are experts on radiation treatment of tumors and tissue [28,29,42].
What has not been mentioned in any detail so far is the aspect of localization, i.e., visualization of the paramagnetic probes in-vivo, or EPR imaging. This technique, which follows the NMR/MRI approach by inducing a magnetic field gradient in the region to be imaged followed by back-projection reconstruction did not develop quite as rapidly as NMR. Since the linewidths of the individual radical lines are much broader than those of e.g., protons in an MRI experiment, the magnitude of the field gradients required were engineering challenges and were, of course, limited. The first in-vivo reports came from our laboratory at Ohio State in the mid-1980s on celery, then with other plant species and eventually a tumor implanted in the tail of a mouse [34]. While that work described fairly primitive, one-dimensional gradient imaging experiments with relatively long sampling times, it was the first time a tumor image in a living animal was reported by EPR.
The field later advanced tremendously with the advent of 3-D imaging and spectral-spatial imaging where more than one radical or lineshape exist within the sample, and especially pulsed FT EPR imaging methods [66,71,72,87]. (We proposed exploring this approach in our laboratory in the 1990s but were throttled by our NIH review group on the basis of feasibility, yet a year or so later the intramural NIH group cited above reported exceptionally successful results with pulsed FT EPR imaging.) As mentioned earlier, notable accomplishments were the imaging of oxygen in an isolated beating heart by Zweier and Kuppusamy [109] first using spin probes or glucose chars, imaging of tumors in small rodents, working the way up to larger species. The real future and impact on human health and medicine lies in the applications of clinical in-vivo EPR. While animal studies are informative and powerful, they can never totally guide the surgeon or physician in the clinic. A comprehensive volume on in-vivo EPR was edited earlier [9] with the generous help of Harold Swartz. In the next year or two we will publish a coedited volume on clinical in-vivo EPR [93].
5.Spin labeling: A general approach to applying EPR to biomedical/bimolecular systems
As mentioned in the introduction, due to the lack of strong paramagnetic centers in the majority of biological systems in general, one needed a general labeling technique with EPR modeled after extrinsic fluorescence labeling. And, in fact for EPR, this would have taken a lot longer to develop (or possibly never happened) without the coincidence of major synthetic organic chemistry development. Specifically, much of the chemistry of aminoxyl (aka nitroxide) radicals originated from the pioneering synthetic work of Eduardo Rozantsev and several colleagues at the USSR Academy of Sciences, Institute of Chemical Physics (Moscow) and the Institute of Organic Chemistry (Novosibirsk) [80,100]. The concept of spin labeling using these radicals originated from and the vision of Professor Harden M. McConnell at Stanford University, in discussions with several biochemists (particularly Lubert Stryer). That is to say, the spin labeling method was ‘born’ at Stanford in 1964 with the first publication a year later [85]. Yet, it is noteworthy that the ‘first’ spin label experiment did not actually involve aminoxyl/nitroxide radicals but rather utilized a natural acridine drug analog, namely the cation radical of the antidepressant chlorpromazine. McConnell had conceived the chlorpromazine experiment to probe whether acridine molecules were isolated from one another when bound to DNA, or stacked. Chlorpromazine, which mimics acridine in its interaction with DNA, could be studied in its paramagnetic radical anion state. In this experiment, done solely by visiting Professor Shun-ichi Ohnishi from the Biophysics Department, Kyoto University, DNA fibers/strands complexed with this radical were flowed through a very narrow capillary placed in a modified Q-band (35 GHz) cavity so that the direction of flow was either parallel or perpendicular to the applied magnetic field direction. The resulting ‘oriented’ spectra displayed the major axes of the hyperfine interaction for this nitrogen centered radical, which was believed to intercalate the major groove of DNA. The result geometrically confirmed this intercalation [73].
5.1.The spin label reporter group method
The diagnostic advantage of the aminoxyl radical/nitroxide probes comes from the electron-nuclear hyperfine interaction, in particular that between the free electron and the N-14 nucleus. This hyperfine interaction is anisotropic; that is the orientation of the N–O group in the applied magnetic field shows a different hyperfine coupling constant when the N–O group orients parallel with the applied field (of the order of 30–32 Gauss) vs. perpendicular to the applied field (of the order of 5–6 Gauss). However, this is observed only in cases where the sample is rigid, non-tumbling and all coaligned in a crystal matrix host. If the sample is random, but frozen (immobilized), one observes an envelope (powder) spectrum of all of the possible orientations and hyperfine couplings between 5 to 32 Gauss. As the sample starts to tumble (i.e., the effective ‘viscosity’ decreases) these hyperfine interaction slowly begin to average out and the spectrum eventually transforms to a narrow three line 15–16 Gauss hyperfine isotropic spectrum where the anisotropy is totally averaged to zero, leaving what’s called the Fermi contact isotropic hyperfine interaction. Fortunately, these spectra can be simulated and quantitatively analyzed to derive a rotational correlation time for the tumbling radical, including the anisotropy of this tumbling. Hence the spin label technique probes the biological system by observing the motion of the spin label probe, which is a reflection of the overall tumbling rate of the macromolecule, if the radical is rigidly held via a covalent linkage or strong noncovalent association. One can analyze domain motion and/or the resultant ‘free’ tumbling of the label tethered to a protein. One can also observe changes occurring when the protein undergoes structural changes in the vicinity of the label that frees up conformationally restricted motion and/or disrupts attractive noncovalent interactions with a protein. For noncovalently bound labels, the strength of the interaction is reflected in the motion of the label as well as the size, depth and hydrophobicity of the cavity in which the label resides. The particular advantage of the aminoxyl radical/nitroxide probes is their versatility in organic synthesis with the ability to incorporate almost any functional group or determinant into the structure to fit the system of interest. Overall the method is exquisitely sensitive, particularly to protein structure and dynamics, but also to nucleic acids, lipids and membranes and more complex biological structures [6 –8,14,16,45].
For applications to proteins and enzymes, one can study the dynamic conformational properties of an enzyme or protein during function, denaturation, folding and unfolding, as well as complexation/aggregation with other proteins. If one can work with a protein single crystal, even more detailed structural information can be derived. For example, in McConnell’s group and later at Ohio State, we developed spin label substrates for α-chymotrypsin which were reporters of the orientation (and specificity) of these molecule complexing with the enzyme [13]. A particularly interesting experiment compared two enantiomers of the same spin labeled substrate with the enzyme, compared their kinetics and then derived a detailed structural/mechanistic explanation of their behavior from the orientation(s) derived from the single crystal EPR experiment with the known X-ray crystal structure of the enzyme [5].
Needless to say, a plethora of very informative and clever spin label studies ensued, for both proteins and enzymes, nucleic acids [16] and membranes [45]. The work on membranes was also pioneered by the McConnell group, where they contributed to major principles and aspects of membrane structure and dynamics that were not known up to then [50,51,58,59,67]. Certainly the spin label technique is now one of the ‘textbook’ biophysical methods for studying bimolecular systems; yet for proteins this frequently involved chemical modification of amino acid side chains which was somewhat limited in its scope.
5.2.Site directed spin labeling (SDSL)
Fig. 3.
(A) MTSSL (2,5-dihydro-3,4-bis(methanesulfonylthiomethyl)-2,2,5,5-tetramethyl-1H-pyrrol-1-yloxyl radical) and (B) bis-MTSL (trans-3,4-bis(methanesulfonylthiomethyl)-2,2,5,5-tetramethylpyrrolidin-1-yloxyl radical) spin labels, commercially available from Toronto Research Chemicals, 2 Brisbane Rd., Toronto, Ontario, Canada M3J 2J8.
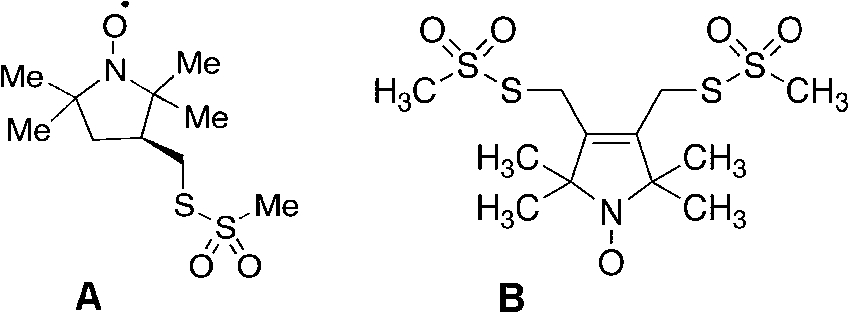
Chemically labeling amino acid side chains was only really specific if one had a free thiol group(s) available and, better yet, if the labeling were reversible when the protein was limited and valuable. It was about this time that two important parallel accomplishments occurred: the developments of a thiol specific reversible spin label and the advent of site specific modification/mutation of proteins. Our group, in collaboration with Kalman Hideg, a clever, prolific synthetic organic chemist from the University of Pecs, Hungary, developed and tested the now famous spin label, MTSL, the methylmethanethiolsulfonate analog of either the pyrrolinyl or pyrrolidinyl spin label. Some sample structures are shown in Fig. 3 [12]. The implementation of the actual experiments to test this label were complicated by the fact that I was on sabbatical in Haifa, Israel and Hideg was in Hungary when there were always concerns about the success of sending and receiving chemicals through the postal system. As it turned out, Kalman Hideg and I were attending different scientific meetings in the Toronto, Canada area. We arranged a clandestine exchange in Toronto airport in the summer of 1980 while Kalman was enroute back to Hungary and I was traveling back with the compound back to Haifa, where, with former postdoctorate Jacob Grunwald, we carried out the feasibility experiments [12]. We showed how this uniquely, stoichiometrically and specifically labeled the protease papain, which contained with a single reactive thiol group at the active site. We showed as well that we could reverse the disulfide labeling with mercaptoethanol or DTT to yield the free enzyme and a thiol containing aminoxyl radical. However, it was the clever insight of Wayne Hubbell to apply this to any protein in general where the DNA could be modified to incorporate Cys groups at any preferred location in the sequence. This was the renaissance of spin labeling and the genesis of the site directed spin labeling technique (SDSL), which blossomed tremendously from the 1990s until present. Some of the key references to the original work on bacteriorhodopsin to some of the more current approaches, as well as sample handling and pitfalls, are well described in reference [49]. In fact, for several years there were sessions devoted solely to just SDSL at magnetic resonance meetings, much like the early days of NMR structures of proteins. The advantages of EPR, spin labeling and SDSL were the fact that the sample need not be optically clear, that MW limits were not the same issue as in NMR and, in fact, the sample could be in the solid or amorphous form as well. In particularly this technique was a real boon to the study of membrane proteins, which were not amenable to X-ray crystallography in general and were a challenge for the NMR spectroscopists as well [49]. Lastly, the whole area of disordered or partially folded proteins: i.e., the molten globule, misfolded proteins, aggregates and fibrils was very applicable to SDSL. A strictly defined, rigid protein structure was not necessary nor the goal since the EPR spin label technique does not necessarily have the atomic resolution capabilities of X-ray or NMR. Yet the method could be powerfully used to probe dynamics, helix structure and intramolecular distances where two labels could be incorporated at two simultaneously introduced Cys sites by virtue of the electron-electron dipolar interaction. Another approach was to incorporate a paramagnetic cation binding locus as the second site, where the metal ion was a point dipole [101]. By using sophisticated pulsed EPR techniques (called DEER or PELDOR) these distances were measurable over the ca. 10 to 50 Å range of what is normally long-range interactions that are not easily accessed in NMR [17]. A recent book on the pulsed DEER/PELDOR techniques, including their history and development as well as applications to spin label metalloprotein systems is well described in ref [96]. Thus it became possible to map intramolecular distances and by trilaterating to derive partial structures. Hence the double labeling approach became quite popular, despite its two drawbacks: resultant tumbling motion of the label meant that the position of the free electron on the N–O group was ambiguous and could only be estimated by modeling this motion; second (but an obstacle that can be overcome in several cases) is the immense amount of work involved in incorporating two pairs of Cys residues to obtain just one intramolecular distance. That is the real challenge comes with intermolecular distance measurements by SDSL where one would need to engineer 4 Cys groups into a protein and allow it to fold correctly without scrambling. The need for labels that would bind to a protein at two points, such that the nitroxide ring motion on the tether was severely restricted, would reduce the distance ambiguities and yield a more accurate estimate of domain motion as well. This was solved by the synthesis of bis-MTSL spin labels, commercially available from Toronto Research Chemicals, containing two methythiolmethanesulfonate functionalities on the same nitroxide ring that would covalently attach to two Cys groups that were coincidently within the structural reach and dimensions of this bifunctional label (also shown in Fig. 3(B)). A recent application with the bis-MTSL spin label comes from the work of Stevens et al. [84], which also reviews pulsed DEER/PELDOR applications and history. A typical EPR experiment by the author is depicted in Fig. 4.
Fig. 4.
Larry Berliner working at the Bruker L-band/X-band in-vivo imaging spectrometer at the Centre for Advanced Imaging, University of Queensland. He is holding a coffee bean which was imaged at X-band on the basis of its paramagnetic radical content.

6.Metalloproteins and metal cluster containing systems
Some of the early work in metalloproteins focused on iron containing proteins such as hemoglobin and cytochrome C. These were done at liquid nitrogen or helium temperatures as mentioned earlier, as many of these paramagnetic metals had extremely short relaxation times, hindering sensitivity. Much of the pioneering work on hemoglobin and myoglobin was done by the group of Masao Kotani [60,61]. In fact, it appears that early EPR spectroscopy studying hemoglobin and other iron-containing proteins (some isolated from cadavers and tissue) actually detected nitric oxide NO in their samples coordinated to the iron in hemoglobin; however, it was assumed to be related to nitrate and nitrite metabolism [1] and were not aware of what was going to transpire in the late 1990s when the Nobel Prize was awarded for the discovery of NO in muscle relaxation. To the author this seemed an amazing coincidence to have this phenomenal discovery right under the noses of the EPR spectroscopists, not realizing at the time the complex chemistry involved in nitric oxide synthase and its relationship with cyclic GMP, all involved in muscle relaxation.
The major advances in this area, particularly in the study of multi-centered metal complexes and clusters has been the advent of pulsed EPR instrumentation, which has increased sensitivity and information content significantly. Some of these are electron nuclear double resonance (ENDOR) electron-spin-echo-envelope-modulation (ESEEM) spectroscopy which are methods that provide high resolution by directly measuring nuclear spins, providing information on both strong and very weak interactions with the electron(s). Hyperfine sub-level correlation (HYSCORE) spectroscopy is a 2D spectroscopy method based on the same principles. While some of these methods were developed in the 1960s [70], only a small number of groups worked the field, because of the expensive instrumentation, the lack of suitable microwave components and slow digital electronics. It was not until commercial development, first by Varian Associates and then extensively by Bruker BioSpin, that this powerful technology was exploited to the fullest. As mentioned in Section 1.1 coverage of the vast area of biochemical and model metal containing systems and the relevant pulse methods is beyond the scope and intent of this article. However, very good description and review of these methodologies comes from the former Schweiger group [18] as well as the recent book referenced earlier [96].
7.Prognosis for the future
ESR/EPR has developed rapidly since its inception, with even more strides starting in the late 1980s with the advent of site specific spin labeling and major progress with in-vivo EPR and imaging. The interest and cooperation of the major vendors in providing commercialized instrumentation has also been a major asset to progress in the field. From the biomedical/clinical viewpoint, we look forward to many advances in human EPR and methods of non-invasive (or minimally invasive) diagnosis that exceed competing technologies in sensitivity and characterization. Advances are being made in developing aminoxyl (i.e., nitroxide) radicals that are more resistant to biological reduction. An excellent book on these radicals is found in ref [48]. Several of the design principles for radical stability to bioreduction will stem from the work of Villamena and colleagues mentioned earlier [4,57,77,99].
The spin label method is now a ‘fixture’ in the armament of the biomolecular structure researcher and shows great promise for membrane proteins and very large systems that are not easily attacked by X-ray crystallography or NMR. The synthetic chemistry has been advanced, particularly for nucleic acid labels, utlilizing bio-orthogonal labeling techniques, such as ‘click chemistry’ as outlined in a chapter in [96]. This book also reviews the use of Gd(III) labels and combined aminoxyl-metal ion labels as unique distance probes [96]. I have been guilty, more than once, of concluding that both spin labeling and in-vivo EPR had ‘run their course’. However, it is clear that spin labeling and in-vivo EPR continue to advance, with a forthcoming volume on Clinical In-vivo EPR to be published in 2016 [93]. In fact, a fifth Spin Labeling: Theory and Applications volume in the Biological Magnetic Resonance series will be written over the next one to two years as a resurrection of the original, now out-of-print, textbooks [6,7] with both the original fundamentals as well as the latest state-of-the-art approaches and methods and details.
Notes
Acknowledgements
It was impossible to cover the entire history comprehensively and I am sure that my references to other important work is incomplete. I apologize to the numerous major contributors to ESR/EPR studies who are not properly acknowledged here. I owe thanks to many funding agencies for my ability to carry out research over the years, particularly the National Science Foundation, the National Institutes of Health and especially the American Heart Association. Most important, I have been incredibly fortunate to work with numerous outstanding graduate students and post-docs, colleagues, collaborators, and visitors, many of whom were not possible to mention here. Lastly, the author had the honor and privilege of being mentored by Harden M. McConnell who was an inspirational teacher, friend and mentor.
References
1 | [[1]] Y. Azhipa, L.P. Kaiushin and E.I. Nikiski, EPR spectra of iron-containing complexes arising in animal tissues during some types of hypoxia, Biofizika 14: ((1969) ), 852–857. |
2 | [[2]] G. Bacic, M.J. Nilges, T. Walczak and H.M. Swartz, The use of in vivo EPR to follow the pharmacokinetics of drugs, Phys. Med. 5: ((1989) ), 307–313. |
3 | [[3]] G. Bacic, T. Walczak, F. Demsar and H.M. Swartz, Electron spin resonance imaging of tissues with lipid-rich areas, Magn. Reson. Med. 8: ((1988) ), 209–219. |
4 | [[4]] X. Bao, P. Tao, F.A. Villamena and C.M. Hadad, Spin trapping of hydroperoxyl radical by a cyclic nitrone conjugated to β-cyclodextrin: A computational study, Theoretical Chemistry Accounts 131: ((2012) ), 1171–1207. |
5 | [[5]] R.S. Bauer and L.J. Berliner, Spin label investigations of chymotrypsin active site structure in single crystals, J. Mol. Biol. 128: ((1979) ), 1–19. |
6 | [[6]] L.J. Berliner (ed.), Spin Labeling: Theory and Applications, Academic Press, New York, (1976) . |
7 | [[7]] L.J. Berliner (ed.), Spin Labeling II: Theory and Applications, Academic Press, New York, (1979) . |
8 | [[8]] L.J. Berliner (ed.), Spin Labeling: The Next Millennium, Biological Magnetic Resonance, Vol. 14: , Plenum Publishing Corp., New York, (1998) . |
9 | [[9]] L.J. Berliner (ed.), In Vivo EPR (ESR): Theory and Applications, Biological Magnetic Resonance, Vol. 18: , Kluwer Academic/Plenum Publishing Corp., New York, (2003) . |
10 | [[10]] L.J. Berliner and H. Fujii, Magnetic resonance imaging of biological specimens by electron paramagnetic resonance of nitroxide spin labels, Science 227: ((1985) ), 517–519. |
11 | [[11]] L.J. Berliner, H. Fujii, X. Wan and S.J. Lukiewicz, Feasibility study of imaging a living murine tumour by electron paramagnetic resonance, Magn. Reson. Med. 4: ((1987) ), 380–384. |
12 | [[12]] L.J. Berliner, J. Grunwald, H.O. Hankovszky and K. Hideg, A novel reversible thiol specific spin label: Papain active site labeling and inhibition, Anal. Biochemistry 119: ((1982) ), 450–455. |
13 | [[13]] L.J. Berliner and H.M. McConnell, A spin labeled substrate for α-chymotrypsin, Proc. Nat. Acad. Sci. USA 55: ((1966) ), 708–712. |
14 | [[14]] L.J. Berliner and J. Reuben (eds), Spin Labeling: Theory and Applications, Biological Magnetic Resonance, Vol. 8: , Plenum Publishing Corp., New York, (1989) . |
15 | [[15]] L.J. Berliner and X. Wan, In vivo pharmacokinetics by electron paramagnetic resonance spectroscopy, Magn. Reson. Med. 9: ((1989) ), 430–434. |
16 | [[16]] A.M. Bobst, Applications of spin labeling to nucleic acids, in: Spin Labeling II: Theory and Applications, L.J. Berliner, ed., Academic Press, New York, (1979) , pp. 291–346. |
17 | [[17]] P.P. Borbat and J.H. Freed, Multiple-quantum ESR and distance measurement, Chem. Phys. Lett. 313: ((1999) ), 145–154. |
18 | [[18]] C. Calle, A. Sreekanth, M.V. Fedin, J. Forrer, I. Garcia-Rubio, I.A. Gromov, D. Hinderberger, B. Kasumaj, P. Léger, B. Mancosu, G. Mitrikas, M. Santangelo, S. Stoll, A. Schweiger, R. Tschaggelar and J. Harmer, Pulse EPR methods for studying chemical and biological samples containing transition metals, Helvetica Chimica Acta 89: ((2006) ), 2495–2521. |
19 | [[19]] I.D. Campbell, The evolution of protein NMR, Biomedical Spectroscopy and Imaging 2: ((2013) ), 245–264. |
20 | [[20]] S.M. Chacko, M. Khan, M.L. Kuppusamy, R.P. Pandian, S. Varadharaj, K. Selvendiran and P. Kuppusamy, Myocardial oxygenation and functional recovery in infarct rat hearts transplanted with mesenchymal stem cells, American Journal of Physiology – Heart and Circulatory Physiology 296: ((2009) ), H1263–H1273. |
21 | [[21]] B. Commoner, Electron-spin resonance (ESR) studies of biochemical and biological systems, Acad. Royal. Belg., Classe Sci. Mem. 33: ((1961) ), 114–161. |
22 | [[22]] B. Commoner, J.J. Heise and J. Townsend, Light-induced paramagnetism in chloroplasts, Proc. Natl. Acad. Sci. USA 42: ((1956) ), 710–718. |
23 | [[23]] B. Commoner and T.C. Hollocher Jr., Free radicals in heart muscle mitochondrial particles: General characteristics and localization in the electron transport system, Proc. Natl. Acad. Sci. USA 46: ((1960) ), 405–416. |
24 | [[24]] B. Commoner and J.L. Ternberg, Free radicals in surviving tissues, Proc. Natl. Acad. Sci. USA 47: ((1961) ), 1374–1384. |
25 | [[25]] B. Commoner, J. Townsend and G.E. Pake, Free radicals in biological materials, Nature 174: ((1954) ), 689–691. |
26 | [[26]] F. Demsar, H.M. Swartz and M. Schara, Use of field gradient EPR to measure diffusion of nitroxides in tissues, Magn. Reson. Med. Biol. 1: ((1988) ), 17–24. |
27 | [[27]] F. Demsar, T. Walczak, P. Morse II, G. Bacic, Z. Zolnai and H.M. Swartz, Detection of diffusion of oxygen by fast scan EPR imaging, J. Magn. Reson. 76: ((1988) ), 224–231. |
28 | [[28]] M. Elas, R. Bell, D. Hleihel, E.D. Barth, C. McFaul, C.R. Haney, J. Bielanska, K. Pustelny, K.-H. Ahn, C.A. Pelizzari, M. Kocherginsky and H.J. Halpern, Electron paramagnetic resonance (EPR) oxygen image hypoxic fraction plus radiation dose strongly correlates with tumor cure in FSa fibrosarcomas, International Journal Radiation Oncology, Biology, Physics 71: ((2008) ), 542–549. |
29 | [[29]] M. Elas, J.M. Magwood, B. Butler, C. Li, R. Wardak, E.D. Barth, B. Epel, S. Rubinstein, C.A. Pelizzari, R.R. Weichselbaum and H.J. Halpern, EPR oxygen images predict tumor control by a 50 percent tumor control radiation dose, Cancer Research 73: ((2013) ), 5328–5335. |
30 | [[30]] A. Feldman, E. Wildman, G. Bartolinini and L.H. Piette, In vivo electron spin resonance in rats, Phys. Med. Biol. 20: ((1975) ), 602–612. |
31 | [[31]] W. Froncisz and J.S. Hyde, The loopgap resonator, a new microwave lumped circuit ESR sample structure, J. Magn. Reson. 47: ((1982) ), 515–521. |
32 | [[32]] W. Froncisz, C.S. Lai and J.S. Hyde, Spin-label oximetry: Kinetic study of cell respiration using a rapid-passage |
33 | [[33]] J. Fuchs, N. Groth and T. Herrling, Applications of in vivo EPR spectroscopy and imaging to skin, in: In Vivo EPR (ESR): Theory and Applications, L.J. Berliner, ed., Biological Magnetic Resonance, Vol. 18: , Kluwer Academic/Plenum Publishing Corp., New York, (2003) , pp. 483–513. |
34 | [[34]] H. Fujii and L.J. Berliner, One- and two-dimensional EPR imaging studies on phantoms and plant specimens, Magn. Reson. Med. 2: ((1985) ), 275–282. |
35 | [[35]] H. Fujii and L.J. Berliner, Nitric oxide: Prospects and perspectives of in-vivo detection by L-band EPR spectroscopy, Phys. Med. Biol. 43: ((1998) ), 1–8. |
36 | [[36]] H. Fujii, J. Koscielniak and L.J. Berliner, Determination and characterization of nitric oxide generation in mice by in-vivo L-band EPR spectroscopy, Magn. Reson. Med. 38: ((1997) ), 565–568. |
37 | [[37]] H. Fujii, X. Wan, J. Zhong, K. Yoshikawa and L.J. Berliner, In vivo imaging of spin-trapped nitric oxide in rats with septic shock: MRI spin trapping, Magn. Reson. Med. 42: ((1999) ), 235–239. |
38 | [[38]] M. Giordano, F. Momo and A. Sotgiu, On the design of a re-entrant square cavity as resonator for low-frequency ESR spectroscopy, Journal of Physics E: Scientific Instruments 16: ((1985) ), 774. |
39 | [[39]] M. Gomberg, An instance of trivalent carbon: Triphenylmethyl, J. Am. Chem. Soc. 22: ((1900) ), 757–771. |
40 | [[40]] O. Grinberg and L.J. Berliner (eds), Very High Field (VHF) ESR/EPR, Biological Magnetic Resonance, Vol. 22: , Kluwer Academic/Plenum Publishing Corp., New York, (2004) . |
41 | [[41]] H.J. Halpern, M.K. Bowman, D.P. Spencer, J. van Polen, A.C. Nelson, E.M. Dowey and B.A. Teicher, Imaging radio frequency electron-spin-resonance spectrometer with high resolution and sensitivity for in vivo measurements, Rev. Sci. Instr. 60: ((1989) ), 1040–1050. |
42 | [[42]] H.J. Halpern, C. Yu, M. Peric, E.D. Barth, G.S. Karczmar, J.N. River, D.J. Grdina and B.A. Teicher, Measurement of differences in pO2 in response to perfluorocarbon/carbogen in FSa and NFSa murine fibrosarcomas with low-frequency electron paramagnetic resonance oximetry, Radiat. Res. 145: ((1996) ), 610–618. |
43 | [[43]] J. He, N. Beghein, P. Ceroke, R.B. Clarkson, H.M. Swartz and B. Gallez, Development of biocompatible oxygen-permeable films holding paramagnetic carbon particles: Evaluation of their performance and stability in EPR oximetry, Magn. Reson. Med. 46: ((2001) ), 1522–2594. |
44 | [[44]] R.J. Heckley, Free radicals in dry tissues, in: Biological Applications of Electron Spin Resonance, H.M. Swartz, J.R. Bolton and D.C. Borg, eds, Wiley, New York, (1972) , p. 197. |
45 | [[45]] M.A. Hemminga and L.J. Berliner (eds), ESR Spectroscopy in Membrane Biophysics, Biological Magnetic Resonance, Vol. 27: , Springer Publishing Corp., New York, (2007) . |
46 | [[46]] T. Herrling, J. Fuchs, J. Rehberg and N. Groth, UV-induced free radicals in the skin detected by ESR spectroscopy and imaging using nitroxides, Free Radic. Biol. Med. 35: ((2003) ), 59–67. |
47 | [[47]] T. Herrling, L. Zastrow and N. Groth, Classification of cosmetic products – The radical protection factor (RPF), Seifen Öle Fette Wachse J. 124: ((1998) ), 282–284. |
48 | [[48]] R. Hicks (ed.), Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, Wiley, Chichester, (2010) . |
49 | [[49]] W.L. Hubbell and C. Altenbach, Investigation of structure and dynamics in membrane proteins using site-directed spin labeling, Current Opinion in Structural Biology 4: ((1994) ), 566–573. |
50 | [[50]] W.L. Hubbell and H.M. McConnell, Spin-label studies of the excitable membranes of nerve and muscle, Proc. Natl. Acad. Sci. 61: ((1968) ), 12–16. |
51 | [[51]] W.L. Hubbell and H.M. McConnell, Motion of steroid spin labels in membranes, Proc. Natl. Acad. Sci. 63: ((1969) ), 16–22. |
52 | [[52]] J.M.S. Hutchison, ESR spectrometry on the whole mouse in-vivo: Optimum frequency considerations, Journal of Physics E: Scientific Instruments 4: ((1971) ), 703–704. |
53 | [[53]] I. Isenberg and S.L. Baird Jr., Electron-spin resonance study of Escherichia coli, Acad. Royal. Belg., Classe Sci. Mem. 33: ((1962) ), 170–173. |
54 | [[54]] K. Kaneko, K. Itoh, L.J. Berliner, K. Miyasaka and H. Fujii, Consequences of nitric oxide generation in epileptic-seizure rodent models as studied by in vivo EPR, Magn. Reson. Med. 48: ((2002) ), 1051–1056. |
55 | [[55]] N. Khan, H. Hou, P. Hein, R.J. Comi, J.C. Buckey, O. Grinberg, I. Salikhov, S.Y. Lu, H. Wallach and H.M. Swartz, Black magic and EPR oximetry: From lab to initial clinical trials, Advances Experimental Medicine Biology 566: ((2005) ), 119–125. |
56 | [[56]] N. Khan, B.B. Williams, H. Hou, H. Li and H.M. Swartz, Repetitive tissue pO2 measurements by electron paramagnetic resonance oximetry: Current status and future potential for experimental and clinical studies, Antioxid. Redox Signal. 9: ((2007) ), 1169–1182. |
57 | [[57]] S.U. Kim and F.A. Villamena, Reactivities of superoxide and hydroperoxyl radicals with disubstituted cyclic nitrones: A DFT study, J. Physical Chemistry A 116: ((2012) ), 886–898. |
58 | [[58]] R.D. Kornberg and H.M. McConnell, Lateral diffusion of phospholipids in a vesicle membrane, Proc. Natl. Acad. Sci. 68: ((1971) ), 2564–2568. |
59 | [[59]] R.D. Kornberg and H.M. McConnell, Inside-outside transitions of phospholipids in vesicle membranes, Biochemistry 10: ((1971) ), 1111–1120. |
60 | [[60]] M. Kotani, On the electronic state of iron in hemoglobins, with a short introduction to problems of quantum biophysics, Rev. Mod. Phys. 35: ((1963) ), 717–720. |
61 | [[61]] M. Kotani, Paramagnetic properties and electronic structure of iron in heme proteins, Advances in Quantum Chemistry 4: ((1968) ), 227–266. |
62 | [[62]] L.N. Kubrina, W.S. Caldwell, P.I. Mordvintcev, I.V. Malenkova and A.F. Vanin, EPR evidence for nitric oxide production from guanidino nitrogens of L-arginine in animal tissues in vivo, Biochimica Biophysica Acta 1099: ((1992) ), 233–237. |
63 | [[63]] C.S. Lai and A.M. Komarov, Spin trapping of nitric oxide produced in vivo in septic-shock mice, FEBS Lett. 345: ((1994) ), 120–124. |
64 | [[64]] K.J. Liu, P. Gast, M. Moussavi, S.W. Norby, N. Vahidi, T. Walczak, M. Wu and H.M. Swartz, Lithium phthalocyanine: A probe for electron paramagnetic resonance oximetry in viable biological systems, Proc. Natl. Acad. Sci. USA 90: ((1993) ), 5438–5442. |
65 | [[65]] S.J. Lukiewicz and S.G. Lukiewicz, in: XIIth International Pigment Cell Conference, Giessen, FRG, (1983) , p. 133. |
66 | [[66]] M. Matsuo, S. Matsumoto, J.B. Mitchell, M.C. Krishna and K. Camphausen, Magnetic resonance imaging of the tumor microenvironment in radiotherapy: Perfusion, hypoxia, and metabolism, Semin. Radiat. Oncol. 24: ((2014) ), 210–217. |
67 | [[67]] H.M. McConnell and B.G. McFarland, The flexibility gradient in biological membranes, Ann. NY Acad. Sci. 195: ((1972) ), 207–217. |
68 | [[68]] G. Meenakshisundara, E. Eteshola, A. Blank, S.C. Lee and P. Kuppusamy, A molecular paramagnetic spin-doped biopolymeric oxygen sensor, Biosensors and Bioelectronics 25: ((2010) ), 2283–2289. |
69 | [[69]] L. Michaelis, Theory of the reversible two-step oxidation, J. Biol. Chem. 96: ((1932) ), 703–715. |
70 | [[70]] W.B. Mims, K. Nassau and J.D. McGee, Spectral diffusion in electron resonance lines, Physical Review 123: ((1961) ), 2059–2069. |
71 | [[71]] R. Murugesan, M. Afeworki, J.A. Cook, N. Devasahayam, R. Tschudin, J.B. Mitchell, S. Subramanian and M.C. Krishna, A broadband pulsed radio frequency electron paramagnetic resonance spectrometer for biological applications, Rev. Sci. Instr. 69: ((1998) ), 1869–1876. |
72 | [[72]] R. Murugesan, J.A. Cook, N. Devasahayam, M. Afeworki, S. Subramanian, R. Tschudin, J.H. Larsen, A.J.B. Mitchell, A. Russo and M.C. Krishna, In vivo imaging of a stable paramagnetic probe by pulsed-radiofrequency electron paramagnetic resonance spectroscopy, Magn. Reson. Med. 38: ((1997) ), 409–414. |
73 | [[73]] S.I. Ohnishi and H.M. McConnell, Interaction of the radical ion of chlorpromazine with deoxyribonucleic acid, J. Am. Chem. Soc. 87: ((1965) ), 2293–2295. |
74 | [[74]] S.J. Opella, The development of solid-state NMR of membrane proteins, Biomedical Spectroscopy and Imaging 3: ((2014) ), 81–105. |
75 | [[75]] R.P. Pandian, N.L. Parinandi, G. Ilangovan, J.L. Zweier and P. Kuppusamy, Novel particulate spin probe for targeted determination of oxygen in cells and tissues, Free Radic. Biol. Med. 35: ((2003) ), 1138–1148. |
76 | [[76]] B.K. Rivera, S.K. Naidu, K. Subramanian, M. Joseph, H. Hou, N. Khan, H.M. Swartz and P. Kuppusamy, Real-time, in vivo determination of dynamic changes in lung and heart tissue oxygenation using EPR oximetry, Advances Experimental Medicine Biology 812: ((2014) ), 81–86. |
77 | [[77]] M. Rosselin, F. Choteau, K. Zéamari, K.M. Nash, A. Das, R. Lauricella, E. Lojou, B. Tuccio, F.A. Villamena and G. Durand, Reactivities of substituted α-phenyl-N-tert-butyl nitrones, J. Organic Chemistry 79: ((2014) ), 6615–6626. |
78 | [[78]] J.R. Rowlands, ESR studies of biochemical effects of atmospheric pollutants, Report 1,0299, in: Environmental Protection Research Catalog, Part 1, U.S. Environmental Protection Agency, Office of Research and Monitoring, Research Information Division, 1972, p. I-44. |
79 | [[79]] J.R. Rowlands, E.M. Cause, C.F. Rodriguez and H.C. McKee, Electron spin resonance studies of vegetation damage, Final report, SWRI Project 05-2622-01, Southwest Research Institute, San Antonio, TX, 1970, 67. |
80 | [[80]] E.G. Rozantsev, Free Nitroxyl Radicals, Plenum Press, New York, (1970) . |
81 | [[81]] E.K. Ruuge, T.M. Kerimov and A.V. Panemanglor, Effect of lyophilization on the free-radical state of animal cells, Biofizika 21: ((1976) ), 124–128. |
82 | [[82]] K. Salikhov, Modern development of magnetic resonance (Zavoisky 100), Applied Magnetic Resonance 35: ((2009) ), 361–362. |
83 | [[83]] A. Sotgiu, Resonator design for in vivo EPR spectroscopy, J. Magn. Reson. 65: ((1985) ), 206–214. |
84 | [[84]] M.A. Stevens, J.E. McKay, J.L.S. Robinson, H. El Mkami, G.M. Smith and D.G. Norman, The use of the Rx spin label in orientation measurement on proteins by EPR, Phys. Chem. Chem. Phys. ((2015) ). doi:10.1039/C5CP04753F. |
85 | [[85]] T.J. Stone, T. Buckman, P.L. Nordio and H.M. McConnell, Spin-labeled biomolecules, Proc. Natl. Acad. Sci. 54: ((1965) ), 1010–1017. |
86 | [[86]] W.K. Subczynski and J.S. Hyde, The diffusion-concentration product of oxygen in lipid bilayers using the spin label |
87 | [[87]] S. Subramanian, R. Murugesan, N. Devasahayam, J.A. Cook, M. Afeworki, T. Pohida, R.G. Tschudin, J.B. Mitchell and M.C. Krishna, High-speed data acquisition system and receiver configurations for time-domain radiofrequency electron paramagnetic resonance spectroscopy and imaging, J. Magn. Reson. 137: ((1999) ), 379–388. |
88 | [[88]] W.K. Subszynski, S. Lukiewicz and J.S. Hyde, Murine in vivo L-band ESR spin-label oximetry with a loop-gap resonator, Magn. Reson. Med. 3: ((1986) ), 747–754. |
89 | [[89]] H.M. Swartz, ESR studies of cells and tissues, in: Biological Applications of Electron Spin Resonance, H.M. Swartz, J.R. Bolton and D.C. Borg, eds, Wiley, New York, (1972) , pp. 155–158. |
90 | [[90]] H.M. Swartz, Electron spin resonance studies of carcinogenesis, Adv. Canc. Res. 15: ((1972) ), 227–252. |
91 | [[91]] H.M. Swartz, In vivo EPR, in: Foundations of Modern EPR, S. Eaton, G. Eaton and K. Salikhov, eds, World Scientific Publishing, Singapore/New Jersey/London, (1998) , pp. 451–458. |
92 | [[92]] H.M. Swartz and L.J. Berliner, History of in vivo EPR, in: In Vivo EPR (ESR): Theory and Applications, L.J. Berliner, ed., Biological Magnetic Resonance, Vol. 18: , Kluwer Academic/Plenum Publishing Corp., New York, (2003) , pp. ix–xviii. |
93 | [[93]] H.M. Swartz and L.J. Berliner (eds), Clinical In-Vivo EPR, Biological Magnetic Resonance, Vol. 34: , Springer Science+Business, Media, New York, (2016) , in press. |
94 | [[94]] H.M. Swartz, K.J. Liu, F. Goda and T. Walczak, India ink: A potential clinically applicable EPR oximetry probe, Magn. Reson. Med. 31: ((1994) ), 229–232. |
95 | [[95]] H.M. Swartz and R.P. Molenda, Electron spin resonance spectra of some normal tissues: Effect of microwave power, Science 148: ((1965) ), 94–95. |
96 | [[96]] C. Timmel and J. Harmer (eds), Structural Information from Spin-Labels and Intrinsic Paramagnetic Centres in the Biosciences, Structure and Bonding, Vol. 152: , Springer-Verlag, Berlin/Heidelberg, (2014) . |
97 | [[97]] K. Ulbert, Mechanical damage of keratin proteins observed by electron spin resonance (E.P.R.), Nature 195: ((1962) ), 175. |
98 | [[98]] D.S. Vikram, A. Bratasz, R. Ahmad and P. Kuppusamy, A comparative evaluation of EPR and OxyLite oximetry using a random sampling of pO2 in a murine tumor, Radiat. Res. 168: ((2007) ), 308–315. |
99 | [[99]] F.A. Villamena, A. Das and K.M. Nash, Potential implication of the chemical properties and bioactivity of nitrone spin traps for therapeutics, Future Med. Chem. 4: ((2012) ), 1171–1207. |
100 | [[100]] L.B. Volodarsky, V.A. Reznikov and V.I. Ovcharenko, Synthetic Chemistry of Stable Nitroxides, CRC Press, Boca Raton, (1993) . |
101 | [[101]] J. Voss, W.L. Hubbell and H.R. Kaback, Distance determination in proteins using designed metal ion binding sites and site-directed spin labeling: Application to the lactose permease of Escherichia coli, Proc. Natl. Acad. Sci. USA 92: ((1995) ), 12300–12303. |
102 | [[102]] R. Webb, Milestone 8: New resonance, in: Nature Milestones: Spin, Nature Publishing Group, London, (2008) , pp. 6–7. |
103 | [[103]] B.B. Williams, N. Khan, B. Zaki, A. Hartford, M.S. Ernstoff and H.M. Swartz, Clinical electron paramagnetic resonance (EPR) oximetry using India ink, Advances Experimental Medicine Biology 662: ((2010) ), 149–156. |
104 | [[104]] H. Yokoyama, T. Ogata, N. Tsuchihashi, M. Hiramatsu and N. Mori, A spatiotemporal study on the distribution of intraperitoneally injected nitroxide radical in the rat head using an in vivo ESR imaging system, Magn. Reson. Imaging 14: ((1996) ), 559–563. |
105 | [[105]] T. Yoshimura, H. Yokoyama, S. Fujii, F. Takayama, K. Oikawa and H. Kamada, In vivo EPR detection and imaging of endogenous nitric oxide in lipopolysaccharide-treated mice, Nat. Biotechnol. A 14: ((1996) ), 992–994. |
106 | [[106]] E. Zavoisky, Paramagnetic relaxation of liquid solutions for perpendicular fields, J. Phys. E(USSR) 9: ((1945) ), 211–216. |
107 | [[107]] E. Zavoisky, Spin-magnetic resonance in paramagnetics, J. Phys. E (USSR) 9: ((1945) ), 245–249. |
108 | [[108]] E. Zavoisky, Spin magnetic resonance in the decimetre-wave region, J. Phys. E (USSR) 10: ((1946) ), 197–198. |
109 | [[109]] J.L. Zweier and P. Kuppusamy, Electron paramagnetic resonance measurements of free radicals in the intact beating heart: A technique for detection and characterization of free radicals in whole biological tissues, Proc. Natl. Acad. 85: ((1988) ), 5703–5707. |


